

Abstract
DNA conformation may deviate from the classical B-form in ∼13% of the human genome. Non-B DNA regulates many cellular processes; however, its effects on DNA polymerization speed and accuracy have not been investigated genome-wide. Such an inquiry is critical for understanding neurological diseases and cancer genome instability. Here, we present the first simultaneous examination of DNA polymerization kinetics and errors in the human genome sequenced with Single-Molecule Real-Time (SMRT) technology. We show that polymerization speed differs between non-B and B-DNA: It decelerates at G-quadruplexes and fluctuates periodically at disease-causing tandem repeats. Analyzing polymerization kinetics profiles, we predict and validate experimentally non-B DNA formation for a novel motif. We demonstrate that several non-B motifs affect sequencing errors (e.g., G-quadruplexes increase error rates), and that sequencing errors are positively associated with polymerase slowdown. Finally, we show that highly divergent G4 motifs have pronounced polymerization slowdown and high sequencing error rates, suggesting similar mechanisms for sequencing errors and germline mutations.
The three-dimensional conformation of DNA at certain sequence motifs may deviate from the canonical double-stranded B-DNA (the right-handed helix with 10 nt per turn) (Watson and Crick 1953) in helix orientation and strand number (Bacolla and Wells 2004; Mirkin 2008; Zhao et al. 2010). Approximately 13.2% of the human genome (394.2 Mb) has the potential to form non-B DNA structures (Supplemental Table S1), which are implicated in a myriad of cellular processes, and are associated with cancer and neurological diseases (Bacolla and Wells 2004; Mirkin 2007; Wang and Vasquez 2007; Zhao et al. 2010; Maizels 2015). For instance, adjacent runs of guanines can form G-quadruplex (G4) structures (Fig. 1A; Sen and Gilbert 1988) that participate in telomere maintenance (Parkinson et al. 2002), replication initiation (Huppert and Balasubramanian 2005; Besnard et al. 2012), and transcriptional regulation (Siddiqui-Jain et al. 2002). Consequently, G4 structures have emerged as attractive anti-cancer therapeutic targets (Balasubramanian et al. 2011). Additional non-B DNA structures associated with transcriptional regulation include left-handed Z-DNA duplexes formed within alternating purine-pyrimidine sequences (Wittig et al. 1991), A-phased repeats with helix bending formed within A-rich tracts (Jansen et al. 2012), and H-DNA triplexes formed within polypurine/polypyrimidine tracts and mirror repeats (Fig. 1A; Mirkin et al. 1987; Belotserkovskii et al. 2010). Finally, Short Tandem Repeats (STRs), which also affect gene expression (Sawaya et al. 2013), can adopt slipped-strand (Sinden et al. 2007) and other non-B DNA conformations (Mirkin and Mirkin 2007). Expansions of STRs are associated with numerous neurological and muscular degenerative diseases (Castel et al. 2010). Notably, expansions of the hexanucleotide STR forming a G4 structure within the C9orf72 gene is the most common genetic cause of amyotrophic lateral sclerosis (ALS) (Renton et al. 2011). Moreover, STRs are enriched in cancer-related genes and participate in their functions (Haberman et al. 2008). Thus, growing evidence indicates that non-B DNA plays a pivotal role in several cellular pathways impacting health and disease.
Figure 1.

Non-B DNA motifs and Inter-Pulse Duration (IPD) analysis pipeline. (A) Non-B DNA motif types: patterns, putative structures, and counts of nonoverlapping 100-bp windows containing one (and only one) motif with IPD measurements on the reference or reverse-complement strand. (B) During SMRT sequencing, IPDs are recorded for each nucleotide in each subread (each pass on the circular DNA template). Subreads are aligned against the genome, and an IPD value is computed averaging ≥3 subread IPDs for each genome coordinate. We form 100-bp windows around annotated non-B motifs, extract their IPD values, and pool windows containing motifs of the same type to produce a distribution of IPD curves over the motif and its flanks (represented via 5th, 25th, 50th, 75th, and 95th quantiles along the 100 window positions). We also form a set of nonoverlapping 100-bp windows free from any known non-B motif and pool them to produce a “motif-free” distribution of IPD curves. Each motif type is then compared to motif-free windows through Interval-Wise Testing (IWT). (G4) G-quadruplexes; (G4+/G4−) G4 annotated on the reference/reverse-complement strand; (DR) direct repeats.
Whereas the transient ability of non-B DNA motifs to form noncanonical structures regulates many cellular processes (Zhao et al. 2010), these structures can also affect DNA synthesis and lead to genome instability, and thus can be viewed as both a blessing and a curse (Valton and Prioleau 2016). In vitro and ex vivo studies of individual loci showed that non-B DNA formation inhibits prokaryotic and eukaryotic DNA polymerases, causing their pausing and stalling of a replication fork (Kang et al. 1995; Samadashwily et al. 1997; Krasilnikova and Mirkin 2004; Voineagu et al. 2008; Eddy et al. 2015). These processes have been postulated to underlie non-B DNA-induced genome instability, that is, increase in chromosomal rearrangements, including those observed in cancer (Bacolla et al. 2004; Wang et al. 2008). Moreover, the increased occurrence of point mutations at non-B DNA was demonstrated at individual loci in plasmid constructs (for review, see Zhao et al. 2010; Bacolla et al. 2011; Inagaki et al. 2013), at disease-associated genes (Kamat et al. 2016), and among genetic variants from the 1000 Genomes Project (Du et al. 2014). Because the effect of non-B DNA on mutagenesis is driven by both the inherent DNA sequence and polymerase fidelity (Ananda et al. 2014), we hypothesize that these structures can impact the efficiency and accuracy of DNA synthesis. Despite the critical importance of non-B DNA structures, ours is the first genome-wide study of their joint impact on polymerization speed and errors.
To evaluate whether DNA polymerization speed (i.e., polymerization kinetics) and polymerase errors are affected by non-B DNA, we utilized data from Single-Molecule Real-Time (SMRT) sequencing. In addition to determining the primary nucleotide sequence, this technology, which uses an engineered bacteriophage phi29 polymerase (Eid et al. 2009), records Inter-Pulse Durations (IPDs) (Fig. 1B), that is, the times between two fluorescent pulses corresponding to the incorporation of two consecutive nucleotides (Flusberg et al. 2010). We used IPDs as a measure of polymerization kinetics. SMRT sequencing allows a direct, simultaneous investigation of the genome-wide effects of several non-B DNA motif types on polymerization kinetics and errors. We also contrasted SMRT polymerization kinetics and sequencing error rates in highly mutable versus invariant non-B DNA motifs, finding a potential link between polymerization in sequencing instruments and in living cells.
Results
Non-B DNA motifs influence polymerization kinetics
We considered 92 different motif types potentially forming non-B DNA (Fig. 1A; Supplemental Tables S1–S3; Zhao et al. 2010), including predicted motifs from the non-B DNA DataBase (Cer et al. 2012) and annotated STRs (Fungtammasan et al. 2015). We constructed motif-containing genomic windows taking ±50 bp from the center of each motif (most were shorter than 100 bp) (Supplemental Fig. S1) and excluded overlapping windows (Supplemental Tables S2, S3). For controls, we constructed 100-bp motif-free windows to represent genomic background, that is, putative B-DNA. We populated each motif-containing and motif-free window with 100 single-nucleotide resolution IPDs (Fig. 1B) from a human genome previously sequenced with SMRT at 69× (Zook et al. 2016). This was performed separately for the reference and reverse-complement strands, because each strand is used separately as a template in SMRT sequencing (Fig. 1B). For each motif type, we aligned the centers of all motifs and aggregated IPD curves across windows, producing a distribution of IPD curves per strand (Fig. 1B).
To evaluate whether non-B motifs present polymerization kinetics patterns different from B-DNA, we used Interval-Wise Testing (IWT) (Cremona et al. 2018), a novel Functional Data Analysis (FDA) approach, and identified genomic bases or intervals at which IPD curve distributions significantly differ between motif-containing and motif-free 100-bp windows (Fig. 2A–E; two-sided test, see Methods). We indeed found altered polymerization kinetics in and/or around several non-B DNA motifs. Below, we describe results for the reference strand (a total of 2,916,328 motif-containing and 2,524,489 motif-free windows) (Fig. 2A–D, upper panels; Fig. 2E; Supplemental Figs. S2–S10, S11A,C,E); results for the reverse complement serve as a biological replicate (Fig. 2A–D, lower panels; Supplemental Fig. S11B,D,F).
Figure 2.

