

Abstract
Genetic disorders caused by cilia dysfunction, termed ciliopathies, frequently involve the intraflagellar transport (IFT) system. Mutations in IFT subunits—including IFT‐dynein motor DYNC2H1—impair ciliary structures and Hedgehog signalling, typically leading to “skeletal” ciliopathies such as Jeune asphyxiating thoracic dystrophy. Intriguingly, IFT gene mutations also cause eye, kidney and brain ciliopathies often linked to defects in the transition zone (TZ), a ciliary gate implicated in Hedgehog signalling. Here, we identify a C. elegans temperature‐sensitive (ts) IFT‐dynein mutant (che‐3; human DYNC2H1) and use it to show a role for retrograde IFT in anterograde transport and ciliary maintenance. Unexpectedly, correct TZ assembly and gating function for periciliary proteins also require IFT‐dynein. Using the reversibility of the novel ts‐IFT‐dynein, we show that restoring IFT in adults (post‐developmentally) reverses defects in ciliary structure, TZ protein localisation and ciliary gating. Notably, this ability to reverse TZ defects declines as animals age. Together, our findings reveal a previously unknown role for IFT in TZ assembly in metazoans, providing new insights into the pathomechanism and potential phenotypic overlap between IFT‐ and TZ‐associated ciliopathies.
Keywords: C. elegans, cilia, dynein, IFT, transition zone
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton; Membrane & Intracellular Transport
Introduction
Cilia are evolutionarily conserved eukaryotic organelles that perform motility and/or sensory functions in protists, plants and multicellular animals (metazoans) 1, 2, 3, 4. The microtubule‐based axonemes of motile and non‐motile (primary) cilia are templated from a basal body and harbour within their proximal‐most region a transition zone (TZ) characterised by Y‐shaped structures that connect the microtubules to the overlying ciliary membrane 5, 6, 7, 8, 9. The TZ consists of numerous proteins, most of which are membrane‐associated, that form a diffusion barrier or “ciliary gate” 10, 11, 12, 13, 14, 15, 16. The function of this gate is important for the correct localisation of signalling proteins within the organelle, including components of the Hedgehog signalling pathway in vertebrates 17, 18, 19, 20. Mobilisation of ciliary proteins across this barrier and within the compartment depends on an intraflagellar transport (IFT) trafficking machinery 21, 22, 23.
Discovered by the Rosenbaum Lab in 1993 24, IFT has a well‐established role in building functional cilia 21, 22, 25. Five IFT modules participate in moving ciliary cargo, such as tubulin building blocks and receptors, into or out of cilia 22, 26, 27, 28. Anterograde trafficking from the basal body to the ciliary tip is carried out by kinesin‐2 family motors, namely heterotrimeric kinesin‐II and homodimeric KIF17/OSM‐3 29, 30. Retrograde trafficking from the ciliary tip to the base depends on a multi‐protein IFT‐dynein assembly that includes cytoplasmic dynein‐2 heavy chain (DYNC2H1), as well as light, intermediate and light intermediate chains 31, 32, 33, 34, 35, 36. The kinesin and dynein molecular motors mobilise two cargo‐binding subcomplexes, termed IFT‐A (containing 7 or more proteins) and IFT‐B (at least 15 proteins), which play important roles in retrograde and anterograde trafficking, respectively. Disruption of IFT‐dynein or IFT‐A subunits in mammals, Caenorhabditis elegans and Chlamydomonas reinhardtii leads to short cilia with accumulations of IFT proteins at expanded (“bulbous”) ciliary tips 8, 32, 33, 37, 38, 39. In contrast, mutations in IFT‐B subunits typically result in near‐complete loss of the ciliary axoneme 8, 40, 41. Finally, an accessory complex containing at least 8 Bardet–Biedl syndrome (BBS) proteins, termed BBSome, associates with the IFT machinery and helps to transport specific ciliary cargo 42, 43, 44. Notably, several components of the Hedgehog signalling pathway are dependent on IFT‐associated proteins to regulate their dynamic ciliary localisation 18, 19, 45.
The vast majority of known TZ and IFT proteins (over 60 in total) are linked to human disorders termed ciliopathies 46, 47. Disruption of an IFT‐A, IFT‐B or IFT‐dynein subunit typically causes so‐called “skeletal” ciliopathies, such as Jeune asphyxiating thoracic dystrophy (JATD), cranioectodermal dysplasia (CED) and Ellis‐van Creveld syndrome (EVC) 47, 48, 49, 50. In more rare cases, mutations in the same IFT proteins can be associated with ciliopathies that principally affect the eye and kidney—including retinitis pigmentosa (RP), cone‐rod dystrophy (CRD) and nephronophthisis (NPHP) 47, 51, 52, 53. The reason for such different phenotypic presentations is unclear. However, eye‐ and/or kidney‐associated ciliopathies, sometimes coupled with liver and brain anomalies as well as polydactyly, are the hallmarks of TZ‐associated ciliopathies; these include NPHP, Meckel syndrome, Joubert syndrome and Senior‐Løken syndrome 46, 47, 54. Ciliopathies are therefore complex—exhibiting multigenic and multiallelic traits, and a spectrum of partially overlapping clinical phenotypes. A central question relevant to ciliary biology and human disease is how specific mutations in particular components of different ciliary structures/processes correlate with discrete clinical ailments. Moreover, whether ciliary defects can be reversed in ciliopathy patients remains virtually unknown.
In this study, we identify the first metazoan temperature‐sensitive (ts) cilia mutant, which we show affects the function of CHE‐3, the C. elegans orthologue of the retrograde IFT‐dynein‐2 motor DYNC2H1 33. We use this novel ts‐che‐3 mutant to reveal roles for retrograde IFT in maintaining the ciliary axoneme, anterograde IFT, as well as correct assembly and function of the TZ ciliary gate. Notably, the ts‐che‐3 mutant enables us to show post‐developmental regrowth of cilia and restoration of TZ gate function. Our findings suggest a functional interplay between IFT and the TZ, whereby disruption of IFT can manifest in defects in TZ assembly and function. This suggests a potential mechanism for how IFT mutations could cause typical IFT‐associated skeletal ciliopathies, TZ‐associated renal/retinal/brain ciliopathies or, potentially, a combination of both. Furthermore, the ability to rebuild the ciliary axoneme and TZ post‐developmentally in our model system raises the possibility of therapeutic intervention for some post‐natal aspects of IFT‐ and TZ‐associated ciliopathies.
Results
Genetic screen uncovers a temperature‐sensitive dynein‐2 heavy chain mutant
To provide new insights into retrograde IFT, we conducted an unbiased genetic screen to obtain mutants affecting this process (Fig 1A). A C. elegans paraquat‐resistant mutant library, known to be enriched for mutants with defective cilia 55, was subjected to two sequential screens. The first screen identified animals deficient in their uptake of a fluorescent dye into their head and tail sensory neurons (Fig 1A), a process that depends on intact chemosensory cilia which are normally exposed to the environment 8, 56, 57. Such dye‐filling (dyf) mutants frequently have cilia structure defects caused by mutations in core IFT or BBS proteins 8, 37, 42, 58, 59, 60, 61. The second screen involved introducing GFP‐tagged IFT markers (IFT‐A subunit CHE‐11/IFT140, IFT‐B subunit OSM‐5/IFT88 and IFT‐dynein‐associated XBX‐1/DYNC2LI1) in the dyf mutants and visualising them by microscopy. This helped reveal IFT protein accumulations (Fig 1A) typical of retrograde IFT mutants 33, 37, 60.
Figure 1. Screen for retrograde IFT defects identifies a temperature‐sensitive allele of the dynein heavy chain CHE‐3, the orthologue of mammalian DYNC2H1.

- Genetic screen for retrograde IFT defects uncovers a temperature‐sensitive (ts) mutation in C. elegans CHE‐3 (dynein‐2 heavy chain). Paraquat‐resistant strains were first screened for dye‐filling defects (Dyf phenotype) typically indicative of cilia structure anomalies (examples of normal and defective DiI dye‐filling are shown). Three different IFT reporters were then introduced into the dyf mutants (CHE‐11::GFP shown as an example) to reveal candidate retrograde IFT mutants showing strong IFT protein accumulation at cilia tips. Whole‐genome sequencing revealed the likely causative mutation in one of the strains, namely in the retrograde IFT‐dynein motor CHE‐3 (DYNC2H1 human orthologue). A variable Dyf phenotype at 20°C suggested the possibility of a ts mutation. Scale bar, 20 μm.
- The Dyf phenotype of the ts‐che‐3 mutant was confirmed, with animals grown at the permissive temperature (15°C) showing the ability to uptake dye, while those raised at the restrictive temperature (25°C) are Dyf. Scale bar, 20 μm.
- Using the homodimeric kinesin OSM‐3 as a marker for IFT, proper localisation is seen in the ts‐che‐3 mutant at the permissive temperature, with the strongest signal at the basal body (bb). At the restrictive temperature, most of the OSM‐3 signal is found within the axoneme of the short, bulbous cilia. Scale bar is 4 μm.
- The ts‐che‐3 mutant shows normal osmotic avoidance at permissive temperature but not at the restrictive temperature, indicating defective chemosensation. The osm‐9 mutant is included as a positive control. n = 50 animals; error bars are standard error.
Several candidate retrograde IFT mutants were uncovered, one of which was subjected to whole‐genome sequencing and found to contain a likely deleterious mutation in the gene encoding the dynein‐2 heavy chain, CHE‐3. A genetic complementation assay with an existing che‐3 null allele (e1124) confirmed that che‐3 is indeed the gene responsible for the Dyf phenotype. The mutation, G1997E, alters a glycine residue conserved across all protist, plant and metazoan DYNC2H1 orthologues (Fig EV1A). It is located immediately adjacent to an essential aspartic acid residue that contacts Mg2+‐ATP in the second AAA ATPase domain of the motor protein (Fig EV1B) 62, 63, 64, 65. The replacement of glycine with glutamate likely interferes with the binding or affinity of ATP in the AAA2 domain, which is required for dynein motor activity 62, 63, 64, 65.
Figure EV1. The temperature‐sensitive mutation in C. elegans CHE‐3 (orthologue of human DYNC2H1) is in a glycine residue next to the ATP binding site and immediately adjacent to an evolutionarily conserved aspartic acid (Asp; D) likely required for ATP binding/hydrolysis in the AAA2 domain.

