

Abstract
Patient: Female, newborn
Final Diagnosis: Anomalous fusion of right pulmonary artery-to-aortic arch
Symptoms: Respiratory failure
Medication: —
Clinical Procedure: —
Specialty: Cardiology
Objective:
Congenital defects/diseases
Background:
We present a report of a rare cardiac malformation case as well as a review of the literature. In addition, the diagnostic features are discussed.
Case Report:
The case of a female newborn who died on her third day of life was studied at the Institute of Legal Medicine, University of Chieti-Pescara (Italy). The investigations around her death revealed a cardiac congenital malformation, seen as a rare variant of a common arterial trunk, in which the aorta was fused with the right branch of the pulmonary artery. The ascending aorta showed hypoplasia, while the coronary arteries were free of any pathological findings. The atrial septum showed a closed foramen ovale and the ventricular septum did not show any defect. Only an isolated right ventricular hypertrophy and dilation with no other cardiac abnormalities was found. The cause of death was acute respiratory failure on the third day of extrauterine life when the ductus Botalli closed. The karyotype analysis performed in this case was normal, and the fluorescent in situ hybridization analysis did not show the 22q11.2 microdeletion suggestive of the DiGeorge syndrome.
Conclusions:
These findings underline the value of 3-dimensional/4-dimensional ultrasound imaging when added to a fetal cardiology screening program, and the need for improvements in postnatal screening routines by using pulse oximetry in order to discover isolated vascular defects before circulatory collapse occurs, as well as to reduce the medico-legal disputes in cases of missed diagnosis. We found the relevant literature search lacked a description of this congenital malformation, which supports our deeper perinatal investigation.
MeSH Keywords: Heart Defects, Congenital; Prenatal Diagnosis; Pulmonary Artery
Background
Common truncus arteriosus (CAT) is a rare congenital cardiac malformation in which a single arterial trunk that emerges from the heart gives rise to the systemic, pulmonary, and coronary arteries [1]. This occurs in 1% to 4% of all cases of congenital heart disease, and without intervention it is typically fatal during the first year of life. Other common cardiac-associated defects include: large, maligned outlet ventricular septal defects; anomalies of the coronary arteries (15%); interruption to the aortic arch (11% to 15%); right-sided aortic arch (33%), persistent left superior caval vein, atrial septal defect; and an anomalous subclavian artery [2,3]. The early identification of these defects is imperative to reduce infant mortality and to address the best approach to their surgical correction. In particular, the combination of an interrupted aortic arch or truncal valve surgery with CAT carries high early mortality (29.8%, 30.0%, respectively) with high risk of reintervention in survivors [4–7]. The association with chromosomal anomalies is consistent (8.7%), and up to 30% of patients with a CAT have the 22q11.2 deletion, which should be ruled out before surgical repair [8–10]. Additional associated anomalies of 22q11.2 deletion syndrome include renal anomalies, anal atresia, tethered cord syndrome, retrognathia, laryngotracheomalacia, vascular rings formed by a right aortic arch and aberrant left subclavian artery, with left-sided ligamentum arteriosum [11].
We report here a case of CAT with fusion of the aorta with the right pulmonary artery, accompanied by hypoplasia of the ascending aorta without associated cardiac anomaly, which caused fatal congestive heart failure in a 3-day-old female infant.
Case Report
History
A woman with her first pregnancy delivered a female newborn at the 41st week of gestational age. There was no history of medication use during pregnancy. The infant weighed 2960 g at birth, and was 49 cm in length, with a head circumference of 31 cm. Based on anamnestic data, at birth the newborn was considered to be a healthy child. The clinical examination of the cardiopulmonary apparatus excluded any pathological findings. Her cardiac frequency was 130 beats per minute (bpm).
The physical examination of the newborn the day after birth revealed a decreased cardiac frequency (90–100 bpm). An urgent electrocardiogram (ECG) was requested to evaluate the QT tract. This ECG was considered to be normal. On the same day, the physicians booked an ECG examination for 5 days later, and they discharged the child in good clinical condition. The day after the discharge (on the third day of life), the newborn was brought to the pediatric emergency department. She was cyanotic, hypotonic, and without any effective cardiopulmonary activity. After unsuccessful cardiopulmonary resuscitation, the physicians recorded her death.