Polymerization kinetics at non-B DNA. (A–D) IPD curve distributions in motif-containing (red) versus motif-free (blue) 100-bp windows, on reference (top) and reverse-complement (bottom) strands. Thick lines designate the medians; dark-shaded areas show the 25th–75th quantiles; light-shaded areas show the 5th–95th quantiles. Red/blue marks (below top and above bottom plots) show positions with IPDs in motif-containing windows higher/lower than in motif-free windows (IWT-corrected P-values ≤0.05). Heatmaps (between top and bottom plots) show sequencing depth of motif-containing relative to motif-free windows (in percentages, can be >100%) on reference (Depth ref) and reverse-complement (Depth rev) strands, and percentage of windows with the motif (Motif) at each position. (A) G-quadruplexes; (B–D) STRs with disease-linked repeat number variation; (E) IWT results for IPD curve distributions in motif-containing versus motif-free windows (reference strand). Each row shows significance levels (−log of corrected P-values) along 100 window positions for one motif type. (White) nonsignificant (corrected P-value >0.05); (red/blue) significant with IPDs in motif-containing windows higher/lower than in motif-free windows. STRs are grouped according to putative structure. (F) Comparison between observed mean IPDs in motif-containing windows and predictions from a dinucleotide compositional regression fitted on motif-free windows (reference strand). Bonferroni-corrected t-test P-values for differences: (****) P ≤ 0.0001; (***) P ≤ 0.001; (**) P ≤ 0.01; (*) P ≤ 0.05. (Black) nonsignificant (corrected P-value >0.05); (red/blue) significant with observed mean IPDs higher/lower than composition-based predictions; (box plot whiskers) 5th and 95th quantiles of the differences.
Two lines of evidence are consistent with G4 motifs hindering polymerase progression. First, they decreased polymerization speed. Compared to motif-free windows, G4-containing windows had significantly higher IPDs near their centers, that is, near the motifs (up to 1.7-fold IPD increase at the 95th quantile) (Fig. 2A). All G4 motif types exhibited this elevation, although the IPD curve shapes differed depending on the motif sequence (Supplemental Fig. S3). Furthermore, the shape of the IPD distribution encompassing all G4 motif types remained the same (Supplemental Fig. S4) when we limited our analysis to motifs forming the most stable G4 quadruplexes, as identified by in vitro ion concentration manipulations (Chambers et al. 2015). Second, sequencing depth was lower at G4 motifs than at motif-free windows (86% of motif-free depth) (Fig. 2A), suggesting that the former hindered polymerization, resulting in fewer reads covering the motif (lower depth could in part be attributed to lower sequencing quality, but not to read terminations, within G4 motifs) (Supplemental Note S1). Polymerization slowdown and decreased sequencing depth were evident on the reference strand where G4s were annotated (“G4+”; Fig. 2A, upper panel), consistent with G-quadruplex structures forming only on the guanine-rich strand (Maizels 2015). Elevated IPDs were observed in all sequencing passes through the same G4+ containing circular template (Fig. 1B; Supplemental Fig. S5), suggesting that the structure is not resolved during sequencing. In contrast, the corresponding opposite strand (Fig. 2A, lower panel), as well as the reference strand where G4s were annotated on the reverse-complement strand (Fig. 2E, “G4−”)—both cytosine-rich—showed a significant overall polymerization acceleration and displayed a smaller decrease in sequencing depth (92% of motif-free depth).
We observed that several other non-B DNA motifs, for example, A-phased repeats, inverted repeats, mirror repeats, and Z-DNA, had significantly altered polymerization kinetics—both slower (higher IPD) and faster (lower IPD) (Fig. 2E; Supplemental Fig. S6). In contrast to the G4 motifs, the effects on polymerization kinetics were similar on the two sequenced strands (Fig. 2E; Supplemental Fig. S11B) suggesting that, for these motifs, non-B DNA can be formed with similar probability on each strand during the sequencing reaction (Fig. 1B).
Additionally, we found that STRs altered polymerization kinetics in a length- and sequence-dependent manner (Fig. 2B–E; Supplemental Figs. S7–S10); these variables impact the types and stability of non-B DNA structures that can form in addition to slipped structures (Supplemental Table S4). For STRs with ≥2-nt repeated units, the variation in polymerization kinetics was periodic, with the period (in bases) matching the length of the repeated unit (Supplemental Note S2)—consistent with effects of strand slippage. This pattern was evident for trinucleotide STRs whose expansions at some loci are associated with neurological diseases (Fig. 2B–D), for example, (CGG)n, (CAG)n, and (GAA)n implicated in Fragile X syndrome, Huntington's disease, and Friedreich's ataxia, respectively (Castel et al. 2010). Genome-wide, (CGG)n repeats showed a strong periodic decrease in polymerization speed (elevated IPDs) on the annotated strand (up to ninefold IPD increase at the 95th quantile) (Fig. 2C), consistent with their ability to form G4-like structures and hairpins (Nadel et al. 1995). The pattern for (CAG)n repeats, also capable of forming hairpins (Mirkin and Mirkin 2007), was similar (Fig. 2B). Globally, STRs capable of forming hairpins (Supplemental Table S4) presented the most striking polymerization deceleration and periodicity (Fig. 2B,C,E; Supplemental Fig. S7). In contrast, STRs forming H-DNA (Supplemental Table S4), including (GAA)n, accelerated polymerization (Fig. 2D,E; Supplemental Fig. S8). For many STRs, significant deviations from background IPD levels were shifted 5′ to the annotated motif (Fig. 2E; Supplemental Fig. S11), possibly due to polymerase stalling caused by difficulty in accommodating the alternative DNA structure within the polymerase active site.
The alterations in polymerization kinetics at non-B DNA motifs are not readily explained by either base modifications or by nucleotide composition. First, IPD patterns for most non-B DNA motifs were still clearly detectable in amplified DNA (Supplemental Fig. S12), suggesting that they were not due to base modifications in the original template DNA (Flusberg et al. 2010). Second, compositional regressions with either single-nucleotide or dinucleotide composition explained only a relatively small portion of mean IPD variation among motif-free windows—11.5% for single nucleotides and 20.8% for dinucleotides. Moreover, the mean IPDs in most motif-containing windows were significantly different from those predicted by such regressions (Fig. 2F; Supplemental Fig. S13). Thus, nucleotide composition falls far short of explaining IPD variations at non-B DNA motifs (Supplemental Figs. S13, S14). In particular, the mere presence of guanines in G4+ motifs cannot explain the overall substantial deceleration of polymerization observed at these sites.
Polymerization kinetics and biophysical characteristics of G-quadruplexes correlate
To experimentally test whether non-B DNA structures can form at predicted motifs, we investigated the relationship between polymerization kinetics and biophysical characteristics of the 10 G4 motifs most common in the human genome (Supplemental Table S5). According to circular dichroism spectroscopy (CD) and native polyacrylamide gel electrophoresis (PAGE) analyses, all 10 motifs quickly formed stable quadruplexes at low potassium concentrations, suggesting that they have a high propensity to form such structures (Kypr et al. 2009), albeit with different molecularity (intra- or intermolecular) and strand orientations (parallel or antiparallel) (Supplemental Table S5). Using regressions for intramolecular G4s, we found a significant positive relationship between mean IPD and delta epsilon (P < 2 × 10−16, R2 = 32.3%) (Fig. 3), a measure of structure organization quality obtained by CD, and between mean IPD and melting temperature (P < 2 × 10−16, R2 = 5.7%) (Supplemental Fig. S15B, solid line in cyan), a measure of thermostability and structure denaturation obtained by light absorption (Supplemental Table S5; results for intermolecular G4s are shown in Supplemental Fig. S15). Thus, polymerization slowdown and the biophysical characteristics of G4 formation are correlated, strongly suggesting that the motifs indeed form G4 structures during the SMRT sequencing reaction (intramolecular G4 structures are only a few nanometers in diameter) (Neidle and Balasubramanian 2006) and thus can fit within the 60 × 100 nm wells of Pacific Biosciences (PacBio) instruments (Turner et al. 2017).
Figure 3.