- Multiple sequence alignment showing a portion of the AAA2 domain containing the glycine (G1997) residue mutated to glutamic acid (Glu; E) in CHE‐3 (G2056 residue in human DYNC2H1).
- Close‐up of Gly and Asp amino acids in relation to ATP‐Mg2+ within the AAA2 domain of human DYNC2H1 (crystal structure PDB accession number 4RH7).
Although a che‐3 null mutant has already been characterised 33, 66, we discovered that the G1997E mutation in CHE‐3 causes a temperature‐sensitive (ts) Dyf phenotype (Fig 1). We therefore sought to study this mutant further, as it represents the first metazoan temperature‐sensitive cilia mutant to be uncovered.
The ts‐che‐3 mutant exhibits temperature‐sensitive cilia defects
At the permissive temperature of 15°C, the ts‐che‐3 mutant features no dye‐filling defective phenotype and no overt ciliary defects, as judged by the correct localisation of IFT markers (Fig 1B and C). In contrast, 100% of ts‐che‐3 animals are dye‐filling defective at the restrictive temperature of 25°C (Fig 1B). This dye‐filling phenotype can be explained by severe cilia structure anomalies, since the ts‐che‐3 mutant has short cilia with large bulbous accumulations of IFT reporters (Fig 1C), similar to the previously reported phenotype for the che‐3 null allele 33.
Consistent with these structural ciliary defects, the ts‐che‐3 animals display a temperature‐dependent sensory/behavioural phenotype. At permissive temperature, ts‐che‐3 mutants avoid a high‐osmolarity barrier, similar to wild‐type animals (Fig 1D). However, at restrictive temperature, the ts‐che‐3 animals largely fail to sense and avoid the barrier. This phenotype is comparable to that of a mutant with defects in the osmosensing ciliary TRPV ion channel (osm‐9) (Fig 1D) and mutants (e.g., IFT) with ciliogenic defects 42, 60, 67, 68. Thus, our genetic screen for IFT retrograde mutants has uncovered a novel dynein‐2 temperature‐sensitive mutation that exhibits largely normal ciliary structure and function at the permissive temperature, while resembling the null mutant at the restrictive temperature.
Dynein‐2 is required for correct assembly of transition zone proteins
There is growing evidence for physical and functional connections between the IFT system and the TZ ciliary gate (see Discussion). Yet, in C. elegans, the function of IFT is largely unimpaired when the TZ is completely disrupted 12. Whether IFT is required for the correct assembly and function of the TZ has not been specifically investigated in metazoans.
As part of our molecular analyses of the ts‐che‐3 mutant, we discovered that at the restrictive temperature, a fluorescently tagged TZ protein (MKS‐6; orthologue of human CC2D2A/MKS6/JBTS9) displays, in addition to seemingly normal TZ localisation, ectopic distribution to more distal regions of the short and bulbous cilia (Fig 2A). We imaged additional TZ proteins in the ts‐che‐3 mutant at restrictive temperature (25°C), namely NPHP‐4 (NPHP4) and CEP‐290 (CEP290). We found similar ectopic localisation with NPHP‐4 (Fig 2A). Despite CEP‐290 appearing similar at the restrictive and permissive temperatures, there is a small, yet significant amount of ectopic TZ protein in the ciliary middle segment. We confirmed that the TZ defects are not exclusive to the ts‐che‐3 mutant, since similar ectopic localisation of several TZ proteins (NPHP‐1, NPHP‐4, MKS‐1, MKSR‐1 and MKS‐6) is observed in the che‐3 null mutant (Fig EV2A). Furthermore, to rule out any gain‐of‐function or dominant‐negative effects, we looked at animals heterozygous for the ts‐che‐3 allele grown at the restrictive temperature. We found that they appear wild‐type in terms of MKS‐6 protein localisation (Fig EV2B).
Figure 2. Loss of IFT‐dynein function at restrictive temperature results in TZ protein localisation defects that are reversed at permissive temperature.

- Three TZ markers (NPHP‐4, CEP‐290 and MKS‐6) are shown at both the permissive (15°C) and restrictive (25°C) temperatures in the ts‐che‐3 mutant. At the permissive temperature, all three markers localise to the TZ, while at the restrictive temperature exhibit ectopic localisation (leakage) distally into the ciliary axoneme. While NPHP‐4 and MKS‐6 show significant accumulations, CEP‐290 is grossly normal. At the permissive temperature, both IFT co‐markers (CHE‐13 and XBX‐1) display strong localisation to the basal body (bb) and along the axoneme. At the restrictive temperature, the IFT markers show accumulation in the axoneme. Accumulations emphasised using asterisks.
- To determine whether ciliary compartmentalisation could be restored, young ts‐che‐3 mutant adults were transferred to the permissive temperature after being raised at the restrictive temperature. The TZ proteins reassembled at the TZ, with loss of the ectopic accumulations, and IFT protein localisation was also restored—strongest at the basal body, similar to animals grown at the permissive temperature.
- After 24 h at the restrictive temperature, TZ proteins appear to begin to accumulate distally within the axoneme in ts‐che‐3 mutants. This is seen, in small amounts, for NPHP‐4 and CEP‐290 but with stronger ectopic localisation for MKS‐6. The IFT proteins CHE‐13 and XBX‐1 also show accumulations in the axoneme after 24 h at the restrictive temperature.
Figure EV2. The null and ts‐che‐3 mutant, but not the heterozygous ts mutant show ectopic TZ protein localisation, which recovers after FRAP in the ts mutant.

- In the null allele che‐3(e1124), five transition zone (TZ) proteins show distal ectopic accumulation (marked by asterisks) in the bulbous axonemes of the mutant cilia, something not observed in wild‐type animals. TZs are indicated with an arrowhead. Dotted lines and arrows indicate direction and location of either cilia or dendrites (den). Scale bar is 4 μm.
- When grown at the restrictive temperature the cilia of animals heterozygous for the ts‐che‐3 allele resemble those found in wild‐type, with no apparent ectopic TZ (MKS‐6) or IFT (XBX‐1) protein accumulations seen in the homozygous mutant. Scale bar is 4 μm.
- After photobleaching the distal end of a truncated axoneme in a ts‐che‐3 mutant grown at the restrictive temperature, MKS‐6 shows fluorescence recovery within seconds. White boxes indicate pixels analysed for the graph, which represents a ratio of fluorescence intensity between the area photobleached (indicated by the red box) to an area not photobleached. Scale bar is 0.5 μm.
Interestingly, unlike previous reports which determined that TZ proteins are immobile at the TZ 69, FRAP (fluorescence recovery after photobleaching) analysis shows that ectopic MKS‐6 exhibits fast, diffusion‐like mobility (Fig EV2C). This suggests that the protein is not correctly assembled/tethered in this distal ciliary region, compared to its normal localisation at the TZ. Importantly, the temperature‐sensitive nature of our che‐3 mutant allowed us to test for the reversibility of this phenotype. After shifting the ts‐che‐3 mutant from restrictive temperature (25°C) to permissive temperature (15°C) for 24 h, the accumulation of TZ proteins within the distal ciliary region was lost, and the TZ proteins became largely confined to the correct TZ region (Fig 2B).
Next, we wondered whether IFT is required to maintain the proper localisation of TZ proteins, and if turning “off”, IFT would result in ectopic TZ protein localisation. To answer this, we shifted the ts‐che‐3 mutant raised at the permissive (15°C) to restrictive (25°C) temperature for 24 h and assessed NPHP‐4, CEP‐290 and MKS‐6 localisation. At the permissive temperature, these proteins localise correctly to the TZ (Fig 2A). Upon temperature upshift (young adults) for 24 h, a small but significant amount of TZ protein localises ectopically distally within the ciliary axoneme (Fig 2C), with MKS‐6 being most visibly affected.
Our findings reveal that TZ proteins show ectopic localisation when the animals are raised at restrictive temperature (Fig 2A); this ectopic localisation can be removed by restoring IFT function (Fig 2B). Furthermore, TZ proteins show delocalisation distally when IFT is turned “off” by a temperature shift to 25°C (Fig 2C). Altogether, this provides evidence that IFT is required for retrieving ectopically distributed TZ proteins and maintaining their correct localisation/assembly in the TZ region.
Dynein‐2 is required for correct function of the transition zone “ciliary gate”
Based on our observation that TZ proteins show ectopic localisation in the ts‐che‐3 mutant, we queried whether the function of the ciliary gate is also compromised. Two different membrane‐associated proteins that normally localise at the base of cilia (more specifically the periciliary membrane compartment or PCMC), namely RPI‐2 (human RP2 orthologue) and TRAM‐1 (TRAM1), were used to test for the integrity of the TZ or “ciliary gate” as previously done 10. At the restrictive temperature, the ts‐che‐3 mutant shows abnormal ciliary accumulation of both markers (Fig 3A), a phenotype observed in most TZ mutants 10, 12, 16. The ts‐che‐3 strain permitted us to test the reversibility of this phenotype. Upon shifting from restrictive to permissive temperature for 24 h, ts‐che‐3 animals re‐established their ability to exclude RPI‐2 and TRAM‐1 from the ciliary compartment (Fig 3A). Hence, loss of CHE‐3 function causes a partial displacement of TZ proteins, which correlates with impaired TZ (ciliary gate) activity. Interestingly, only a small proportion of the TRAM‐1 appears to “leak” into the cilium at restrictive temperature, whereas a majority of the RPI‐2 accumulates in the cilium when IFT is impaired. This suggests that RPI‐2 is targeted to cilia in an IFT‐independent manner and might subsequently be removed from cilia by IFT, thus resulting in its final localisation at the ciliary base.
Figure 3. Abrogating CHE‐3 function causes defects in transition zone gate function and ultrastructure.

