

Abstract
Canonical Wnt/β‐catenin signaling is an essential regulator of various cellular functions throughout development and adulthood. Aberrant Wnt/β‐catenin signaling also contributes to various pathologies including cancer, necessitating an understanding of cell context‐dependent mechanisms regulating this pathway. Since protein–protein interactions underpin β‐catenin function and localization, we sought to identify novel β‐catenin interacting partners by affinity purification coupled with tandem mass spectrometry in vascular smooth muscle cells (VSMCs), where β‐catenin is involved in both physiological and pathological control of cell proliferation. Here, we report novel components of the VSMC β‐catenin interactome. Bioinformatic analysis of the protein networks implies potentially novel functions for β‐catenin, particularly in mRNA translation, and we confirm a direct interaction between β‐catenin and the fragile X mental retardation protein (FMRP). Biochemical studies reveal a basal recruitment of β‐catenin to the messenger ribonucleoprotein and translational pre‐initiation complex, fulfilling a translational repressor function. Wnt stimulation antagonizes this function, in part, by sequestering β‐catenin away from the pre‐initiation complex. In conclusion, we present evidence that β‐catenin fulfills a previously unrecognized function in translational repression.
Keywords: β‐catenin, FMRP, mRNA translation, pre‐initiation complex, Wnt signaling
Subject Categories: Protein Biosynthesis & Quality Control
Introduction
The β‐catenin protein fulfills fundamental roles in a variety of cellular processes, both at the cell membrane and in the nucleus, and its malfunction has been linked to developmental disorders and diseases, including cancer 1, 2, 3, 4. Underlying β‐catenin's pleiotropic functions is its complex domain structure, consisting of an N‐terminal domain which is targeted by glycogen synthase kinase 3 (GSK3) 5, a middle region comprised of 12 armadillo repeats mediating various protein–protein interactions, an adjacent “helix C” which packs together the armadillo repeats 6, and a C‐terminal transactivation domain 7.
Under basal conditions, β‐catenin interacts with α‐catenin and cadherins to promote cell adhesion, while activation of the canonical Wnt signaling pathway results in its accumulation, nuclear translocation, and interaction with various transcription factors to enhance target gene expression 8, 9. These seemingly disparate roles of β‐catenin are tightly regulated by signaling pathways through protein–protein interactions to finely balance β‐catenin stability and subcellular localization and provide diverse phenotypic responses 10, 11. Since the control of Wnt/β‐catenin signaling is a potential target for restricting cellular proliferation, migration, and apoptosis in various pathological contexts, key β‐catenin interactors are under investigation as possible therapeutic targets 12, 13, 14. Although the regulation of β‐catenin function by a number of different protein–protein interactions has been documented 15, 16, the diverse roles of β‐catenin in a myriad of different cell types and tissues suggest that multiple context‐dependent protein networks are likely involved in β‐catenin function and remain uncharacterized.
One particular context of interest is β‐catenin functions during development and in post‐natal physiology and pathology in vascular smooth muscle cells (VSMCs). Since VSMCs are, unlike other muscle types, uniquely able to maintain a high degree of phenotypic plasticity without terminal differentiation, β‐catenin signaling and localization are crucial determinants of the VSMC phenotype in response to external stimuli 10, 12, 13. VSMC phenotype modulation has been also implicated in a variety of vascular pathologies such as atherosclerosis and restenosis following angioplasty. In view of the potent role of β‐catenin in the vascular context, we further characterized its regulation in VSMCs.
Since a primary determinant of β‐catenin function relies on protein interaction networks, our initial goal was to characterize the β‐catenin interactome in VSMCs. To address this, we utilized immunoaffinity purification coupled with LC‐MS/MS to identify 131 potential β‐catenin interacting partners in VSMCs. While well‐known interactors such as α‐catenin were identified, a number of the interacting partners were novel and previously uncharacterized. Gene ontology (GO) enrichment analysis revealed a highly significant β‐catenin interactome involvement in mRNA translation, a previously unidentified connection with β‐catenin function. In view of this potential association with translation, we were intrigued by the identification of the fragile X mental retardation protein (FMRP), a translational regulator, as a β‐catenin interacting protein along with several other proteins involved in translational control. Further experimental evidence supports the idea that β‐catenin interacts with FMRP at the messenger ribonucleoprotein (mRNP) and translational pre‐initiation complex to repress translation, and thus, we present evidence suggesting a non‐canonical and novel role for β‐catenin. These observations may have profound implications for our understanding of Wnt/β‐catenin signaling in various cellular processes and in a number of disease states.
Results
Characterization of β‐catenin interacting partners
β‐Catenin is a multifunctional protein and has been found at a variety of subcellular locations where it interacts with specific binding partners in a spatio‐temporal manner 16. However, identification of interacting partners with β‐catenin in a VSMC context has not been systematically carried out. Therefore, we initially documented the β‐catenin interactome using an affinity purification approach coupled to tandem LC‐MS/MS (Fig 1A). The β‐catenin containing protein complexes were immunoprecipitated (IP) with a polyclonal β‐catenin antibody or an unprogrammed rabbit serum as control (representative immunoblot shown in Appendix Fig S1). The proteins found in the immune‐complex were subsequently identified by LC‐MS/MS analysis. The criteria used for the inclusion of a protein as a potential interactor was the presence of at least one single unique peptide in two or more of the five β‐catenin IP replicates and none in any of the five controls. The utility of this approach was justified by the absence of β‐catenin in all unprogrammed rabbit serum eluates and its presence in all anti‐β‐catenin column eluates. Protein identifications from anti‐β‐catenin column eluates were confirmed to be sample‐specific based on their absence in SEQUEST results from the preceding blank runs and therefore did not result from HPLC column carryover.
Figure 1. Schematic of experimental workflow and top 50 sorted β‐catenin interactors shown with GO cellular component analysis.
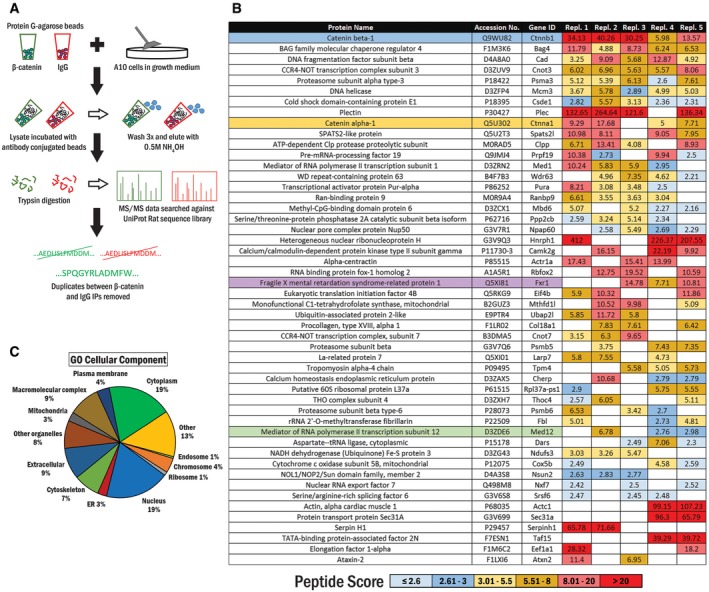
- Workflow for identification of the β‐catenin interactome in A10 cells.
- The list of potential β‐catenin interacting partners. β‐Catenin (blue) is present in all replicates. Previously reported cytoplasmic and nuclear interacting partners, α‐catenin and Mediator 12, are highlighted in yellow and green, respectively. FXR1, which shares a high sequence and domain homology with FMRP, is highlighted in purple.
- A first‐level gene ontology (GO) domain analysis of the top 50 candidates in the GO domain “cellular component”. The percentage of proteins associated with each GO term of the respective domain is represented in the pie chart.
Using this pipeline, we identified 131 potential β‐catenin interacting proteins. We ranked them according to the number of replicates in which they were present and then by the average peptide score (defined in Materials and Methods). The top 50 interacting partners are displayed in Fig 1B, and the full list is shown in Table EV1. Of the top 50 interactors, 88% were found in 3 or more of the biological replicates. We confirmed that well‐known interactors such as α‐catenin 17 and Mediator 12 18 were present in the list indicating that the remainder might thus contain novel bona fide β‐catenin interactors.
To categorize the β‐catenin interactome, we initially performed a top‐level gene ontology (GO) term analysis of these interactors for the “cellular component” domain. This analysis ascertained that β‐catenin interacting partners were present in various cellular compartments, for instance in the cytoplasm (19%) or nucleus (19%) (Fig 1C). Of note was the presence of the ribosome as a cellular component of a number of β‐catenin interactors.
In order to further interrogate the identified interactors, we performed a preliminary analysis on a candidate protein that was ranked similarly to and shared comparable properties with the known interactor α‐catenin. We chose the intermediate filament binding protein Plectin, which is a cytoplasmic protein that anchors intermediate filaments and microtubules to specific locations such as cell junctions, intracellular structures, and various organelles 19. Immunofluorescence of endogenous β‐catenin (antibody validation for immunofluorescence is shown in Appendix Fig S2) and Plectin in A10 cells revealed distinct co‐localization throughout and exclusively in the cytoplasm but not in the nucleus. We analyzed an RGB line scan of a cell indicated in the figure which shows that the two highest peaks of the red signal (green arrows) corresponding to FMRP were not matched with β‐catenin peaks. However, two other peaks of the red signal (red arrows) are tightly matched with the corresponding green peaks (Appendix Fig S3). Interestingly, these proteins also co‐localized at cytoskeletal fiber structures in some cells (Appendix Fig S4). The general approach depicted above for Plectin was utilized as an initial screen to confirm or reject a number of AP‐MS/MS identified interactors as interactions of interest for further study.
