

Abstract
Sirtuins (SIRTs) are a class of lysine deacylases that regulate cellular metabolism and energy homeostasis. Although sirtuins have been proposed to function in nutrient sensing and signaling, the underlying mechanism remains elusive. SIRT7, a histone H3K18‐specific deacetylase, epigenetically controls mitochondria biogenesis, ribosomal biosynthesis, and DNA repair. Here, we report that SIRT7 is methylated at arginine 388 (R388), which inhibits its H3K18 deacetylase activity. Protein arginine methyltransferase 6 (PRMT6) directly interacts with and methylates SIRT7 at R388 in vitro and in vivo. R388 methylation suppresses the H3K18 deacetylase activity of SIRT7 without modulating its subcellular localization. PRMT6‐induced H3K18 hyperacetylation at SIRT7‐target gene promoter epigenetically promotes mitochondria biogenesis and maintains mitochondria respiration. Moreover, high glucose enhances R388 methylation in mouse fibroblasts and liver tissue. PRMT6 signals glucose availability to SIRT7 in an AMPK‐dependent manner. AMPK induces R388 hypomethylation by disrupting the association between PRMT6 and SIRT7. Together, PRMT6‐induced arginine methylation of SIRT7 coordinates glucose availability with mitochondria biogenesis to maintain energy homeostasis. Our study uncovers the regulatory role of SIRT7 arginine methylation in glucose sensing and mitochondria biogenesis.
Keywords: arginine methylation, glucose sensing, mitochondria biogenesis, PRMT6, SIRT7
Subject Categories: Metabolism; Post-translational Modifications, Proteolysis & Proteomics
Introduction
Sirtuins are a class of lysine deacylases that remove various acylation modifications from target proteins 1. With the help of NAD+, sirtuins remove acetyl groups and other acyl groups from modified lysine residues, hence modifying the function of substrate proteins 1, 2. The catalytic nature of sirtuins implies an intimate relationship with cellular metabolism. In fact, sirtuins have a fundamental role in regulating metabolic pathways and energy homeostasis 3, 4, 5. As such, deregulation of sirtuins has been implicated in multiple metabolic diseases, including aging, cancers, obesity, and diabetes mellitus 6, 7, 8, 9. Notably, sirtuins have been proposed as nutrient sensors 10. In response to the fluctuations of intracellular NAD+ level, acylation of downstream target proteins is well‐controlled by sirtuins, thereby fine‐tuning cellular metabolism 11. However, the regulatory mechanism of nutrient‐sensing function of sirtuins remains largely unknown.
Mammalian sirtuin family contains seven different members, SIRT1‐7 3. Different from other sirtuins, SIRT7 shows weak deacetylase activity in vitro 12, 13. SIRT7 has a nucleolar localization signal and redistributes within the nucleus in response to nutritional stress 14. These observations indicate that the deacetylase function of SIRT7 is under tight metabolic control within cells. Multiple studies using genetic knockout models suggest that SIRT7 regulates mitochondria biogenesis, ribosomal biogenesis, glucose homeostasis, and genotoxic stress response 8, 15, 16, 17, 18. Through connecting nutritional status and transcriptional program of biogenesis, SIRT7 functions as an important regulatory node to maintain the metabolic balance. Histone is a major substrate of SIRT7 19. SIRT7 removes histone H3K18 acetylation (H3K18ac) mark from the promoter of its target genes and induces transcription regression 19, 20. As an H3K18 deacetylase, SIRT7 epigenetically controls the transcription of genes involved in mitochondria biogenesis, ribosomal biosynthesis, and DNA damage response 15, 16, 20. The notion that SIRT7 functions as a key player in glucose sensing is supported by at least three lines of evidence: (i) Sirt7 ablation resulted in disorders of glucose homeostasis and lipid metabolism 8, 15; (ii) SIRT7 modulated the expression of gluconeogenic genes to maintain blood glucose level 21; and (iii) SIRT7 inhibition suppressed glycolytic activity of hepatocellular cancer cells 22. As an epigenetic modulator of glucose metabolism, SIRT7 connects glucose availability to cellular biogenesis. But still, the mechanism by which cells signal glucose availability to SIRT7 remains obscure.
Arginine methylation has been emerging as a key post‐translational modification that regulates signal transduction and cellular metabolism 23, 24, 25. Protein arginine methyltransferases (PRMTs) catalyze methylation reactions on arginine residue of the substrate proteins 26. Mammalian cells express at least nine different PRMTs (PRMT1‐PRMT9) 27. Arginine can be mono‐methylated by all PRMTs. Depending on the catalytic property of PRMTs, mono‐methylarginine is further converted to asymmetric di‐methylarginine or symmetric di‐methylarginine, by type I PRMT (PRMT1‐4, PRMT6, and PRMT8) and type II PRMT (PRMT5 and PRMT9), respectively 28. Recent protein arginine methylome studies have identified hundreds of arginine‐methylated proteins, among which is SIRT7 29, 30. We thus speculate that arginine methylation serves as a regulatory mechanism in SIRT7‐mediated glucose sensing and signaling.
Results
SIRT7 is methylated at arginine 388
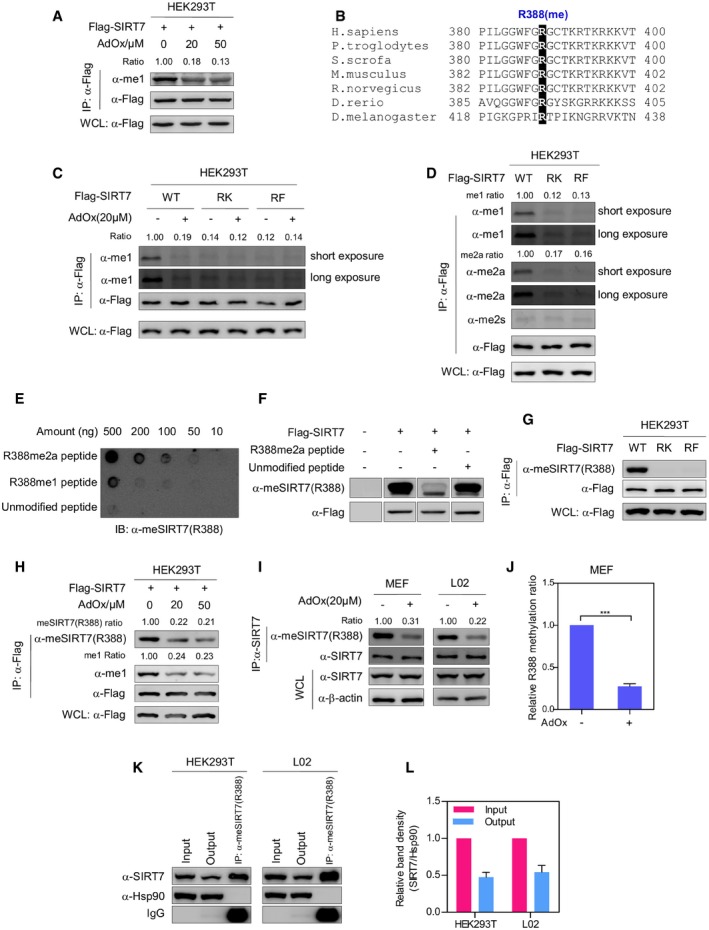
In recent proteomic studies 29, 30, SIRT7 was identified as an arginine‐methylated protein. To validate this finding, Flag‐tagged SIRT7 was ectopically expressed in HEK293T cells. Western blotting using an anti‐mono‐methylarginine antibody (α‐me1) showed that immunopurified SIRT7 was indeed methylated (Fig 1A). Furthermore, SIRT7 methylation decreased by more than fivefold after treating cells with adenosine dialdehyde (AdOx), a PRMT inhibitor (Fig 1A). Previous methylome studies indicated that arginine 388 (R388), a highly conserved residue, was the only putative methylation site of SIRT7 (Fig 1B). To test whether R388 is methylated, we mutated R388 to lysine (K) or phenylalanine (F). The R‐to‐K mutant is used as an unmethylatable mimetic, while R‐to‐F mutation mimics the methylated state of arginine 31, 32. Strikingly, both R388K (RK) and R388F (RF) mutants showed a dramatic reduction in arginine methylation, compared to wild‐type SIRT7 (Fig 1C). AdOx treatment decreased the methylation level of wild‐type SIRT7 but not RK/RF mutants (Fig 1C). These observations indicate that SIRT7 is methylated at R388.
Figure 1. SIRT7 is methylated at arginine 388.
-
AArginine methylase inhibitor decreases SIRT7 methylation. HEK293T cells expressing Flag‐tagged SIRT7 were treated with increasing doses of AdOx as indicated for 24 h. After immunoprecipitated with Flag‐beads, SIRT7 was analyzed by Western blot and detected by an antibody against mono‐methylarginine (α‐me1). Relative methylation ratio (Ratio) was calculated by normalizing arginine methylation level against the Flag‐SIRT7 protein. WCL, whole cell lysate.
-
BR388 of SIRT7 is evolutionarily conserved. Amino acid sequences corresponding to R388 of human SIRT7 were aligned.
-
CPRMT inhibitor reduces arginine methylation of wild‐type SIRT7, but not its RK/RF mutants. HEK293T cells expressing wild‐type SIRT7 or its RK/RF mutants were treated with or without AdOx for 24 h. Arginine methylation of immunopurified SIRT7 was determined by Western blot.
-
DSIRT7 is mono‐ and asymmetrically di‐methylated at R388. Flag‐tagged SIRT7 and its RK/RF mutants were expressed in HEK293T cells. Immunopurified SIRT7 was blotted with antibodies against mono‐methylarginine (α‐me1), asymmetric di‐methylarginine (α‐me2a), and symmetric di‐methylarginine (α‐me2s), respectively.
-
ER388 site‐specific methylation antibody recognizes R388‐methylated peptide, but not the unmodified peptide. The unmodified peptide corresponding to R388, R388‐mono‐methylated (R388me1) peptide, and R388‐asymmetrically di‐methylated (R388me2a) peptide were blotted with α‐meSIRT7(R388) antibody.
-
FR388‐methylated peptide, but not the unmodified peptide, blocks α‐meSIRT7(R388) antibody. After incubation with unmodified peptide or R388‐methylated peptide (R388me2a), α‐meSIRT7(R388) antibody was used to detect immunopurified SIRT7.
-
GRK and RF mutations disrupt SIRT7 methylation. Flag‐tagged wild‐type SIRT7 and its RK/RF mutants were immunoprecipitated and blotted with α‐meSIRT7(R388) antibody.
-
HSIRT7 is methylated at R388. HEK293T cells ectopically expressing Flag‐tagged SIRT7 were treated with increasing concentrations of AdOx for 24 h as indicated. SIRT7 protein was pulled down by Flag‐beads. Arginine methylation of SIRT7 was detected with a site‐specific antibody against R388 methylation [α‐meSIRT7(R388)].
-
I, JAdOx treatment decreases R388 methylation of SIRT7. MEF and L02 cells were treated with AdOx for 24 h. Methylation of immunoprecipitated endogenous SIRT7 was determined by Western blot (I). The band intensity of Western blots was measured to quantify relative R388 methylation ratio (J) (mean ± SD, n = 3 experimental replicates, ***P < 0.001, unpaired two‐tailed t‐test).