Autopsy findings
At autopsy, the study of the heart revealed a severe congenital cardiac malformation. The aorta was fused with the right branch of the pulmonary artery, just after the origin of the epiaortic vessels, giving rise to a common trunk (Figure 1). The infant also had a hypertrophic right myocardium (Figure 2), ascending aorta hypoplasia, signs of pulmonary edema, and coronary arteries free of any pathological findings.
Figure 1.
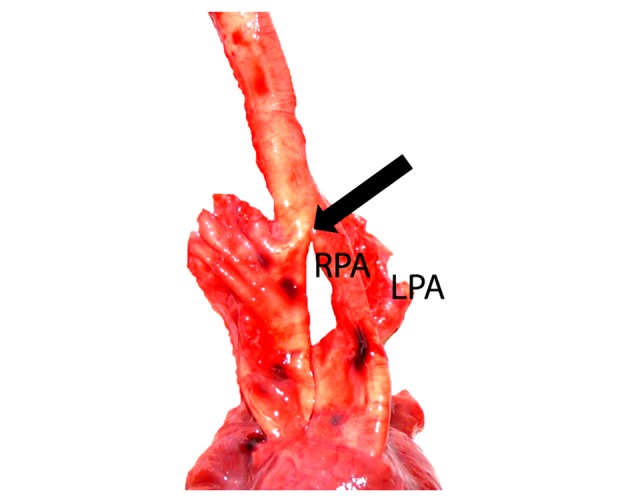
Aorta fused with the right pulmonary artery branch (arrow). RPA – right pulmonary artery; LPA – left pulmonary artery.
Figure 2.
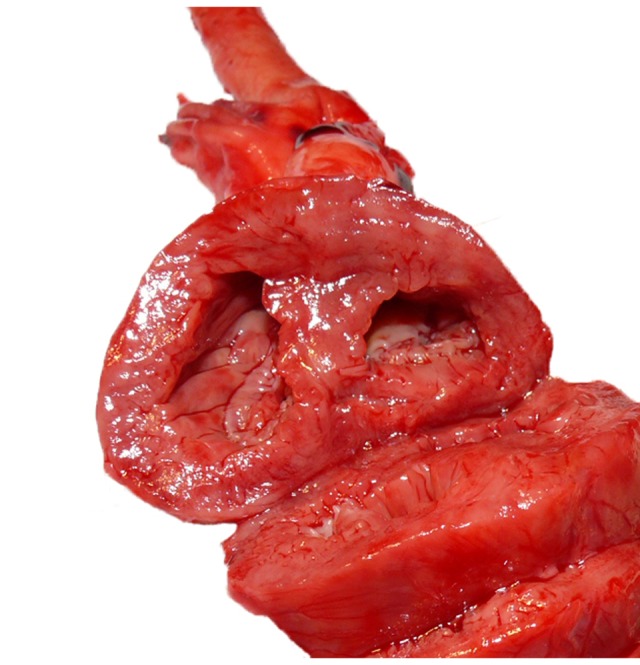
Transverse section of the heart. Note the markedly thickened right myocardium.
There was nothing else of note according to the macroscopic examination. The microscopic study of the brain and lungs showed diffuse signs of blood congestion.
Death was due to respiratory arrest secondary to deep anoxia. The closure of the ductus Botalli (also called the ductus arteriosus) had worsened the effects of the pathological shunt; indeed, this closure precipitated cardiac failure. The consequent lower cardiac output led to progressive arterial desaturation and death.
The postnatal karyotype of the infant was normal (46 XX) and the fluorescent in situ hybridization (FISH) for the DiGeorge syndrome critical region (22q11) was undertaken; it did not show the 22q11.2 deletion.
Discussion
The best classification of CAT is still under debate. According to Collett and Edwards [12], CAT malformations can be subdivided into different anatomical types, with respect to the origin of the pulmonary arteries. In type I, a short main pulmonary trunk arises from the truncus and divides into the right and left pulmonary branches. In types II and III, the pulmonary trunk is absent, the 2 pulmonary branches arise posteriorly, they are separated from the truncus, and either close to each other (type II) or more laterally and widely separated (type III). In type IV, there are no pulmonary arteries, and this should be considered a type of pulmonary atresia with ventricular septal defect.