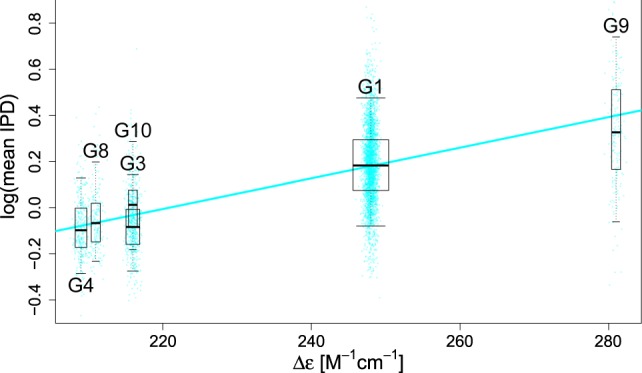
Relationship between G-quadruplex stability and polymerization kinetics. For the 10 most common G-quadruplex motif types (G1 through G10, in order), we measured circular dichroism (delta epsilon) and light absorption (melting temperature [Tm]) and computed average IPD values for each of thousands of motif occurrences in the genome (Supplemental Table S5). For intramolecular G4s, average IPDs were regressed on delta epsilon (R2 = 32.3%). (Box plot whiskers) 5th and 95th quantiles; (box plot width) proportional to the square root of the sample size for each motif; (points) individual occurrences used in the regressions, with horizontal jittering for visualization (results for delta epsilon in intermolecular G4s and for Tm are shown in Supplemental Fig. S15).
Our experiments also showed that statistical FDA techniques applied to polymerization kinetics data can enable non-B DNA structure discovery. Although not possessing a canonical G4 motif, the (GGT)n STR has an IPD profile similar to that of G4+ (Fig. 2E; Supplemental Fig. S10E) and its reverse complement (ACC)n has an IPD profile similar to that of G4− (Fig. 2E; Supplemental Fig. S10B), suggesting that (GGT)n may fold into a G4-like structure. Remarkably, biophysical analyses (CD, native PAGE, and thermal denaturation) showed that (GGT)n motifs indeed adopt quadruplex conformation (Supplemental Fig. S16; Supplemental Table S6).
Non-B DNA motifs affect sequencing error rates
To examine whether phi29 polymerase accuracy is affected during synthesis of non-B DNA motifs of different types in the genome, we contrasted SMRT sequencing error rates between such motifs and motif-free regions. Error rates were computed using the same human genome sequenced at 69× (Zook et al. 2016) that was used for the IPD analysis above. Because of the potential for inaccurate typing of STRs (Fungtammasan et al. 2015) and for motif misalignments in repetitive loci, we restricted our attention to six non-STR motif types present on the reference strand of the nonrepetitive portion of the genome (Table 1; Supplemental Table S7). We focused on motifs themselves (as opposed to 100-bp motif-containing windows), and for controls we identified motif-free regions matched to motifs in number and length. We excluded motifs and motif-free regions with fixed differences between sequenced and reference genomes (i.e., with germline variants present in the sequenced genome compared to the reference), and computed sequencing error rates as the proportion of variants (relative to hg19) within the total number of nucleotides sequenced for the motif or motif-free region—including errors supported even by a single read (Methods). Because we were interested in a detailed analysis of errors made by the polymerase, we used raw SMRT reads and not the circular consensus sequences. Below, we present results for errors on the newly synthesized strand that uses the template strand annotated with non-B DNA motifs.
Table 1.
Error rates at non-B DNA motifs during SMRT sequencing

We observed a strong effect of G4 motifs on SMRT error rates. Mismatches were markedly elevated (1.79-fold) on the newly synthesized strand when G4s were present on the template strand (Table 1). Deletions were increased in both G4+ and G4− (1.49- and 1.11-fold, respectively) (Table 1). Insertions, the most common error type for SMRT sequencing, were depressed when the templates encoded G4+ and particularly G4− motifs (Table 1). In contrast to G4 motifs, Z-DNA displayed depressed mismatches and deletions, but increased insertions (Table 1). In summary, the rates of all three types of SMRT sequencing errors differed between non-B motifs and motif-free regions, with strong elevations of mismatches and deletions at G4− motifs.
We next tested whether SMRT mismatch error rates could be explained by sequence composition. Only 4.1% of the variability in SMRT error rates in motif-free windows could be explained by single-nucleotide composition (Supplemental Fig. S17A). Among the four nucleotides, the content of guanines was the most correlated to SMRT errors, and its increase led to elevated SMRT error rates (Supplemental Fig. S17A). Dinucleotide compositional regression also explained a rather small proportion of variability in SMRT error rates in motif-free windows (R2 = 5.6%). Furthermore, SMRT error rates in most types of motifs (in all but A-phased repeats) were significantly different than those predicted by such compositional regressions (Supplemental Fig. S17B). Thus, nucleotide composition does not suffice to explain SMRT error rate variation at both motif-free windows and non-B DNA motifs. In particular, the high concentration of guanines in G4+ motifs cannot explain the increase in SMRT error rates observed at such sites.
Increased SMRT error rates are associated with polymerase slowdown, particularly at non-B DNA
We next studied whether SMRT error rates are associated with polymerization speed. We focused on G4+ and G4− motifs, which had the strongest effect on SMRT error rates among the non-B motif types examined (Table 1), and used motif-free windows for comparison. We fitted a regression expressing SMRT mismatch error rates as a function of mean IPD values corrected for nucleotide composition (residual mean IPDs, i.e., the difference between observed mean IPDs and those predicted using single-nucleotide composition) (Methods). The model also accounted for the three groups of regions—G4+ motifs, G4− motifs, and motif-free windows—and had an overall R2 of 35.4% (Fig. 4; Supplemental Fig. S18). We found a significantly positive linear relationship between SMRT mismatch rates and residual mean IPDs in motif-free windows (slope = 0.11, P = 2.9 × 10−10). Interestingly, the slope of the regression line was significantly steeper for G4+ than for motif-free windows (slope = 0.26, P = 1.9 × 10−12 for difference with motif-free), while G4− had a slope similar to motif-free windows (slope = 0.08, P = 0.17 for difference with motif-free). Thus, SMRT mismatch errors are positively associated with polymerase slowdown, and this association is particularly strong in G4+.
Figure 4.

Errors are linked to kinetic variation. Sequencing through templates containing G-quadruplex motifs (G4+) increases the positive relationship between mismatches in SMRT sequencing and IPD values (corrected for sequence composition). This does not occur when sequencing through the reverse complement of G-quadruplex motifs (G4−). The regression model accounting for three groups of regions (G4+, G4−, and motif-free windows) has R2 equal to 35.4%. Slopes are 0.26, 0.08, and 0.11 for G4+ motifs (N = 5937), G4− motifs (N = 5695), and motif-free windows (N = 11,632, which is the sum of samples sizes for G4+ and G4−), respectively.
Polymerase speed and mutation occurrence
Mutation rates are known to be nonuniform across the genome; however, the mechanisms leading to such regional variation are not entirely understood (Hodgkinson and Eyre-Walker 2011). Our results on sequencing errors for the SMRT technology, as well as previous in vitro polymerase studies (Kang et al. 1995; Usdin and Woodford 1995; Delagoutte et al. 2008; Eddy et al. 2015) demonstrating the effects of non-B DNA on DNA synthesis by phage, prokaryotic and eukaryotic polymerases, raise an intriguing question: Are mutation rates in vivo also affected by these motifs via polymerase slowdown? Environmental influences apart, mutations are the net result of polymerase errors and the lack of repair in the cell. Here, we are making a simplifying assumption that mutations result primarily from polymerase errors (Makova and Li 2002). Examining the speed of polymerases at the nucleotide level in eukaryotic cells is a challenging endeavor, but can the polymerization kinetics and error profile of the phi29 enzyme provide us with a hint of how non-B DNA motifs might be affecting mutations? To address this question, we contrasted SMRT error rates and mean IPDs between G4+ motifs with high and low human germline mutation rates approximated by the level of human-orangutan divergence or by the level of intraspecific diversity inferred from the 1000 Genomes Project (1000 Genomes Project Consortium 2015). In more detail, we compared SMRT mismatch error rates and mean IPDs (corrected for single-nucleotide composition; see above) between G4+ motifs in the top 3% versus bottom 3% of human-orangutan divergence, as well as between G4+ motifs in the top 3% versus bottom 3% of diversity from the 1000 Genomes Project (Supplemental Table S9; Supplemental Note S3). Human and orangutan genomes (Locke et al. 2011) used for the divergence calculations were generated with the highly accurate Sanger technology. For the diversity calculation, we considered only variants with minor allele frequency equal to or above 0.05 in the 1000 Genomes Project; this minimizes false positive variants due to sequencing errors. Using simulations, we demonstrated that variants with such minor allele frequency are very unlikely to be generated by the increased error rate of Illumina sequencing at G4 motifs (Supplemental Note S4). Therefore, both the divergence and the diversity data used here are expected to be highly accurate.