- In the ts‐che‐3 mutant grown at the permissive temperature (15°C), both RPI‐2/RP2 and TRAM‐1/TRAM1 are localised to the PCMC, just proximal to the ciliary axoneme. At the restrictive temperature, TRAM‐1 shows leakage into the axoneme with its strongest localisation at the PCMC. Intriguingly, RPI‐2 now displays strong localisation to the axoneme, indicating that in the absence of IFT, RPI‐2 may be targeted into the cilium. When ts‐che‐3 mutant animals grown at the restrictive temperature (25°C) are shifted at young adult to the permissive (15°C) for 24 h, both TRAM‐1 and RPI‐2 exhibit restoration of their PCMC localisation. Consistent with other TZ markers, both MKS‐2 and MKSR‐1 show ectopic localisation in the axoneme when grown at the restrictive temperature compared to the permissive temperature. Also consistent with what was observed for other markers, after 24 h at the permissive temperature, animals grown at the restrictive temperature show loss of the ectopic localisation. Fluorescence quantification is shown for each marker at the indicated temperature in the heat maps on the right. Each point in the plot represents one pixel along the centre of the basal body (BB), transition zone (TZ) and middle segment (MS) regions. Dotted areas (three pixels) in the MS were used to quantify ectopic localisation (MKS‐2 and MKSR‐1) or accumulation (RPI‐2 and TRAM‐1) for statistical analyses. n = 10 cilia (4–6 animals), Kruskal–Wallis test, *P < 0.05, **P < 0.01; scale bars are 4 μm.
- Loss of CHE‐3 function results in short, bulbous cilia containing electron‐dense accumulations and abnormal membrane–microtubule connections. Shown are transmission electron micrograph (TEM) cross sections of an amphid channel cilium in wild‐type and che‐3(e1124) null mutant animals. Representative images of wild‐type cilia show intact middle segment (containing doublet microtubules) and transition zone (with Y‐shaped links) (left top and bottom panels) regions. Representative images of che‐3(e1124) cilia reveal apparently intact transition zones, with visible Y‐link structures, but enlarged ciliary ends filled with electron‐dense accumulations, which often appear vesicular. The bulbous structures reveal doublet microtubules associated with the membrane, and occasionally ectopic microtubule‐to‐membrane connections, which sometimes appear similar to transition zone Y‐links in the region just distal to the seemingly “normal” TZ structure. Schematics show longitudinal (left images) and cross section (right images) representations of wild‐type and che‐3(e1124) cilia. tz, transition zone; pcmc, periciliary membrane compartment; scale bars are as indicated in (nm) for TEM images.
Given the improper localisation of TZ proteins in the ts‐che‐3 mutant, we wondered whether there are ultrastructural defects within the TZ compartment, which could explain our observed ciliary gate phenotypes. Interestingly, both the che‐3 null and the ts‐che‐3 mutant (grown at restrictive temperature) animals appear to have, from Transmission Electron Microscopy (TEM) analysis, all of the ultrastructural features expected of the TZ; namely, the TZ regions feature doublet microtubules connected to a constricted ciliary membrane via Y‐links (Figs 3B and EV3) 8. Whether more subtle TZ ultrastructure defects are present cannot be excluded. Based on fluorescent reporters, the largely intact TZ of ts‐che‐3 animals (Figs 2A and 3A) is consistent with the presence of Y‐links at the TZ. However, the ts‐che‐3 mutant (at restrictive temperature) and null che‐3 mutant animals have doublet microtubules in the expanded (distal) region of the axoneme that remain in close association with the membrane (Fig 3B and EV3). Some of these doublets exhibit connections—both Y‐shaped links and “stubs”—that are not normally observed in the middle segments of wild‐type cilia. Most interestingly, the Y‐links found in the expanded membrane are just distal to the seemingly normal TZ, indicating either that the Y‐links extend distally into the middle segment or that the distal end of the TZ is structurally compromised (expanded) in the che‐3 mutants. Also, it is possible that these ectopic microtubule‐to‐membrane connections result from the presence of the mislocalised TZ proteins we observe (Figs 2A and EV2A).
Figure EV3. Ultrastructural defects observed in cilia of che‐3 null and temperature sensitive mutants.

At the restrictive temperature of 25°C, the temperature‐sensitive che‐3 mutant, che‐3(nx159ts), has ultrastructural features similar to those of the che‐3(e1124) null mutant at 20°C. Left panels show transmission electron micrograph (TEM) cross sections of amphid channel cilia. Schematics show the amphid channel in longitudinal and cross‐section orientations (only 3 of 10 cilia shown), indicate the section positions (in microns, relative to position “0”), and denote the phenotypes observed (summarised in point form). Scale bars are 500 nm (all images identically scaled). Some images placed on a grey background. PCMC, periciliary membrane compartment. Arrowheads indicate transition zones, and asterisks show middle and distal ciliary segments.
Altogether, our findings reveal that in C. elegans, disruption of the IFT retrograde motor results in the mislocalisation of TZ proteins, malfunction of the TZ “ciliary gate” and disorganisation of the distal end of the TZ ultrastructure. To our knowledge, this represents the first evidence for a specific role for IFT in building a functional TZ. Furthermore, the use of our temperature‐sensitive mutant confirms that restoring the function of retrograde IFT (and potentially anterograde IFT; see below) can re‐establish the correct localisation of the TZ proteins and proper gate function.
Dynein‐2 is required for maintenance of primary cilia
Our discovery of a temperature‐sensitive dynein‐2 mutant affecting retrograde IFT allows us to probe the requirement of the IFT transport system in not only the formation, but also the maintenance, of primary cilia.
To determine whether disrupting dynein‐2 function impairs ciliary maintenance, we used BBS‐7, which is part of the IFT‐associated BBS complex, as a cilium length reporter. At permissive temperature (15°C), the average length of phasmid cilia in wild‐type and the ts‐che‐3 mutant is 6.2 μm (Fig 4A). Upon shifting the animals on the first day of adulthood to restrictive temperature (25°C), a progressive loss of cilium length is observed in the ts‐che‐3 mutant, to approximately 3.8 μm after 18 h and 3.4 μm after 24 h (Fig 4A). Despite observing near full‐length cilia after 12 h, which is not significantly different from wild‐type or ts mutant incubated at the permissive temperature, there is often accumulation of BBS‐7 present in the cilia (Fig 4A, asterisks). This indicates that despite no significant reduction in ciliary length, IFT assembly/function within the cilium is impaired. From these experiments, we conclude that dynein‐2 is required for the maintenance of metazoan non‐motile (primary) cilia, consistent with observations obtained from motile cilia in the unicellular green alga, C. reinhardtii 70, 71, 72. Such a function may depend on the cell type, however, as IFT is required for ciliogenesis but not for axonemal maintenance in Trypanosoma brucei motile cilia and mouse spermatozoa 73, 74.
Figure 4. Ciliary maintenance and IFT function upon shift to the restrictive temperature (25°C).

- Using the IFT marker BBS‐7, cilia lengths were measured for wild‐type and the ts‐che‐3 mutant animals raised at the permissive (15°C) and shifted to the restrictive temperature (25°C) for three time points. After 12 h, some accumulations (indicated by asterisks in the fluorescence images) are observed within the cilia, but there is not a significant reduction in cilium length. After 18 and 24 h, significant reduction in ciliary length is observed and large accumulations of BBS‐7 is seen within short bulbous cilia, similar to what is observed in ts‐che‐3 animals raised at the restrictive temperature. n = 30–34 cilia, one per animal; Kruskal–Wallis test, **P < 0.01; error bars are mean absolute deviation; scale bars are 4 μm.
- IFT velocities were measured for wild‐type or ts‐che‐3 mutant animals (grown at permissive temperature) and imaged at the permissive temperature, restrictive temperature or after 3 h at the restrictive temperature. Compared to wild‐type, anterograde velocities in the ts‐che‐3 dynein mutant are similar, while IFT retrograde rates are highly reduced for both the middle and distal ciliary segments. Interestingly, the ts‐che‐3 IFT‐dynein mutant appears to not only have slow retrograde IFT trains, but the trains also appear much larger and less frequent. Initially upon the temperature shift (< 15 min), IFT rates are much faster for both wild‐type and ts‐che‐3 dynein mutant, although retrograde IFT remains much slower in the mutant. After 3 h at the restrictive temperature, IFT velocities are intermediate between the rates seen at 15°C and those observed at the initial shift to 25°C. Error bars are 95% CI. Wild‐type (10–15 animals): anterograde n = 141–326, retrograde n = 100–271; for che‐3(nx159ts) (18–25 animals): anterograde n = 234–312, retrograde n = 64–103; y‐axis scale bar is 2 s; x‐axis scale bar is 1 μm.
Dynein‐2‐mediated retrograde IFT is required for anterograde IFT
Considering the nature of the CHE‐3(G1997E) temperature‐sensitive mutation—which we hypothesise affects the motor activity of the IFT‐dynein, and the observed IFT protein ciliary accumulation phenotype—we anticipated that the mutation results in a specific defect in retrograde IFT. We initially sought to answer two questions regarding the IFT system in the ts‐che‐3 mutant: (i) How quickly is IFT‐dynein function affected upon shifting to restrictive temperature? and (ii) What specific effect does the mutation have on IFT‐dynein behaviour?
To address these questions, we used an IFT reporter to image IFT in the cilia of animals grown at the permissive temperature (15°C), and after shifting to the restrictive temperature (25°C) for defined periods. At the permissive temperature, the ts‐che‐3 mutant displays anterograde velocities similar to wild‐type in the middle and distal ciliary segments. However, our kymograph analyses reveal that the ts‐che‐3 mutant harbours retrograde IFT trains that are slower (traces have altered slope) and larger (traces are wider) compared to wild‐type; furthermore, the ratio of detectable retrograde‐to‐anterograde train particles observed is much reduced at 15°C: 0.273 for che‐3(nx159ts) versus 0.926 for wild‐type, P < 0.001 using the chi‐square test (Figs 4B and EV4A). These data suggest a potentially specific defect in retrograde transport at the permissive temperature, namely fewer retrograde particles that move more slowly.
Figure EV4. IFT behaviour in ts‐che‐3 mutant.

- Anterograde IFT velocities of CHE‐11/IFT140 are quite similar for wild‐type and che‐3(nx159ts) when animals raised at the permissive temperature are imaged at the permissive or restrictive temperatures (< 30 min). However, retrograde velocities are much reduced in che‐3(nx159ts) mutants compared to wild‐type animals. All velocities observed were markedly increased when the imaging was performed at 25°C compared to cilia imaged at 15°C, indicating a temperature dependence on IFT rates. Error bars are 95% CI. For wild‐type (13–15 animals): anterograde n = 301–493, retrograde n = 309–440; for che‐3(nx159ts) (18–48 animals): anterograde n = 727–776, retrograde n = 401–407. Y‐axis scale bars are 2 s; x‐axis scale bars are 1 μm.