Russel et al. [13] reinforced the advantages of a new nomenclature and classification based on the simplified and rationalized scheme of Jacobs [14]..This scheme classifies the heart on the dominance of either the systemic aortic, with a mortality risk during repair of 14.1%, or the pulmonary components if associated with hypoplastic aortic trunk, with a mortality risk during repair of the common trunk of 29.8%, reconciling the existing classification with recent findings relating to cardiac development [15,16]. The Russel classification and the relative variants are given in Table 1.
Table 1.
Russel Classification and the relative CAT variants.
| Classification dominance | Variant |
|---|---|
| I. Primary CAT lesions | |
| Pulmonary dominant | Common trunk bifurcates into right and left |
| Branched pulmonary arteries and ductal continuation to descending aorta | |
| Ascending aorta emerges from common trunk as a side branch | |
| Interrupted aortic arch | |
| Hypoplastic aortic arch (coarctation) | |
| Aortic dominant | Common trunk resembles an ascending aorta continuing on to a normal aortic arch |
| Branched pulmonary arteries emerge from the left posterior aspect of the trunk | |
| Adjacent origins of pulmonary arteries | |
| Origin of one pulmonary artery via arterial duct | |
| Sinusal origin of pulmonary arteries | |
| Balanced pattern (rarely encountered) | |
| II. Lesion modifiers | Interventricular communication |
| Muscular postero-inferior rim, perimembranous, intact septum | |
| Truncal valvar override | |
| Balanced, exclusively from right ventricle, exclusively from left ventricle | |
| Truncal valvar leaflets: two, three, four, five, or more | |
| Truncal valvar stenosis: mild, moderate, severe | |
| Truncal valvar insufficiency: mild, moderate, severe | |
| Crossed pulmonary arteries | |
According to the Russel classification, the anomaly of the case we reported here, which consisted of fusion of the aorta with the right pulmonary artery and concomitant hypoplasia of the aorta, led us to believe that this was a CAT variant with pulmonary dominance. This isolated abnormality was responsible for the onset of the pulmonary vascular congestion that caused the cardiac failure symptoms and death of the infant. A thorough review of the literature revealed some previous reports of CAT variants and their surgical repair [17–32] (Table 2), but to date, no similar case as we described here has been reported in the literature.
Table 2.
CAT variants reported in the literature after the onset of Russel Classification in 2011.
| Variant | Source reference |
|---|---|
| Prenatal diagnosis of anomalous origin of the right pulmonary artery from the ascending aorta | Jung MJ et al. 2002 [17] |
| Anomalous origin of the right pulmonary artery from the ascending aorta | Prifti E et al. 2004 [18] |
| Anomalous origin of the left coronary artery from the right pulmonary artery in an infant with coarctation of the aorta | Radha AS et al. 2004 [19] |
| Aortic origin of the left pulmonary artery in an infant with Fallot’s tetralogy | Carretero J et al. 2005 [20] |
| Surgical results of anomalous origin of the right pulmonary artery from the ascending aorta, including reoperation for infrequent complications. | Kajihara N et al. 2008 [21] |
| Prenatal diagnosis of anomalous origin of the right pulmonary artery from the ascending aorta, with hypoplastic right ventricle and pulmonary stenosis | Oztunc F et al. 2008 [22] |
| Anomalous origin of the right pulmonary artery from the ascending aorta: 64-slice MDCT findings | Kwon SH et al. 2009 [23] |
| Anomalous origin of the left coronary artery from the right pulmonary artery presenting following relief of left heart obstruction: a distinct and predictable clinicopathological syndrome | Morgan G et al. 2010 [24] |
| Anomalous left coronary artery from the right pulmonary artery with aortic fusion | Kumar KS et al. 2012 [25] |
| Unusual combination of hypoplastic left ventricle, atrioventricular septal defect with restrictive ventricular septal defect, and common arterial trunk | Tripathi RR et al. 2012 [26] |
| Case of congenital aneurysm of sinus of Valsalva with common arterial trunk | Nakamura Y et al. 2014 [27] |
| The trunk with a twist: right sinus origin of pulmonary arteries in a child with common arterial trunk | Gupta SK et al. 2014 [28] |
| A rare type of common arterial trunk with interrupted aortic arch, partial anomalous pulmonary venous connection, and phenylketonuria | Ayyildiz P et al. 2015 [29] |
| Surgical correction of truncus arteriosus with unusual origin of the right coronary artery | Rodríguez H et al. 2016 [30] |
| Prenatal diagnosis of the rare association of common arterial trunk and double aortic arch | Rock A et al. 2016 [31] |
| An unusual combination of truncus arteriosus, interrupted aortic arch, and hypoplastic left ventricle | Marathe SP et al. 2017 [32] |
It should be considered that up to 10% of cases with CAT are associated with genetic syndromes [33]. One of the frequent causes of CAT is the 22q11.2 microdeletion involved in the DiGeorge syndrome [34]. The CAT defect can also be due to GATA6 mutation (member of a small family of zinc finger transcription factors) present on chromosome 18q11.2, which is accompanied by agenesis or pancreatic hypoplasia [35]. Mutations of the CHD7 gene (member of the chromodomain helicase DNA protein family) were detected in over 75% of patients with CHARGE syndrome (coloboma, heart defect, atresia choanae, retarded growth and development, genital hypoplasia, and ear anomalies/deafness), a syndromic disease that has been also linked to the presence of CAT abnormalities [36]. In our case, FISH analysis ruled out the association of the CAT with microdeletion on chromosome 22q11.2, while the GATA6 and CHD7 mutations were not associated with the reported defect since the autopsy procedure failed to find any of the related malformations linked to these 2 gene mutations.