-
K, LQuantification of R388‐methylated endogenous SIRT7 in HEK293T and L02 cells. R388‐methylated endogenous SIRT7 was captured from HEK293T and L02 cells by using R388 site‐specific methylation antibody (K). Input, output, and immunoprecipitate were analyzed by Western blot to quantify the percentage of R388‐methylated protein (L) (mean ± SD, n = 3 experimental replicates).
Arginine residue is methylated in a processive manner, resulting in mono‐methylarginine (me1), asymmetric or symmetric di‐methylarginine (me2a or me2s). Interestingly, wild‐type SIRT7 was efficiently recognized by the mono‐methylarginine antibody (α‐me1) and asymmetric di‐methylarginine antibody (α‐me2a), but not symmetric di‐methylarginine antibody (α‐me2s; Fig 1D). RK and RF mutations disrupted mono‐methylation and asymmetric di‐methylation of SIRT7 (Fig 1D). These results further support R388 as a major methylation site of SIRT7. To precisely detect R388 methylation of SIRT7, we generated a site‐specific antibody against methylated R388 [α‐meSIRT7(R388)]. This antibody showed high specificity in dot blot assay and antigen peptide competition assay (Fig 1E–F). Of note, dot blotting showed that R388 methylation antibody recognized both mono‐methylated and di‐methylated peptides, suggesting that this antibody reflects both R388‐mono‐methylated and R388‐di‐methylated SIRT7 proteins (Fig 1E). R388 site‐specific methylation antibody efficiently recognized wild‐type SIRT7, but not RK or RF mutants (Fig 1G). Furthermore, R388 methylation of ectopically expressed SIRT7 was decreased by AdOx treatment in HEK293T cells (Fig 1H). AdOx treatment significantly reduced R388 methylation of endogenous SIRT7 protein in L02 cells and mouse embryonic fibroblasts (MEF, P = 2.5 × 10−6) (Fig 1I–J). To quantify R388‐methylated SIRT7 in cultured cells, we performed immunoprecipitation using R388 methylation‐specific antibody to capture methylated SIRT7. Hsp90 was included as a loading control. Interestingly, R388‐methylated SIRT7 accounted for 52.3% and 45.7% of total SIRT7 in HEK293T and L02 cells, respectively (Fig 1K–L). Together, these results suggest that R388 is a major, if not the only, methylation site of SIRT7. The occurrence of R388 methylation on endogenous SIRT7 protein strongly indicates that R388 methylation is a physiologically relevant modification.
PRMT6 directly methylates SIRT7 at R388
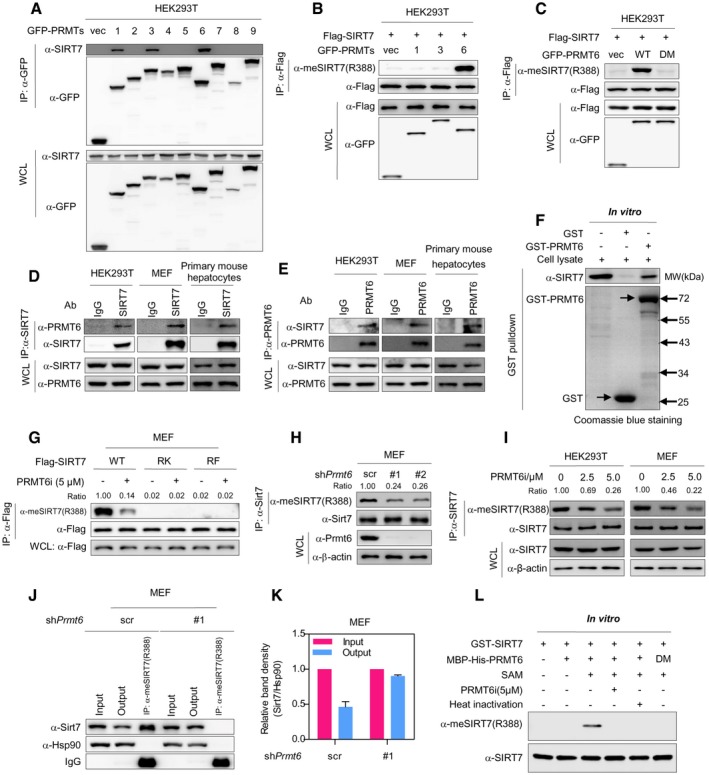
Arginine methylation of proteins is tightly controlled by PRMTs 26, 27. To identify the PRMT(s) responsible for SIRT7 methylation, GFP‐tagged PRMTs (PRMT1‐9) were individually expressed in HEK293T cells. Co‐immunoprecipitation (co‐IP) assay showed that SIRT7 interacted with PRMT1, PRMT3, and PRMT6 (Fig 2A). Co‐expression of PRMT6, but not PRMT1 or PRMT3, remarkably increased arginine methylation of SIRT7 (Fig 2B). More importantly, wild‐type PRMT6, but not its catalytic‐deficient mutant (V86K/D88A, DM) 33, dramatically increased SIRT7 methylation (Fig 2C). These data suggest that PRMT6 methylates SIRT7. In line with these findings, reciprocal co‐immunoprecipitation indicated that endogenous SIRT7 interacted with PRMT6 in HEK293T, MEF, and mouse primary hepatocytes (Fig 2D–E). Glutathione S‐transferase (GST) pull‐down assay further demonstrated that SIRT7 directly bound to PRMT6 in vitro (Figs 2F and EV1A).
Figure 2. PRMT6 directly methylates SIRT7 at R388.
-
ASIRT7 binds to PRMT1, PRMT3, and PRMT6. GFP‐tagged PRMTs (PRMT1‐9) were expressed in HEK293T cells. PRMT protein was immunoprecipitated with GFP antibody and analyzed by Western blot.
-
BCo‐expression of PRMT6, but not PRMT1 or PRMT3, increases R388 methylation of SIRT7. Flag‐tagged SIRT7 and GFP‐tagged PRMT1, PRMT3, or PRMT6 were co‐expressed in HEK293T cells. R388 methylation of immunopurified SIRT7 was analyzed by Western blot.
-
CWild‐type PRMT6, but not its catalytic‐deficient mutant, upregulates SIRT7 R388 methylation. Wild‐type PRMT6 or its V86K/D88A mutant (DM) was co‐expressed with Flag‐tagged SIRT7 in HEK293T cells. R388 methylation of immunopurified SIRT7 was detected by Western blot.
-
D, EPRMT6 physically interacts with endogenous SIRT7. Endogenous SIRT7 (D) or PRMT6 (E) was pulled down using corresponding antibody from HEK293T, MEF, and primary mouse hepatocytes, respectively. Input and immunoprecipitates were analyzed by Western blot.
-
FSIRT7 associates with PRMT6 in vitro. Recombinant GST‐PRMT6 was incubated with HEK293T lysates. After GST pulldown, the interaction of recombinant protein with endogenous SIRT7 was analyzed by Western blot. The arrows indicate GST and GST‐PRMT6 protein, respectively.
-
GPRMT6 inhibitor reduces arginine methylation of wild‐type SIRT7, but not RK/RF mutants. Wild‐type SIRT7 or its RK/RF mutants were stably expressed in MEF cells. After treating cells with or without PRMT6‐specific chemical inhibitor (PRMT6i) for 24 h, SIRT7 was immunoprecipitated and blotted with R388‐specific methylation antibody.
-
HPrmt6 depletion decreases R388 methylation of endogenous Sirt7. Scramble control cells (scr) or MEF cells stably expressing two different shRNAs (#1 and #2) against Prmt6 were established. R388 methylation of immunopurified endogenous Sirt7 was determined by Western blot.
-
ITreatment with PRMT6 inhibitor reduces R388 methylation of endogenous SIRT7 in a dose‐dependent manner. MEF and HEK293T cells were treated with increasing doses of PRMT6i for 24 h as indicated. R388 methylation of immunoprecipitated endogenous SIRT7 was determined by Western blot.
-
J, KPrmt6 depletion reduces the percentage of R388‐methylated endogenous Sirt7 in MEF cells. R388‐methylated endogenous Sirt7 was captured from scramble control cells (scr) and Prmt6‐knockdown MEF cells by using R388 site‐specific methylation antibody (J). Input, output, and immunoprecipitate were analyzed by Western blot to quantify the percentage of R388‐methylated protein (K) (mean ± SD, n = 3 experimental replicates).
-
LRecombinant PRMT6 directly methylates SIRT7 at R388 in vitro. Bacterially expressed PRMT6 and SIRT7 proteins were incubated with SAM or PRMT6i as indicated. Heat‐inactivated PRMT6 was included as negative control. R388 methylation of recombinant SIRT7 protein was determined by Western blot.
Figure EV1. PRMT6 directly methylates SIRT7 at R388.
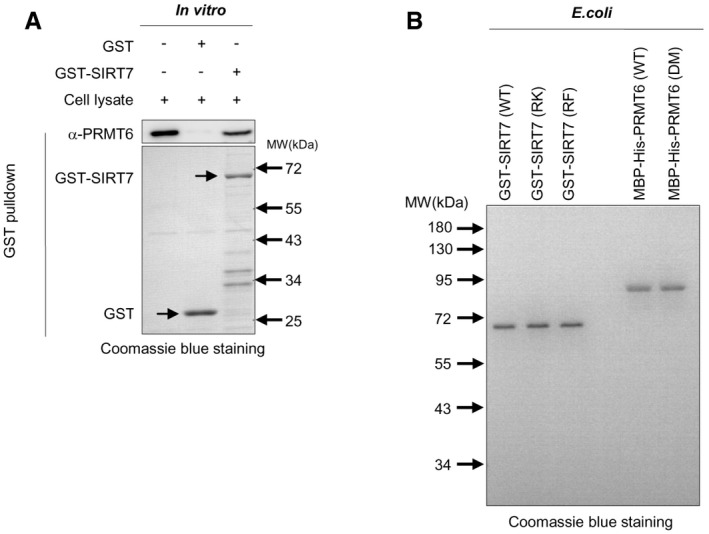
- PRMT6 associates with SIRT7 in vitro. Recombinant SIRT7 was incubated with HEK293T lysates. After GST pulldown, the interaction of recombinant protein with endogenous PRMT6 was analyzed by Western blot. The arrows indicate GST and GST‐SIRT7 protein, respectively.
- Purification of recombinant GST‐SIRT7, MBP‐His‐PRMT6, and their mutants. Recombinant SIRT7 and PRMT6 were expressed in E. coli. Purified recombinant proteins were resolved with SDS–PAGE and analyzed by Coomassie blue staining.