Highly divergent (or diverse) G4+ motifs had higher IPD values (i.e., experienced polymerase slowdown), compared with G4+ motifs having low divergence (or diversity; P = 4 × 10−4 and P = 0.046 for divergence and diversity, respectively; t-test for difference in means) (Supplemental Table S9). Moreover, highly divergent (or diverse) G4+ motifs had higher error rates than did G4+ motifs with low divergence (or diversity; P = 0.040 and P = 0.014 for divergence and diversity, respectively; t-test for difference in means) (Supplemental Table S9). Therefore, indeed, we found divergence (diversity) to be negatively related to polymerization speed and positively related to SMRT sequencing errors, suggesting that G-quadruplexes affect not only sequencing errors, but also germline mutations in vivo.
Discussion
SMRT sequencing polymerization kinetics data are produced during every SMRT sequencing experiment, but are rarely analyzed by researchers, except for studies of DNA modifications (e.g., Schadt et al. 2013). Our genome-wide study exemplifies the usefulness of such data in four additional scientific domains: (1) studies of polymerization kinetics; (2) discovery of novel non-B DNA structures; (3) analysis of sequencing errors; and (4) correlating polymerase kinetics with error rate. With the increasing popularity of SMRT sequencing and growing publicly available data, we expect an acceleration of progress in these areas.
Analyzing hundreds of thousands of non-B DNA motif occurrences genome-wide, we observed polymerization slowdown or acceleration during SMRT sequencing for the majority of motif types considered. Particularly striking patterns were noted for G4s and STRs, for which we found strong slowdown and periodic alterations in polymerization speed, respectively. Our study corroborates a previous analysis of 27 occurrences of (CGG)n—a motif capable of forming a G4 structure and a hairpin—which indicated polymerization speed alterations in the E. coli genome (Sawaya et al. 2015). It was suggested that non-B DNA acts as a polymerization speed modifier along the genome not only in a sequencer, but also under natural conditions, that is, in the cell (Sawaya et al. 2015). Backing of this hypothesis also comes from ex vivo analyses of a handful of loci capable of non-B DNA formation (Samadashwily et al. 1997; Krasilnikova and Mirkin 2004). Future experiments should examine this intriguing possibility on a genome-wide basis. Also, our results lend support to the hypothesis that periodic polymerization kinetics patterns at disease-associated STRs contribute to their instability (Loomis et al. 2013).
We demonstrate that analyzing polymerization kinetics data with FDA statistical techniques can enable discovery of novel motifs forming non-B DNA structures. We identified (GGT)n motifs as potentially forming a G4-like structure based on their polymerization speed patterns and used biophysical profiling to validate the formation of such structure. The genomic distribution and potential function of such newly identified non-B motifs should be studied further. Moreover, our study shows that statistical analyses of IPD patterns can characterize non-B DNA in a way that is orthogonal to conventional biophysical profiling.
Elevated SMRT sequencing errors in many non-B DNA motifs should be taken into account when evaluating sequencing results. Many such errors are likely corrected during circular sequencing, but biases might remain in the sequence consensus and when using the long sequencing read mode (as opposed to the circular consensus sequencing mode). Of particular interest is our result concerning the nonrandomness of insertions, which are the most common type of SMRT errors. We observe that insertions increase in Z-DNA, but in fact decrease in other non-B DNA motif types. In light of this, recalibrating base quality scores in SMRT sequencing reads should be considered in future work. Additionally, the effect of non-B DNA on the speed and error rates of other sequencing technologies, particularly the recently released Oxford Nanopore Sequencer, should be evaluated. The Nanopore sequencer uses an enzyme that controls movement through the nanopore and thus might also be affected by non-B DNA structures.
To our knowledge, our analyses are the first to document an association between polymerization speed and accuracy on a genome-wide scale. We find that the phi29 polymerase makes more errors when the IPD is high, that is, when polymerization is slowed down. This effect is significant for B-DNA, and further magnified in G-quadruplexes, even after correcting for nucleotide composition. Thus, polymerase accuracy may be affected by DNA sequence and structural features that hinder processive synthesis. Consistent with our findings here, previous in vitro studies showed a positive correlation between DNA polymerase error rates and pausing during synthesis (e.g., slow synthesis). Notably, misincorporation errors by the replicative DNA polymerase alpha are associated with polymerase pausing (Fry and Loeb 1992), and polymerase alpha errors within STRs are positively correlated with polymerase pausing and non-B DNA formation (Hile and Eckert 2004).
Our study indicates a significant effect of non-B DNA on the fidelity of DNA synthesis. With a genome-wide analysis of data generated by a sequencing instrument, we substantially expanded prior knowledge of this phenomenon gained by examination of plasmid constructs, disease-associated genes, and human genetic diversity (Zhao et al. 2010; Bacolla et al. 2011; Inagaki et al. 2013; Du et al. 2014). Our most prominent observation, the high incidence of mismatches and deletion errors during sequencing of G4 motifs, is in line with these motifs harboring excessive disease-causing point mutations (Kamat et al. 2016) and nucleotide variants (point mutations and indels combined) (Du et al. 2014) based on 1000 Genomes Project data. Using the level of divergence (or diversity) as a proxy for germline mutation rates, we observed significantly larger SMRT polymerase slowdown and error rates within G4s with very high divergence compared to G4s with very low divergence (diversity). Selection is expected to decrease divergence and diversity levels; however, we have no reason to predict that G4s undergoing stronger selection should have faster rates of DNA synthesis and lower sequencing error rates. Thus, a link between polymerase accuracy and germline mutation rates is more plausible. Our results, taken together with previously published studies, argue for the pervasive role of G4s in affecting polymerase fidelity and germline mutation rates in the genome. It remains, however, a caveat of our study that selection may be affecting the levels of divergence (or diversity) in G4 motifs. Deconvoluting the effects of selection and mutation rates in living cells will be a challenging but important future study.
Compared with errors in sequencing instruments, mutations in the cell are the net result of DNA synthesis errors by more than 15 different polymerases and enzymes from numerous DNA repair pathways (Sweasy et al. 2006). Furthermore, mutation occurrence is influenced by additional factors (e.g., chromatin, etc.) (Makova and Hardison 2015). Notwithstanding all these caveats, our results, together with published results of Du and colleagues (Du et al. 2014), lend support to the notion that non-B DNA is one among the “local DNA environment” factors affecting mutation rates and patterns (Cooper et al. 2011). Future studies should specifically investigate what share of local variation in mutation rates along the genome can be explained by the presence of non-B DNA. Our findings, together with observations on the transient nature of non-B DNA conformations (Zhao et al. 2010), portray non-B DNA as an effective modulator of genome structure. This is particularly significant in view of recent evidence broadening the spectrum of mechanisms through which non-B DNA may modulate the cell, encompassing, for example, epigenetic instability (Valton and Prioleau 2016) and noncoding RNA regulation (Simone et al. 2015).
Methods
Non-B DB and STR annotations
Annotations of A-phased, direct, inverted and mirror repeats, G-quadruplexes, and Z-DNA motifs were downloaded from the non-B DataBase (DB) (https://nonb-abcc.ncifcrf.gov). Because this annotation was provided on the human reference hg19, we used that reference for the whole study. Additionally, we annotated STRs on the human reference (hg19) using STR-FM (Fungtammasan et al. 2015). We only considered mono-, di-, tri-, and tetranucleotide STRs with ≥8, ≥4, ≥3, and ≥3 repeats, respectively (Fungtammasan et al. 2015). We then collapsed STR motifs that could be matched by changing their reading frame (Supplemental Table S8). For instance, (AGC)n, (CAG)n, and (GCA)n were collapsed into the (AGC)n group. We restricted our attention to non-B motifs and STRs annotated on autosomes.
Constructing genomic windows
Polymerization kinetics was studied in 100-bp windows (Fig. 1B). Motif-containing windows were centered at the middle coordinates of the annotated motifs in our list (Supplemental Fig. S1). The centers of STRs with different repeat numbers were shifted to ensure their alignment (Supplemental Table S8). Overlapping motif-containing windows (with motifs of the same or different type) were filtered out, leaving a total of 2,926,560 windows. All windows not containing motifs and not overlapping motif-containing windows were labeled as motif-free (a total of 3,649,152 windows).