- After 3 h at the restrictive temperature, some ts dynein mutants raised at the permissive temperature begin to show very slow retrograde IFT, whereas others exhibit no IFT in either direction. This suggests that in the ts‐che‐3 mutant, IFT stochastically shuts down over time after the shift to the restrictive temperature. Y‐axis scale bars are 2 s; x‐axis scale bars are 1 μm.
Initially, after shifting wild‐type animals from permissive (15°C) to restrictive (25°C) temperature (for < 15 min), both anterograde and retrograde IFT velocities increase substantially, by approximately twofold (Figs 4B and EV4A). This is consistent with previous observations in Trypanosoma, where IFT velocities increase following a temperature upshift 75, thus extending this phenomenon to metazoan primary cilia. The ts‐che‐3 mutant animals also exhibit these altered, temperature‐dependent IFT dynamics, although retrograde velocities are still much reduced in the mutant (Figs 4B and EV4A). After 3 h at the restrictive temperature (25°C), some ts‐che‐3 cilia show no IFT in either direction, lack retrograde IFT, or display extremely reduced retrograde IFT velocities (Fig EV4B). In contrast, wild‐type cilia showed frequent anterograde and retrograde IFT in all animals, for all conditions.
Together, our findings reveal that the ts‐che‐3 mutant exhibits, at permissive temperature and shortly following a shift to restrictive temperature, anterograde IFT that is comparable to that of wild‐type animals. Retrograde IFT, on the other hand, is slower in the mutant at both permissive and restrictive temperatures. After a more prolonged period of time at restrictive temperature, a proportion of mutant animals begin to show defects in retrograde IFT (missing or much slower), as well as anterograde IFT. From these results, we suggest that the temperature‐sensitive mutation in the dynein‐2 motor specifically affects retrograde IFT. Retrograde IFT function is sufficient to maintain ciliary structure and function at the permissive temperature, but at restrictive temperature, retrograde IFT eventually halts, and this is likely to impair anterograde IFT, leading to cilium degeneration and IFT protein accumulations. Importantly, the reversible nature of the ts‐che‐3 mutant (Figs 2B and 3A) provides us with a valuable tool to study the role of IFT on the formation and functions of cilia.
Cilia repair potential decreases in older animals
Despite displaying a strong ciliogenic phenotype at the restrictive temperature of 25°C (short cilia with prominent IFT protein accumulations at the tip), the ts‐che‐3 mutant retains the capacity to rebuild cilia (and properly assemble TZ proteins) following a 24‐h return to permissive temperature (15°C) at the first day of adulthood (Figs 2B, 5A and B, and EV5A and B). We therefore wondered whether older adults were also able to regenerate cilia. Unexpectedly, we initially observed an apparent enhancement of TZ protein assembly in older ts‐che‐3 animals compared to younger adults when continuously grown at the restrictive temperature (Figs 5A and B, and EV5A and B). At days 4 and 8 of adulthood for MKS‐6 (“MKS module” TZ protein), and day 4 for NPHP‐4 (“NPHP module” TZ protein), ectopic TZ protein localisation is reduced compared to 1‐day‐old adults (Figs 5A and B, and EV5A and B). While perhaps surprising, our findings are consistent with those of Cornils and colleagues 76, who reported an improvement in cilia morphology (TZ was not looked at) for some hypomorphic IFT mutants during ageing, a process requiring IFT. However, we also discovered a significant, age‐dependent difference in the ability of ts‐che‐3 mutant animals to restore their ciliary structures when shifted from restrictive to permissive temperature (Figs 5A and B, and EV5A and B). Specifically, the reduction in ectopic TZ ciliary protein localisation seen upon restoring IFT‐dynein function, by shifting the animals to the permissive temperature, decreased as the animals aged (Figs 5A and B, and EV5A and B). These findings reveal that in nematode primary cilia, TZ incorporation of MKS and NPHP proteins (“regeneration”) is possible post‐developmentally, but that this ability declines as the animal age.
Figure 5. Transition zone repair potential decreases with age while older adults show reduced TZ protein localisation defects.

-
A, BTo assess the effects of IFT restoration on TZ assembly, ts‐che‐3 mutant animals were shifted from the restrictive to the permissive temperature at different time points during their development (days 1, 4 and 8 of adulthood). The TZ markers MKS‐6 (A) and NPHP‐4 (B) show strong ectopic ciliary accumulations when raised at the restrictive temperature until day 1 of adulthood. After a shift to the permissive temperature for 24 h, day 2 adults show “repaired” TZ protein localisation resembling that of wild‐type animals. For both markers, day 4 adults (maintained at the restrictive temperature) show less ectopic TZ protein localisation than day 1 adults. The correction of ectopic localisation following shift to the permissive temperature for 24 h was reduced in day 4 (to day 5) of adulthood compared to that observed in day 1 (to day 2) adults. For MKS‐6, but not NPHP‐4, day 8 adults (maintained at the restrictive temperature) also display less ectopic accumulation than day 1 adults. The correction of ectopic TZ protein localisation in animals shifted to the permissive temperature after 8 days of adulthood is also much smaller in magnitude for both markers, indicating that this repair is most effective in day 1 adults. In summary, as the adults age, ectopic TZ protein localisation is reduced, while the ability to correct this phenotype by restoring IFT function also reduces. tz, transition zone; scale bars are 4 μm. Heat map graphs corresponding to the bar graphs are located in Fig EV5. n = 10 cilia (4–7 animals), Kruskal–Wallis test, *P < 0.05, **P < 0.01, error bars are mean absolute deviation.
Figure EV5. TZ and IFT repair potential decreases with age while older adults show reduced localisation defects.
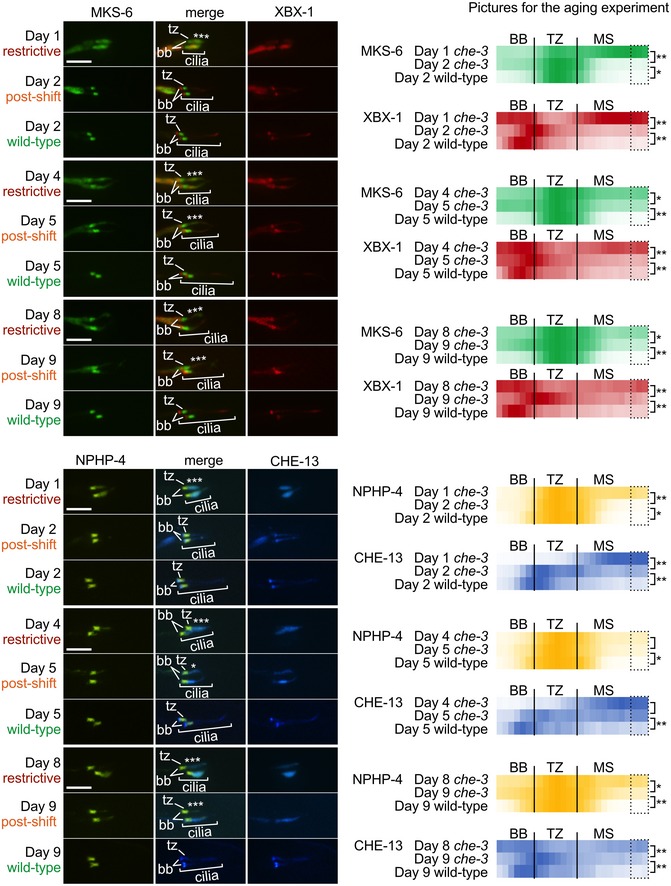
Shown are animals grown at restrictive temperature and shifted at days 1, 4 or 8 of adulthood to the permissive for 24 h. For both TZ markers—MKS‐6 and NPHP‐4—there is significant reduction in ectopic protein localisation (marked by asterisks) after a 24‐h shift to the permissive temperature except for NPHP‐4 at day 4, which shows a reduction but is not statistically significant. For both IFT markers (XBX‐1 and CHE‐13), the same trend is observed. After a 24‐h shift to the permissive temperature, there is significant reduction in ectopic accumulation except at day 4 for CHE‐13, which shows a reduction but is not statistically significant. Despite a reduction in ectopic localisation for all markers, as the animals age, the “repair” seen after 24 h at the permissive temperature (i.e., IFT function is restored) is reduced as the animals age. Fluorescence quantification is shown for each marker at the indicated temperature in the heat maps on the right. Each point in the plot represents one pixel along the centre of the basal body (BB), transition zone (TZ) and middle segment (MS) regions. Dotted areas (three pixels) in the MS were used to quantify ectopic localisation (TZ markers) or accumulation (IFT markers) for statistical analyses. n = 10 cilia (4–7 animals), Kruskal–Wallis test, *P < 0.05, **P < 0.01. tz, transition zone; bb, basal body; scale bars are 4 μm.
Discussion
Methods used to manipulate gene function and uncover biological insights typically involve obtaining gene knockouts or knockdowns (e.g., RNA interference) followed by phenotypic analyses. Such approaches often have limitations, such as the lack of options to reverse cellular defects that may be pertinent to alleviating or curing human diseases. Alternative methods, such as targeting a protein for degradation, inducing ectopic localisation or dimerisation, provide temporal control but not reversibility. Here, we describe the discovery and analysis of the first temperature‐sensitive (ts) ciliary gene mutant for any metazoan. Our C. elegans genetic screen uncovered a ts mutation in the catalytic subunit of the intraflagellar transport (IFT)‐dynein‐2 motor (CHE‐3; orthologue of human DYNC2H1) (Figs 1A and EV1A). This ts mutant dynein appears to completely lose its function at the restrictive temperature of 25°C, mimicking a null mutant, and can regain function after shifting to 15°C, the permissive temperature. We use this ts‐CHE‐3 mutant to reveal the temporal requirements for IFT‐dynein in retrograde and anterograde IFT, as well as in the proper assembly and maintenance of the ciliary axoneme and transition zone (TZ).
Notably, temperature‐sensitive mutations in C. elegans DHC‐1, the cytoplasmic dynein‐1 motor subunit, were previously studied by Schmidt and colleagues 77. Dynein‐1 is thought to act independently from that of the IFT‐associated dynein‐2, and consistent with this notion, the disruption of DHC‐1 in C. elegans results in a centriole translocation defect 78 that is not observed in the ts‐che‐3 mutant we describe herein.