Gene ontology enrichment analysis
To further categorize and analyze the β‐catenin interactome, we performed a gene ontology (GO) biological process enrichment analysis to determine biological pathways over‐represented among the list of interacting proteins (Fig 2A). Terms that were shared among a set of common genes were evaluated using the Kappa statistic, and interrelatedness among nodes exceeding a Kappa score of 0.5 was connected to each other. Among the processes depicted here, we confirmed several terms were previously associated with canonical β‐catenin function, including tube morphogenesis 20, actin filament organization 21, and cell growth 22. Interestingly, we also noted that several GO terms that were not previously associated with β‐catenin function, such as mRNA regulation and translation, were identified. Surprisingly, the largest number of genes and the lowest P‐values within the network were associated with some of these “novel” processes, when compared with more canonical β‐catenin‐related terms (Fig 2B). Since the terms “translation” and “mRNA processing” were novel processes for β‐catenin and their P‐values suggested that these terms were not coincidental, we decided to focus on characterizing a β‐catenin interaction with proteins involved in these processes.
Figure 2. GO biological process enrichment analysis.
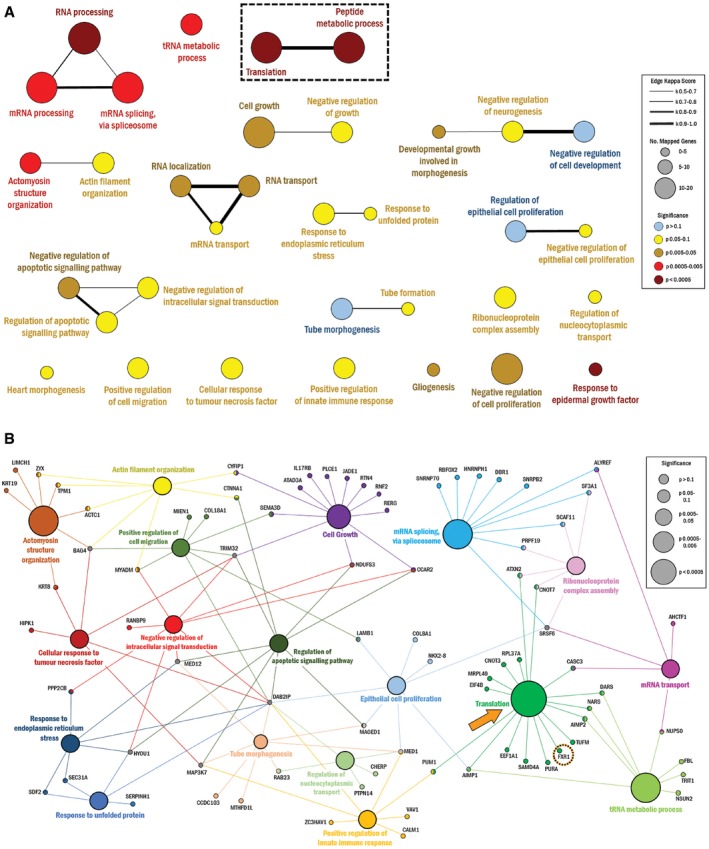
- All 131 candidate proteins were used to identify over‐represented GO biological process terms, which are represented in the following network. Novel processes “translation” and “peptide metabolic process” are outlined. The node size corresponds to the number of candidate proteins associated with a particular term. The node color corresponds to the associated term's P‐value. The line thickness corresponds to the kappa score value to show interrelatedness as defined in the legend. Node position and line length were created arbitrarily.
- Leading terms from (A) are represented by their associated proteins. “Translation” node and FXR1 protein are highlighted. The node size corresponds to the P‐value of a particular term, as defined in the legend. Node position and line legth were created arbitrarily.
Data information: Statistical method: a right‐sided hypergeometric test, corrected using the Bonferroni step‐down method.
β‐Catenin co‐localizes and directly interacts with FMRP
First, we sought to verify the interactions between β‐catenin and a potential interactor involved in translational regulation—the fragile X mental retardation protein (FMRP). Our initial MS/MS analysis revealed that FMRP was present in five out of six biological replicates and correspondingly absent in all of the IgG control samples. FMRP was again identified in the next experimental dataset, although we did also identify it in some IgG controls in the subsequent analyses. In addition, the closely related fragile X‐related protein (FXR1) was also identified in this analysis (highlighted in Figs 1B and 2B). On balance, we reasoned that the data indicated that the interaction merited further analysis of fragile X proteins and further biochemical and imaging data supported the FMRP:β‐catenin interaction (see below). Immunofluorescence analysis of endogenous β‐catenin and ectopically expressed Flag‐FMRP in A10 VSMC cells revealed co‐localization in distinct perinuclear puncta. A line scanned RGB profile revealed that FMRP and β‐catenin fluorescence intensity peaks were matched, and an orthogonal projection showed co‐localization in the XY, XZ, and YZ planes above and around the nucleus (Fig 3A). Furthermore, we confirmed endogenous β‐catenin and ectopically expressed Flag‐FMRP formed a protein complex by co‐immunoprecipitation (coIP) analysis (Fig 3B‐i). We also found that the two proteins interact at endogenous levels in primary vascular smooth muscle cells, based on coIP analysis (Fig 3B‐ii).
Figure 3. β‐Catenin interaction with FMRP.
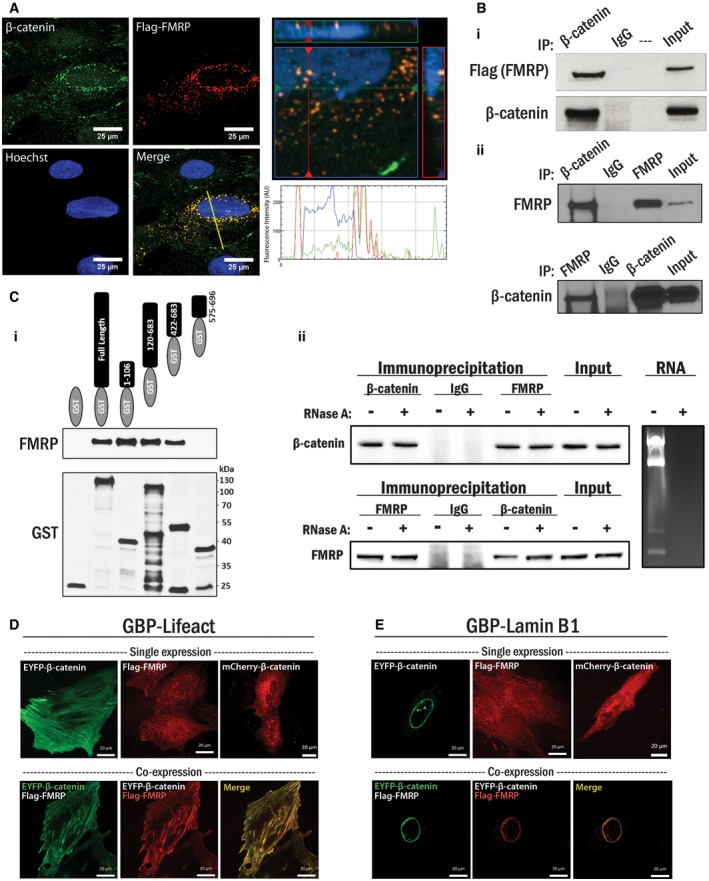
- (Left panel) A10 cells were transfected with Flag‐FMRP for 3 h then cultured for 24 h prior to fixation and staining for β‐catenin (green), Flag (FMRP, red), and nucleus (blue). (Bottom right panel) A line was drawn through a representative cell to indicate relative intensity of RGB signals. (Top right panel) An orthogonal projection was generated by a Z‐stack (240 nm interval) image set using the ZEN program (Zeiss) depicting the region surrounding the nucleus in the XY, XZ, and YZ planes.
- (i) HEK 293T cells were transfected with Flag‐FMRP as in (A), and endogenous β‐catenin protein complexes were immunoprecipitated with β‐catenin antibody. Co‐immunoprecipitated Flag‐FMRP protein was detected by Western blot analysis with the Flag antibody. Non‐programmed rabbit IgG was used as a control. (ii) Primary VSMCs were harvested and subjected to an endogenous coIP analysis with either β‐catenin or FMRP antibody. The corresponding protein was detected in the precipitated immunocomplex by Western blot. Non‐programmed rabbit IgG was used as a control.
- (i) Purified 6x‐His‐FMRP and various regions of β‐catenin proteins fused to GST were incubated with glutathione‐agarose beads. Purified GST or GST‐fusion proteins were visualized by Western blotting using the GST antibody, and sizes of corresponding GST‐fusion proteins were confirmed. Pulled‐down FMRP proteins by GST fused β‐catenin were detected by immunoblotting with the FMRP antibody. (ii) Interaction between FMRP and β‐catenin was assessed in the presence or absence of RNA. HEK 293T cells were subjected to an endogenous coIP analysis using either β‐catenin or FMRP antibody, in the presence or absence of RNase A (10 μg/ml). The corresponding protein was detected in the precipitated immunocomplex by Western blot, and non‐programmed rabbit IgG was used as a control. RNA was extracted from parallel cultured cells, and total RNA content was analyzed by agarose gel electrophoresis in the presence or absence of RNase A.
- (Top panel) A10 cells were transfected with GBP‐Lifeact and either EYFP‐β‐catenin, Flag‐FMRP, or mCherry‐β‐catenin. The cells were fixed and subjected to either fluorescence microscopy or immunofluorescent staining for Flag (red), as indicated. (Bottom panel) A10 cells were transfected with GBP‐Lifeact similar to above, but with both EYFP‐β‐catenin and Flag‐FMRP, and the cells were subjected to fluorescent microscopy and immunofluorescent staining for Flag (red). Scale bars, 20 μm.
- A10 cells were transfected as in (D) with GBP‐lamin B1. Scale bars, 20 μm.