IPDs
We used publicly available PacBio resequencing data (69×) from an individual male (HG002; NA24385) belonging to the Genome in a Bottle Ashkenazim trio (Zook et al. 2016). We analyzed 228 SMRT cells sequenced with P6-C4 chemistry in a mode maximizing the subread length and not the number of passes (Rhoads and Au 2015). On average, each molecule was sequenced in 2.12 passes, with the majority of the molecules sequenced only in a single pass resulting in a single subread (74.76% of the molecules). Sequencing reads were aligned to hg19 with pbalign (smrtanalysis-2.3.0), resulting in an ∼52× average read depth, and IPDs were computed at nucleotide resolution with ipdSummary.py (https://github.com/PacificBiosciences/kineticsTools/tree/master/kineticsTools)—this produces one IPD value per site averaging among at least three subreads, normalizing for intermolecule variability and trimming for outliers. The resulting IPDs, which are strand-specific (any observed slowdown or acceleration of the polymerization concerns the strand used as template), were then used to populate motif-containing and motif-free 100-bp windows according to their coordinates (Fig. 1B); each window thus contains an IPD curve comprising 100 values or less (if some nucleotides lack IPDs). All windows with no IPD values were filtered out, and only motifs with ≥15 windows with IPDs on both strands were retained for subsequent analyses. This left us with a total of 2,916,328 motif-containing and 2,524,489 motif-free windows on the reference strand, and 2,916,377 motif-containing and 2,524,612 motif-free windows on the reverse-complement strand. Next, for each motif type (Supplemental Tables S2, S3), and separately for each strand, we aligned the 100-bp windows. This resulted in strand-specific IPD curve distributions for each motif type. An IPD curve distribution was visualized plotting quantiles (5th, 25th, 50th, 75th, and 95th) of the IPD values at each of the 100 nt along the aligned windows (Figs. 1B, 2A–D; Supplemental Figs. S3, S4, S6–S10). IPD distributions were visually unaffected by variants between the sequenced and the reference genomes.
Interval-Wise Testing for differences in IPDs
To detect statistically significant differences between IPD curve distributions in motif-containing and motif-free windows, separately for each motif type and strand, we used the Interval-Wise Testing procedure for “omics” data implemented in the R Bioconductor package and Galaxy tool IWTomics (Campos-Sánchez et al. 2016; Pini and Vantini 2017; Cremona et al. 2018). IWT treats the IPD values in a 100-bp window as a curve (Fig. 1B) and assesses differences between two groups of curves (containing a given motif, and motif-free) performing a nonparametric (permutation) test at all possible scales, from the individual nucleotides to the whole 100 bp. When IWT detects a significant difference at a particular scale, it also identifies the locations (window coordinates) that lead to the rejection of the null hypothesis (for details, see Supplemental Text). Because IWT is computationally expensive, we ran it on a maximum of 10,000 curves for each motif type and strand (sample sizes are listed in Supplemental Tables S2, S3). For motif types with n ≥ 10,000 windows, we randomly subsampled 10,000 windows and tested against a random set of 10,000 motif-free windows; this was repeated 10 times to ensure robust results. For motif types with n ≤ 10,000 windows, we tested both against a random set of 10,000 motif-free windows and against a random set of n motif-free windows; in both cases we repeated the comparison against 10 random sets, again to ensure robust results. IWT was performed using three test statistics: the mean difference, the median difference, and the multiquantile difference (i.e., the sum of the 5th, 25th, 50th, 75th, and 95th quantile differences). Results for the latter, which most effectively captures differences in curve distributions, are presented in Figure 2E and Supplemental Figure S11A,B, and those for mean and median are presented in Supplemental Figure S11C,D and S11E,F, respectively. P-values were computed using 10,000 random permutations (independent samples, two-tailed test). The procedure produces an adjusted P-value curve (comprising 100 P-values, one for each nucleotide, adjusted up to the selected scale) for each comparison (Supplemental Fig. S2). We summarized results for all motif types in adjusted P-value heat maps (Fig. 2E; Supplemental Fig. S11, multiquantile difference; Supplemental Fig. S11, mean and median). Red/blue indicate positive/negative observed differences and are shown only for significant locations (adjusted P-value ≤0.05 in each of the 10 repetitions).
Effect of sequence composition on IPDs
To investigate whether differences in IPD values depend on incorporation of different nucleotides, we computed mean IPD, a single-nucleotide composition vector PSi = (pA, pT, pC, pG) (pA + pT + pC + pG = 100%), and a dinucleotide composition vector PDi = (pAA, pAC, pAG, …, pTT) (pAA + pAC + pAG + · · · + pTT = 100%) in each 100-bp window. We considered only motif-free windows and combined data from both strands. First, we measured the marginal effect of each nucleotide j = A,C,G,T as the correlation between log(mean IPD) and pj. Next, we used compositional regression models (Aitchison 1986; Pawlowsky-Glahn et al. 2015) to quantitate the overall effect of single-nucleotide and dinucleotide composition on IPDs. The single-nucleotide sequence composition vector PSi was mapped to a three-dimensional Euclidean vector XSi = (x1, x2, x3) using the isometric log-ratio transform, and a multiple regression model was fitted for log(mean IPD) on x1, x2, x3. Model assumptions and validity were checked with standard multiple regression diagnostic plots and tests, and the R2 was used to evaluate composition effect strength. Similarly, the dinucleotide composition vector PDi was mapped to a 15-dimensional Euclidean vector XDi = (x1, x2, …, x15), and a multiple regression model was fitted for log(mean IPD) on x1, x2, …, x15. The dinucleotide compositional regression model fitted on motif-free windows, which had higher R2 (Results), was then used to predict the mean IPD values of motif-containing windows based on their composition, separately on each strand. For each motif type, we computed the differences between these predictions and observed mean IPDs (on logarithmic scale), created their box plots, and performed two-sided t-tests for the mean difference being equal to zero—using a Bonferroni correction to adjust for multiple motif testing (Fig. 2F; Supplemental Fig. S13).
Experimental characterization of G-quadruplexes
The 10 most common G-quadruplex motifs (Supplemental Table S5) from non-B DB annotations, as well as the (GGT)n motifs, were studied by circular dichroism (CD), native polyacrylamide gel electrophoresis (PAGE), and UV absorption melting profiles, as described previously (Kejnovská et al. 2017). Single-stranded oligos were used in structure characterization of G-quadruplexes for three reasons. First, G-quadruplexes often play a regulatory role in molecular processes where DNA is single-stranded, such as replication, transcription, and repair (Dolinnaya et al. 2016). Second, single-strandedness allows better characterization of quadruplex formation and thus is most often used in experimental studies (Dailey et al. 2010). Third, the analysis of single-stranded structures is most relevant for SMRT sequencing where, even though sequencing starts with a double-stranded template, the two strands are dissociated during sequencing process.
Initially, we considered only intramolecular G-quadruplexes, computed the mean IPD in each occurrence of the motifs, and fitted a simple regression for log(mean IPD) on delta epsilon (for each motif, delta epsilon was measured once, and mean IPD was computed for hundreds or thousands of occurrences) (Supplemental Table S5; Fig. 3; Supplemental Fig. S15A). Next, we considered both intra- and intermolecular G-quadruplexes and fitted a multiple regression for log(mean IPD) on delta epsilon, the molecularity of the G-quadruplexes (either intra or intermolecular; a binary predictor), and their interaction. We fitted similar single and multiple regressions (considering only intramolecular G-quadruplexes, and both intra- and intermolecular G-quadruplexes) replacing delta epsilon with melting temperature (Tm) (Supplemental Fig. S15B). In both cases we identified final models with backward selection.
SMRT sequencing errors
Data are again those from PacBio sequencing of HG002; NA24385 (Zook et al. 2016). Errors were analyzed restricting attention to motif occurrences (not motif-containing 100-bp windows). Due to potential misalignments at motifs in the repetitive parts of the genome, motifs and motif-free windows overlapping with RepeatMasker (Smit et al. 2004) annotations (rmsk track obtained at https://genome.ucsc.edu) were excluded from this analysis. To focus on errors and not on fixed differences, all motifs and motif-free windows overlapping variants between HG002 and hg19 were also excluded—we used high confidence calls from a benchmarking data set generated in Zook et al. (2016). For each motif type, control sets were constructed picking a filtered motif-free 100-bp window at random from within 0.5 Mb upstream of or downstream from each motif occurrence and trimming it to produce a motif-free region of the same length of the motif occurrence itself. This matches motif occurrences and motif-free regions in number and length (which guarantees the same measurement resolution for errors), as well as in broad genomic location. We note that results are virtually unchanged if we do not match broad genomic location and select controls completely at random from the genome.
Error rates (the number of mismatches, insertions, or deletions relative to hg19, divided by the total number of nucleotides from all subreads in a given region and expressed as a percentage) were calculated for the newly synthesized strand that used six non-STR motif types and corresponding motif-free regions as a template. Since our purpose was detecting polymerase errors, we calculated the error rates based on individual subreads by accessing the alignment files directly and considering also low-frequency variants, including those supported by a single subread.