We next set out to further test whether PRMT6 methylates R388 of SIRT7 in physiological setting. Treatment of cells with a PRMT6‐specific inhibitor (PRMT6i) remarkably reduced R388 methylation of wild‐type SIRT7, but not its RK or RF mutants (Fig 2G). We further transduced MEF cells with two different short hairpin RNAs (shRNAs) targeting Prmt6. Prmt6 depletion significantly decreased R388 methylation of endogenous Sirt7 (Fig 2H). In addition, PRMT6i treatment dose‐dependently decreased R388 methylation of endogenous SIRT7 in HEK293T and MEF cells (Fig 2I). Quantification of R388‐methylated Sirt7 further indicated that the fraction of methylated Sirt7 decreased from 53.7% to 9.7% after depleting Prmt6 from MEF cells (Fig 2J–K). These results suggest that PRMT6 is necessary for R388 methylation. To interrogate whether PRMT6 directly methylates SIRT7 at R388, we purified bacterially expressed PRMT6 and SIRT7 proteins to homogeneity (Fig EV1B) and performed in vitro methylation assay. After incubation with recombinant PRMT6, SIRT7 showed a strong increase in R388 methylation in the presence of SAM, the methyl donor (Fig 2L). Notably, R388 methylation was undetectable in the absence of SAM or PRMT6 (Fig 2L). Similarly, no increase in R388 methylation signal was observed in the presence of PRMT6 inhibitor. Heat‐inactivated PRMT6 or catalytically inactive mutant PRMT6 (DM) lost the ability to upregulate R388 methylation (Fig 2L). Together, these data clearly demonstrate that PRMT6 directly modifies SIRT7. PRMT6 is both sufficient and necessary for R388 methylation.
R388 methylation of SIRT7 suppresses its H3K18 deacetylase activity
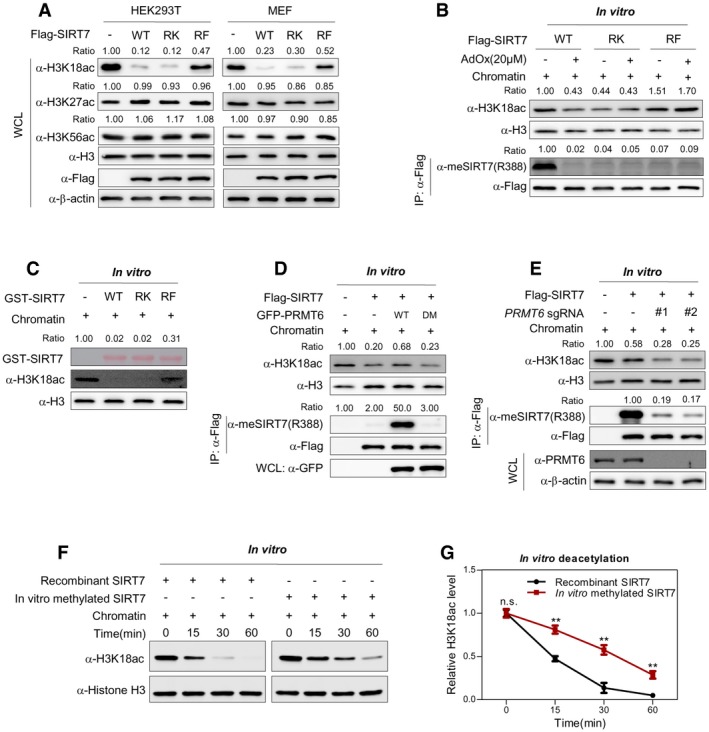
R388 locates close to carboxyl‐terminus of SIRT7, a region critical for its H3K18 deacetylase activity 34. We thus speculate that R388 methylation possibly regulates the deacetylase activity of SIRT7. As expected, ectopic expression of wild‐type SIRT7 and its RK mutant decreased the global acetylation level of H3K18 (Fig 3A). In contrast, RF mutant showed negligible effect on H3K18 acetylation (Fig 3A). To confirm this finding, we extracted chromatin from HEK293T cells and performed deacetylation assay in vitro. When we incubated immunopurified SIRT7 with chromatin in vitro, RF mutant of SIRT7 displayed decreased H3K18 deacetylase activity, compared to wild‐type SIRT7. RK mutation showed negligible effect on H3K18 deacetylase activity when cells were treated with AdOx (Fig 3B). More importantly, the H3K18 deacetylase activity of wild‐type SIRT7, but not that of RK/RF mutant, was remarkably increased by AdOx treatment (Fig 3B). Furthermore, in vitro deacetylation assay clearly demonstrates that recombinant RF mutant, but not RK mutant, is deficient in removing H3K18ac from chromatin (Fig 3C). These data suggest that R388 methylation potentially suppresses the H3K18 deacetylase activity of SIRT7. To our surprise, although considerable fraction of SIRT7 was R388‐methylated under basal conditions (Figs 1K–L and 2J–K), the RK mutant of SIRT7 was as active as the wild‐type protein in vivo. This observation suggests that R388K mutant cannot faithfully mimic unmethylated SIRT7, potentially due to the conformational difference in the side chains of arginine and lysine.
Figure 3. R388 methylation of SIRT7 suppresses its H3K18 deacetylase activity.
-
ARF, but not RK mutant, suppresses H3K18 deacetylase activity of SIRT7. Flag‐SIRT7 and its RK/RF mutants were expressed in HEK293T and MEF cells, respectively. Cell lysates were subjected to Western blot to detect histone H3K18 acetylation. H3K27ac and H3K56ac were included as negative control. The level of each histone acetylation marker was normalized against histone H3 (Ratio).
-
BR388 methylation of SIRT7 decreases its H3K18 deacetylase activity. HEK293T cells expressing wild‐type SIRT7 or its RK/RF mutants were treated with or without AdOx for 24 h. The SIRT7 enzyme was immunopurified with Flag‐beads and further incubated with extracted chromatin in vitro. R388 methylation of immunopurified SIRT7 and H3K18ac of in vitro deacetylated chromatin were determined by Western blot.
-
CRF, but not RK mutant, shows reduced H3K18 deacetylase activity in vitro. Chromatin was extracted from HEK293T cells and incubated with recombinant GST‐tagged SIRT7 (wild‐type and RK/RF mutants) in vitro for 2 h. H3K18ac was determined by Western blot.
-
DWild‐type PRMT6, but not its methylase‐dead mutant, decreases the H3K18 deacetylase activity of SIRT7. Flag‐tagged SIRT7 was co‐expressed with wild‐type PRMT6 or its catalytic‐inactive mutant (DM) in HEK293T cells. The H3K18 deacetylase activity of immunopurified SIRT7 was assayed in vitro.
-
EPRMT6 deficiency promotes the H3K18 deacetylase activity of SIRT7. Control sgRNA or two independent sgRNAs (#1 and #2) against PRMT6 was introduced into HEK293T cells. Flag‐tagged SIRT7 was ectopically expressed in these cells. The histone deacetylase activity of immunoprecipitated SIRT7 was assayed in vitro using chromatin as a substrate. R388 methylation of immunopurified SIRT7 and H3K18 acetylation level of in vitro deacetylated chromatin were determined by Western blot.
-
F, GMethylated recombinant SIRT7 shows reduced H3K18 deacetylase activity. In vitro methylated SIRT7 from Fig 2L was subjected to in vitro deacetylation assay. H3K18 acetylation level of in vitro deacetylated chromatin was determined by Western blot (F). Relative H3K18ac level was quantified from three independent experiments (G) (mean ± SD, n = 3 experimental replicates, **P < 0.01, n.s. = not significant, unpaired two‐tailed t‐test).
We further co‐expressed SIRT7 with PRMT6 and determined how PRMT6 modulated the H3K18 deacetylase activity of SIRT7. Co‐expression of wild‐type PRMT6, but not its methylase‐dead mutant (DM), inhibited the in vitro deacetylase activity of SIRT7 (Fig 3D). Additionally, we genetically disrupted PRMT6 using two different short guide RNA (sgRNA) and ectopically expressed SIRT7 in HEK293T cells. In vitro activity assay showed that PRMT6 deletion increased the H3K18 deacetylase activity of immunoprecipitated SIRT7 (Fig 3E). These results suggest that PRMT6‐induced R388 methylation suppresses the H3K18 deacetylase activity of SIRT7.
We next asked through what mechanism R388 methylation reduced SIRT7 activity. Although R388 is close to a nucleolar localization sequence, RF mutant showed similar distribution pattern to wild‐type protein (Fig EV2A). Besides, immunofluorescence of SIRT7 showed that PRMT6i treatment did not alter SIRT7 localization (Fig EV2B). These results suggest that R388 methylation does not modulate SIRT7 subcellular localization. We thus speculate that R388 methylation directly modulates the intrinsic activity of SIRT7. By using extracted chromatin as the substrate, we determined the deacetylase activity of recombinant SIRT7 after in vitro methylation reaction (Fig 2L). Compared to the unmethylated control SIRT7, methylated recombinant SIRT7 showed a significant decrease in its H3K18 deacetylase activity (Fig 3F–G). These data indicate that R388 methylation directly suppresses the histone deacetylase activity of SIRT7.
Figure EV2. R388 methylation of SIRT7 suppresses its H3K18 deacetylase activity.
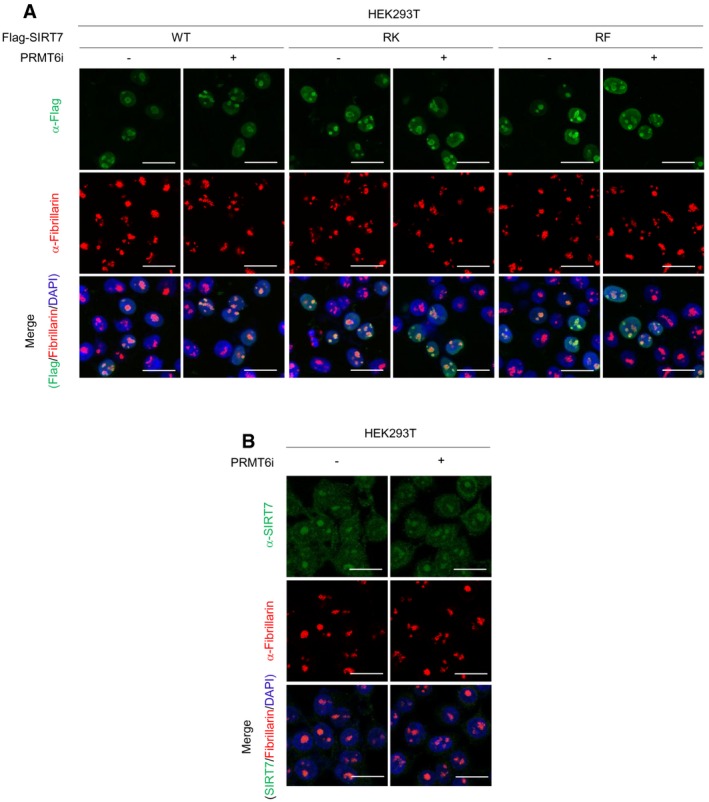
- R388K and R388F mutations do not alter subcellular distribution of SIRT7. HEK293T cells expressing Flag‐tagged wild‐type SIRT7 (WT) or its R388K(RK)/R388F(RF) mutants were treated with or without 5 μM PRMT6i for 24 h. Cells were stained with Flag antibody (green), fibrillarin antibody (red), and DAPI (blue). Scale bars = 25 μm.