To further document this interaction, we performed a series of GST pull‐down assays using purified, bacterially produced GST‐β‐catenin as bait, and purified 6xHis‐FMRP as prey. Since GST‐β‐catenin but not GST alone interacted with FMRP, the physical interaction is direct. Both the N terminus (aa 1–106) and armadillo repeat region (aa 120–683) of β‐catenin were sufficient to interact with FMRP independently (Fig 3C‐i). As FMRP is an RNA interacting protein, we wanted to ascertain whether RNA scaffolding might mediate the interaction. To test this, we performed a series of coIP assays with and without the addition of RNase A. The addition of RNase A to the cell lysates (10 ng/μl) during overnight incubation with the antibody‐conjugated beads did not affect either β‐catenin or FMRP immunoprecipitation, or their capacity to interact with each other (Fig 3C‐ii). RNA was extracted from parallel cell lysates to confirm complete RNA degradation by addition of RNase. Based on these data, we conclude that β‐catenin directly interacts with FMRP and that the protein interaction is RNA independent.
Next, to further assess the β‐catenin/FMRP interaction, we used an interaction trap in a cellular system using a 3‐way fluorescence hybridization assay based on GFP‐nanotrap technology 23. Briefly, the general principle of this system is that a GFP binding protein (GBP), a fragment of llama single‐chain antibody (13 kDa) specific for GFP 23, is anchored to a distinctive subcellular site by fusing it to a specific anchor protein (e.g., lamin B1 to the nuclear lamina or Lifeact peptide to cytosolic F‐actin). GFP/EYFP fused to the protein of interest is introduced into the cell as a “bait”, and the GFP/EYFP‐fusion protein is then recruited to the specific subcellular location by a high‐affinity interaction between GBP‐GFP/EYFP, thus establishing the protein “trap”. If a candidate protein interacts with the bait, the candidate protein is therefore enriched at the specific anchored site. This can be visualized by labeling the candidate protein with a fluorescent tag for in vivo live‐cell imaging or by immunofluorescence using an antibody targeting the candidate. This system provides a highly robust assay of the protein–protein interaction in a cellular system. We used two different anchor sites for GBP: fused with Lifeact for cytosolic F‐actin and lamin B1 for the nuclear lamina. Lifeact is a 17 amino acid peptide fragment from the actin binding protein 140 (Abp140) of S. cerevisiae 24. The Lifeact peptide has been previously shown to bind to F‐actin with high affinity while having a minimal effect on actin and microtubule dynamics 25. Lamin B1 is an integral component of the nuclear lamina 26.
Next, we ectopically expressed GBP‐Lifeact and EYFP‐β‐catenin and checked for EYFP localization (green) to actin filaments. Lifeact localized to the actin filaments marked by Alexa Fluor 633 tagged phalloidin (blue), and we concomitantly observed staining with anti‐β‐catenin antibody (Alexa Fluor 546, red) indicating that it was recruited to the GBP‐Lifeact. A line scan for RGB profile reveals matching peaks for all three fluorescent signals (Appendix Fig S5). Consistent with these data, A10 cells transfected with GBP‐Lifeact and EYFP‐β‐catenin resulted in distinct β‐catenin localization to F‐actin, while Flag‐FMRP or mCherry‐β‐catenin was not recruited to the F‐actin when expressed alone (Fig 3D, top panels). However, ectopic expression of all three factors together—GBP‐Lifeact, EYFP‐β‐catenin, and Flag‐FMRP—resulted in a readily observable FMRP localization to F‐actin, and precise overlap with EYFP‐β‐catenin (Fig 3D, bottom panels). These data indicate a robust interaction between β‐catenin and FMRP in live cells.
Next, we anchored EYFP‐β‐catenin to the inner nuclear membrane using GBP‐lamin B1. In A10 cells, EYFP‐β‐catenin was recruited to the nuclear envelope where GBP‐lamin B1 was anchored. Ectopic expression of GBP‐lamin B1 and Flag‐FMRP or mCherry‐β‐catenin alone resulted in a dispersed localization (Fig 3E, top panels). However, similar to the results observed with Lifeact, when co‐expressed together, GBP‐lamin B1, EYFP‐β‐catenin, and Flag‐FMRP resulted in a striking re‐localization of Flag‐FMRP to the nuclear membrane (Fig 3E, bottom panels). We also performed control experiments to ensure that EYFP and Flag themselves were not responsible for this observed interaction (described in Appendix Fig S6). Taken together, these data indicate a robust interaction between β‐catenin and FMRP.
β‐Catenin represses translation and associates with the pre‐initiation complex
Based on the FMRP:β‐catenin interaction and perinuclear co‐localization, we tested the novel possibility that β‐catenin might regulate FMRP function at the translation machinery. We therefore tested two possibilities: (i) whether β‐catenin is a part of the pre‐initiation complex with FMRP and (ii) whether global translation was affected by manipulation of β‐catenin protein levels.
To test the first possibility, we examined the distribution of β‐catenin using polysome profiling by sucrose density gradient centrifugation (10–50%) in HEK 293T cells with and without FMRP depletion. Under normal conditions, β‐catenin protein, indeed, co‐fractionated with messenger ribonucleoprotein complexes (mRNPs) with sedimentation values ranging from 40S to 60S, while FMRP and eukaryotic initiation factor 4E (eIF4E) both co‐sediment in the 40S to 80S range (Fig 4A, left panel). siRNA‐mediated depletion of FMRP results in a corresponding decrease in its protein level in the polysomes along with a reduction in β‐catenin co‐sedimentation, as it is no longer present in fractions containing mRNPs (Fig 4A, right panel). Based on these results, we hypothesized that β‐catenin, FMRP, and eIF4E may reside in a common complex.
Figure 4. β‐Catenin and FMRP are co‐localized in the pre‐initiation complex.
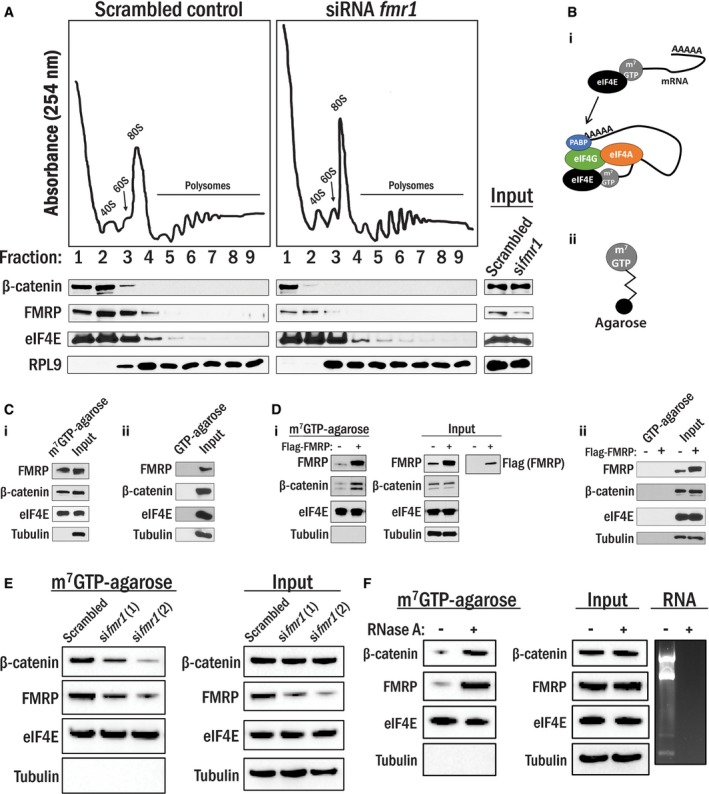
- Co‐sedimentation of protein/RNA complexes on a 10‐50% sucrose gradient. Cell lysates generated from the cells transfected with either siRNA targeting fmr1 or its scrambled control were ultracentrifuged in sucrose gradients; peaks corresponding to the 40S and 60S subunits, 80S monosome, and polysomes were detected by UV absorbance at 254 nm, and indicated proteins in these fractions were detected by Western blot. Corresponding total cell lysate was used as input.
- (i) eIF4E preferentially binds to the 5′cap (m7GTP) of mRNAs and recruits the pre‐initiation complex to that site. (ii) m7GTP‐agarose beads.
- (i) Precipitated proteins in A10 cell lysates with m7GTP‐agarose beads were identified by Western blot analysis. eIF4E and tubulin were shown as positive and negative controls, respectively. Total lysates were used as input. (ii) A10 cell lysates were incubated with GTP‐agarose beads, and none of the proteins tested were precipitated with the beads. Total lysates were used as input control.
- (i) HEK 293T cells were transfected with empty vector or Flag‐FMRP, and lysates were subjected to m7GTP‐agarose pull‐down as in (C). (ii) HEK 293T cells were transfected with empty vector or Flag‐FMRP, and lysates were subjected to GTP‐agarose pull‐down as in (i).
- HEK 293T cells were transfected with either siRNAs targeting fmr1 or scrambled control and were subjected to m7GTP‐agarose pull‐downs as in (C).
- HEK 293T cells lysates were subjected to m7GTP‐agarose pull‐downs as in (C) in the presence or absence of RNase A (10 μg/ml). RNA was extracted from parallel lysates, and RNA content was analyzed by agarose gel electrophoresis.
Next, we isolated the pre‐initiation complex using a well‐characterized m7GTP‐agarose bead pull‐down assay. Since eIF4E interacts with the 7‐methylguanylate cap (m7G) of mRNA with high affinity to initiate translation, cell lysates can be incubated with m7GTP‐agarose beads to enrich for the eIF4E complex and other associated proteins (Fig 4B) 27; this technique has previously been used to assess FMRP in the pre‐initiation complex 28. In our analysis with A10 smooth muscle cells, endogenous β‐catenin but not tubulin was, in fact, detected in the pre‐initiation complex along with FMRP. Furthermore, a control experiment using GTP‐agarose beads without the m7G modification did not produce any interactions between the beads and indicated proteins (Fig 4C‐i and ii, respectively). Moreover, in HEK 293T cells, forced expression of Flag‐FMRP results in an increased association of endogenous β‐catenin with the eIF4E pre‐initiation complex without affecting β‐catenin expression, while again, no proteins interacted with the GTP‐agarose control beads (Fig 4D‐i and ii, respectively). Loss of FMRP protein by siRNA‐mediated silencing resulted in a corresponding reduction in the association of β‐catenin to the complex, again without affecting β‐catenin expression (Fig 4E). Interestingly, when we treated lysates with RNase A, there was an increase in both β‐catenin and FMRP association with the m7GTP beads (Fig 4F). Therefore, in agreement with above, β‐catenin and FMRP specifically associate with the m7GTP‐agarose beads without RNA but not with GTP‐agarose beads. A potential reason for the increasing amounts of β‐catenin and FMRP associated with the m7GTP beads with RNase A might be due to liberating FMRP from mRNA and allowing more FMRP to interact with the beads, while also recruiting β‐catenin. These results indicate that β‐catenin associates with the pre‐initiation complex in an FMRP‐dependent manner, possibly contributing to regulation of mRNA translation.