Comparison of errors between motifs and motif-free regions
To compare error rates between motif occurrences and matching motif-free regions, we used a two-part test (Lachenbruch 1976; Taylor and Pollard 2009) that contrasts both the heights of spikes at error rate 0 (corresponding to regions without errors) and the distributions of positive error rates. The compound null hypothesis is that both the spike at 0 (proportion of 0 rates) and the distribution on positive values (continuous component on non-0 rates) are the same in the two groups, versus the two-sided alternative that either or both differ between the groups. We considered the two-part statistic, V2 = B2 + T2, where B2 is the continuity-corrected binomial test statistic (contrasting the proportions of 0 rates) and T2 is the square of the t-test statistic (contrasting the non-0 rates). P-values were generated approximating the distribution of the test statistic V2 under the null hypothesis with a χ2(2), in order to overcome the computational burden of estimating its distribution using permutations. For several cases, we also computed P-values based on 10,000 random permutations and obtained almost indistinguishable results. For robustness, each test was repeated 10 times, using separate sets of randomly generated matching motif-free regions, and significance was assessed based on the maximum P-value (maximum P-values ≤0.10 are coded by standard stars-and-dots representation in Table 1) (Extended Data File 1).
In addition to running the tests, we computed rate fold differences (the numbers in Table 1) as follows. For each motif type, we considered the whole portion of the genome covered by its occurrences. For comparison, we considered the portion of the genome covered by all nonrepetitive, without fixed differences 100-bp motif-free windows (note: not matching motif-free regions). SMRT error rates were estimated dividing the total number of errors by the total number of bases sequenced in the portion of the genome under consideration. Rate fold differences were then computed, for each motif type and each error type, as motif rate over motif-free rate if the former is larger, and motif-free rate over motif rate otherwise.
Effect of sequence composition on errors
To investigate whether differences in SMRT sequencing values depend on the presence of different nucleotides, we computed a single-nucleotide composition vector PSi = (pA, pT, pC, pG) (pA + pT + pC + pG = 100%), and a dinucleotide composition vector PDi = (pAA, pAC, pAG, …, pTT) (pAA + pAC + pAG + · · · + pTT = 100%) in each nonrepetitive, without fixed differences 100-bp motif-free windows (note: not matching motif-free regions; this choice permits us to investigate sequence composition effect on the portion of the genome covered by all 100-bp motif-free windows). First, we measured the marginal effect of each nucleotide j = A,C,G,T on SMRT mismatch error rates as the correlation between log(errorRate) and pj. Next, we used compositional regression models (Aitchison 1986; Pawlowsky-Glahn et al. 2015) for log(errorRate) to quantitate the overall effect of single-nucleotide and dinucleotide composition on SMRT mismatch error rate. This analysis mirrored the one performed to study the effect of sequence composition on IPD values (see above). The single-nucleotide compositional regression model fitted on motif-free windows was then used to predict the SMRT error rates of motif occurrences based on their composition (some motifs are quite short; hence, the dinucleotide composition could not be accurately estimated). For each motif type, we computed the differences between these predictions and observed mean IPDs (on logarithmic scale), created their box plots, and performed two-sided t-tests for the mean difference being equal to zero—using a Bonferroni correction to adjust for multiple motif testing (Supplemental Fig. S17).
Relationship between errors and IPDs
We considered G4+ and G4− occurrences, as well as nonrepetitive, without fixed differences 100-bp motif-free windows (note: not matching motif-free regions), in order to obtain three independent groups of regions. 100-bp motif-free windows were randomly subsampled to a number equal to the sum of G4+ and G4− occurrences. We fitted a linear regression model for log(errorRate) (the SMRT mismatch rates on logarithmic scale) with predictors: (1) residual log(mean IPD), (2) region type (G4+ motifs, G4− motifs, baseline motif-free windows), and (3) interaction between (1) and (2). The residual log(mean IPD) in each region was computed as the difference between the observed mean IPD (on logarithmic scale) and the corresponding prediction using single-nucleotide composition (compositional regression model fitted on nonrepetitive, without fixed differences 100-bp motif-free windows; single-nucleotide composition was used because many G4 motifs are short; hence, the dinucleotide composition could not be accurately estimated).
Variants from human-orangutan divergence
We downloaded the 46 species Vertebrate Multiz Alignment (Blanchette et al. 2004; Harris 2007) from the UCSC Genome Browser (Multiple Alignment Format [MAF] files from https://genome.ucsc.edu/index.html) and considered nucleotide substitutions between human and orangutan. These variants were intersected with our motif occurrences. To obtain an approximate measure of divergence, we divided the number of variants in each motif occurrence by their length. Motifs overlapping with RepeatMasker (Smit et al. 2004) annotations were excluded also from this analysis.
Variants from the 1000 Genomes Project
We acquired all annotated variants from the 1000 Genomes Project (Variant Call Format [VCF] files from http://www.internationalgenome.org/) and intersected the coordinates of those with a global minor allele frequency (across all populations) equal to or above 0.05 with our motif occurrences. To obtain an approximate measure of diversity, we divided the number of SNPs in each motif by their length. Motifs overlapping with RepeatMasker (Smit et al. 2004) annotations were excluded also from this analysis.
Relationship between mutations and IPDs
We compared log(errorRate) (the SMRT mismatch rates on logarithmic scale) and residual log(mean IPD) (correcting for single-nucleotide composition, see above) between G4+ occurrences with divergence (diversity) levels smaller or equal to the 3rd percentile and larger or equal to the 97th. Since variants are rare events, a large proportion of motifs have null divergence (diversity), that is, no genetic variants. As a consequence, there were many more than 3% of the G4+ occurrences with divergence (diversity) equal to 0 (the 3rd percentile). In fact, such occurrences were much more abundant than those with divergence (diversity) above the 97th percentile. We subsampled the former 1000 times to a size equal to the number of the latter. A two-sample, two-sided t-test was performed each time to test for differences in mean between low and high divergence (diversity) G4+ occurrences. Median P-values (across 1000 tests) are reported in Table 1, together with median log(errorRate) and median residual log(mean IPD) for the same sets of G4+ occurrences.
Data and code availability
All scripts are available as Supplemental Code as well as in the public GitHub repository (https://github.com/makovalab-psu/nonBKinetics). The latest versions of the scripts may be downloaded directly from this repository.
Supplementary Material
Acknowledgments
We thank J. Korlach and S. Kingan (Pacific Biosciences Inc.) for comments on the manuscript, and B. Chen, R. Vegesna, F. Cumbo, M. Tomaszkiewicz, and M. Ferguson-Smith for assistance. Funding was provided by Penn State Eberly College of Sciences, The Huck Institute of Life Sciences at Penn State, and the Penn State Institute for CyberScience, as well as, in part, under grants from the Pennsylvania Department of Health using Tobacco Settlement and CURE Funds. The department specifically disclaims any responsibility for any analyses, responsibility, or conclusions. Additional funding was provided by the Czech Science Foundation (Grant 18-00258S).
Author contributions: W.M.G., M.A.C., M.C., and R.S.H. performed the computational and statistical analyses; I.K. and E.K. performed the biophysical experiments; and W.M.G., M.A.C., K.E., F.C., and K.D.M. wrote the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.241257.118.
Freely available online through the Genome Research Open Access option.