It is well known that the CAT malformation is associated with a high risk of early mortality if it is not surgically treated [37]. This underlines the importance of prenatal diagnosis to decrease neonatal morbidity and mortality. Moreover, this allows parents and physicians to determine whether to continue a pregnancy, to plan elective delivery at a tertiary care center to avoid the need for postnatal transport, to reduce the risk of compromising the neonatal hemodynamic stability and costs, as well as to reduce the inevitable future medico-legal disputes in cases where there is no prenatal diagnosis.
Through antenatal screening it is possible to use an ECG to obtain the complete diagnosis of CAT by 18 weeks gestation, with 95% confidence [38]. In CAT, the 4-chamber views are normal, and therefore an analysis of the great arteries and the outflow is necessary for diagnosis. In addition, a functional analysis can predict prenatal and postnatal prognosis. Particular attention should be given to the occurrence of arch anomalies, which can increase the risk of a 22q11.2 microdeletion (e.g., a right aortic arch, interruption of the aortic arch), which will worsen the long-term prognosis and increase the risk of recurrence [33]. Although the frequency of intra-uterine diagnosis has increased in the last few years, the CAT detection rates reported remain low: 32.0% to 76.8% [39,40] because the outflow tracts are only visualized in 57% of cases, with a variation between regions from 25% to 84%.
Recently, we have witnessed an evolution in the prenatal assessment of the fetal heart and great vessels defects by the use of new imaging modalities. The 3-dimensional/4-dimensional ultrasound multiplanar imaging is a display modality that allows the simultaneous visualization of up to 8 parallel anatomical planes [41]. These multiple axial views increase the detection rate of cardiac anomalies [42], thus simplifying the examination of the fetal heart and ultimately reducing the dependence of the examination on the skill of the individual operator, which will thus minimize potential errors [43].
Also, in the newborn period, detailed echocardiography that evaluates all of the anatomical components and usual and unusual cardiac anomalies that are associated with CAT remain the primary, and in most cases the only, mode of pre-operative and intra-operative evaluation of this malformation [44]. Recent studies have assessed the pulse oximetry as a form of screening for serious forms of congenital heart disease in the neonatal period. This imaging method adds value to the existing screening by identifying critical congenital heart defects that are not detected prenatally or by neonatal screening [45].
Conclusions
Although management of patients with unconventional physiologies remains challenging, such as for those with d-transposition of the great arteries and the common arterial trunk, advances in prenatal diagnosis and pre-operative and postoperative management have significantly improved survival in these patient populations. However, there remains significant room for improvement, particularly with regard to morbidity. Future studies should focus on optimizing and standardizing in-hospital and out of hospital care, with the goal being to improve morbidity, mortality, and functional outcomes. Most CATs can be diagnosed prenatally with a high degree of accuracy. However, the correct assessment of the interventricular septum, the right outflow tract, the distal aortic arch, and the anatomy of the pulmonary artery branch is often difficult to perform prenatally, although sequential echocardiography might help to obtain a correct diagnosis. Early diagnosis of babies with lesions that can result in cardiovascular collapse and death might improve their survival, as well as reducing morbidity. With a late diagnosis, some of these babies can present in a very poor clinical state and with a compromised outcome. At the same time, a late diagnosis might determine the beginning of medico-legal disputes.