- PRMT6i does not change the localization of endogenous SIRT7. HEK293T cells were treated with or without 5 μM PRMT6i for 24 h. Cells were stained with SIRT7 antibody (green), fibrillarin antibody (red), and DAPI (blue). Scale bars = 25 μm.
R388 methylation of SIRT7 promotes mitochondria biogenesis
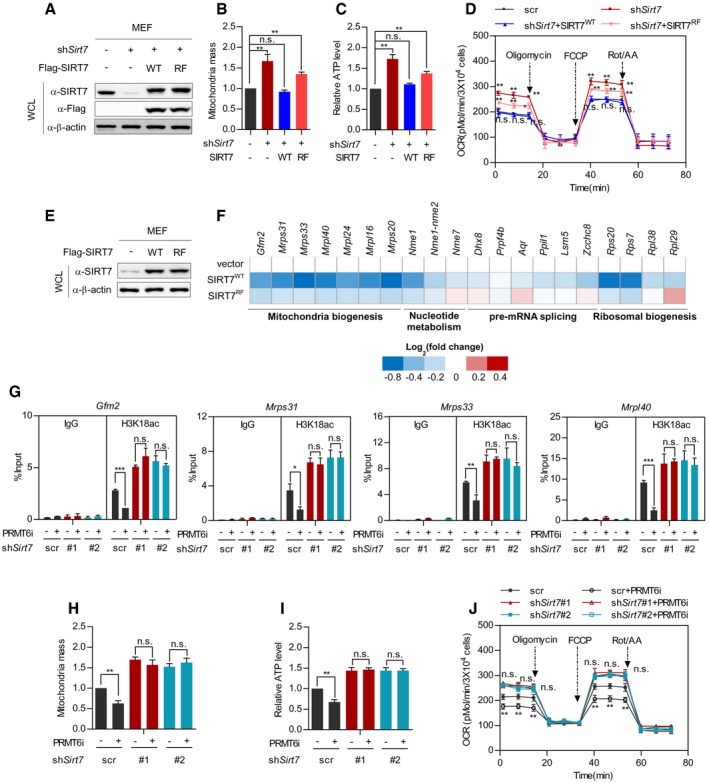
As an H3K18 deacetylase, SIRT7 induces transcriptional repression of its target genes and modulates mitochondria biogenesis 16. Interestingly, PRMT6 transcriptionally modulates hepatic glucose metabolism 35, suggesting PRMT6 as a regulator of cellular metabolism. To this end, we speculated whether PRMT6 interacts with SIRT7 to modulate mitochondria function. We established Sirt7‐knockdown/putback MEF cells (Fig 4A) and quantified mitochondria mass by using MitoTracker Red, a fluorescent dye of mitochondria. While Sirt7 depletion increased mitochondria mass, re‐introduction of wild‐type SIRT7 restored mitochondria quantity (Fig 4B). Meanwhile, re‐expressing RF mutant of SIRT7 had a mild effect on mitochondria mass (Fig 4B). This result indicates that R388 methylation potentially promotes mitochondria biogenesis. As mitochondria functions as the powerhouse for energy production, we further quantified cellular adenosine triphosphate (ATP), an indicator of energy metabolism. In agreement, silencing Sirt7 increased ATP level (Fig 4C). Reconstitution of wild‐type SIRT7, but not its RF mutant, restored cellular ATP production (Fig 4C). Furthermore, we employed metabolic flux assay to evaluate mitochondria function. Depleting Sirt7 enhanced oxygen consumption rate (OCR) of MEF cells (Fig 4D). Re‐introduction of wild‐type SIRT7 rescued the decreased mitochondria respiration in Sirt7‐deficient cells, while re‐expressing RF mutant partially restored oxygen consumption in Sirt7‐depleted cells (Fig 4D). Interestingly, extracellular acidification rate (ECAR) showed a neglectable change in Sirt7‐knockdown/rescue cells (Fig EV3A). Together, these data suggest that SIRT7 methylation promotes mitochondria function.
Figure 4. R388 methylation of SIRT7 promotes mitochondria biogenesis.
-
A–DWild‐type SIRT7, but not its RF mutant, restores mitochondria mass, ATP level, and cellular respiration in Sirt7‐deficient MEF cells. Wild‐type SIRT7 or its RF mutant was re‐expressed in Sirt7‐knockdown MEF cells (A). Mitochondria mass was determined by MitoTracker Red staining (B). Cellular ATP was quantified and normalized to cell number (C). Respiration flux was determined as indicated (D). (mean ± SD, n = 3 experimental replicates, *P < 0.05; **P < 0.01; n.s. = not significant, unpaired two‐tailed t‐test).
-
E, FWild‐type SIRT7, but not its RF mutant, downregulates SIRT7‐target genes involved in mitochondria biogenesis. Wild‐type SIRT7 and its RF mutant were stably expressed in MEF cells (E). SIRT7‐target gene expression was determined and presented as a heatmap on a log2 scale (F).
-
G–JInhibition of Prmt6 decreases mitochondria mass, ATP level, and the respiration flux of control cells, but not Sirt7‐knockdown MEF cells. Control cells and stable Sirt7‐knockdown MEF cells were treated with or without 5 μM PRMT6i for 24 h. The H3K18ac level at the promoter of SIRT7‐target genes (Gfm2, Mrps31, Mrps33, and Mrpl40) was determined by ChIP‐qPCR, and Rabbit IgG was used as a negative control (G). Mitochondria mass was determined by MitoTracker Red staining (H). Cellular ATP was quantified and normalized to cell number (I). Respiration flux was determined as indicated (J). (Data shown are mean ± SD, n = 3 experimental replicates, *P < 0.05; **P < 0.01; ***P < 0.001; n.s. = not significant, unpaired two‐tailed t‐test)
Figure EV3. R388 methylation of SIRT7 promotes mitochondria biogenesis.
- Extracellular acidification rate of SIRT7‐knockdown/putback cells. Wild‐type SIRT7 or its RF mutant was re‐expressed in Sirt7‐knockdown MEF cells. Cells were treated with glucose, oligomycin, and 2DG as indicated. The extracellular acidification rate was determined using seahorse metabolic flux analyzer (data shown are mean ± SD, n = 3 experimental replicates).
- Characterization of Sirt7‐knockdown efficiency. The knockdown efficiencies of two independent shRNAs (#1 and #2) targeting Sirt7 in MEF cells were determined by Western blot.
SIRT7 epigenetically regulates the expression of genes involved in mitochondria biogenesis, nucleotide metabolism, pre‐mRNA splicing, and ribosomal biogenesis 16, 19. To test whether SIRT7 methylation modulates target gene expression, we stably expressed wild‐type SIRT7 and its RF mutant in MEF cells (Fig 4E). While wild‐type SIRT7 selectively suppressed the mRNA expression of target genes involved in mitochondria biogenesis, RF mutant had a marginal effect on the expression of these genes (Fig 4F). This observation indicates that PRMT6 potentially modulates mitochondria biogenesis through regulating SIRT7 methylation. To test this, we performed chromatin immunoprecipitation‐coupled quantitative PCR (ChIP‐qPCR) assay to determine the effect of PRMT6 on H3K18 acetylation at the promoter of SIRT7‐target genes. PRMT6i treatment led to a dramatic decrease in H3K18 acetylation at the promoter of Gfm2, Mrps31, Mrps33, and Mrpl40 in control cells, but not Sirt7‐knockdown cells (Figs 4G and EV3B). We next determined whether PRMT6 modulated mitochondria function. Chemical inhibition of Prmt6 decreased mitochondria mass in control MEF cells, but not Sirt7‐depleted cells (Fig 4H). Prmt6 suppression also reduced cellular ATP level in a Sirt7‐dependent manner (Fig 4I). Importantly, PRMT6 inhibition decreased mitochondria respiration in control MEF cells, but not in Sirt7‐depleted cells (Fig 4J). Collectively, these results strongly suggest that PRMT6‐mediated R388 methylation promotes mitochondria biogenesis and respiration.
PRMT6 modulates SIRT7 methylation in an AMPK‐dependent manner
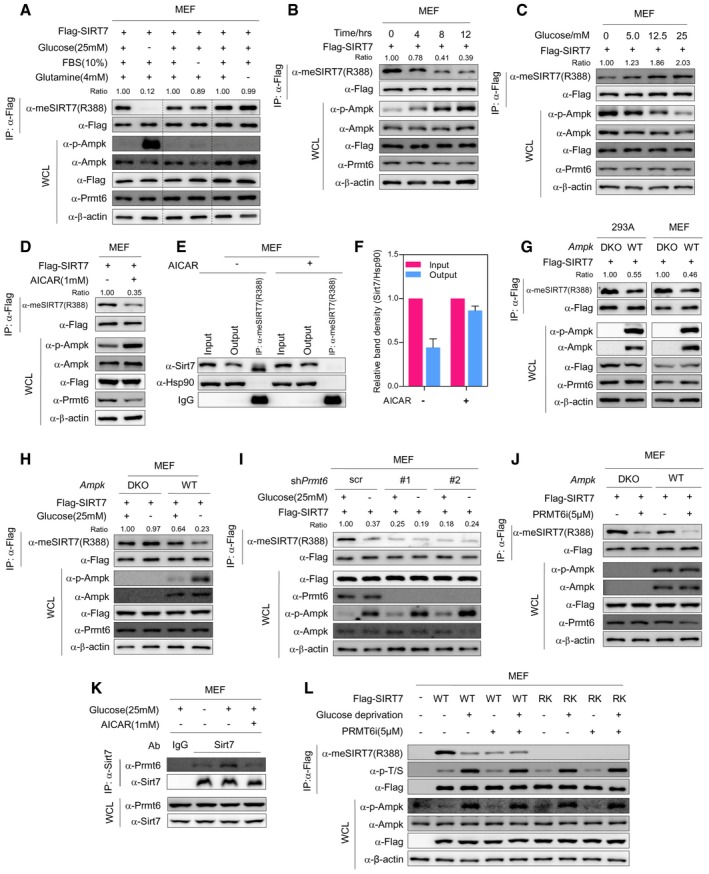
Sirtuins have been proposed as nutrient sensors 10, 36. As SIRT7 methylation modulates mitochondria function, we hypothesize that PRMT6‐induced SIRT7 methylation is involved in nutrient sensing and couples nutrient availability with mitochondria biogenesis. To test this hypothesis, we depleted different nutrients from culture media. Strikingly, removal of glucose, but not serum or glutamine, diminished R388 methylation of SIRT7 (Fig 5A). This effect was accompanied by the activation of AMP‐activated protein kinase (AMPK), as AMPK phosphorylation was dramatically increased (Fig 5A). Importantly, glucose starvation decreased R388 methylation in a time‐dependent manner (Fig 5B). When cells were treated with increasing concentrations of glucose, R388 methylation was dose‐dependently increased (Fig 5C). Of note, R388 methylation level was reversely correlated with AMPK phosphorylation (Fig 5A–C). These results raise the possibility that glucose availability modulates SIRT7 methylation through AMPK.
Figure 5. PRMT6 modulates SIRT7 methylation in an AMPK‐dependent manner.