The primary description of a che‐3 null mutant in C. elegans by Signor and colleagues noted a ciliary truncation/bulge with IFT protein accumulations 32, consistent with our analyses of the ts‐che‐3 mutant. Their study did not specifically look for TZ defects, due to the lack of specific fluorescently tagged TZ reporters available at the time (in fact, the region labelled as the “transition zone” or “TZ” is now known to represent the periciliary membrane 7, 10). Here, we show that both the null and ts‐che‐3 mutants (the latter grown at the restrictive temperature) share the ectopic TZ protein distribution (Figs 2A and EV2A) using TZ marker proteins and observe ectopic Y‐links by TEM (Figs 3B and EV3).
Role for IFT‐dynein in the maintenance of metazoan primary cilia
Our findings show that dynein‐2 is required for maintaining the structure of primary cilia in C. elegans, a metazoan model system. Interestingly, our ts‐che‐3 mutant is partially defective at the permissive temperature, indicating that while dynein‐2 is required for ciliary maintenance, ciliary structure is maintained despite the reduced dynein‐2 activity. A similar requirement for retrograde IFT trafficking was observed by analysing temperature‐sensitive dynein‐2 mutants in Chlamydomonas 70, 71, although IFT is not required for the maintenance of Trypanosoma or mouse sperm flagella 73, 74. It will be interesting to determine whether mammalian primary cilia show a dependence on IFT, similar to what we observe.
Although IFT persists for some time in our ts‐che‐3 mutant, retrograde IFT eventually halts. Anterograde IFT also ceases, indicating that anterograde IFT trafficking in C. elegans depends on continued retrograde IFT transport. Interestingly, this is contrary to what is seen in Chlamydomonas, where anterograde IFT persists in the absence of retrograde IFT. This may be due to intrinsic differences between IFT in Chlamydomonas and metazoans. For example, in green algae, IFT‐kinesin diffuses back to the base 79, 80, 81, potentially allowing for additional rounds of IFT in the absence of the IFT retrograde motor. In C. elegans and presumably other metazoans, retrieval of IFT kinesins from the cilium requires retrograde IFT 82. Indeed, our kymograph analyses reveal that anterograde IFT eventually halts when retrograde IFT is abrogated. Remarkably, some ciliated plant species encode IFT subunits and IFT‐kinesin, but lack a dynein motor altogether, again revealing key differences between cilium formation and maintenance in non‐metazoan and metazoan species 83.
Role of IFT in building a functional TZ gate: significance for ciliopathies
Our observation that disruption of IFT in the ts‐che‐3 mutant impacts TZ protein localisation and function is novel in metazoans and potentially of significance in relation to ciliopathies. Interestingly, the connection between IFT and TZ biogenesis was first observed in Trypanosoma, where the absence of IFT (by RNAi) results in shorter TZs 84.
The mechanism by which the IFT‐dynein machinery may facilitate TZ assembly and function is unclear. One possibility is that during ciliogenesis, TZ proteins begin their assembly within the proximal‐most region of the axoneme, but a portion of these can become mislocalised to distal ciliary regions and potentially create ectopic axoneme–membrane connections (Figs 2A, 3B and 6A). Such an incomplete assembly of TZ proteins at the base of cilia may result in a ciliary gate that is not (fully) functional. Consistent with this possibility, we observe mislocalised TZ proteins and ciliary gate dysfunction when the IFT‐dynein motor is impaired (ts‐che‐3 at restrictive temperature and che‐3 null; Figs 2A, EV2A, and 3A). The role of the IFT retrograde machinery may therefore be to assist the retrieval of the mis‐assembled TZ proteins (Fig 6A). Evidence for this hypothesis was obtained with our ts‐che‐3 mutant: TZ proteins within the bulbous ciliary tip were reassembled after restoring IFT‐dynein function (return to permissive temperature) (Fig 2B).
Figure 6. Model for the role of intraflagellar transport (IFT) in the correct assembly and function of the ciliary transition zone (TZ), and relationship to ciliopathies.

- Kinesin‐ and dynein‐dependent anterograde and retrograde particles (1 and 2, respectively) are shown harbouring IFT‐A, IFT‐B and BBS subcomplexes involved in ciliary cargo transport. Under normal conditions, any misassembly or partial disassembly of proteins from the TZ (3) can be recovered by the retrograde IFT machinery (4). Disruption of retrograde transport (and consequently, anterograde transport) leads to an accumulation of TZ proteins in a short, bulbous cilium as well as impaired TZ assembly and function (as observed in our temperature‐sensitive IFT‐dynein mutant at the restrictive temperature), phenotypes that are reversible when IFT is restored at the permissive temperature.
- Cross‐talk between different ciliary modules and distinct classes of ciliopathies. Most ciliary modules are associated with a specific class of ciliopathies; for example, IFT‐A and IFT‐dynein are linked to skeletal ciliopathies, BBS proteins are associated with Bardet–Biedl syndrome, and transition zone proteins are associated with Joubert syndrome and related disorders (JSRDs), MKS, NPHP and eye‐related disorders, but not skeletal ciliopathies. However, a subset of proteins are associated with ciliopathies generally typical of a different module, indicating potential overlap in function. The module–disease connections are shown as solid lines, with the width correlating to the number of causative genes currently identified (1 to 10+, as indicated). MKS, Meckel syndrome; JBTS, Joubert syndrome; COACH, cerebellar vermis hypoplasia/oligophrenia/ataxia/coloboma/hepatic fibrosis; OFD, oral‐facial‐digital syndrome; SLSN, Senior‐Løken syndrome; NPHP, nephronophthisis; RP, retinitis pigmentosa; CRD, cone‐rod dystrophy; LCA, Leber congenital amaurosis; BBS, Bardet–Biedl syndrome; CED, cranioectodermal dysplasia; SRTD, short‐rib thoracic dysplasia; JATD, Jeune asphyxiating thoracic dystrophy.
Deleterious mutations in IFT genes are well‐known causes of skeletal ciliopathies such as Jeune asphyxiating thoracic dystrophy (Jeune syndrome; Fig 6B) 46, 47, 50, 85. Intriguingly, however, some IFT gene variants cause phenotypically distinct ciliopathies, which in some cases are reminiscent of those caused by TZ dysfunction 53. For example, mutations in WDR19 (encoding the IFT‐A subunit IFT144) can cause a skeletal ciliopathy such as Jeune syndrome, whereas other mutations in WDR19 result in isolated NPHP or Senior‐Løken syndrome 86, which are most commonly associated with TZ‐associated ciliopathies (Fig 6B). Similarly, mutations in TTC21B (IFT‐A subunit IFT139) can result in distinct ciliopathies, namely Jeune syndrome or NPHP 53, 87. Disruption of the IFT‐B subcomplex can also give rise to disparate phenotypic presentations: TRAF3IP1/IFT54 mutations were first reported to cause NPHP with retinal degeneration 88, but a new study identified mutations in the same gene that cause the skeletal ciliopathies asphyxiating thoracic dystrophy (ATD) or short‐rib polydactyly syndrome (SRPS) 49.
One possible explanation is that with certain mutations, the functionality of the IFT proteins is largely maintained but their roles in forming a functional TZ are impaired (Fig 6). Thus far, however, components of the IFT‐dynein machinery, including the heavy chain (DYNC2H1), have all been specifically associated with skeletal ciliopathies 49. Further sequencing of IFT‐associated genes in patients with a variety of ciliopathies will help reveal the extent to which IFT‐associated genes, including DYNC2H1, may be linked to one or more particular types of ciliopathies. For instance, mutations in IFT172 not only cause skeletal ciliopathies, but are also associated with isolated retinal degeneration or Bardet–Biedl syndrome (BBS) 51, 89. In the latter case, the “core” IFT172 function may be largely maintained, but a specific physical/functional connection between the IFT protein and the BBSome may be lost, leading to BBS rather than a skeletal ciliopathy.
While our manuscript focusses on the role of CHE‐3 (IFT‐dynein DYNC2H1) in building a functional TZ, C. elegans strains with mutations in IFT‐B subunits, which have very short cilia, have been observed by TEM to possess ectopic doublet microtubules with Y‐links at the distal ends of dendrites, proximal to the TZ 8. Since anterograde IFT eventually halts when retrograde IFT is disrupted, it is possible that anterograde IFT—perhaps together with retrograde IFT—is essential for fully assembling a functional TZ in metazoans (similar to that shown in Trypanosomes 81).
Furthermore, there is precedence for mislocalised TZ proteins in mammalian cells. Disruption of the centrosomal protein CEP162 causes swellings and ectopic accumulation of TZ proteins (including TMEM67/MKS3) at the ciliary tip 90. Also, overexpression of TZ proteins results in ectopic TZ protein localisation 91. Hence, there are likely multiple factors—including IFT and CEP162—necessary to correctly assemble a TZ at the base of the ciliary axoneme.
Functional interactions between IFT and TZ macromolecular complexes and potential link to Hedgehog signalling
While a role for IFT in building a functional TZ has not previously been reported, several studies suggest functional connections between IFT and the TZ. For example, Zhao and Malicki showed that at least one IFT protein (IFT74) binds a TZ component (B9D2) and provided evidence for a genetic interaction between B9D2 and IFT52 in zebrafish 92. Additional genetic interactions between TZ genes and IFT or BBS genes have been reported in C. elegans and mouse 93, 94.
Our observation that IFT is required for the correct assembly and TZ ciliary gating provides additional evidence for such functional interactions. It will be important to investigate these mechanisms in a mammalian system, especially in relation to Hedgehog signalling. Several IFT as well as BBS proteins are directly implicated in Hedgehog signalling 18, 45, 95, 96, 97. Defects in Hedgehog component ciliary transport are thought to cause many of the observed ciliopathy phenotypes. However, the TZ is also directly implicated in Hedgehog signalling, ostensibly acting as a trafficking gateway that can be modulated 14,98, 99,100. Our work suggests that IFT may also influence the function of the gate, such that the Hedgehog signalling defects underlying ciliopathy phenotypes could at least partly result from TZ dysfunction, i.e., represent a combination of IFT and TZ defects. However, this hypothesis will need to be investigated in vertebrate/mammalian systems, as C. elegans does not have a canonical Hedgehog signal transduction pathway.
Post‐developmental regain of ciliary structure and function
Our use of the temperature‐sensitive IFT‐dynein mutant provides evidence that ciliary axoneme and TZ defects have the potential to be rescued post‐developmentally. This may be informative in regard to human therapeutic intervention. While restoration of developmental defects (e.g., skeletal anomalies) may only be possible at an early stage of human development, other phenotypic presentations, such as retinal degeneration or kidney function, may have a longer window of opportunity for intervention. Encouragingly, progress towards such therapies is being made: For example, “rescues” of mutations in the CEP290 gene, which encodes a TZ protein, have been demonstrated in a murine ciliopathy model, as well as in patient cells 101, 102, 103.