To test the second possibility, we utilized a technique known as surface sensing of translation (SUnSET), which measures global mRNA translation using puromycin incorporation into newly synthesized peptides coupled with their detection by a monoclonal puromycin antibody (Fig EV1). This method of measuring protein synthesis is comparable to classical radioactive methods 29. The utility of this method is depicted in Fig EV2, in which we pre‐treated HEK 293T cells with various concentrations of cycloheximide for 4 h to inhibit protein translation, followed by a pulse treatment with puromycin (0.5 μM) for 15 min which incorporates puromycin into the actively elongating peptides which are then detected by Western blot analysis. We then performed a series of β‐catenin silencing experiments using two different siRNAs in primary VSMCs, A10, and HEK 293T cells. We depleted β‐catenin expression by siRNA or scrambled siRNA as a control, and then, the cells were subjected to a 15 min pulse of puromycin treatment (0.5 μM). Results in each experiment indicated that depletion of β‐catenin discernibly increased translation in all three cell types tested compared to the control condition (Fig 5A), suggesting that β‐catenin has a previously uncharacterized function in translational regulation.
Figure EV1. Depiction of the SUnSET technique.
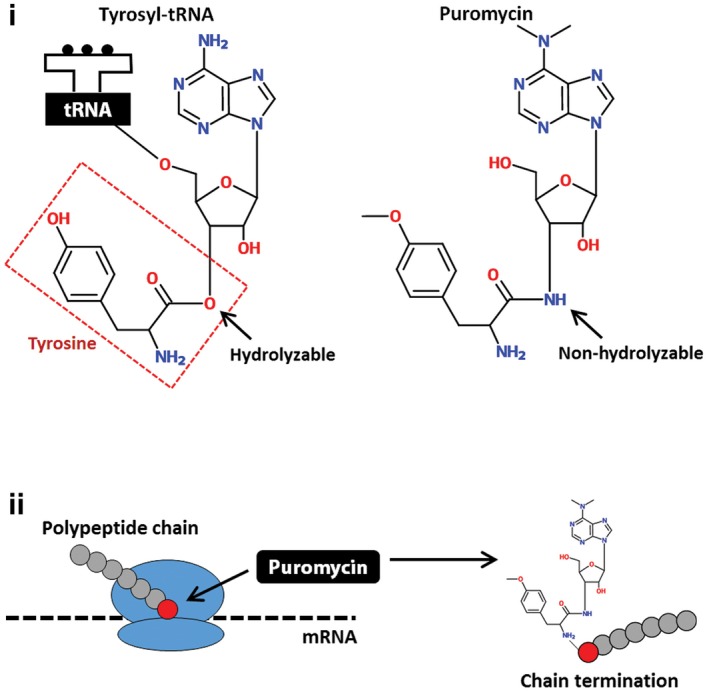
(i) Puromycin is structurally similar to tyrosyl‐tRNA, but contains a non‐hydrolyzable amide bond where tyrosine is normally cleaved from the tRNA complex and incorporated into a polypeptide chain. (ii) Puromycin is added to a growing polypeptide chain and terminates elongation of the peptide chain synthesis.
Figure EV2. Inhibition of translation by cycloheximide assessed by SUnSET assay.
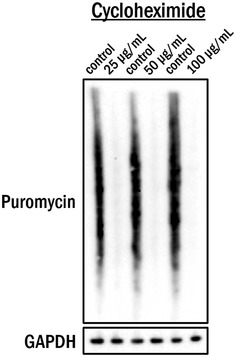
HEK 293T cells were treated with indicated concentrations of cycloheximide or solvent (water) for 4 h prior to incubation with 0.5 μM puromycin for 15 min. Cell lysates were subjected to Western blot analysis for detection of incorporated puromycin. GAPDH was used as a loading control.
Figure 5. β‐Catenin inhibits translation.
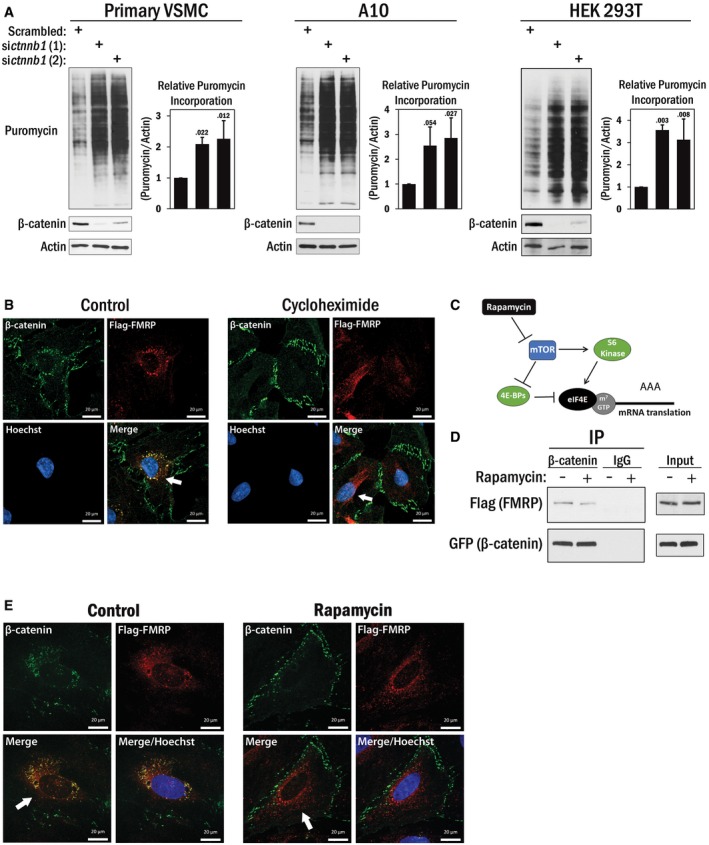
- Primary VSMCs (left panel), A10 (middle panel), and HEK 293T cells (right panel) were transfected with siRNAs targeting ctnnb1 (β‐catenin) or scrambled RNA. After 36 h, the cells were pulsed with 0.5 μM puromycin for 15 min and then harvested. The puromycin incorporated peptides were detected by Western blot analysis with puromycin antibody. β‐Catenin blots indicated efficacy of siRNA, and actin was used as a loading control. The average puromycin/actin ratio (n = 3) is shown in the graphs to the right of each cell type, and the error bars indicate standard deviation. A one‐way ANOVA and Tukey post hoc test were performed, with P‐values indicated above the error bars.
- A10 cells were transfected with Flag‐FMRP then treated with either cycloheximide or its solvent (DMSO) for 4 h prior to fixation. β‐Catenin (green) and Flag (FMRP, red) were visualized by immunofluorescence. Nucleus (blue) was identified by DNA staining with Hoechst 33342. Arrows indicate a loss of β‐catenin signal from the perinuclear region that was seen in the control following cycloheximide treatment. Scale bars, 20 μm.
- Schematic depiction of rapamycin‐mediated inhibition of cap‐dependent translation.
- A10 cells were transfected with Flag‐FMRP as above and then pre‐treated with rapamycin or solvent (DMSO) for 1 h in serum‐free media. Serum was added for 2 h before the cells were harvested and subjected to coIP analysis using β‐catenin antibody. Non‐programmed rabbit IgG was used as a control. Existence of Flag‐FMRP in the immunocomplex was detected by Flag antibody and β‐catenin by GFP antibody.
- A10 cells were transfected with Flag‐FMRP followed by rapamycin treatment similar to (D). Cells were fixed and stained for β‐catenin (green), Flag (FMRP, red), and nucleus (blue). A loss of β‐catenin signal from the perinuclear region that was seen in the control is indicated by the arrows. Scale bars, 20 μm.
Source data are available online for this figure.
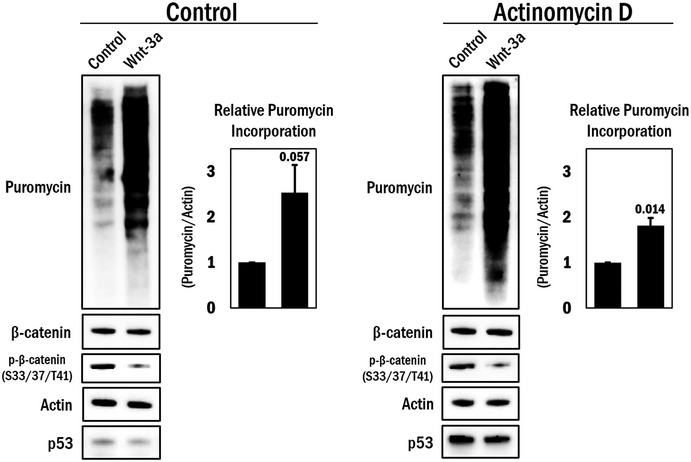
Finally, since β‐catenin's function as a transcriptional co‐regulator is well established, we attempted to clarify whether the increased puromycin incorporation following β‐catenin loss may be partly due to changes in transcription rather than a translational de‐repression. To address this, we performed the SUnSET assay under conditions in which we transiently inhibited transcription using actinomycin D, in conjunction with β‐catenin silencing. As was indicated in Fig 5A, loss of β‐catenin noticeably enhanced translation in the control (solvent) condition as previously observed; however, concurrent transcriptional inhibition did not substantially alter this effect (Fig EV3). P53 was used as an indicator of actinomycin D activity, which is stabilized due to transcriptional repression of mdm2 30, 31. Based on these data, we conclude that, at least under the conditions of this assay, the changes in translation when β‐catenin is depleted are primarily due to a de‐repression of translation and are not appreciably due to β‐catenin's transcriptional function.