-
AGlucose depletion reduces SIRT7 methylation. MEF cells stably expressing Flag‐SIRT7 were cultured in medium depleted with glucose (12 h), FBS (12 h), or glutamine (24 h). R388 methylation of immunopurified SIRT7 and AMPK phosphorylation were determined by Western blot. R388 methylation level was normalized to Flag‐SIRT7 (Ratio).
-
BGlucose starvation decreases R388 methylation of SIRT7 in a time‐dependent manner. MEF cells stably expressing Flag‐SIRT7 were cultured in glucose‐free media as indicated. R388 methylation level of immunopurified SIRT7 was determined by Western blot.
-
CGlucose dose‐dependently upregulates R388 methylation of SIRT7. MEF cells stably expressing Flag‐SIRT7 were cultured with increasing concentrations of glucose as indicated for 12 h. R388 methylation of SIRT7 and AMPK phosphorylation were determined by Western blot.
-
DAICAR treatment decreases R388 methylation of SIRT7. MEF cells stably expressing Flag‐SIRT7 were treated with or without AICAR for 12 h. R388 methylation of immunoprecipitated SIRT7 was determined by Western blot.
-
E, FAICAR reduces the percentage of R388‐methylated endogenous Sirt7 in MEF cells. R388‐methylated endogenous Sirt7 was captured from control and AICAR‐treated MEF cells by using R388 site‐specific methylation antibody (E). Input, output, and immunoprecipitate were analyzed by Western blot to quantify the percentage of R388‐methylated protein (F) (mean ± SD, n = 3 experimental replicates).
-
GAMPK deletion increases SIRT7 R388 methylation. Flag‐tagged SIRT7 was stably expressed in AMPK wild‐type (WT) and AMPK α1/α2 double‐knockout (DKO) MEF cells and 293A cells. SIRT7 protein was immunopurified with Flag‐beads, and R388 methylation was determined by Western blot.
-
HAMPK is required for glucose starvation‐induced R388 hypomethylation of SIRT7. AMPK wild‐type (WT) and AMPK α1/α2 double‐knockout (DKO) MEF cells stably expressing Flag‐SIRT7 were cultured with or without glucose for 12 h. SIRT7 was purified with Flag‐beads, and R388 methylation was determined by Western blot.
-
IPRMT6 is necessary for AMPK‐induced SIRT7 hypomethylation. Flag‐tagged SIRT7 was stably expressed in scramble control MEF cells or cells expressing two independent shRNAs targeting Prmt6 (#1 and #2). Cells were cultured with or without glucose for 12 h. R388 methylation of immunopurified SIRT7 was determined by Western blot.
-
JPRMT6 is necessary for AMPK‐induced SIRT7 hypomethylation. Wild‐type MEFs and AMPK DKO cells were treated with PRMT6i for 24 h. SIRT7 was purified by Flag‐beads, and R388 methylation of SIRT7 was determined by Western blot.
-
KBoth AICAR treatment and glucose depletion impair the association between Prmt6 and Sirt7. MEF cells were cultured with or without glucose and treated with AICAR as indicated. Co‐immunoprecipitation was performed using SIRT7 antibody. Input and immunoprecipitates were analyzed by Western blot.
-
LR388 methylation of SIRT7 does not modulate its phosphorylation. MEF cells stably expressing Flag‐tagged wild‐type SIRT7 or RK mutant were treated with PRMT6i (24 h) and cultured with or without glucose (12 h) as indicated. SIRT7 protein was immunopurified with Flag‐beads, and R388 methylation and phosphorylation of SIRT7 were determined by Western blot.
The AMPK functions as a nutrient and energy sensor to maintain mitochondria homeostasis 37, 38. We next set out to test whether SIRT7 methylation is under the control of AMPK. MEF cells stably expressing Flag‐tagged SIRT7 were treated with AICAR, a chemical activator of AMPK. AICAR markedly decreased R388 methylation of SIRT7 (Fig 5D). A subsequent quantification assay indicated that the percentage of R388‐methylated endogenous Sirt7 decreased from 55.7% to 13.7% by AICAR treatment in MEF cells (Fig 5E–F). Besides, double knockout of the catalytic subunit α1/α2 of AMPK (DKO) resulted in an elevation of R388 methylation in MEF and 293A cells (Fig 5G). Glucose starvation‐induced R388 hypomethylation was abrogated in AMPK‐deficient MEFs (Fig 5H). These data clearly show that glucose starvation decreases R388 methylation of SIRT7 in an AMPK‐dependent manner.
To elucidate the role of PRMT6 in AMPK‐induced SIRT7 hypomethylation, control MEF cells and Prmt6‐knockdown cells were cultured with or without glucose. Interestingly, glucose depletion failed to modulate R388 methylation of SIRT7 in the absence of Prmt6 (Fig 5I). Consistently, genetic ablation of AMPK had no effect on R388 methylation when cells were treated with PRMT6i (Fig 5J). Endogenous co‐immunoprecipitation further indicated that glucose depletion or AICAR treatment weakened the association between Sirt7 and Prmt6 (Fig 5K). Interestingly, recent studies suggest that SIRT7 is phosphorylated by AMPK 14. We ask whether a crosstalk between SIRT7 methylation and phosphorylation happens within cells. We expressed wild‐type SIRT7 and its RK mutant in MEF cells, and treated the cells with PRMT6i in the presence or absence of glucose. When cells underwent glucose starvation, RK mutant showed a phosphorylation pattern similar to wild‐type SIRT7 (Fig 5L). Interestingly, PRMT6 inhibitor reduced R388 methylation, but not serine/threonine phosphorylation of SIRT7 (Fig 5L). These observations suggest that there is no crosstalk between SIRT7 methylation and phosphorylation. Taken together, PRMT6 is necessary for AMPK to signal glucose availability to SIRT7.
SIRT7 methylation connects glucose sensing with mitochondria biogenesis
We next interrogated the connection between glucose sensing and SIRT7 methylation in vivo. For fasted animals, intraperitoneal administration of glucose increased Sirt7 R388 methylation in hepatocytes in a time‐dependent manner, with a concomitant decrease in AMPK phosphorylation (Fig 6A). As the R388 methylation antibody showed high specificity in whole cell extract immunoblotting and immunohistochemistry (IHC) (Fig EV4A–C), we performed IHC staining using this antibody. R388 methylation of Sirt7 was dramatically increased after glucose administration (Fig 6B), indicating that R388 methylation is a physiologically relevant modification in vivo. To determine whether SIRT7 methylation coupled glucose sensing with mitochondria biogenesis, MEF cells stably expressing Flag‐tagged SIRT7 were treated with AICAR and PRMT6 inhibitor either individually or in combination. Both PRMT6 inhibitor and AICAR resulted in R388 hypomethylation, decreased H3K18 acetylation at the promoter of SIRT7‐target genes, and reduced target gene expression (Fig 6C–E). More importantly, combined treatment with PRMT6i and AICAR did not lead to further reduction in R388 methylation of SIRT7, H3K18 acetylation at target gene promoters, or target gene expression (Fig 6C–E). These results strongly suggest that AMPK and PRMT6 function in the same pathway to modulate SIRT7 methylation and to epigenetically regulate mRNA expression of SIRT7‐target genes. In accordance, quantification of mitochondria demonstrated that treatment with either AICAR or PRMT6i downregulated mitochondria mass, while combined treatment did not lead to further reduction in mitochondria mass (Fig 6F). Taken together, PRMT6 signals glucose availability to SIRT7 methylation and regulates mitochondria biogenesis in an AMPK‐dependent manner.
Figure 6. SIRT7 methylation connects glucose sensing with mitochondria biogenesis.
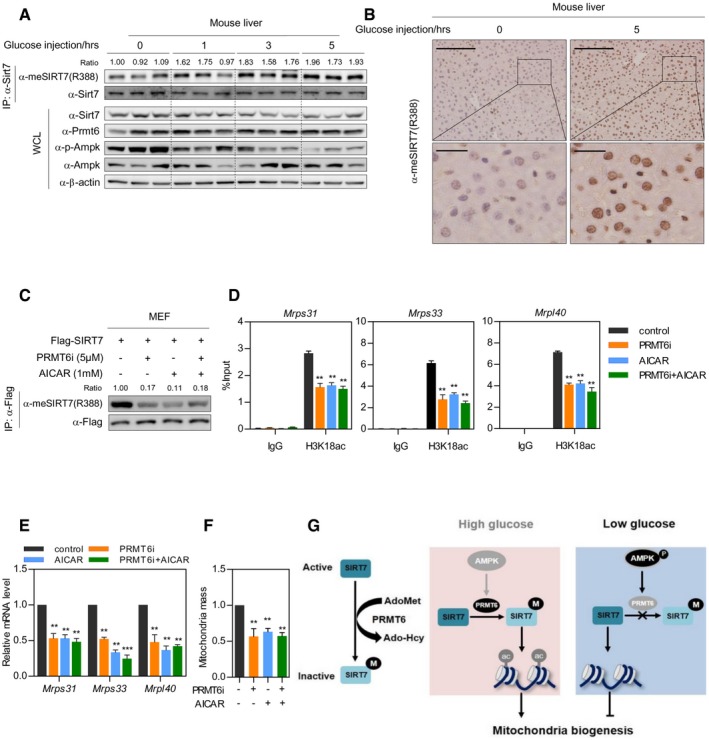
-
A, BGlucose administration leads to R388 hypermethylation of Sirt7 in mouse hepatocyte. After fasting for 16 h, mice were injected with glucose. Hepatic tissue was harvested in a time‐dependent manner as indicated (n = 3 for each time point). SIRT7 protein was immunoprecipitated from whole liver extracts. R388 methylation of Sirt7 was determined by Western blot (A) and immunohistochemical staining (B). Scale bar in the upper panel, 500 μm; scale bar in the lower panel, 100 μm.
-
C–FAICAR and PRMT6 inhibitor decrease R388 methylation and epigenetically downregulate SIRT7‐target genes. MEF cells stably expressing Flag‐SIRT7 were incubated with or without PRMT6i for 12 h, followed by treatment with 1 mM AICAR for 12 h. R388 methylation of SIRT7 was determined by Western blot (C). H3K18ac level at SIRT7‐target gene promoters was determined by ChIP‐qPCR. Normal rabbit IgG was used as a negative control (D). mRNA expression was determined by qPCR analysis (E). Mitochondria mass was determined by MitoTracker Red staining (F). (Data shown are mean ± SD, n = 3 experimental replicates, **P < 0.01; ***P < 0.001, unpaired two‐tailed t‐test)
-
GThe model illustrating regulation of SIRT7 by arginine methylation. R388 methylation inhibits the H3K18 deacetylase activity of SIRT7 to maintain mitochondria homeostasis in an AMPK‐dependent manner.
Figure EV4. SIRT7 methylation connects glucose sensing with mitochondria biogenesis.