References
- The 1000 Genomes Project Consortium. 2015. A global reference for human genetic variation. Nature 526: 68–74. 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitchison J. 1986. The statistical analysis of compositional data. Chapman and Hall, New York. [Google Scholar]
- Ananda G, Hile SE, Breski A, Wang Y, Kelkar Y, Makova KD, Eckert KA. 2014. Microsatellite interruptions stabilize primate genomes and exist as population-specific single nucleotide polymorphisms within individual human genomes. PLoS Genet 10: e1004498 10.1371/journal.pgen.1004498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacolla A, Wells RD. 2004. Non-B DNA conformations, genomic rearrangements, and human disease. J Biol Chem 279: 47411–47414. 10.1074/jbc.R400028200 [DOI] [PubMed] [Google Scholar]
- Bacolla A, Jaworski A, Larson JE, Jakupciak JP, Chuzhanova N, Abeysinghe SS, O'Connell CD, Cooper DN, Wells RD. 2004. Breakpoints of gross deletions coincide with non-B DNA conformations. Proc Natl Acad Sci 101: 14162–14167. 10.1073/pnas.0405974101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacolla A, Wang G, Jain A, Chuzhanova NA, Cer RZ, Collins JR, Cooper DN, Bohr VA, Vasquez KM. 2011. Non-B DNA-forming sequences and WRN deficiency independently increase the frequency of base substitution in human cells. J Biol Chem 286: 10017–10026. 10.1074/jbc.M110.176636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian S, Hurley LH, Neidle S. 2011. Targeting G-quadruplexes in gene promoters: a novel anticancer strategy? Nat Rev Drug Discov 10: 261–275. 10.1038/nrd3428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belotserkovskii BP, Liu R, Tornaletti S, Krasilnikova MM, Mirkin SM, Hanawalt PC. 2010. Mechanisms and implications of transcription blockage by guanine-rich DNA sequences. Proc Natl Acad Sci 107: 12816–12821. 10.1073/pnas.1007580107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besnard E, Babled A, Lapasset L, Milhavet O, Parrinello H, Dantec C, Marin JM, Lemaitre JM. 2012. Unraveling cell type–specific and reprogrammable human replication origin signatures associated with G-quadruplex consensus motifs. Nat Struct Mol Biol 19: 837–844. 10.1038/nsmb.2339 [DOI] [PubMed] [Google Scholar]
- Blanchette M, Kent WJ, Riemer C, Elnitski L, Smit AF, Roskin KM, Baertsch R, Rosenbloom K, Clawson H, Green ED, et al. 2004. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res 14: 708–715. 10.1101/gr.1933104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos-Sánchez R, Cremona MA, Pini A, Chiaromonte F, Makova KD. 2016. Integration and fixation preferences of human and mouse endogenous retroviruses uncovered with functional data analysis. PLoS Comput Biol 12: e1004956 10.1371/journal.pcbi.1004956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castel AL, Cleary JD, Pearson CE. 2010. Repeat instability as the basis for human diseases and as a potential target for therapy. Nat Rev Mol Cell Biol 11: 165–170. 10.1038/nrm2854 [DOI] [PubMed] [Google Scholar]
- Cer RZ, Donohue DE, Mudunuri US, Temiz NA, Loss MA, Starner NJ, Halusa GN, Volfovsky N, Yi M, Luke BT, et al. 2012. Non-B DB v2.0: a database of predicted non-B DNA-forming motifs and its associated tools. Nucleic Acids Res 41: D94–D100. 10.1093/nar/gks955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers VS, Marsico G, Boutell JM, Di Antonio M, Smith GP, Balasubramanian S. 2015. High-throughput sequencing of DNA G-quadruplex structures in the human genome. Nat Biotechnol 33: 877–881. 10.1038/nbt.3295 [DOI] [PubMed] [Google Scholar]
- Cooper DN, Bacolla A, Férec C, Vasquez KM, Kehrer-Sawatzki H, Chen JM. 2011. On the sequence-directed nature of human gene mutation: the role of genomic architecture and the local DNA sequence environment in mediating gene mutations underlying human inherited disease. Hum Mutat 32: 1075–1099. 10.1002/humu.21557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremona MA, Pini A, Cumbo F, Makova KD, Chiaromonte F, Vantini S. 2018. IWTomics: testing high-resolution sequence-based ‘Omics’ data at multiple locations and scales. Bioinformatics 34: 2289–2291. 10.1093/bioinformatics/bty090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dailey MM, Miller MC, Bates PJ, Lane AN, Trent JO. 2010. Resolution and characterization of the structural polymorphism of a single quadruplex-forming sequence. Nucleic Acids Res 38: 4877–4888. 10.1093/nar/gkq166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delagoutte E, Goellner GM, Guo J, Baldacci G, McMurray CT. 2008. Single-stranded DNA-binding protein in vitro eliminates the orientation-dependent impediment to polymerase passage on CAG/CTG repeats. J Biol Chem 283: 13341–13356. 10.1074/jbc.M800153200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolinnaya NG, Ogloblina AM, Yakubovskaya MG. 2016. Structure, properties, and biological relevance of the DNA and RNA G-quadruplexes: overview 50 years after their discovery. Biochemistry (Mosc) 81: 1602–1649. 10.1134/S0006297916130034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X, Gertz EM, Wojtowicz D, Zhabinskaya D, Levens D, Benham CJ, Schäffer AA, Przytycka TM. 2014. Potential non-B DNA regions in the human genome are associated with higher rates of nucleotide mutation and expression variation. Nucleic Acids Res 42: 12367–12379. 10.1093/nar/gku921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy S, Maddukuri L, Ketkar A, Zafar MK, Henninger EE, Pursell ZF, Eoff RL. 2015. Evidence for the kinetic partitioning of polymerase activity on G-quadruplex DNA. Biochemistry 54: 3218–3230. 10.1021/acs.biochem.5b00060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, Peluso P, Rank D, Baybayan P, Bettman B, et al. 2009. Real-time DNA sequencing from single polymerase molecules. Science 323: 133–138. 10.1126/science.1162986 [DOI] [PubMed] [Google Scholar]
- Flusberg BA, Webster DR, Lee JH, Travers KJ, Olivares EC, Clark TA, Korlach J, Turner SW. 2010. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat Methods 7: 461–465. 10.1038/nmeth.1459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry M, Loeb LA. 1992. A DNA polymerase α pause site is a hot spot for nucleotide misinsertion. Proc Natl Acad Sci 89: 763–767. 10.1073/pnas.89.2.763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fungtammasan A, Ananda G, Hile SE, Su MS, Sun C, Harris R, Medvedev P, Eckert K, Makova KD. 2015. Accurate typing of short tandem repeats from genome-wide sequencing data and its applications. Genome Res 25: 736–749. 10.1101/gr.185892.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberman Y, Amariglio N, Rechavi G, Eisenberg E. 2008. Trinucleotide repeats are prevalent among cancer-related genes. Trends Genet 24: 14–18. 10.1016/j.tig.2007.09.005 [DOI] [PubMed] [Google Scholar]
- Harris RS. 2007. “Improved pairwise alignment of genomic DNA.” PhD thesis, Pennsylvania State University. [Google Scholar]
- Hile SE, Eckert KA. 2004. Positive correlation between DNA polymerase α-primase pausing and mutagenesis within polypyrimidine/polypurine microsatellite sequences. J Mol Biol 335: 745–759. 10.1016/j.jmb.2003.10.075 [DOI] [PubMed] [Google Scholar]
- Hodgkinson A, Eyre-Walker A. 2011. Variation in the mutation rate across mammalian genomes. Nat Rev Genet 12: 756–766. 10.1038/nrg3098 [DOI] [PubMed] [Google Scholar]
- Huppert JL, Balasubramanian S. 2005. Prevalence of quadruplexes in the human genome. Nucleic Acids Res 33: 2908–2916. 10.1093/nar/gki609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki H, Ohye T, Kogo H, Tsutsumi M, Kato T, Tong M, Emanuel BS, Kurahashi H. 2013. Two sequential cleavage reactions on cruciform DNA structures cause palindrome-mediated chromosomal translocations. Nat Commun 4: 1592 10.1038/ncomms2595 [DOI] [PubMed] [Google Scholar]
- Jansen A, van der Zande E, Meert W, Fink GR, Verstrepen KJ. 2012. Distal chromatin structure influences local nucleosome positions and gene expression. Nucleic Acids Res 40: 3870–3885. 10.1093/nar/gkr1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamat MA, Bacolla A, Cooper DN, Chuzhanova N. 2016. A role for non-B DNA forming sequences in mediating microlesions causing human inherited disease. Hum Mutat 37: 65–73. 10.1002/humu.22917 [DOI] [PubMed] [Google Scholar]
- Kang S, Ohshima K, Shimizu M, Amirhaeri S, Wells RD. 1995. Pausing of DNA synthesis in vitro at specific loci in CTG and CGG triplet repeats from human hereditary disease genes. J Biol Chem 270: 27014–27021. 10.1074/jbc.270.45.27014 [DOI] [PubMed] [Google Scholar]