Figure EV3. De‐repression of β‐catenin‐mediated translation repression is independent of transcription.
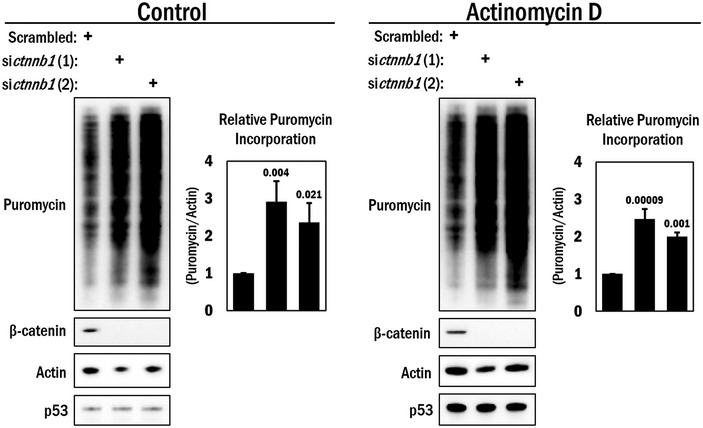
A10 cells were transfected with siRNAs targeting ctnnb1 (β‐catenin) or scrambled RNA (control) and 36 h later treated with either actinomycin D (0.5 μg/ml) or DMSO for 4 h. Before harvesting, the cells were pulsed with 0.5 μM puromycin for 15 min and subjected to Western blot analysis for detection of the puromycin incorporated peptides. β‐Catenin blots indicated efficacy of siRNA. Actin was used as a loading control, and p53 was used as a control for actinomycin D activity. The average puromycin/actin ratio is shown in the graphs to the right (n = 3). A one‐way ANOVA and Tukey post hoc test were performed, with P‐values indicated above the error bars.Source data are available online for this figure.
Cycloheximide treatment disrupts cytoplasmic β‐catenin and FMRP co‐localization
Previous studies have linked FMRP to translational regulation by interaction with eIF4E through a unique 4E binding protein, cytoplasmic FMRP‐interacting protein 1 (CYFIP1) 28. Interestingly, CYFIP1 was also identified in our list of β‐catenin interacting proteins (Table EV1), further supporting the idea that β‐catenin might play a role in translation through FMRP complex formation. Next, we determined whether pharmacological inhibition of translation might affect the β‐catenin:FMRP interaction. In order to do this, we used the protein synthesis inhibitor cycloheximide, which reversibly prevents translational elongation in eukaryotes 32. To optimize the assay conditions, we transfected A10 cells with Flag‐FMRP, and 24 h later treated with cycloheximide (50 ng/μl) or solvent (DMSO) for a further period of up to 16 h. We documented relative protein levels of Flag‐FMRP and β‐catenin to determine at which time point translation was inhibited but ectopically expressed protein was not yet degraded (Appendix Fig S7). Based on these results, we then incubated Flag‐FMRP expressing cells with cycloheximide or its control (DMSO) for 4 h and then performed an immunofluorescence assay to determine the localization of Flag‐FMRP and β‐catenin. When compared to control conditions where Flag‐FMRP and β‐catenin were co‐localized at the perinuclear regions, cells in which translation was blocked resulted in a loss of co‐localization between endogenous β‐catenin and Flag‐FMRP (Fig 5B), suggesting that pharmacological interference with translation disassembles the FMRP:β‐catenin complex.
The β‐catenin: FMRP interaction is modulated by mTOR signaling
To address the β‐catenin:FMRP interaction from a different perspective, we reasoned that their interaction might be dependent on upstream mTOR signaling, which is known to be a potent regulator of cap‐dependent translation (Fig 5C) 33. Since FMRP functions in the pre‐initiation complex to regulate mRNA translation, and studies have suggested that mTOR signaling is hyperactive in fmr1‐deficient mice 34, we used the mTOR inhibitor rapamycin to determine whether the β‐catenin:FMRP interaction is altered when mTOR signaling is suppressed. Treatment with rapamycin resulted in a reduced interaction of β‐catenin with FMRP by coIP, without a change in total β‐catenin and FMRP expression levels (Fig 5D), and also a loss of perinuclear co‐localization of β‐catenin with FMRP (Fig 5E), further supporting the idea that pharmacological manipulation of cap‐dependent translation modulates the β‐catenin:FMRP interaction.
Wnt‐mediated nuclear translocation of β‐catenin enhances translation
It is well known that numerous inputs such as nutrient levels, mitogens and energy status can modulate pre‐initiation complex (PIC) assembly. It is therefore of note that multiple cellular signaling pathways converge on PIC regulation as reported for EGF, insulin‐stimulated S6K1. PIC is therefore a platform for signaling events on the ribosome. We therefore postulated that Wnt signaling, which fulfills a central role in the biology of many cell types, might also impinge on translational control. In particular, we hypothesized that sequestering β‐catenin away from the cytoplasm to the nucleus might thus de‐repress translation. This is a tractable hypothesis since Wnt treatment promotes an extensive re‐localization of β‐catenin to the nucleus 8. Therefore, to test this idea, we used purified Wnt‐3a to stimulate cells that were serum deprived and again assessed translation using puromycin incorporation. Upon activation of Wnt signaling, β‐catenin accumulation in the nucleus was confirmed by indirect immunofluorescence analysis. In conjunction, global translation increased by > twofold compared to the control; this effect was observed both in primary VSMCs and in A10 cells (Fig 6A and B, respectively). Repeating this experiment under conditions of transcriptional inhibition (actinomycin D) did not alter the translational de‐repression by Wnt stimulation (Fig EV4).
Figure 6. Wnt‐dependent cytoplasmic to nuclear translocation of β‐catenin de‐represses translation.
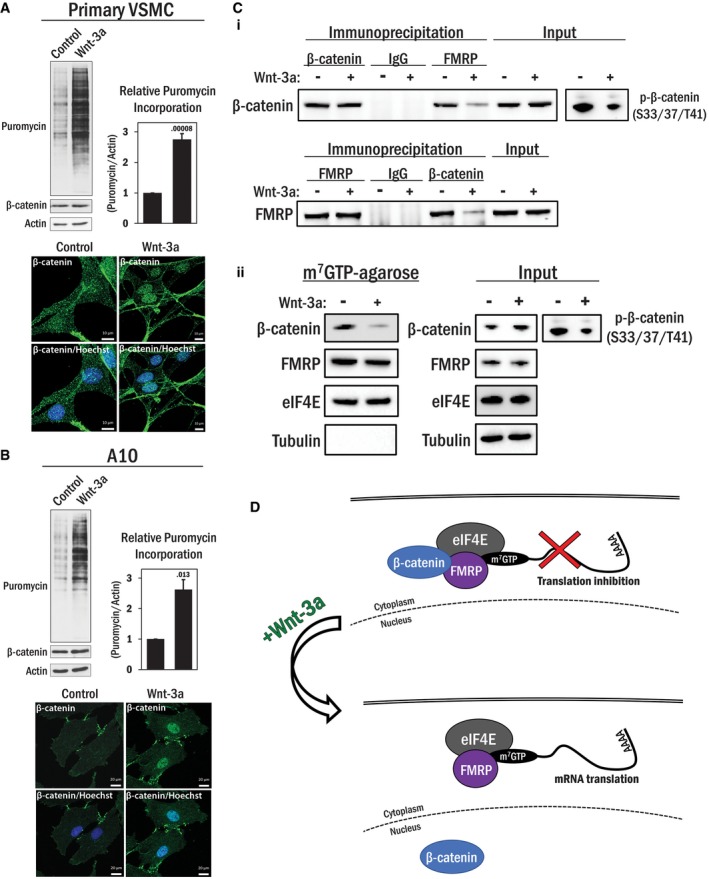
- (Top panel) Primary VSMCs were serum deprived overnight prior to stimulation with Wnt‐3a (100 ng/ml) or its solvent (0.1% BSA in PBS) for 4 h. The cells were treated with 0.5 μM puromycin for 15 min and harvested for Western blot analysis. Total β‐catenin levels are indicated, and actin was used as a loading control. The average puromycin/actin ratio (n = 3) was graphed. Error bars indicate standard deviation. An independent samples t‐test was performed (2‐tailed), with the P‐value indicated above the error bar. (Bottom panel) Cells were treated similarly to above and fixed and stained for β‐catenin (green) and nucleus (blue). Scale bars, 10 μm.
- The experiment in (A) was repeated using A10 cells. Scale bars, 20 μm.
- (i) HEK 293T cells were serum deprived for 1 h and then stimulated with Wnt‐3a (100 ng/ml) or its solvent (0.1% BSA in PBS) for 4 h. The cells were harvested and subjected to an endogenous coIP analysis using either β‐catenin or FMRP antibody. The corresponding proteins in the precipitated immunocomplex were detected by Western blot analysis. Non‐programed rabbit IgG was used as a control. Efficacy of Wnt‐3a treatment was assessed by inhibition of GSK3‐mediated phosphorylation of β‐catenin (S33/37/T41) by Wnt‐3a. (ii) HEK 293T cells were serum deprived for 1 h and then stimulated with Wnt‐3a (100 ng/ml) or its solvent (0.1% BSA in PBS) for 4 h. Cell lysates were then subjected to m7GTP‐agarose pull‐down analysis. eIF4E and tubulin were used as positive and negative controls, respectively, and phospho‐β‐catenin (S33/37/T41) Western blots were used as controls for Wnt‐3a stimulation.