In conclusion, we propose that the disentanglement of functional interactions between the TZ ciliary gate and the IFT system will become important for understanding how disruption of IFT and/or the TZ can abrogate signalling pathways—potentially beyond Hedgehog signalling—and can manifest as specific ciliopathy phenotypes that may be amenable to therapeutic intervention.
Materials and Methods
Construction and maintenance of C. elegans strains
All C. elegans strains used in this study are listed in Table EV1. Strains were cultured using standard techniques and maintained at 20°C unless otherwise noted; for some experiments, strains were incubated at 15 or 25°C and are indicated as such. Standard genetic crosses were used to generate double mutants and introduce transgenes into different genetic backgrounds.
Dye‐filling and behavioural assays
Dye‐filling assays were conducted by incubating animals for 30 min in a solution of lipophilic dye (DiI) diluted with M9 buffer 1:200 for the screen and 1:1,000 for imaging, followed by recovery (feeding) on seeded NGM plates to minimise gut fluorescence, as detailed 56. Dye uptake into amphid and phasmid neurons was assessed by fluorescence stereomicroscopy. Osmolarity avoidance assays were carried out by measuring the proportion of adult animals that cross a ring of high osmotic strength (8M glycerol) within a 10 min timespan, as described.
Identification of temperature‐sensitive che‐3 mutant
A library of paraquat‐resistant C. elegans strains obtained from Dai Ayusawa was screened in triplicate for dye‐filling defective mutants 55. IFT reporters (CHE‐11::GFP; OSM‐5::GFP and XBX‐1::GFP) were introduced into Dyf mutants by genetic crossing. Epifluorescence microscopy of the IFT reporters revealed several mutants with potential defects in retrograde IFT, including mev‐52. Whole‐genome sequencing of the mev‐52 strain revealed a missense mutation in the dynein‐2 heavy chain subunit gene (DYNC2H1), che‐3, which alters a glycine residue to a glutamic acid (G1997E). A variable dyf phenotype at 20°C led us to test for a potential temperature‐sensitive effect of the mutation; this was confirmed, with the mev‐52 mutant (renamed ts‐che‐3) dye‐fill positive at 15°C and dye‐fill defective at 25°C. A standard complementation test was performed as previously described 104. Heterozygous animals were created by crossing wild‐type males to ts‐che‐3 hermaphrodites and imaging the progeny. Outcrossing was completed for three generations by following the Dyf phenotype at the restrictive temperature followed by three generations, following the mutation using PCR and restriction digest (the mutation creates an MboII site).
Imaging cilia and intraflagellar transport (IFT) assays
To visualise fluorescently tagged IFT proteins, animals were immobilised with 10 mM levamisole and 0.1‐μm‐diameter polystyrene microspheres (Polysciences 00876‐15, 2.5% w/v suspension) on 10% agarose pads. Microscopy was performed using a spinning disc confocal system (WaveFX system from Quorum Technologies) equipped with a 100× oil (N.A. 1.4) objective and Hamamatsu C9100‐13 EMCCD camera. Volocity software (PerkinElmer) was used to acquire images and time‐lapse movies. 15 or 25°C was achieved on the microscope by temperature control of the objective (via circulating water bath, a peristaltic pump, a brass objective ring) and a 0.22‐mm thermocouple probe mounted on an agarose pad similar to the worms. Imaging for Fig EV4A was completed on a Zeiss Axioskop 2 mot with a 100× oil (N.A. 1.3) objective via a Hamamatsu Orca ER C4742‐80 CCD camera and Open Lab (V5) (Agilent) software), in walk‐in incubators set at 15 or 25°C as indicated. Imaging for Fig EV2C was performed on a Zeiss LSM 800 confocal microscope, and analysis was performed using ImageJ.
Kymograph analyses to quantify IFT velocities and fluorescent intensity profiles were generated by using the ImageJ plugin kymographclear 82. Cilium length measurement was performed using the Volocity software (PerkinElmer).
Transmission electron microscopy
For TEM analyses, the che‐3(nx159ts) strain was maintained at 25°C, and the che‐3(e1124) strain at 20°C. Young adult (day 1) nematodes were then fixed, sectioned, and imaged as previously reported. One animal was sectioned for each che‐3 allele and one for wild‐type 57.
Author contributions
VLJ, NJL, CL, SM, CLW, PNI, BKY, OEB and MRL conceived experiments. VLJ, NJL, CL, SM, CLW, PNI and OEB performed experiments. VLJ, OEB and MRL wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Review Process File
Acknowledgements
We thank Navin Bhopal for assistance with the genetic screen for retrograde IFT mutants, Dai Ayusawa for providing the paraquat‐resistant (mev) strains, and the Caenorhabditis Genetics Center (CGC) for strains. M.R.L. acknowledges funding from the Canadian Institutes of Health Research (Gouvernement du Canada | CIHR | Institute of Genetics (IG); grants MOP‐82870, MOP‐142243 and PJT‐156042) and a senior scholarship award from Michael Smith Foundation for Health Research (MSFHR). V.J. holds postdoctoral fellowships from MSFHR and KRESCENT. O.E.B. acknowledges funding from Science Foundation Ireland (SFI; grant 16/BBSRC/3394). We thank Dimitri Scholz and Tiina O'Neill of the UCD Conway Institute imaging facility, and Sofia Tsiropoulou (OEB lab), for microscopy assistance. C.L.W. and B.K.Y. were supported by Foundation for the National Institutes of Health (FNIH) R01DK06565.
EMBO Reports (2018) 19: e45862
References
- 1. Satir P (2017) CILIA: before and after. Cilia 6: 1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sung C‐H, Leroux MR (2013) The roles of evolutionarily conserved functional modules in cilia‐related trafficking. Nat Cell Biol 15: 1387–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Satir P, Mitchell DR, Jékely G (2008) How did the cilium evolve? Curr Top Dev Biol 85: 63–82 [DOI] [PubMed] [Google Scholar]
- 4. Cavalier‐Smith T (2009) Predation and eukaryote cell origins: a coevolutionary perspective. Int J Biochem Cell Biol 41: 307–322 [DOI] [PubMed] [Google Scholar]
- 5. Garcia‐Gonzalo FR, Reiter JF (2016) Open sesame: how transition fibers and the transition zone control ciliary composition. Cold Spring Harb Perspect Biol 9: a028134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ishikawa H, Marshall WF (2011) Ciliogenesis: building the cell's antenna. Nat Rev Mol Cell Biol 12: 222–234 [DOI] [PubMed] [Google Scholar]
- 7. Reiter JF, Blacque OE, Leroux MR (2012) The base of the cilium: roles for transition fibres and the transition zone in ciliary formation, maintenance and compartmentalization. EMBO Rep 13: 608–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Perkins LA, Hedgecock EM, Thomson JN, Culotti JG (1986) Mutant sensory cilia in the nematode Caenorhabditis elegans . Dev Biol 117: 456–487 [DOI] [PubMed] [Google Scholar]
- 9. Hedgecock EM, Culotti JG, Thomson JN, Perkins LA (1985) Axonal guidance mutants of Caenorhabditis elegans identified by filling sensory neurons with fluorescein dyes. Dev Biol 111: 158–170 [DOI] [PubMed] [Google Scholar]
- 10. Williams CL, Li C, Kida K, Inglis PN, Mohan S, Semenec L, Bialas NJ, Stupay RM, Chen N, Blacque OE et al (2011) MKS and NPHP modules cooperate to establish basal body/transition zone membrane associations and ciliary gate function during ciliogenesis. J Cell Biol 192: 1023–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huang L, Szymanska K, Jensen VL, Janecke AR, Innes AM, Davis EE, Frosk P, Li C, Willer JR, Chodirker BN et al (2011) TMEM237 is mutated in individuals with a joubert syndrome related disorder and expands the role of the TMEM family at the ciliary transition zone. Am J Hum Genet 89: 713–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jensen VL, Li C, Bowie RV, Clarke L, Mohan S, Blacque OE, Leroux MR (2015) Formation of the transition zone by Mks5/Rpgrip1L establishes a ciliary zone of exclusion (CIZE) that compartmentalises ciliary signalling proteins and controls PIP2 ciliary abundance. EMBO J 34: 2537–2556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sang L, Miller JJ, Corbit KC, Giles RH, Brauer MJ, Otto EA, Baye LM, Wen X, Scales SJ, Kwong M et al (2011) Mapping the NPHP‐JBTS‐MKS protein network reveals ciliopathy disease genes and pathways. Cell 145: 513–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Garcia‐Gonzalo FR, Corbit KC, Sirerol‐Piquer MS, Ramaswami G, Otto EA, Noriega TR, Seol AD, Robinson JF, Bennett CL, Josifova DJ et al (2011) A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat Genet 43: 776–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schouteden C, Serwas D, Palfy M, Dammermann A (2015) The ciliary transition zone functions in cell adhesion but is dispensable for axoneme assembly in C. elegans . J Cell Biol 210: 35–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li C, Jensen VL, Park K, Kennedy J, Garcia‐Gonzalo FR, Romani M, De Mori R, Bruel A‐L, Gaillard D, Doray B et al (2016) MKS5 and CEP290 dependent assembly pathway of the ciliary transition zone. PLoS Biol 14: e1002416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bangs F, Anderson KV (2016) Primary cilia and mammalian hedgehog signaling. Cold Spring Harb Perspect Biol 9: a028175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Haycraft CJ, Banizs B, Aydin‐Son Y, Zhang Q, Michaud EJ, Yoder BK (2005) Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet 1: e53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Caspary T, Larkins CE, Anderson KV (2007) The graded response to sonic hedgehog depends on cilia architecture. Dev Cell 12: 767–778 [DOI] [PubMed] [Google Scholar]