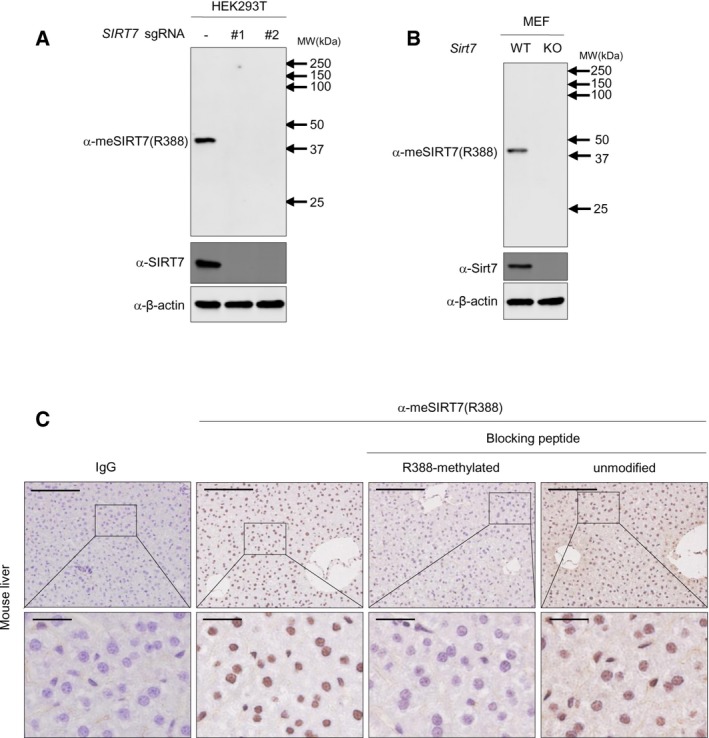
-
A, Bα‐meSIRT7(R388) antibody shows high specificity in blotting whole cell extract of HEK293T (A) and MEF (B). Control sgRNA or two independent sgRNAs (#1 and #2) against SIRT7 were introduced into HEK293T cells. Whole cell lysates were subjected to Western blot analysis.
-
Cα‐meSIRT7(R388) antibody shows high specificity in IHC staining. After incubating with unmethylated peptide or R388‐methylated peptide, the α‐meSIRT7(R388) antibody was used for IHC staining. R388 methylation was detected using the formalin‐fixed, paraffin‐embedded mice liver tissue sections. Shown were representative photographs from triplicated experiments. Scale bars = 500 μm in the upper panels and 100 μm in the lower panels.
Discussion
In this study, we delineated an AMPK‐PRMT6‐SIRT7 pathway in maintaining mitochondria homeostasis (Fig 6G). We find that PRMT6 signals glucose availability to SIRT7 methylation and epigenetically modulates mitochondria biogenesis. PRMT6‐mediated SIRT7 methylation serves as an important regulatory mechanism in coordinating glucose availability and mitochondria function.
To determine R388 methylation status of SIRT7, we successfully generated a R388 methylation‐specific antibody. Due to its high specificity in detecting SIRT7 in whole cell lysate and IHC, meSIRT7(R388) antibody served as a useful tool to monitor and quantify SIRT7 methylation. Of note, anti‐meSIRT7(R388) antibody reacted with both mono‐methylated and di‐methylated R388 peptides in dot blot assay (Fig 1E). This result suggests that meSIRT7(R388) antibody recognizes both mono‐methylated and di‐methylated SIRT7 proteins. To more precisely detect SIRT7 methylation, mass spectrometry is no doubt the most powerful tool. Proteomic profiling of SIRT7 methylation would not only determine what fraction of SIRT7 is methylated, but also address the physiological significance of SIRT7 methylation across different cell lines and tissue types.
Based on SIRT7 structure 34, 39, R388 does not locate in the catalytic center of SIRT7. To elucidate how R388 methylation modulates SIRT7 activity, we subjected methylated recombinant SIRT7 to in vitro deacetylation assay. Methylated SIRT7 showed a striking decrease in its H3K18 deacetylase activity. Although R388 locates closely to a nucleolar localization signal sequence, SIRT7 distribution remained unchanged when cells were treated with PRMT6 inhibitor. These data strongly support that R388 methylation directly modulates the intrinsic activity of SIRT7. Further insights into SIRT7‐histone interaction and catalytic kinetics would help to explain how R388 methylation delays the deacetylation reaction.
Sirtuins have been proposed as nutrient sensors and master regulators of cell metabolism, with the regulatory mechanism of sirtuins poorly understood. Previous studies have demonstrated that SIRT7, as a histone H3K18 deacetylase, epigenetically modulates multiple anabolic processes, including mitochondria biogenesis. However, the regulatory mechanism of SIRT7 in cellular metabolism remains to be defined. Here, we demonstrate that arginine methylation of SIRT7 suppresses its deacetylase activity to maintain mitochondria biogenesis. Of note, SIRT7 also participates in ribosomal biogenesis and DNA damage repair 15, 20, 40, at least in part through its desuccinylase activity 41. Thus, R388 methylation possibly modulates the deacylase activity of SIRT7 and plays a broader regulatory role in cellular metabolism and stress response. Interestingly, AMPK has been shown to directly phosphorylate SIRT7 at threonine 153 (T153) and modulate rDNA transcription to maintain energy homeostasis 14. Here, we demonstrate that R388 methylation regulates mitochondria biogenesis, but not rDNA transcription, in response to glucose availability. These results suggest that different post‐translational modification of SIRT7 modulates diverse biosynthetic processes. SIRT7 potentially functions as a hub in the decision‐making process during nutrient sensing and signaling.
Although PRMT6 had been implicated in glucose metabolism 35, its function in cellular metabolism is yet to be defined. We identified PRMT6 as the methylase of SIRT7. PRMT6 methylates SIRT7 to control its deacetylase activity and to modulate H3K18 acetylation level at SIRT7‐target gene promoters. Through SIRT7 methylation, PRMT6 regulates mitochondria function by modulating H3K18 acetylation level of genes involved in mitochondria biogenesis. Besides, the interaction of PRMT6 and SIRT7 is controlled by glucose availability or AMPK. By modulating SIRT7 methylation, PRMT6 signals glucose availability to SIRT7 and epigenetically regulates mitochondria biogenesis and respiration. PRMT6 functions as a critical regulator of mitochondria biology. Together, our study uncovers an AMPK‐PRMT6‐SIRT7 pathway in coupling glucose sensing with mitochondria function.
Materials and Methods
Reagents
Antibodies against SIRT7 (Abcam, #ab62748), fibrillarin (Abcam, #ab5421), PRMT6 (Proteintech, #15395‐1‐AP), GFP (Proteintech, #66002‐1‐Ig), Flag (Aogma, #9622), Flag (Sigma, #F3165), mono‐methyl arginine (Cell Signaling, #8015), asymmetric di‐methyl arginine (Cell Signaling, #13522), symmetric di‐methyl arginine (Cell Signaling, #13222), Hsp90 (Cell Signaling, #4877), Phospho‐(Ser/Thr) (Cell Signaling, #9631S), and β‐actin (Aogma, #9601) were purchased commercially. GFP‐beads (ChromoTek, #gta‐20) and Flag‐beads (Sigma, #A2220) were commercially obtained. To generate a site‐specific antibody to detect the R388‐methylated SIRT7 [α‐meSIRT7(R388)], synthesized peptide GGWFGR(me)GCTKRTK (GL Biochem) was coupled to KLH as an antigen to immunize the rabbit. Anti‐serum was collected after five doses of immunization. Adenosine dialdehyde (AdOx) (Sigma, #A7154), PRMT6 inhibitor (MedChemExpress, #EPZ020411), and glucose (Sigma, #G7021) were commercially obtained.
Cell culture
HEK293T and L02 cells were purchased from the Cell Bank of Type Culture Collection of Chinese Academy of Sciences, Shanghai. HEK293T, AMPK wild‐type and AMPK α1/α2 double‐knockout (DKO) MEF cells, and AMPK wild‐type and AMPK α1/α2 double‐knockout (DKO) 293A cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen, #128000017); L02 cells were maintained in RPMI 1640 Medium (Invitrogen, #31800022). The medium was supplemented with 10% fetal bovine serum (Invitrogen, #10099‐141) in the presence of 1% penicillin and streptomycin. DMEM without glucose was purchased from Invitrogen (#11966025). For glucose starvation, cells were cultured in the medium containing different concentrations of glucose as indicated.
Plasmids
The cDNA encoding full‐length human SIRT7 was cloned into Flag, GST‐tagged vectors (pcDNA3.1, pGEX‐4T1). Plasmids encoding PRMTs were kindly provided by Dr. Yanzhong Yang (City of Hope Cancer Center, USA). Point mutations of SIRT7 and PRMT6 were generated by using Platinum Pfx DNA Polymerase (Invitrogen, #11708‐013). Flag‐tagged wild‐type SIRT7 or its mutants (RK and RF) were subcloned into the pQCXIH vector. All expression constructs were verified by DNA sequencing.
Generation of stable cell pools
To generate stable PRMT6‐knockout HEK293T cell pools, two independent sgRNAs were designed as described previously 42, 43. sgRNAs targeting PRMT6 were inserted in pX459, and sgRNA hairpin sequences are listed in Table EV1. HEK293T cells were selected by 2 μg/mL puromycin for 3 days after transfected with the vectors for 36 h. To generate stable Prmt6‐ or Sirt7‐knockdown MEF cell, shRNAs targeting Prmt6 or Sirt7 were inserted into pMKO.1‐puro vector, and targeting sequences are listed in Table EV1. Retrovirus was produced by using a two‐plasmid packaging system as previously described 44. MEF cells were infected with the retrovirus and selected with 2 μg/ml puromycin for 1 week. To generate SIRT7‐knockdown/rescue stable cell pools, Flag‐tagged wild‐type human SIRT7 or its RF mutant was cloned into the retroviral pQCXIH vector. Retrovirus was produced by using a two‐plasmid packaging system. Stable Sirt7‐knockdown MEF cells were transduced with the virus, followed by a selection in 200 μg/ml hygromycin B for 1 week. AMPK DKO MEF cells were from Dr. Wei Liu (Zhejiang University, China). AMPK DKO 293A cells were from Dr. Kunliang Guan (University of California, San Diego, USA).
Immunoprecipitation
Cells were lysed in 0.5% NP‐40 lysis buffer (50 mM Tris–HCl, pH 8.0, 150 mM NaCl, 0.5% NP‐40, 1 mM DTT) with protease inhibitors cocktail (Selleck). After centrifugation at 16,200 g for 15 min, the supernatants were collected and incubated with Flag‐beads at 4°C for 4 h or incubated with primary antibody at 4°C for 8 h followed by incubating with Protein‐A beads (Millipore) for another 4 h at 4°C. After incubation, samples were washed with lysis buffer for four times. Proteins were re‐suspended in SDS loading buffer and subjected to SDS–PAGE.
Recombinant protein expression and purification
Wild‐type SIRT7 and its mutants (RK and RF) were cloned into the pGEX‐4T1 vector. Wild‐type PRMT6 and its catalytic‐deficient mutant (V86K/D88A, DM) were cloned into pMAL vector. The plasmids were transformed into BL21 cells. Cells were cultured at 37°C in LB medium. Isopropyl‐β‐D‐1‐thiogalactopyranoside (IPTG) was added to a final concentration of 0.2 mM when OD600 was between 0.5 and 0.7. The culture was grown for 14 h at 16°C.