- A working model for the relief of β‐catenin‐mediated translational repression by Wnt stimulation.
Source data are available online for this figure.
Figure EV4. De‐repression of the β‐catenin‐mediated translation repression by Wnt stimulation is independent of transcription.
A10 cells were serum deprived overnight prior to 1 h pre‐treatment with either actinomycin D (0.5 μg/ml) or its solvent (DMSO), and the cells were stimulated with Wnt‐3a (100 ng/ml) or its solvent (0.1% BSA in PBS) for 4 h. Before harvesting for Western blot analysis, the cells were further treated with 0.5 μM puromycin for 15 min. Total β‐catenin and phospho‐β‐catenin (S33/37/T41) levels indicate effective Wnt‐3a stimulation. P53 was used as a control for actinomycin D activity, and actin was used as a loading control. The average puromycin/actin ratio was graphed (n = 3). Error bars indicate standard deviation. An independent samples t‐test was performed (two‐tailed), with the P‐value indicated above the error bar.Source data are available online for this figure.
In addition, parallel biochemical analysis revealed that Wnt‐3a stimulation reduced the interaction between β‐catenin and FMRP, compared to unstimulated cells despite a moderate increase in total β‐catenin expression levels (Fig 6C‐i). β‐Catenin association with the pre‐initiation complex was also diminished with Wnt‐3a treatment, while there was no change in FMRP association with m7GTP beads (Fig 6C‐ii). Therefore, while part of the effect of Wnt‐3a mediated translational increases that we observe may be due to transcriptional effects, a substantial fraction of the observed effects are directly attributed to translational de‐repression.
Collectively, these data demonstrate that sequestration of cytoplasmic β‐catenin into the nucleus by Wnt signaling contributes to a de‐repression of translation. We contend that this observation could account for a previously unrecognized amplification of the effect of Wnt signaling since it would influence the cellular proteotype by not only enhancing β‐catenin‐mediated transcription (canonical effect) but also generally increasing translation of the RNA complement of the cell.
Discussion
In this β‐catenin interactome study, we have identified a novel cytoplasmic β‐catenin binding partner, FMRP. This interaction underpins a previously unknown function of β‐catenin at the pre‐initiation complex as a translational regulator. β‐Catenin has previously been extensively characterized and established as an indispensable multifunctional protein that has no paralog 35. For example, it is a key member of the cadherin cell adhesion complex and provides a link between the cytoplasmic tail of cadherin and the actin cytoskeleton 36. In addition, β‐catenin's C‐terminal transactivation domain makes it a critical nuclear transcriptional co‐activator as a downstream effector of canonical Wnt signaling 5. The divergent roles and localization of β‐catenin make it one of a few “jack‐of‐all‐trades” proteins, and one whose function is primarily determined by which proteins it interacts with. Studies have suggested that the major mechanism of β‐catenin localization is retention by protein interactions rather than by regulated shuttling, which is corroborated by the fact that β‐catenin does not contain any well‐characterized localization signals in its primary sequence 8, 37, 38. Here, we present evidence indicating a hitherto unrecognized role for β‐catenin in translational repression, which may have important implications for our mechanistic understanding of canonical Wnt signaling.
A hallmark of Wnt signaling is the dramatic nuclear translocation of β‐catenin and subsequent transcriptional induction of a variety of Wnt‐dependent target genes. This results in substantive cellular phenotypic changes in many developmental systems as well as altered properties of some cancer cells 39, 40, 41. Our data suggest that in addition to promoting the transcription of Wnt‐dependent target genes, β‐catenin translocation into the nucleus may, in so doing, de‐repress the translational machinery. Thus, we suggest a revised model for β‐catenin's function in baseline conditions and in response to Wnt signaling which involves translational regulation in addition to its well‐known role in transcriptional induction. In brief, we propose that in the absence of Wnt stimulation, β‐catenin functions both at the membrane, in association with cadherins, to regulate cell adhesion and concomitantly at the translational machinery to repress translation in association with FMRP. Upon Wnt activation, β‐catenin re‐localizes to and accumulates in the nucleus to activate target gene transcription and, by its sequestration to the nucleus, simultaneously dissociates from the pre‐initiation complex to de‐repress translation (Fig 6D). Our model purports a dual function on both transcriptional induction and translational de‐repression that results in a compound impact on Wnt‐dependent gene expression. The subcellular localization of β‐catenin, which is dictated by protein–protein interactions, is therefore an important determinant of its impact on transcription and translation. Thus, we conclude that the cytoplasmic pool of β‐catenin/FMRP impacts the regulation of translation. It is noteworthy that several studies have indicated that some neurodegenerative effects of fragile X syndrome (FXS), the genetic disease resulting from suppression of FMRP expression, can be ameliorated by pharmacological inhibition of GSK3, which ordinarily mediates the APC dependent degradation of β‐catenin 42. Whether these observations are causally linked remains to be tested.
While the cytoplasmic accumulation of β‐catenin in response to GSK3 inhibition by Wnt simulation has been mechanistically implicated in the increased nuclear levels of β‐catenin in which a “spillover” mechanism is proposed, we recently reported that this model, at least in one context, is over‐simplified since β‐catenin nuclear accumulation is dependent on a p38 MAPK‐dependent phospho‐interaction with MEF2 in vascular smooth muscle cells 8. In the studies reported here, the mechanism by which β‐catenin dissociates from the pre‐initiation complex in response to Wnt stimulation remains to be fully determined. Based on our previous studies, the dissociation of the β‐catenin/FMRP complex may be dependent on post‐translational modifications of β‐catenin and/or FMRP that leads to disruption of the interaction interface.
Characterization of FMRP function is as yet incomplete, and two well‐characterized RNA motifs mediate FMRP‐dependent translational repression. More recently, FMRP has also been implicated as a positive translational regulator of superoxide dismutase 1 mRNA through a novel RNA stem loop motif 43. The actual number of direct RNA cargoes with which FMRP and the related FXR1 protein interact has been a subject of some debate and a detailed discussion of this topic is provided elsewhere 44. Moreover, it has been speculated that the FMRP/FXR1 RNA cargoes may also vary from one cell type to another. The effects of the β‐catenin:FMRP/FXR1 interaction on protein synthesis, as we have documented here, may be far more profound since many of the protein products of the direct RNA binding targets of FMRP/FXR1 are involved in a myriad of cellular processes, in particular cellular signaling and gene/protein expression 44. Thus, alteration in the protein level expression of these direct FMRP mRNA cargoes will ultimately result in a potentially vast change in the cellular proteome due to the compound effects of the direct RNA binding along with the downstream consequences on gene expression of those changes. If β‐catenin participates in an FMRP/FXR1 interaction with its primary RNA cargo, then the impact on protein expression, as we have monitored by the SUnSET assay in β‐catenin‐depleted cells, might extend much further than the sum of the direct FMRP RNA binding targets. In support of this idea is a published quantitative proteomics study using SILAC which reported that the levels of 5,023 proteins were altered in fmr1 KO cell lines 45. Thus, the impact of alteration in the direct FMRP target RNAs (reportedly approximately 4% of the RNAs in neurons 46) has a potentially much larger downstream effect on the cellular proteome, possibly affecting the protein levels of a large fraction of the proteome (this could possibly be as high as 40—50% of the proteome). A more general mechanism of FMRP‐mediated translational repression has been implicated in a study in which an FMRP/CYFIP1 complex was shown to bind the translation initiation factor eIF4E 28. Interestingly, we also identified CYFIP1 in the β‐catenin interactome. In addition, FMRP has also been characterized to directly associate with polyribosomes causing reversible ribosome stalling in order to regulate translation 47. This is a complex issue that awaits more detailed characterization of how β‐catenin might influence FMRP target RNAs and, importantly, the downstream consequences of those altered translational events.
In summary, we have provided evidence of a protein–protein interaction between β‐catenin and FMRP that may serve to regulate translation. In VSMCs, although the modulation from quiescent to proliferative phases is essential for wound repair, mis‐regulation can also lead to vascular disease. Since β‐catenin plays an important role in this process, determining the mechanisms involved in this phenotypic switch could provide novel therapeutic targets in some vascular diseases such as atherosclerosis in which VSMC switching is fundamental. Furthermore, these observations may not be limited to a specific cell lineage due to the relatively ubiquitous expression of both β‐catenin and FMRP. Our data so far indicate that this interaction can occur in a number of different cell types, suggesting that β‐catenin might play an integral role in translational regulation. Our observation that the β‐catenin/FMRP complex can exert effects on translational control highlights a potentially important advance in our understanding of Wnt/β‐catenin signaling and also extends possibilities for therapeutic targeting of both Wnt‐ and FMRP‐dependent pathologies.
Materials and Methods
Cell culture and immunoprecipitation for LC‐MS/MS
Cell culture
A10 rat aorta vascular smooth muscle cells (ATCC CRL‐1476) were grown at 37°C in 5% CO2 in high glucose DMEM with 100 U/ml penicillin, 100 μg/ml streptomycin, 1.5 g/l sodium bicarbonate, and 10% (v/v) fetal bovine serum. Cells were harvested after two washes in PBS and immediately lysed in NP‐40 lysis buffer (0.5% NP‐40 v/v, 50 mM Tris–HCl pH 7.6, 150 mM NaCl, 100 mM NaF, 10 mM sodium pyrophosphate, 2 mM EDTA, 1 mM of Na3VO4, 1 mM phenylmethylsulfonyl fluoride, 1 μg/ml leupeptin, 1 μg/ml aprotinin, 1 μg/ml pepstatin A). Lysates were centrifuged for 10 min at 21,000 g at 4°C to remove insoluble pellets and the supernatants collected. Cell culture, collection, and lysis were performed in quintuplicate.