Footnotes
Conflicts of interest
None.
References:
- 1.Lev M, Saphir O. Truncus arteriosus communis persistens. J Pediatr. 1942;20:74–88. [Google Scholar]
- 2.Elzein C, Ilbawi M, Kumar S, et al. Severe truncal valve stenosis: Diagnosis and management. J Card Surg. 2005;20:589–93. doi: 10.1111/j.1540-8191.2005.00124.x. [DOI] [PubMed] [Google Scholar]
- 3.Tlaskal T, Hucin B, Kucera V, et al. Repair of persistent truncus arteriosus with interrupted aortic arch. Eur J Cardiothorac Surg. 2005;28:736–41. doi: 10.1016/j.ejcts.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 4.Jacobs ML, O’Brien SM, Jacobs JP, et al. An empirically based tool for analyzing morbidity associated with operations for congenital heart disease. J Thorac Cardiovasc Surg. 2013;145:1046–57. doi: 10.1016/j.jtcvs.2012.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Russell HM, Pasquali SK, Jacobs JP, et al. Outcomes of repair of common arterial trunk with truncal valve surgery: A review of the society of thoracic surgeons congenital heart surgery database. Ann Thorac Surg. 2012;93:164–69. doi: 10.1016/j.athoracsur.2011.04.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Konstantinov IE, Karamlou T, Blackstone EH, et al. Truncus arteriosus associated with interrupted aortic arch in 50 neonates: A congenital heart. Surgeons Society Study Ann Thorac Surg. 2006;81:214–23. doi: 10.1016/j.athoracsur.2005.06.072. [DOI] [PubMed] [Google Scholar]
- 7.Henaine R, Azarnoush K, Belli E, et al. Fate of the truncal valve in truncus arteriosus. Ann Thorac Surg. 2008;85:172–78. doi: 10.1016/j.athoracsur.2007.07.039. [DOI] [PubMed] [Google Scholar]
- 8.Goldmuntz E, Emanuel BS. Genetic disorders of cardiac morphogenesis. The DiGeorge and velocardiofacial syndromes. Circ Res. 1997;80:437–43. doi: 10.1161/01.res.80.4.437. [DOI] [PubMed] [Google Scholar]
- 9.Carotti A, Digilio MC, Piacentini G, et al. Cardiac defects and results of cardiac surgery in 22q11.2 deletion syndrome. Dev Disabil Res Rev. 2008;14:35–42. doi: 10.1002/ddrr.6. [DOI] [PubMed] [Google Scholar]
- 10.Ryan AK, Goodship JA, Wilson DI, et al. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: A European collaborative study. J Med Genet. 1997;34:798–804. doi: 10.1136/jmg.34.10.798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garavelli L, Rosato S, Wischmeijer A, et al. 22q11.2 distal deletion syndrome: Description of a new case with truncus arteriosus type 2 and review. Mol Syndromol. 2011;2:35–44. doi: 10.1159/000334262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Collett RW, Edwards JE. Persistent truncus arteriosus; A classification according to anatomic types. Surg Clin North Am. 1949;29(4):1245–70. doi: 10.1016/s0039-6109(16)32803-1. [DOI] [PubMed] [Google Scholar]
- 13.Russell HM, Jacobs ML, Anderson RH, et al. A simplified categorization for common arterial trunk. J Thorac Cardiovasc Surg. 2011;141:645–53. doi: 10.1016/j.jtcvs.2010.08.022. [DOI] [PubMed] [Google Scholar]
- 14.Jacobs ML. Congenital heart surgery nomenclature and database project: Truncus arteriosus. Ann Thorac Surg. 2000;69:S50–55. doi: 10.1016/s0003-4975(99)01320-x. [DOI] [PubMed] [Google Scholar]
- 15.Jacobs ML, O’Brien SM, Jacobs JP, et al. An empirically based tool for analyzing morbidity associated with operations for congenital heart disease. J Thorac Cardiovasc Surg. 2013;145:1046–57. doi: 10.1016/j.jtcvs.2012.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O’Brien SM, Clarke DR, Jacobs JP, et al. An empirically based tool for analyzing mortality associated with congenital heart surgery. J Thorac Cardiovasc Surg. 2009;138:1139–53. doi: 10.1016/j.jtcvs.2009.03.071. [DOI] [PubMed] [Google Scholar]