- Kejnovská I, Bednárová K, Renciuk D, Dvoráková Z, Školáková P, Trantírek L, Fiala R, Vorlícková M, Sagi J. 2017. Clustered abasic lesions profoundly change the structure and stability of human telomeric G-quadruplexes. Nucleic Acids Res 45: 4294–4305. 10.1093/nar/gkx191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasilnikova MM, Mirkin SM. 2004. Replication stalling at Friedreich's Ataxia (GAA)n repeats in vivo. Mol Cell Biol 24: 2286–2295. 10.1128/MCB.24.6.2286-2295.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kypr J, Kejnovská I, Renciuk D, Vorlícková M. 2009. Circular dichroism and conformational polymorphism of DNA. Nucleic Acids Res 37: 1713–1725. 10.1093/nar/gkp026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachenbruch PA. 1976. Analysis of data with clumping at zero. Biom Z 18: 351–356. [Google Scholar]
- Locke DP, Hillier LW, Warren WC, Worley KC, Nazareth LV, Muzny DM, Yang SP, Wang Z, Chinwalla AT, Minx P, et al. 2011. Comparative and demographic analysis of orang-utan genomes. Nature 469: 529–533. 10.1038/nature09687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomis EW, Eid JS, Peluso P, Yin J, Hickey L, Rank D, McCalmon S, Hagerman RJ, Tassone F, Hagerman PJ. 2013. Sequencing the unsequenceable: expanded CGG-repeat alleles of the fragile X gene. Genome Res 23: 121–128. 10.1101/gr.141705.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maizels N. 2015. G4-associated human diseases. EMBO Rep 16: 910–922. 10.15252/embr.201540607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makova KD, Hardison RC. 2015. The effects of chromatin organization on variation in mutation rates in the genome. Nat Rev Genet 16: 213–223. 10.1038/nrg3890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makova KD, Li WH. 2002. Strong male-driven evolution of DNA sequences in humans and apes. Nature 416: 624–626. 10.1038/416624a [DOI] [PubMed] [Google Scholar]
- Mirkin SM. 2007. Expandable DNA repeats and human disease. Nature 447: 932–940. 10.1038/nature05977 [DOI] [PubMed] [Google Scholar]
- Mirkin SM. 2008. Discovery of alternative DNA structures: a heroic decade (1979-1989). Front Biosci 13: 1064–1071. 10.2741/2744 [DOI] [PubMed] [Google Scholar]
- Mirkin EV, Mirkin SM. 2007. Replication fork stalling at natural impediments. Microbiol Mol Biol Rev 71: 13–35. 10.1128/MMBR.00030-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirkin SM, Lyamichev VI, Drushlyak KN, Dobrynin VN, Filippov SA, Frank-Kamenetskii MD. 1987. DNA H form requires a homopurine–homopyrimidine mirror repeat. Nature 330: 495–497. 10.1038/330495a0 [DOI] [PubMed] [Google Scholar]
- Nadel Y, Weisman-Shomer P, Fry M. 1995. The fragile X syndrome single strand d(CGG)n nucleotide repeats readily fold back to form unimolecular hairpin structures. J Biol Chem 270: 28970–28977. 10.1074/jbc.270.48.28970 [DOI] [PubMed] [Google Scholar]
- Neidle S, Balasubramanian S. 2006. Quadruplex nucleic acids. Royal Society of Chemistry, London. [Google Scholar]
- Parkinson GN, Lee MP, Neidle S. 2002. Crystal structure of parallel quadruplexes from human telomeric DNA. Nature 417: 876–880. 10.1038/nature755 [DOI] [PubMed] [Google Scholar]
- Pawlowsky-Glahn V, Egozcue JJ, Tolosana-Delgado R. 2015. Modeling and analysis of compositional data. John Wiley & Sons, Chichester, UK. [Google Scholar]
- Pini A, Vantini S. 2017. Interval-wise testing for functional data. J Nonparametr Stat 29: 407–424. [Google Scholar]
- Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, et al. 2011. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72: 257–268. 10.1016/j.neuron.2011.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhoads A, Au KF. 2015. PacBio sequencing and its applications. Genomics Proteomics Bioinformatics 13: 278–289. 10.1016/j.gpb.2015.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samadashwily GM, Raca G, Mirkin SM. 1997. Trinucleotide repeats affect DNA replication in vivo. Nat Genet 17: 298–304. 10.1038/ng1197-298 [DOI] [PubMed] [Google Scholar]
- Sawaya S, Bagshaw A, Buschiazzo E, Kumar P, Chowdhury S, Black MA, Gemmell N. 2013. Microsatellite tandem repeats are abundant in human promoters and are associated with regulatory elements. PLoS One 8: e54710 10.1371/journal.pone.0054710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaya S, Boocock J, Black MA, Gemmell NJ. 2015. Exploring possible DNA structures in real-time polymerase kinetics using Pacific Biosciences sequencer data. BMC Bioinformatics 16: 21 10.1186/s12859-014-0449-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt EE, Banerjee O, Fang G, Feng Z, Wong WH, Zhang X, Kislyuk A, Clark TA, Luong K, Keren-Paz A, et al. 2013. Modeling kinetic rate variation in third generation DNA sequencing data to detect putative modifications to DNA bases. Genome Res 23: 129–141. 10.1101/gr.136739.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen D, Gilbert W. 1988. Formation of parallel four-stranded complexes by guanine-rich motifs in DNA and its implications for meiosis. Nature 334: 364–366. 10.1038/334364a0 [DOI] [PubMed] [Google Scholar]
- Siddiqui-Jain A, Grand CL, Bearss DJ, Hurley LH. 2002. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc Natl Acad Sci 99: 11593–11598. 10.1073/pnas.182256799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simone R, Fratta P, Neidle S, Parkinson GN, Isaacs AM. 2015. G-quadruplexes: emerging roles in neurodegenerative diseases and the non-coding transcriptome. FEBS Lett 589: 1653–1668. 10.1016/j.febslet.2015.05.003 [DOI] [PubMed] [Google Scholar]
- Sinden RR, Pytlos-Sinden MJ, Potaman VN. 2007. Slipped strand DNA structures. Front Biosci 12: 4788–4799. 10.2741/2427 [DOI] [PubMed] [Google Scholar]
- Smit AFA, Hubley R, Green P. 2004. RepeatMasker Open-3.0. http://www.repeatmasker.org
- Sweasy JB, Lauper JM, Eckert KA. 2006. DNA polymerases and human diseases. Radiat Res 166: 693–714. 10.1667/RR0706.1 [DOI] [PubMed] [Google Scholar]
- Taylor S, Pollard K. 2009. Hypothesis tests for point-mass mixture data with application to ‘omics data with many zero values. Stat Appl Genet Mol Biol 8: Article 8 10.2202/1544-6115.1425 [DOI] [PubMed] [Google Scholar]
- Turner S, Kuse R, Kearns G, Monadgemi P, Foquet M, Martinez D. 2017. Nanoscale apertures having islands of functionality. U.S. patent no. US9637380. https://www.google.com/patents/US9637380
- Usdin K, Woodford KJ. 1995. CGG repeats associated with DNA instability and chromosome fragility form structures that block DNA synthesis in vitro. Nucleic Acids Res 23: 4202–4209. 10.1093/nar/23.20.4202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valton AL, Prioleau MN. 2016. G-Quadruplexes in DNA replication: a problem or a necessity? Trends Genet 32: 697–706. 10.1016/j.tig.2016.09.004 [DOI] [PubMed] [Google Scholar]
- Voineagu I, Narayanan V, Lobachev KS, Mirkin SM. 2008. Replication stalling at unstable inverted repeats: interplay between DNA hairpins and fork stabilizing proteins. Proc Natl Acad Sci 105: 9936–9941. 10.1073/pnas.0804510105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Vasquez KM. 2007. Z-DNA, an active element in the genome. Front Biosci 12: 4424–4438. 10.2741/2399 [DOI] [PubMed] [Google Scholar]
- Wang G, Carbajal S, Vijg J, DiGiovanni J, Vasquez KM. 2008. DNA structure-induced genomic instability in vivo. J Natl Cancer Inst 100: 1815–1817. 10.1093/jnci/djn385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson JD, Crick FHC. 1953. Genetical implications of the structure of deoxyribonucleic acid. Nature 171: 964–967. 10.1038/171964b0 [DOI] [PubMed] [Google Scholar]
- Wittig B, Dorbic T, Rich A. 1991. Transcription is associated with Z-DNA formation in metabolically active permeabilized mammalian cell nuclei. Proc Natl Acad Sci 88: 2259–2263. 10.1073/pnas.88.6.2259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Bacolla A, Wang G, Vasquez KM. 2010. Non-B DNA structure-induced genetic instability and evolution. Cell Mol Life Sci 67: 43–62. 10.1007/s00018-009-0131-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zook JM, Catoe D, McDaniel J, Vang L, Spies N, Sidow A, Weng Z, Liu Y, Mason CE, Alexander N, et al. 2016. Extensive sequencing of seven human genomes to characterize benchmark reference materials. Sci Data 3: 160025 10.1038/sdata.2016.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All scripts are available as Supplemental Code as well as in the public GitHub repository (https://github.com/makovalab-psu/nonBKinetics). The latest versions of the scripts may be downloaded directly from this repository.