Antibody‐bead conjugation
5 μg anti‐β‐catenin antibody (Cell Signaling #9562) was immobilized to 50 μl of ImmunoCruz IP/WB Optima F agarose bead substrate (Santa Cruz Biotechnology sc‐45043) overnight at 4°C in 500 μl PBS with 0.1% Tween 20 (v/v). For negative controls, beads were incubated with rabbit serum (Sigma‐Aldrich R9133). Unbound antibody was washed away twice with 500 μl PBS.
Protein binding and elution
Antibody‐conjugated beads were incubated with A10 cell lysates on a nutator overnight at 4°C. The beads were washed twice with 500 μl NP‐40 lysis buffer and then twice with 500 μl of NP‐40 lysis buffer without NP‐40. Bound proteins were eluted for LC‐MS analysis in 500 μl of 0.5 M NH4OH on a nutator for 20 min at room temperature. Separate immunoprecipitation columns were used for each sample, and parallel pre‐immune rabbit serum controls were generated from separate biological replicates of A10 cells. Eluates were dried by a centrifugal concentrator and then dissolved in 100 mM NH4HCO3, and treated with 5 mM dithiothreitol at 60°C then 10 mM iodoacetamide at room temperature in the dark. The alkylated immunoprecipitates were incubated at 37°C overnight with 0.75 μg of trypsin each.
LC‐MS/MS
Sample run
Trypsin digests were dried in a centrifugal concentrator, dissolved in aqueous 0.1% (v/v) formic acid, and separated on a C18 column (15 cm long, 75 μm internal diameter, 3 μm diameter stationary phase, 120 Å pore size) by HPLC over a 60 min 300 nl/min acetonitrile gradient. The HPLC system consisted of a NanoLC‐Ultra 2D pump with a Nanoflex cHiPLC system (AB Sciex, Concord, ON, Canada). Mobile phases A and B were 0.1% formic acid (v/v) in water and acetonitrile, respectively (Honeywell, Morristown, NJ, USA). The following mobile‐phase B concentrations were used: 2% (v/v) at 0 min to 35% at 35 min to 80% from 35.5 to 38.5 min to 2% from 39 to 60 min. Mass spectra of the HPLC eluate were collected on an Orbitrap Elite mass spectrometer (Thermo Scientific, Waltham, MA). Precursor ion scans were collected on the Orbitrap at a resolution of 30,000, and the linear ion trap was used for MS/MS. Tandem mass spectrometry was performed using normalized collision energy of 35, activation Q of 0.250, 2 Da isolation width, and a 10‐ms activation time. Dynamic precursor ion exclusion with a repeat count of 1 and an exclusion mass width of 10 ppm was applied. A 10‐s dynamic exclusion duration and 20‐s repeat duration were used. Between each pair of samples, three blank runs were acquired under identical instrument parameters in order to control analyte carryover.
Peptide score
MS/MS data were searched against the UniProt Rattus norvegicus sequence library (September 18, 2013 edition downloaded from the UniProt Knowledge Base on September 26, 2013) with the SEQUEST algorithm navigated through Proteome Discoverer™ software version 1.3. Search settings were as follows: precursor mass range: 350–5,000 Da; minimum peak count: 1; enzyme: trypsin; maximum missed cleavages: 1; precursor ion tolerance: 10 ppm, product ion tolerance: 0.4 Da, product ion series considered: b and y; dynamic modifications: methionine oxidation (+15.995 Da), phosphorylation (S, T, and Y at +79.966 Da), glutamine deamidation (+0.984 Da) and tryptophan dioxidation (+31.990); static modification: cysteine carbamidomethylation (+57.021 Da). Cutoff scores at the peptide level were determined by automated false discovery rate (FDR) estimation. The source data are provided for the results of the AP‐LC‐MS/MS experiments.
Gene ontology enrichment analysis
Proteins common to anti‐β‐catenin column eluates and pre‐immune rabbit serum controls were removed from potential β‐catenin binding partners. The interactome was summarized by the Software Tool for Researching Annotations of Proteins (STRAP, v1.5) program using each individual protein's annotation in the gene ontology (GO) cellular localization domain 48.
Next, an enrichment analysis of these proteins using the Cytoscape program 49 and ClueGO plug‐in 50 was performed. We employed a right‐sided hypergeometric statistical test, corrected using the Bonferroni step‐down method to determine enriched biological process GO terms from levels 3 to 8. We specified a minimum of five genes comprising at least 2% of a particular GO term to be the cutoff for its inclusion. The proportion of proteins that were shared among pairs of GO terms was determined using the Kappa statistic. This was visualized as edges connecting two nodes together and had a minimum Kappa score of 0.5. Thicker edges signified higher Kappa score values.
We utilized the CluePedia plugin 51 to create a visual network of the enriched GO terms using the terms with the highest number of statistically significant proteins. We then used the most significant GO terms to create another network visualizing the specific proteins associated with particular term(s).
Cell culture, protein extraction, and Western blot analysis
Cells were collected as described above. A10 and HEK 293T cells were obtained from American Type Culture Collection (ATCC, CRL‐1476, and CRL‐3216, respectively). Primary vascular smooth muscle cells were isolated and cultured from mice aortae as previously described 52. Cells were tested for mycoplasma contamination by Hoechst staining. Primary VSMCs and A10 cells were used for the majority of the experiments, where A10 cells were used primarily when it was not possible to achieve the scale required for experimentation by primary cultures. HEK 293T cells were used as a neutral cell background for over‐expression of exogenous proteins, basic biochemical assays, and polysome fractionation (as this procedure was optimized for HEK 293T cells). Cells were grown in serum‐free DMEM overnight for the specified experiments. Extracted proteins were denatured in SDS loading buffer at 95°C for 5 min then run on 10% SDS–PAGE, transferred to a PVDF membrane (Millipore), and blocked in 5% skim milk (w/v) for 1 h prior to primary antibody incubation at 4°C overnight. After brief washes with TBS, the blot was incubated with corresponding HRP‐conjugated secondary antibody in 5% milk in TBS for 1 h at room temp. Expression levels of indicated proteins were recorded by exposing the blot to film (GE Healthcare) or by digital imaging (Bio‐Rad XRS+).
Transfections
For ectopic protein expression and small interfering RNA (siRNA) experiments, cells were transfected with Lipofectamine 2000 (Life Technologies) using instructions provided by the manufacturer and harvested or drug treated 24–48 h later, unless otherwise indicated.
Gene silencing
MISSION siRNA (Sigma‐Aldrich) for rat and mouse ctnnb1 (β‐catenin, SASI_Rn01_00099923 and SASI_Rn01_00099924), human ctnnb1 (SASI_Hs01_00117960 and SASI_Hs01_00117958), human fmr1 (FMRP, SASI_Hs01_00139633 and SASI_Hs01_0013967), and universal scrambled siRNA (SIC001) were used at a concentration of 25 nM.
Antibodies and reagents
Actin (sc‐1616), GFP (sc‐9996), Plectin (sc‐33649), and dsRed (sc‐33354) antibodies were purchased from Santa Cruz. β‐Catenin (#9562), FMRP (#4317), eIF4E (#9742), and tubulin (#3873) antibodies were obtained from Cell Signaling Technology. Flag (F1804) and glutathione‐S‐transferase (GST) (G7781) antibodies were purchased from Sigma‐Aldrich. Puromycin antibody (PNY‐2A4) was obtained from the developmental studies hybridoma bank (DSHB). GAPDH antibody was purchased from Novus Biologicals (NB300‐328H).
Cycloheximide (CYC003) and puromycin dihydrochloride (PUR333) were purchased from BioShop. Rapamycin (#9904) was purchased from Cell Signaling Technologies. Purified Wnt‐3a (315‐20) was purchased from PeproTech. RNase A was purchased from ThermoFisher (EN0531).
Co‐immunoprecipitation
Cells were harvested and protein was extracted as described above. Immunoprecipitation was performed using the ImmunoCruz Optima or protein‐G Plus agarose kit (Santa Cruz Biotechnology), according to the manufacturer's instructions. Eluates were analyzed by Western blot, as described above.
Immunofluorescence
Cells were fixed in 4% paraformaldehyde in PBS for 5 min on ice then 10 min at room temp and then permeabilized with ice cold 90% methanol in water for 1 min on ice. Cells were blocked using 10% goat serum in PBS for 30 min at room temp and incubated overnight with primary antibody diluted (1:100–1:500) in 10% goat serum in PBS at 4°C. Cells were washed three times with PBS (10 min each) and incubated with corresponding secondary antibody conjugated with tetramethylrhodamine (TRITC) or fluorescein isothiocyanate (FITC) (Sigma‐Aldrich) diluted (1:500) in 10% goat serum for 2 h at room temperature. Nucleus was counter‐stained with Hoechst 33342 DNA dye (ThermoFisher, 1 μg/ml) for 15 min. Cells were washed three times with PBS (10 min each) and left in PBS at room temperature for imaging. Images were captured using a Carl Zeiss Axio Observer Z1 with Photometrics Evolve 512 EMCCD Camera. Fluorescence images were acquired under 40× (EC Plan‐Neofluar, 1.30 NA in oil) or 63× (Plan‐Apochromat, 1.40 NA in oil) objectives. Z‐stack images were rendered as orthogonally projected with maximum intensity using ZEN imaging software (Zeiss).
Polysome analysis
Prior to harvesting, cells were pre‐treated with 0.1 mg/ml cycloheximide in water for 5 min, and cell pellets were lysed in hypotonic buffer [5 mM Tris–HCl pH 7.5, 2.5 mM MgCl2, 1.5 mM KCl, EDTA‐free protease inhibitor cocktail (Sigma), 0.1 mg/ml cycloheximide, 2 mM DTT, 100 units RNase inhibitor (Sigma), 0.5% Triton X, and 0.5% sodium deoxycholate] then centrifuged at 21,000 g for 5 min at 4°C. Supernatants were added to sucrose gradients (10–50%) and ultracentrifuged at 30,000 g for 3 h at 4°C. Gradients were then chased with 60% sucrose and absorbance measured at 254 nm, and fractions were subjected to Western blot analysis.
m7GTP‐agarose pull‐down
50 μl immobilized γ‐aminophenyl‐m7GTP(C10‐spacer)‐agarose or γ‐aminooctyl‐GTP(C8‐spacer)‐agarose (Jena Bioscience AC‐155S and AC‐106S, respectively) slurry was washed three times in TBS and incubated with prepared cell lysates overnight at 4°C with gentle agitation. Beads were washed once with TBST (0.1% Tween‐20) then twice with TBS. Samples were heated with SDS loading buffer at 95°C for 5 min to elute from the beads and run on a 10% SDS–PAGE and subjected to Western blot analysis.