- 17.Jung MJ, Yoo SJ. Prenatal diagnosis of anomalous origin of the right pulmonary artery from the ascending aorta. Cardiol Young. 2002;12(2):186–88. doi: 10.1017/s1047951102000392. [DOI] [PubMed] [Google Scholar]
- 18.Prifti E, Bonacchi M, Murzi B, et al. Anomalous origin of the right pulmonary artery from the ascending aorta. J Card Surg. 2004;19(2):103–12. doi: 10.1111/j.0886-0440.2004.04023.x. [DOI] [PubMed] [Google Scholar]
- 19.Radha AS, Dharan BS, Kumar RK, Rao SG. Anomalous origin of left coronary artery from right pulmonary artery in an infant with coarctation of the aorta. Ann Thorac Surg. 2004;78(1):324–36. doi: 10.1016/S0003-4975(03)01469-3. [DOI] [PubMed] [Google Scholar]
- 20.Carretero J, Rissech M, Mortera C, et al. Aortic origin of the left pulmonary artery in an infant with Fallot’s tetralogy. Rev Esp Cardiol. 2005;58(9):1124–26. [PubMed] [Google Scholar]
- 21.Kajihara N, Imoto Y, Sakamoto M, et al. Surgical results of anomalous origin of the right pulmonary artery from the ascending aorta including reoperation for infrequent complications. Ann Thorac Surg. 2008;85(4):1407–11. doi: 10.1016/j.athoracsur.2007.11.081. [DOI] [PubMed] [Google Scholar]
- 22.Oztunç F, Güzeltaş A. Prenatal diagnosis of anomalous origin of the right pulmonary artery from the ascending aorta with hypoplastic right ventricle and pulmonary stenosis. Ultrasound Obstet Gynecol. 2008;32:592–97. doi: 10.1002/uog.4076. [DOI] [PubMed] [Google Scholar]
- 23.Kwon SH, Hyeong J, Han MJ, Kim SC. Anomalous origin of the right pulmonary artery from the ascending aorta: 64-slice MDCT findings. Pediatr Cardiol. 2009;30:208–9. doi: 10.1007/s00246-008-9336-7. [DOI] [PubMed] [Google Scholar]
- 24.Morgan G, Caldarone C, Anderson R, Chaturvedi R. Anomalous origin of the left coronary artery from the right pulmonary artery presenting following relief of left heart obstruction: A distinct and predictable clinico-pathological syndrome. Congenit Heart Dis. 2010;5:327–30. doi: 10.1111/j.1747-0803.2009.00357.x. [DOI] [PubMed] [Google Scholar]
- 25.Kumar KS, Sinha P, Donofrio MT, Jonas RA. Anomalous left coronary artery from the right pulmonary artery with aortic fusion. J Thorac Cardiovasc Surg. 2012;143:505–7. doi: 10.1016/j.jtcvs.2011.07.031. [DOI] [PubMed] [Google Scholar]
- 26.Tripathi RR, Sridhar A, Chidambaram S. Unusual combination of hypoplastic left ventricle, atrioventricular septal defect with restrictive ventricular septal defect, and common arterial trunk. World J Pediatr Congenit Heart Surg. 2012;3:396–98. doi: 10.1177/2150135111434165. [DOI] [PubMed] [Google Scholar]
- 27.Nakamura Y, Aoki M, Hagino I, et al. Case of congenital aneurysm of sinus of Valsalva with common arterial trunk. Ann Thorac Surg. 2014;97(2):710–12. doi: 10.1016/j.athoracsur.2013.06.107. [DOI] [PubMed] [Google Scholar]
- 28.Gupta SK, Kothari SS, Gulati GS, et al. The trunk with a twist: Right sinus origin of pulmonary arteries in a child with common arterial trunk. World J Pediatr Congenit Heart Surg. 2014;5(4):615–19. doi: 10.1177/2150135114537312. [DOI] [PubMed] [Google Scholar]
- 29.Ayyildiz P, Kasar T, Güzeltas A. A rare type of common arterial trunk with interrupted aortic arch, partial anomalous pulmonary venous connection, and phenylketonuria. Cardiol Young. 2015;25(5):996–98. doi: 10.1017/S1047951115000128. [DOI] [PubMed] [Google Scholar]
- 30.Rodríguez H, Montero H, Fernández A, et al. Surgical correction of truncus arteriosus with unusual origin of the right coronary artery. World J Pediatr Congenit Heart Surg. 2016;7(3):407–10. doi: 10.1177/2150135115596585. [DOI] [PubMed] [Google Scholar]