- 20. Goetz SC, Anderson KV (2010) The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet 11: 331–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Taschner M, Lorentzen E (2016) The intraflagellar transport machinery. Cold Spring Harb Perspect Biol 8: a028092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lechtreck KF (2015) IFT‐cargo interactions and protein transport in cilia. Trends Biochem Sci 40: 765–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang J, Deretic D (2014) Molecular complexes that direct rhodopsin transport to primary cilia. Prog Retin Eye Res 38: 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kozminski KG, Johnson KA, Forscher P, Rosenbaum JL (1993) A motility in the eukaryotic flagellum unrelated to flagellar beating. Proc Natl Acad Sci USA 90: 5519–5523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jensen VL, Leroux MR (2017) Gates for soluble and membrane proteins, and two trafficking systems (IFT and LIFT), establish a dynamic ciliary signaling compartment. Curr Opin Cell Biol 47: 83–91 [DOI] [PubMed] [Google Scholar]
- 26. Taschner M, Weber K, Mourão A, Vetter M, Awasthi M, Stiegler M, Bhogaraju S, Lorentzen E (2016) Intraflagellar transport proteins 172, 80, 57, 54, 38, and 20 form a stable tubulin‐binding IFT‐B2 complex. EMBO J 35: 773–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Craft JM, Harris JA, Hyman S, Kner P, Lechtreck KF (2015) Tubulin transport by IFT is upregulated during ciliary growth by a cilium‐autonomous mechanism. J Cell Biol 208: 223–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hao L, Thein M, Brust‐Mascher I, Civelekoglu‐Scholey G, Lu Y, Acar S, Prevo B, Shaham S, Scholey JM (2011) Intraflagellar transport delivers tubulin isotypes to sensory cilium middle and distal segments. Nat Cell Biol 13: 790–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ou GE, Blacque O, Snow JJ, Leroux MR, Scholey JM (2005) Functional coordination of intraflagellar transport motors. Nature 436: 583–587 [DOI] [PubMed] [Google Scholar]
- 30. Pan X, Ou G, Civelekoglu‐Scholey G, Blacque OE, Endres NF, Tao L, Mogilner A, Leroux MR, Vale RD, Scholey JM (2006) Mechanism of transport of IFT particles in C. elegans cilia by the concerted action of kinesin‐II and OSM‐3 motors. J Cell Biol 174: 1035–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Asante D, Stevenson NL, Stephens DJ (2014) Subunit composition of the human cytoplasmic dynein‐2 complex. J Cell Sci 127(Pt 21): 4774–4787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Signor D, Wedaman KP, Orozco JT, Dwyer ND, Bargmann CI, Rose LS, Scholey JM (1999) Role of a class Dhc1b dynein in retrograde transport of Ift motors and Ift raft particles along cilia, but not dendrites, in chemosensory neurons of living Caenorhabditis elegans . J Cell Biol 147: 519–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wicks SR, de Vries CJ, van Luenen HG, Plasterk RH (2000) CHE‐3, a cytosolic dynein heavy chain, is required for sensory cilia structure and function in Caenorhabditis elegans . Dev Biol 221: 295–307 [DOI] [PubMed] [Google Scholar]
- 34. Wilson MJ, Salata MW, Susalka SJ, Pfister KK (2001) Light chains of mammalian cytoplasmic dynein: identification and characterization of a family of LC8 light chains. Cell Motil Cytoskeleton 49: 229–240 [DOI] [PubMed] [Google Scholar]
- 35. Yi P, Li W‐J, Dong M‐Q, Ou G (2017) Dynein‐driven retrograde intraflagellar transport is triphasic in C. elegans sensory cilia. Curr Biol 27: 1448–1461.e7 [DOI] [PubMed] [Google Scholar]
- 36. Rompolas P, Pedersen LB, Patel‐King RS, King SM (2007) Chlamydomonas FAP133 is a dynein intermediate chain associated with the retrograde intraflagellar transport motor. J Cell Sci 120: 3653–3665 [DOI] [PubMed] [Google Scholar]
- 37. Blacque OE, Li C, Inglis PN, Esmail MA, Ou G, Mah AK, Baillie DL, Scholey JM, Leroux MR (2006) The WD repeat‐containing protein IFTA‐1 is required for retrograde intraflagellar transport. Mol Biol Cell 17: 5053–5062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pazour GJ, Dickert BL, Witman GB (1999) The DHC1b (DHC2) isoform of cytoplasmic dynein is required for flagellar assembly. J Cell Biol 144: 473–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Porter ME, Bower R, Knott JA, Byrd P, Dentler W, McIntosh JR (1999) Cytoplasmic dynein heavy chain 1b is required for flagellar assembly in Chlamydomonas . Mol Biol Cell 10: 693–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Qin H, Rosenbaum JL, Barr MM (2001) An autosomal recessive polycystic kidney disease gene homolog is involved in intraflagellar transport in C. elegans ciliated sensory neurons. Curr Biol 11: 457–461 [DOI] [PubMed] [Google Scholar]
- 41. Pazour GJ, Dickert BL, Vucica Y, Seeley ES, Rosenbaum JL, Witman GB, Cole DG (2000) Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene Tg737, are required for assembly of cilia and flagella. J Cell Biol 151: 709–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Blacque OE, Reardon MJ, Li C, McCarthy J, Mahjoub MR, Ansley SJ, Badano JL, Mah AK, Beales PL, Davidson WS et al (2004) Loss of C. elegans BBS‐7 and BBS‐8 protein function results in cilia defects and compromised intraflagellar transport. Genes Dev 18: 1630–1642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peränen J, Merdes A, Slusarski DC, Scheller RH, Bazan JF, Sheffield VC et al (2007) A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell 129: 1201–1213 [DOI] [PubMed] [Google Scholar]
- 44. Liu P, Lechtreck KF (2018) The Bardet‐Biedl syndrome protein complex is an adapter expanding the cargo range of intraflagellar transport trains for ciliary export. Proc Natl Acad Sci USA 115: E934–E943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mukhopadhyay S, Wen X, Chih B, Nelson CD, Lane WS, Scales SJ, Jackson PK (2010) TULP3 bridges the IFT‐A complex and membrane phosphoinositides to promote trafficking of G protein‐coupled receptors into primary cilia. Genes Dev 24: 2180–2193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Baker K, Beales PL (2009) Making sense of cilia in disease: the human ciliopathies. Am J Med Genet C Semin Med Genet 151C: 281–295 [DOI] [PubMed] [Google Scholar]
- 47. Reiter JF, Leroux MR (2017) Genes and molecular pathways underpinning ciliopathies. Nat Rev Mol Cell Biol 18: 533–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Waters AM, Beales PL (2011) Ciliopathies: an expanding disease spectrum. Pediatr Nephrol Berl Ger 26: 1039–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang W, Taylor SP, Ennis HA, Forlenza KN, Duran I, Li B, Sanchez JAO, Nevarez L, Nickerson DA, Bamshad M et al (2018) Expanding the genetic architecture and phenotypic spectrum in the skeletal ciliopathies. Hum Mutat 39: 152–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schmidts M, Arts HH, Bongers EMHF, Yap Z, Oud MM, Antony D, Duijkers L, Emes RD, Stalker J, Yntema J‐BL et al (2013) Exome sequencing identifies DYNC2H1 mutations as a common cause of asphyxiating thoracic dystrophy (Jeune syndrome) without major polydactyly, renal or retinal involvement. J Med Genet 50: 309–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bujakowska KM, Zhang Q, Siemiatkowska AM, Liu Q, Place E, Falk MJ, Consugar M, Lancelot M‐E, Antonio A, Lonjou C et al (2015) Mutations in IFT172 cause isolated retinal degeneration and Bardet‐Biedl syndrome. Hum Mol Genet 24: 230–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dharmat R, Liu W, Ge Z, Sun Z, Yang L, Li Y, Wang K, Thomas K, Sui R, Chen R (2017) IFT81 as a candidate gene for nonsyndromic retinal degeneration. Invest Ophthalmol Vis Sci 58: 2483–2490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Otto EA, Ramaswami G, Janssen S, Chaki M, Allen SJ, Zhou W, Airik R, Hurd TW, Ghosh AK, Wolf MT et al (2011) Mutation analysis of 18 nephronophthisis associated ciliopathy disease genes using a DNA pooling and next generation sequencing strategy. J Med Genet 48: 105–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gonçalves J, Pelletier L (2017) The ciliary transition zone: finding the pieces and assembling the gate. Mol Cells 40: 243–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fujii M, Matsumoto Y, Tanaka N, Miki K, Suzuki T, Ishii N, Ayusawa D (2004) Mutations in chemosensory cilia cause resistance to paraquat in nematode Caenorhabditis elegans . J Biol Chem 279: 20277–20282 [DOI] [PubMed] [Google Scholar]
- 56. Inglis PN, Blacque OE, Leroux MR (2009) Functional genomics of intraflagellar transport‐associated proteins in C. elegans . Methods Cell Biol 93: 267–304 [DOI] [PubMed] [Google Scholar]
- 57. Sanders AAWM, Kennedy J, Blacque OE (2015) Image analysis of Caenorhabditis elegans ciliary transition zone structure, ultrastructure, molecular composition, and function. Methods Cell Biol 127: 323–347 [DOI] [PubMed] [Google Scholar]
- 58. Albert PS, Riddle DL (1983) Developmental alterations in sensory neuroanatomy of the Caenorhabditis elegans dauer larva. J Comp Neurol 219: 461–481 [DOI] [PubMed] [Google Scholar]
- 59. Starich TA, Herman RK, Kari CK, Yeh WH, Schackwitz WS, Schuyler MW, Collet J, Thomas JH, Riddle DL (1995) Mutations affecting the chemosensory neurons of Caenorhabditis elegans . Genetics 139: 171–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schafer JC, Winkelbauer ME, Williams CL, Haycraft CJ, Desmond RA, Yoder BK (2006) IFTA‐2 is a conserved cilia protein involved in pathways regulating longevity and dauer formation in Caenorhabditis elegans . J Cell Sci 119: 4088–4100 [DOI] [PubMed] [Google Scholar]
- 61. Bell LR, Stone S, Yochem J, Shaw JE, Herman RK (2006) The molecular identities of the Caenorhabditis elegans intraflagellar transport genes dyf‐6, daf‐10 and osm‐1. Genetics 173: 1275–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Roberts AJ, Numata N, Walker ML, Kato YS, Malkova B, Kon T, Ohkura R, Arisaka F, Knight PJ, Sutoh K et al (2009) AAA+ Ring and linker swing mechanism in the dynein motor. Cell 136: 485–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schmidt H, Gleave ES, Carter AP (2012) Insights into dynein motor domain function from a 3.3‐Å crystal structure. Nat Struct Mol Biol 19: 492–497, S1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Carter AP, Cho C, Jin L, Vale RD (2011) Crystal structure of the dynein motor domain. Science 331: 1159–1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Schmidt H, Carter AP (2016) Review: structure and mechanism of the dynein motor ATPase. Biopolymers 105: 557–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lewis JA, Hodgkin JA (1977) Specific neuroanatomical changes in chemosensory mutants of the nematode Caenorhabditis elegans . J Comp Neurol 172: 489–510 [DOI] [PubMed] [Google Scholar]