For purification of recombinant SIRT7, cells were harvested and then re‐suspended in lysis buffer (91 mM Na2HPO4, 17 mM NaH2PO4, 1.5 M NaCl, pH 7.40, 20 μg/ml DNase, and 2 mM DTT) with proteinase inhibitors. Cells were lysed by cell disrupter for 10 min at 4°C and then centrifuged at 18,500 g for 20 min at 4°C. The supernatant was loaded onto a glutathione‐agarose column (GST‐Sefinose, Sangon Biotech) pre‐equilibrated with lysis buffer. SIRT7 proteins were eluted with elution buffer (50 mM Tris, pH 8.0, 1 mM DTT, 20 mM GSH). For purification of recombinant PRMT6, cells were harvested and then re‐suspended in binding buffer (5 mM imidazole, 300 mM NaCl, 20 mM Na2HPO4, pH 7.4, 20 μg/ml DNase) with protease inhibitors. Cells were lysed by cell disrupter for 10 min at 4°C and then centrifuged at 18,500 g for 20 min at 4°C. His‐nickel affinity gel (Sigma) precleaned with binding buffer was added into the supernatant. Then, PRMT6 proteins were eluted with elution buffer (150 mM imidazole, 300 mM NaCl, 20 mM Na2HPO4, pH 7.4). Fractions were assayed for purity on SDS–PAGE and concentrated for activity assay and in vitro methylation assay.
GST pulldown
GST‐SIRT7 or GST‐PRMT6 fusion protein was added to HEK293T cell lysate and mixed at 4°C overnight. GST protein was used as a negative control. After incubating the mixture with GST‐beads (Aogma, #90049) for another 2 h at 4°C, beads were harvested by centrifugation and washed four times with 0.3% NP‐40 buffer. Samples were boiled in SDS loading buffer and subjected to Western blotting analysis.
In vitro methylation assay
For in vitro methylation assay, MBP‐His‐tagged PRMT6 was mixed with recombinant substrate (GST‐SIRT7) in methylation reaction buffer (50 mM Tris–HCl, pH 8.0, 20 mM KCl, 5 mM DTT, 4 mM EDTA) in the presence of 200 μM S‐adenosyl‐L‐methionine (Sigma) and incubated at 37°C for 1 h in a final volume of 30 μl. Reactions were stopped by adding SDS–PAGE loading buffer, followed by boiling for 10 min, and subjected to Western blotting analysis.
Chromatin immunoprecipitation and quantitative PCR
ChIP‐qPCR assay was performed as previously described 45. Briefly, 1 × 107 MEF cells were cross‐linked with 1% paraformaldehyde, lysed, and sonicated using the Bioruptor at high‐output power setting for 40 cycles (30 s on and 30 s off). Soluble chromatin was immunoprecipitated with antibodies against H3K18ac (1:200) or rabbit IgG (negative control) at 4°C overnight, followed by incubation with Protein‐A beads for 4 h at 4°C. Beads were washed, and the cross‐linking was reversed. Samples were digested with proteinase K for 1 h at 55°C. The immunoprecipitated DNA was extracted according to the manufacturer's instructions (Qiagen). The DNA fragments were detected by qPCR. All tested primers are listed in Table EV1.
Total RNA was extracted from cultured cells using TRIzol Reagent, and cDNA was synthesized following the manufacturer's instructions (TaKaRa). Real‐time PCR was performed using SYBR Premix ExTaq (TaKaRa, #RR420A). Relative gene expression was calculated by the comparative CT method. β‐actin was included as an endogenous control. The sequence of primers is listed in Table EV1.
Chromatin isolation and in vitro deacetylation
Chromatin was extracted from HEK293T cells as previously described 46. Briefly, cells were re‐suspended at 4 × 107 cells/ml in buffer A (10 mM HEPES, pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 1 mM DTT, 0.1% Triton X‐100) with protease inhibitors. Cells were incubated for 5 min on ice. Nuclei were collected in pellet 1 (P1) by low‐speed centrifugation (1,300 g for 4 min, 4°C). P1 was washed once with buffer A and lysed in buffer B (3 mM EDTA, 0.2 mM EGTA, 1 mM DTT) with protease inhibitors. Insoluble chromatin (P2) was collected by centrifugation (1,700 g for 4 min, 4°C), washed once in buffer B, and then centrifuged again. The final chromatin pellet (P3) was re‐suspended in deacetylation buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 2 mM NAD+, 1 mM DTT), sonicated, and stored at −80°C for further analysis.
In vitro deacetylation assay was performed as previously described 47. Briefly, 1 μg SIRT7 protein and 1 μg extracted chromatin were mixed in deacetylation buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 2 mM NAD+, 1 mM DTT) and incubated at 37°C for 2 h. H3K18 acetylation was determined by Western blot.
Immunoprecipitation‐based quantification of R388‐methylated SIRT7
Immunoprecipitation‐based quantification of arginine‐methylated protein was performed as previously described 48. Briefly, ten million cells were lysed in 1 ml NP‐40 lysis buffer (50 mM Tris–HCl, pH 8.0, 150 mM NaCl, 0.5% NP‐40, 1 mM DTT) with protease inhibitors cocktail (Selleck). The supernatants were collected after centrifugation at 16,200 g for 15 min. 80 μl supernatant was saved as input. The remaining supernatant was incubated with 9 μg α‐meSIRT7(R388) antibody at 4°C for 8 h followed by incubating with Protein‐A beads (Millipore) for another 4 h at 4°C. Samples were centrifugated at 400 g for 5 min. 80 μl supernatant was saved as output. The immunoprecipitated samples were washed with lysis buffer for four times. Samples were re‐suspended in SDS loading buffer and subjected to SDS–PAGE. The fraction of methylated SIRT7 was determined by Western blotting and calculated based on the SIRT7 protein level in input and output samples. Hsp90 was blotted as a loading control.
Immunofluorescence
Cultured cells were washed two times with PBS, fixed in 4% paraformaldehyde for 30 min, permeabilized with 0.2% Triton X‐100 for 30 min, and incubated with 1.0% bovine serum albumin (BSA) for 1 hr. Cells were then incubated with the primary antibodies (mouse anti‐Flag, 1:1,000; rabbit anti‐fibrillarin, 1:1,000; and rabbit anti‐SIRT7, 1:100) overnight at 4°C. Subsequently, cells were washed with PBS and incubated with secondary antibodies for 60 min at room temperature. Nucleus was stained with 4,6‐diamidino‐2‐phenylindole (DAPI). Confocal fluorescence images were captured using a Leica SP5 laser microscope.
Determination of mitochondria mass and ATP level
For quantification of mitochondria mass, MEF cells were stained with 50 nM MitoTracker Red (Invitrogen) for 30 min at 37°C and analyzed by flow cytometry (BD Accuri C6).
The colorimetric assay kit (Sigma, #MAK190) was used for measuring ATP level. Briefly, 1 × 106 cells were snap‐frozen in liquid nitrogen and lysed with 100 μl of ATP assay buffer (provided in the kit). After adding ATP probe in the presence of a developer, samples were incubated at 37°C for 30 min, and ATP level was determined by measuring the absorbance at 570 nm.
Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR)
Oxygen consumption rate and ECAR were determined using the XF96 Extracellular Flux Analyzer (Seahorse Bioscience). Briefly, MEF cells were seeded in 96‐well plates at a density of 2 × 104 cells/well and cultured overnight. Cells were washed with seahorse buffer (DMEM with phenol red containing 25 mM glucose and 2 mM glutamine). Cell Mito Stress Test Kit was used to evaluate mitochondria function. 2 μM oligomycin, 1 μM FCCP, and 0.5 μM rotenone/antimycin A (Rot/AA) were used to determine oxygen consumption. The Glycolysis Stress Test Kit was used to measure glycolytic capacity. 10 mM glucose, 2 μM oligomycin, and 50 mM 2‐deoxyglucose (2DG) were used to determine the ECAR. OCR and ECAR values were normalized to cell number.
Immunohistochemistry (IHC) of mice liver tissue
Staining for R388 methylation was performed with formalin‐fixed paraffin‐embedded mice liver tissue. Following de‐paraffinization, antigen retrieval, and a 30‐min incubation with goat serum, tissue sections were incubated with meSIRT7(R388) antibody (1:100) overnight at 4°C. Secondary biotin‐labeled IgG was then incubated with sections for 30 min at 37°C. Finally, diaminobenzidine (DAB) kit was used for staining.
Animal study
The procedures related to animal experiments were approved by Ethics Committee of Department of Laboratory Animals, Fudan University. Six‐week‐old female Balb/c mice (Sippr‐BK Laboratory, Shanghai) were maintained in cages under controlled temperature and light with ad libitum access to food and water. Animals were fasted for 16 h and then intraperitoneally injected with PBS or 2 mg/g glucose (dissolved in PBS). Three littermates were used for each experiment. Hepatic tissue was harvested in a time‐dependent manner as indicated.
Statistical analysis
Two‐tailed unpaired Student's t‐test was used for all comparisons. All data were presented as mean ± SD for triplicate experiments. P‐values < 0.05 were considered statistically significant (*P < 0.05, **P < 0.01, ***P < 0.001, n.s. = not significant).
Author contributions
W‐WY, Y‐LL, Q‐XZ, DW, and M‐ZL performed the experiments; W‐WY, JQ, X‐HH, Q‐YL, and Y‐PW designed the experiments and analyzed the data; W‐WY, Q‐YL, and Y‐PW co‐wrote the manuscript; and Y‐PW and Q‐YL conceived the idea and supervised the study.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Review Process File
Acknowledgements
We thank members of Cancer Metabolism Laboratory for discussion throughout this study. We thank Dr. Shaoyang Sun for his kind support on seahorse flux analysis. We are grateful to Dr. Wei Liu (Zhejiang University, China) and Dr. Kunliang Guan (UCSD, USA) for providing AMPK DKO cells. We thank Dr. Yanzhong Yang (City of Hope Cancer Center, USA) for sharing PRMTs plasmids. We also thank Biomedical Core Facility, Fudan University, for technical support. This work was supported by MOST (No. 2015CB910401 to Q.‐Y.L.), the Natural Science Foundation of China (Nos. 81790253 and 81430057 to Q.‐Y.L.; Nos. 81790251, 81772946, and 81502379 to Y.‐P.W.), and Innovation Program of Shanghai Municipal Education Commission (No. N173606 to Q.‐Y.L.).