- 31.Rock A, Eltayeb O, Camarda J, Gotteiner N. Prenatal diagnosis of the rare association of common arterial trunk and double aortic arch. Clin Case Rep. 2016;4(7):668–70. doi: 10.1002/ccr3.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marathe SP, Naganur SH, Menon S, et al. An unusual combination of truncus arteriosus, interrupted aortic arch, and hypoplastic left ventricle. World J Pediatr Congenit Heart Surg. 2018;9(6):714–17. doi: 10.1177/2150135117716886. [DOI] [PubMed] [Google Scholar]
- 33.Volpe P, Paladini D, Marasini M, et al. Common arterial trunk in the fetus: Characteristics, associations, and outcome in a multicentre series of 23 cases. Heart. 2003;89:1437–41. doi: 10.1136/heart.89.12.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harris RD. Obstetrics. 22q11 deletion syndrome with truncus arteriosus, hypoplastic left ventricle, VSD, clubfoot, sandal toes, cleft palate, butterfly vertebrae. Ultrasound Q. 2005;21:107–6. [PubMed] [Google Scholar]
- 35.McMillan T, Girgis R, Sellers EA. Neonatal diabetes and protein losing enteropathy: A case report. BMC Med Genet. 2016;17:32. doi: 10.1186/s12881-016-0296-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blake KD, Prasad C. CHARGE syndrome. Orphanet J Rare Dis. 2006;1:34. doi: 10.1186/1750-1172-1-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Knowles R, Griebsch I, Dezateux C, et al. Newborn screening for congenital heart defects: A systematic review and cost-effectiveness analysis. Health Technol Assess. 2005;9(44):1–152. iii–iv. doi: 10.3310/hta9440. [DOI] [PubMed] [Google Scholar]
- 38.Freire G, Miller M, Huhta J. Foetal echocardiography of transposition of the great arteries and common arterial trunk. Cardiol Young. 2012;22:671–76. doi: 10.1017/S104795111200162X. [DOI] [PubMed] [Google Scholar]
- 39.Swanson TM, Selamet Tierney ES, et al. Truncus arteriosus: Diagnostic accuracy, outcomes, and impact of prenatal diagnosis. Pediatr Cardiol. 2009;30:256–61. doi: 10.1007/s00246-008-9328-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Galindo A, Mendoza A, Arbues J, et al. Conotruncal anomalies in fetal life: accuracy of diagnosis, associated defects and outcome. Eur J Obstet Gynecol Reprod Biol. 2009;146(1):55–60. doi: 10.1016/j.ejogrb.2009.04.032. [DOI] [PubMed] [Google Scholar]
- 41.Gotsch F, Romero R, Espinoza J, et al. Prenatal diagnosis of truncus arteriosus using multi planar display in 4D ultrasonography. J Matern Fetal Neonatal Med. 2010;23(4):297–307. doi: 10.3109/14767050903108206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Espinoza J, Hassan SS, Gotsch F, et al. A systematic approach to the use of the multiplanar display in evaluation of abnormal vascular connections to the fetal heart using 4-dimensional ultrasonography. J Ultrasound Med. 2007;26:1461–67. doi: 10.7863/jum.2007.26.11.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Espinoza J, Kusanovic JP, Goncalves LF, et al. A novel algorithm for comprehensive fetal echocardiography using 4-dimensional ultrasonography and tomographic imaging. J Ultrasound Med. 2006;25:947–56. doi: 10.7863/jum.2006.25.8.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nguyen T, Blaine J, Nardell K, et al. Echocardiography of common arterial trunk. Cardiol Young. 2012;22:655–63. doi: 10.1017/S1047951112001497. [DOI] [PubMed] [Google Scholar]
- 45.Ewer AK, Middleton LJ, Furmston AT, et al. PulseOx Study Group Pulse oximetry screening for congenital heart defects in newborn infants (PulseOx): A test accuracy study. Lancet. 2011;378:785–94. doi: 10.1016/S0140-6736(11)60753-8. [DOI] [PubMed] [Google Scholar]