- 67. Colbert HA, Smith TL, Bargmann CI (1997) OSM‐9, a novel protein with structural similarity to channels, is required for olfaction, mechanosensation, and olfactory adaptation in Caenorhabditis elegans . J Neurosci 17: 8259–8269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Schafer JC, Haycraft CJ, Thomas JH, Yoder BK, Swoboda P (2003) XBX‐1 encodes a dynein light intermediate chain required for retrograde intraflagellar transport and cilia assembly in Caenorhabditis elegans . Mol Biol Cell 14: 2057–2070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lambacher NJ, Bruel A‐L, van Dam TJP, Szymańska K, Slaats GG, Kuhns S, McManus GJ, Kennedy JE, Gaff K, Wu KM et al (2016) TMEM107 recruits ciliopathy proteins to subdomains of the ciliary transition zone and causes Joubert syndrome. Nat Cell Biol 18: 122–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Engel BD, Ishikawa H, Wemmer KA, Geimer S, Wakabayashi K, Hirono M, Craige B, Pazour GJ, Witman GB, Kamiya R et al (2012) The role of retrograde intraflagellar transport in flagellar assembly, maintenance, and function. J Cell Biol 199: 151–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lin H, Nauman NP, Albee AJ, Hsu S, Dutcher SK (2013) New mutations in flagellar motors identified by whole genome sequencing in Chlamydomonas . Cilia 2: 14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kozminski KG, Beech PL, Rosenbaum JL (1995) The Chlamydomonas kinesin‐like protein FLA10 is involved in motility associated with the flagellar membrane. J Cell Biol 131: 1517–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Fort C, Bonnefoy S, Kohl L, Bastin P (2016) Intraflagellar transport is required for the maintenance of the trypanosome flagellum composition but not length. J Cell Sci 129: 3026–3041 [DOI] [PubMed] [Google Scholar]
- 74. San Agustin JT, Pazour GJ, Witman GB (2015) Intraflagellar transport is essential for mammalian spermiogenesis but is absent in mature sperm. Mol Biol Cell 26: 4358–4372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Buisson J, Chenouard N, Lagache T, Blisnick T, Olivo‐Marin J‐C, Bastin P (2013) Intraflagellar transport proteins cycle between the flagellum and its base. J Cell Sci 126: 327–338 [DOI] [PubMed] [Google Scholar]
- 76. Cornils A, Maurya AK, Tereshko L, Kennedy J, Brear AG, Prahlad V, Blacque OE, Sengupta P (2016) Structural and functional recovery of sensory cilia in C. elegans IFT mutants upon aging. PLoS Genet 12: e1006325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Schmidt DJ, Rose DJ, Saxton WM, Strome S (2005) Functional analysis of cytoplasmic dynein heavy chain in Caenorhabditis elegans with fast‐acting temperature‐sensitive mutations. Mol Biol Cell 16: 1200–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Li W, Yi P, Zhu Z, Zhang X, Li W, Ou G (2017) Centriole translocation and degeneration during ciliogenesis in Caenorhabditis elegans neurons. EMBO J 36: 2553–2566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Chien A, Shih SM, Bower R, Tritschler D, Porter ME, Yildiz A (2017) Dynamics of the IFT machinery at the ciliary tip. Elife 6: e28606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Liang Y, Pang Y, Wu Q, Hu Z, Han X, Xu Y, Deng H, Pan J (2014) FLA8/KIF3B phosphorylation regulates kinesin‐II interaction with IFT‐B to control IFT entry and turnaround. Dev Cell 30: 585–597 [DOI] [PubMed] [Google Scholar]
- 81. Engel BD, Ludington WB, Marshall WF (2009) Intraflagellar transport particle size scales inversely with flagellar length: revisiting the balance‐point length control model. J Cell Biol 187: 81–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Prevo B, Mangeol P, Oswald F, Scholey JM, Peterman EJG (2015) Functional differentiation of cooperating kinesin‐2 motors orchestrates cargo import and transport in C. elegans cilia. Nat Cell Biol 17: 1536–1545 [DOI] [PubMed] [Google Scholar]
- 83. Wickstead B, Gull K (2007) Dyneins across eukaryotes: a comparative genomic analysis. Traffic Cph Den 8: 1708–1721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Absalon S, Blisnick T, Kohl L, Toutirais G, Doré G, Julkowska D, Tavenet A, Bastin P (2008) Intraflagellar transport and functional analysis of genes required for flagellum formation in trypanosomes. Mol Biol Cell 19: 929–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Beales PL, Bland E, Tobin JL, Bacchelli C, Tuysuz B, Hill J, Rix S, Pearson CG, Kai M, Hartley J et al (2007) IFT80, which encodes a conserved intraflagellar transport protein, is mutated in Jeune asphyxiating thoracic dystrophy. Nat Genet 39: 727–729 [DOI] [PubMed] [Google Scholar]
- 86. Fehrenbach H, Decker C, Eisenberger T, Frank V, Hampel T, Walden U, Amann KU, Krüger‐Stollfuß I, Bolz HJ, Häffner K et al (2014) Mutations in WDR19 encoding the intraflagellar transport component IFT144 cause a broad spectrum of ciliopathies. Pediatr Nephrol Berl Ger 29: 1451–1456 [DOI] [PubMed] [Google Scholar]
- 87. Davis EE, Zhang Q, Liu Q, Diplas BH, Davey LM, Hartley J, Stoetzel C, Szymanska K, Ramaswami G, Logan CV et al (2011) TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat Genet 43: 189–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Bizet AA, Becker‐Heck A, Ryan R, Weber K, Filhol E, Krug P, Halbritter J, Delous M, Lasbennes M‐C, Linghu B et al (2015) Mutations in TRAF3IP1/IFT54 reveal a new role for IFT proteins in microtubule stabilization. Nat Commun 6: 8666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Halbritter J, Bizet AA, Schmidts M, Porath JD, Braun DA, Gee HY, McInerney‐Leo AM, Krug P, Filhol E, Davis EE et al (2013) Defects in the IFT‐B component IFT172 cause Jeune and Mainzer‐Saldino syndromes in humans. Am J Hum Genet 93: 915–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wang W‐J, Tay HG, Soni R, Perumal GS, Goll MG, Macaluso FP, Asara JM, Amack JD, Tsou M‐FB (2013) CEP162 is an axoneme‐recognition protein promoting ciliary transition zone assembly at the cilia base. Nat Cell Biol 15: 591–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Takao D, Wang L, Boss A, Verhey KJ (2017) Protein interaction analysis provides a map of the spatial and temporal organization of the ciliary gating zone. Curr Biol 27: 2296–2306.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Zhao C, Malicki J (2011) Nephrocystins and MKS proteins interact with IFT particle and facilitate transport of selected ciliary cargos. EMBO J 30: 2532–2544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Roberson EC, Dowdle WE, Ozanturk A, Garcia‐Gonzalo FR, Li C, Halbritter J, Elkhartoufi N, Porath JD, Cope H, Ashley‐Koch A et al (2015) TMEM231, mutated in orofaciodigital and Meckel syndromes, organizes the ciliary transition zone. J Cell Biol 209: 129–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Masyukova SV, Landis DE, Henke SJ, Williams CL, Pieczynski JN, Roszczynialski KN, Covington JE, Malarkey EB, Yoder BK (2016) A screen for modifiers of cilia phenotypes reveals novel MKS alleles and uncovers a specific genetic interaction between osm‐3 and nphp‐4. PLoS Genet 12: e1005841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Keady BT, Samtani R, Tobita K, Tsuchya M, San Agustin JT, Follit JA, Jonassen JA, Subramanian R, Lo CW, Pazour GJ (2012) IFT25 links the signal‐dependent movement of Hedgehog components to intraflagellar transport. Dev Cell 22: 940–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Yang N, Li L, Eguether T, Sundberg JP, Pazour GJ, Chen J (2015) Intraflagellar transport 27 is essential for hedgehog signaling but dispensable for ciliogenesis during hair follicle morphogenesis. Dev Camb Engl 142: 2194–2202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Ocbina PJR, Eggenschwiler JT, Moskowitz I, Anderson KV (2011) Complex interactions between genes controlling trafficking in primary cilia. Nat Genet 43: 547–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Chih B, Liu P, Chinn Y, Chalouni C, Komuves LG, Hass PE, Sandoval W, Peterson AS (2012) A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nat Cell Biol 14: 61–72 [DOI] [PubMed] [Google Scholar]
- 99. Shi X, Garcia G, Van De Weghe JC, McGorty R, Pazour GJ, Doherty D, Huang B, Reiter JF (2017) Super‐resolution microscopy reveals that disruption of ciliary transition‐zone architecture cause of joubert syndrome. Nat Cell Biol 19: 1178–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Yee LE, Garcia‐Gonzalo FR, Bowie RV, Li C, Kennedy JK, Ashrafi K, Blacque OE, Leroux MR, Reiter JF (2015) Conserved genetic interactions between ciliopathy complexes cooperatively support ciliogenesis and ciliary signaling. PLoS Genet 11: e1005627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Burnight ER, Wiley LA, Drack AV, Braun TA, Anfinson KR, Kaalberg EE, Halder JA, Affatigato LM, Mullins RF, Stone EM et al (2014) CEP290 gene transfer rescues Leber congenital amaurosis cellular phenotype. Gene Ther 21: 662–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Molinari E, Srivastava S, Sayer JA, Ramsbottom SA (2017) From disease modelling to personalised therapy in patients with CEP290 mutations. F1000Res 6: 669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Garanto A, Chung DC, Duijkers L, Corral‐Serrano JC, Messchaert M, Xiao R, Bennett J, Vandenberghe LH, Collin RWJ (2016) In vitro and in vivo rescue of aberrant splicing in CEP290‐associated LCA by antisense oligonucleotide delivery. Hum Mol Genet 25: 2552–2563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Yook K (2005) Complementation. In WormBook, The C. elegans Research Community (ed) 10.1895/wormbook.1.24.1 [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Review Process File