EMBO Reports (2018) 19: e46377
References
- 1. Greiss S, Gartner A (2009) Sirtuin/Sir2 phylogeny, evolutionary considerations and structural conservation. Mol Cells 28: 407–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang YP, Zhou LS, Zhao YZ, Wang SW, Chen LL, Liu LX, Ling ZQ, Hu FJ, Sun YP, Zhang JY et al (2014) Regulation of G6PD acetylation by SIRT2 and KAT9 modulates NADPH homeostasis and cell survival during oxidative stress. EMBO J 33: 1304–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Blank MF, Grummt I (2017) The seven faces of SIRT7. Transcription 8: 67–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Canto C, Gerhart‐Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J (2009) AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458: 1056–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dominy JE Jr, Lee Y, Jedrychowski MP, Chim H, Jurczak MJ, Camporez JP, Ruan HB, Feldman J, Pierce K, Mostoslavsky R et al (2012) The deacetylase Sirt6 activates the acetyltransferase GCN5 and suppresses hepatic gluconeogenesis. Mol Cell 48: 900–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ramirez T, Li YM, Yin S, Xu MJ, Feng D, Zhou Z, Zang M, Mukhopadhyay P, Varga ZV, Pacher P et al (2017) Aging aggravates alcoholic liver injury and fibrosis in mice by downregulating sirtuin 1 expression. J Hepatol 66: 601–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kugel S, Sebastian C, Fitamant J, Ross KN, Saha SK, Jain E, Gladden A, Arora KS, Kato Y, Rivera MN et al (2016) SIRT6 suppresses pancreatic cancer through control of Lin28b. Cell 165: 1401–1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yoshizawa T, Karim MF, Sato Y, Senokuchi T, Miyata K, Fukuda T, Go C, Tasaki M, Uchimura K, Kadomatsu T et al (2014) SIRT7 controls hepatic lipid metabolism by regulating the ubiquitin‐proteasome pathway. Cell Metab 19: 712–721 [DOI] [PubMed] [Google Scholar]
- 9. Kuang J, Zhang Y, Liu Q, Shen J, Pu S, Cheng S, Chen L, Li H, Wu T, Li R et al (2017) Fat‐specific Sirt6 ablation sensitizes mice to high‐fat diet‐induced obesity and insulin resistance by inhibiting lipolysis. Diabetes 66: 1159–1171 [DOI] [PubMed] [Google Scholar]
- 10. Chalkiadaki A, Guarente L (2012) Sirtuins mediate mammalian metabolic responses to nutrient availability. Nat Rev Endocrinol 8: 287–296 [DOI] [PubMed] [Google Scholar]
- 11. Bonkowski MS, Sinclair DA (2016) Slowing ageing by design: the rise of NAD(+) and sirtuin‐activating compounds. Nat Rev Mol Cell Biol 17: 679–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Michishita E, Park JY, Burneskis JM, Barrett JC, Horikawa I (2005) Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol Biol Cell 16: 4623–4635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, Kim J, Woo J, Kim JH, Choi BH et al (2011) Sirt5 is a NAD‐dependent protein lysine demalonylase and desuccinylase. Science 334: 806–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sun L, Fan G, Shan P, Qiu X, Dong S, Liao L, Yu C, Wang T, Gu X, Li Q et al (2016) Regulation of energy homeostasis by the ubiquitin‐independent REGgamma proteasome. Nat Commun 7: 12497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shin J, He M, Liu Y, Paredes S, Villanova L, Brown K, Qiu X, Nabavi N, Mohrin M, Wojnoonski K et al (2013) SIRT7 represses Myc activity to suppress ER stress and prevent fatty liver disease. Cell Rep 5: 654–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mohrin M, Shin J, Liu Y, Brown K, Luo H, Xi Y, Haynes CM, Chen D (2015) Stem cell aging. A mitochondrial UPR‐mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science 347: 1374–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen S, Seiler J, Santiago‐Reichelt M, Felbel K, Grummt I, Voit R (2013) Repression of RNA polymerase I upon stress is caused by inhibition of RNA‐dependent deacetylation of PAF53 by SIRT7. Mol Cell 52: 303–313 [DOI] [PubMed] [Google Scholar]
- 18. Vazquez BN, Thackray JK, Simonet NG, Kane‐Goldsmith N, Martinez‐Redondo P, Nguyen T, Bunting S, Vaquero A, Tischfield JA, Serrano L (2016) SIRT7 promotes genome integrity and modulates non‐homologous end joining DNA repair. EMBO J 35: 1488–1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Barber MF, Michishita‐Kioi E, Xi Y, Tasselli L, Kioi M, Moqtaderi Z, Tennen RI, Paredes S, Young NL, Chen K et al (2012) SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature 487: 114–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang PY, Li G, Deng ZJ, Liu LY, Chen L, Tang JZ, Wang YQ, Cao ST, Fang YX, Wen F et al (2016) Dicer interacts with SIRT7 and regulates H3K18 deacetylation in response to DNA damaging agents. Nucleic Acids Res 44: 3629–3642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jiang L, Xiong J, Zhan J, Yuan F, Tang M, Zhang C, Cao Z, Chen Y, Lu X, Li Y et al (2017) Ubiquitin‐specific peptidase 7 (USP7)‐mediated deubiquitination of the histone deacetylase SIRT7 regulates gluconeogenesis. J Biol Chem 292: 13296–13311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hu H, Zhu W, Qin J, Chen M, Gong L, Li L, Liu X, Tao Y, Yin H, Zhou H et al (2017) Acetylation of PGK1 promotes liver cancer cell proliferation and tumorigenesis. Hepatology 65: 515–528 [DOI] [PubMed] [Google Scholar]
- 23. Hsu JM, Chen CT, Chou CK, Kuo HP, Li LY, Lin CY, Lee HJ, Wang YN, Liu M, Liao HW et al (2011) Crosstalk between Arg 1175 methylation and Tyr 1173 phosphorylation negatively modulates EGFR‐mediated ERK activation. Nat Cell Biol 13: 174–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jansson M, Durant ST, Cho EC, Sheahan S, Edelmann M, Kessler B, La Thangue NB (2008) Arginine methylation regulates the p53 response. Nat Cell Biol 10: 1431–1439 [DOI] [PubMed] [Google Scholar]
- 25. Wang Y‐P, Zhou W, Wang J, Huang X, Zuo Y, Wang T‐S, Gao X, Xu Y‐Y, Zou S‐W, Liu Y‐B et al (2016) Arginine methylation of MDH1 by CARM1 inhibits glutamine metabolism and suppresses pancreatic cancer. Mol Cell 64: 673–687 [DOI] [PubMed] [Google Scholar]
- 26. Bedford MT, Clarke SG (2009) Protein arginine methylation in mammals: who, what, and why. Mol Cell 33: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang Y, Bedford MT (2013) Protein arginine methyltransferases and cancer. Nat Rev Cancer 13: 37–50 [DOI] [PubMed] [Google Scholar]
- 28. Pahlich S, Zakaryan RP, Gehring H (2006) Protein arginine methylation: cellular functions and methods of analysis. Biochem Biophys Acta 1764: 1890–1903 [DOI] [PubMed] [Google Scholar]
- 29. Guo A, Gu H, Zhou J, Mulhern D, Wang Y, Lee KA, Yang V, Aguiar M, Kornhauser J, Jia X et al (2014) Immunoaffinity enrichment and mass spectrometry analysis of protein methylation. Mol Cell Proteomics 13: 372–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Geoghegan V, Guo A, Trudgian D, Thomas B, Acuto O (2015) Comprehensive identification of arginine methylation in primary T cells reveals regulatory roles in cell signalling. Nat Commun 6: 6758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McBride AE, Cook JT, Stemmler EA, Rutledge KL, McGrath KA, Rubens JA (2005) Arginine methylation of yeast mRNA‐binding protein Npl3 directly affects its function, nuclear export, and intranuclear protein interactions. J Biol Chem 280: 30888–30898 [DOI] [PubMed] [Google Scholar]
- 32. Campbell M, Chang PC, Huerta S, Izumiya C, Davis R, Tepper CG, Kim KY, Shevchenko B, Wang DH, Jung JU et al (2012) Protein arginine methyltransferase 1‐directed methylation of Kaposi sarcoma‐associated herpesvirus latency‐associated nuclear antigen. J Biol Chem 287: 5806–5818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Scaramuzzino C, Casci I, Parodi S, Lievens PM, Polanco MJ, Milioto C, Chivet M, Monaghan J, Mishra A, Badders N et al (2015) Protein arginine methyltransferase 6 enhances polyglutamine‐expanded androgen receptor function and toxicity in spinal and bulbar muscular atrophy. Neuron 85: 88–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tong Z, Wang M, Wang Y, Kim DD, Grenier JK, Cao J, Sadhukhan S, Hao Q, Lin H (2017) SIRT7 is an RNA‐activated protein lysine deacylase. ACS Chem Biol 12: 300–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Han HS, Jung CY, Yoon YS, Choi S, Choi D, Kang G, Park KG, Kim ST, Koo SH (2014) Arginine methylation of CRTC2 is critical in the transcriptional control of hepatic glucose metabolism. Sci Signal 7: ra19 [DOI] [PubMed] [Google Scholar]
- 36. Chalkiadaki A, Guarente L (2012) High‐fat diet triggers inflammation‐induced cleavage of SIRT1 in adipose tissue to promote metabolic dysfunction. Cell Metab 16: 180–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kishton RJ, Barnes CE, Nichols AG, Cohen S, Gerriets VA, Siska PJ, Macintyre AN, Goraksha‐Hicks P, de Cubas AA, Liu T et al (2016) AMPK is essential to balance glycolysis and mitochondrial metabolism to control T‐ALL cell stress and survival. Cell Metab 23: 649–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Herzig S, Shaw RJ (2018) AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol 19: 121–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Priyanka A, Solanki V, Parkesh R, Thakur KG (2016) Crystal structure of the N‐terminal domain of human SIRT7 reveals a three‐helical domain architecture. Proteins 84: 1558–1563 [DOI] [PubMed] [Google Scholar]
- 40. Song C, Hotz‐Wagenblatt A, Voit R, Grummt I (2017) SIRT7 and the DEAD‐box helicase DDX21 cooperate to resolve genomic R loops and safeguard genome stability. Genes Dev 31: 1370–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li L, Shi L, Yang S, Yan R, Zhang D, Yang J, He L, Li W, Yi X, Sun L et al (2016) SIRT7 is a histone desuccinylase that functionally links to chromatin compaction and genome stability. Nat Commun 7: 12235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chakiath CS, Esposito D (2007) Improved recombinational stability of lentiviral expression vectors using reduced‐genome Escherichia coli . Biotechniques 43: 466, 468, 470 [DOI] [PubMed] [Google Scholar]
- 43. Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG et al (2014) Genome‐scale CRISPR‐Cas9 knockout screening in human cells. Science 343: 84–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhao D, Zou SW, Liu Y, Zhou X, Mo Y, Wang P, Xu YH, Dong B, Xiong Y, Lei QY et al (2013) Lysine‐5 acetylation negatively regulates lactate dehydrogenase A and is decreased in pancreatic cancer. Cancer Cell 23: 464–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lan F, Collins RE, De Cegli R, Alpatov R, Horton JR, Shi X, Gozani O, Cheng X, Shi Y (2007) Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1‐mediated gene repression. Nature 448: 718–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mendez J, Stillman B (2000) Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol Cell Biol 20: 8602–8612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tong Z, Wang Y, Zhang X, Kim DD, Sadhukhan S, Hao Q, Lin H (2016) SIRT7 is activated by DNA and deacetylates histone H3 in the chromatin context. ACS Chem Biol 11: 742–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang L, Zhao Z, Meyer MB, Saha S, Yu M, Guo A, Wisinski KB, Huang W, Cai W, Pike JW et al (2014) CARM1 methylates chromatin remodeling factor BAF155 to enhance tumor progression and metastasis. Cancer Cell 25: 21–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Review Process File