GST pull‐down
GST‐β‐catenin and 6xHis‐FMRP fusion proteins were extracted and purified using standard protocols. Briefly, pGEX‐β‐catenin or pET28a‐FMRP vectors were transformed in BL21 E. coli, and the fusion protein expressions were induced with 0.4 mM isopropyl β‐D‐1‐thiogalactopyranoside (IPTG) overnight at 16°C. The bacteria were sonicated and the GST‐ or 6xHis‐fusion proteins were purified with glutathione‐agarose (Sigma‐Aldrich) or nickel‐agarose (Qiagen) beads, respectively. Protein concentrations were determined in SDS–PAGE by a comparative estimation of Coomassie blue staining using the fixed bovine serum albumin (BSA) as a standard. 1 μg GST‐full length β‐catenin protein (or the molar equivalent of the smaller fragments), 2 μg 6xHis‐FMRP, and 25 μl glutathione‐agarose beads (50% slurry) were incubated in 600 μl NETN buffer (100 mM NaCl, 0.5 mM EDTA, 20 mM Tris–HCl (pH 8), 0.5% (v/v) NP‐40) overnight at 4°C. Beads were washed once with TBST and twice with TBS, and eluates were analyzed by Western blot.
Protein synthesis rates by puromycin incorporation and quantification
After initial treatment, cells were incubated with 0.5 μM puromycin for 15 min prior to harvesting or subjected to immunofluorescence analysis. Puromycin incorporation was visualized by Western blot or immunofluorescence using puromycin antibody. Relative puromycin incorporation by Western blot was quantified using ImageJ with the Fiji image processing package. The puromycin bands of a particular condition were traced in their entirety along with the corresponding actin band, and the following was performed: the integrated density of the area directly below the traced lanes was multiplied by the traced area of the protein of interest to calculate background intensity. The integrated density was determined after the background subtraction. The intensity of the puromycin incorporated proteins was normalized relative to the actin protein intensity. Three biological replicates were performed, and the average puromycin intensity was graphed with error bars representing standard deviation. The source data are provided for all puromycin quantifications.
Plasmids
The ORF of the GFP binging peptide (GBP) was PCR‐amplified with Fwd‐primer (ATAAGATTATGGCCGACGTCCAG) and Rev‐primer (ATGGATCCACCTCCACCTCCACCTC) using pCGA‐GBP1‐10gly‐Gal4DBD vector (Addgene #49438) as a template. The amplified ORF was digested with HindIII and BamHI and ligated into the pcDNA3 vector (pcDNA3‐GBP) (Invitrogen). Lamin B1 ORF was PCR‐amplified with Fwd‐primer (ATGGATCCGCGACTGCGACCCCCGTGCCGCCGCGGATG) and Rev‐primer (ATCTCGAGTTACATAATTGCACAGCTTCTATTG) using a C2C12 cDNA library constructed by RT with oligo‐dT. The amplified DNA fragments were digested with BamHI and XhoI and ligated into pcDNA3‐GBP in a same reading frame for GBP‐lamin B1 fusion protein expression. Lifeact‐GBP was constructed by a 3‐way ligation technique with synthesized two double‐stranded DNAs (#1 reading strand; CTAGCATGGGTGTCGCTGACCTG ATCAAAAAG with #1 complimentary strand; CGAACTTTTTGATCAGGTCAGCGACACCGATG), (#2 reading strand; TTCGAGTCCATC TCGAAGGAAGAGC with #2 complimentary strand; TCGAGCTCTTCCTTCGAGATGGACT) and digested pcDNA3 (NheI/XhoI), and PCR‐amplified GBP‐ORF (Xho I/EcoR I) was further ligated for pcDNA3‐Lifeact‐GBP.
pFRT‐TODestFLAGHAhFMROiso1 (Flag‐FMRP) was a gift from Thomas Tuschl (Addgene plasmid #48690). For pet28a‐FMRP, Flag‐FMRP was used as a template and amplified using Fwd‐primer (CGCGGAATTCATGGAGGAGCTGGTGGTGGAAGTGCGGGGCT) and Rev‐primer (CGCGGCGGCCGCTTAGGGTACTCCATTCACGAGTGGTTGCTG), which was digested with EcoRI and NotI then ligated into pet28a vector (Invitrogen).
pGEX‐6P3‐β‐catenin (GST‐β‐catenin) constructs were a gift from Antonio Gracia de Herreros.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE 53 partner repository with the dataset identifier PXD010421.
Author contributions
SE designed and conducted the experiments and co‐wrote the manuscript. TM conducted experiments and provided feedback on the manuscript. MAB and JV collaborated on polysome analysis and interpretation. DW conducted experiments and provided feedback on M‐S analysis. JCM designed experiments, co‐wrote the manuscript, and secured funding.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 5A
Source Data for Figure 6A
Source Data for Figure 6B
Acknowledgements
The authors wish to thank Henrik Zetterberg for providing the space and support to perform additional experiments and Soma Tripathi for her assistance. This work was supported by the Gouvernement du Canada | Canadian Institutes of Health Research (CIHR) (102688 to J.C.M.). JCM is supported by the McLaughlin Research Chair, York University.
EMBO Reports (2018) 19: e45536
References
- 1. Roose J, Huls G, van Beest M, Moerer P, van der Horn K, Goldschmeding R, Logtenberg T, Clevers H (1999) Synergy between tumor suppressor APC and the β‐Catenin‐Tcf4 target Tcf1 . Science 285: 1923–1926 [DOI] [PubMed] [Google Scholar]
- 2. Chan E, Gat U, McNiff JM, Fuchs E (1999) A common human skin tumour is caused by activating mutations in β‐catenin. Nat Genet 21: 410–413 [DOI] [PubMed] [Google Scholar]
- 3. Galceran J, Miyashita‐Lin EM, Devaney E, Rubenstein JLR, Grosschedl R (2000) Hippocampus development and generation of dentate gyrus granule cells is regulated by LEF1. Development 127: 469–482 [DOI] [PubMed] [Google Scholar]
- 4. Logan CY, Nusse R (2004) The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol 20: 781–810 [DOI] [PubMed] [Google Scholar]
- 5. Yost C, Torres M, Miller JR, Huang E, Kimelman D, Moon RT (1996) The axis‐inducing activity, stability, and subcellular distribution of beta‐catenin is regulated in Xenopus embryos by glycogen synthase kinase 3. Genes Dev 10: 1443–1454 [DOI] [PubMed] [Google Scholar]
- 6. Xing Y, Takemaru K‐I, Liu J, Berndt JD, Zheng JJ, Moon RT, Xu W (2008) Crystal structure of a full‐length β‐catenin. Structure 16: 478–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huber AH, Nelson WJ, Weis WI (1997) Three‐dimensional structure of the Armadillo repeat region of β‐catenin. Cell 90: 871–882 [DOI] [PubMed] [Google Scholar]
- 8. Ehyai S, Dionyssiou MG, Gordon JW, Williams D, Siu KW, McDermott JC (2015) A p38 mitogen‐activated protein kinase‐regulated myocyte enhancer factor 2‐beta‐catenin interaction enhances canonical Wnt signaling. Mol Cell Biol 36: 330–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Moon RT, Kohn AD, De Ferrari GV, Kaykas A (2004) Wnt and β‐catenin signalling: diseases and therapies. Nat Rev Genet 5: 691–701 [DOI] [PubMed] [Google Scholar]
- 10. George SJ, Beeching CA (2006) Cadherin: catenin complex: a novel regulator of vascular smooth muscle cell behaviour. Atherosclerosis 188: 1–11 [DOI] [PubMed] [Google Scholar]
- 11. Jin T, George Fantus I, Sun J (2008) Wnt and beyond Wnt: multiple mechanisms control the transcriptional property of beta‐catenin. Cell Signal 20: 1697–1704 [DOI] [PubMed] [Google Scholar]
- 12. Slater SC, Koutsouki E, Jackson CL, Bush RC, Angelini GD, Newby AC, George SJ (2004) R‐cadherin: β‐catenin complex and its association with vascular smooth muscle cell proliferation. Arterioscler Thromb Vasc Biol 24: 1204–1210 [DOI] [PubMed] [Google Scholar]
- 13. Wang X, Xiao Y, Mou Y, Zhao Y, Blankesteijn WM, Hall JL (2002) A role for the β‐catenin/T‐cell factor signaling cascade in vascular remodeling. Circ Res 90: 340–347 [DOI] [PubMed] [Google Scholar]
- 14. Polakis P (2012) Drugging Wnt signalling in cancer. EMBO J 31: 2737–2746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhurinsky J, Shtutman M, Ben‐Ze'ev A (2000) Plakoglobin and β‐catenin: protein interactions, regulation and biological roles. J Cell Sci 113: 3127–3139 [DOI] [PubMed] [Google Scholar]
- 16. Gottardi CJ, Gumbiner BM (2001) Adhesion signaling: how β‐catenin interacts with its partners. Curr Biol 11: R792–R794 [DOI] [PubMed] [Google Scholar]
- 17. Heuberger J, Birchmeier W (2010) Interplay of cadherin‐mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb Perspect Biol 2: a002915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Song LN, Gelmann EP (2008) Silencing mediator for retinoid and thyroid hormone receptor and nuclear receptor corepressor attenuate transcriptional activation by the beta‐catenin‐TCF4 complex. J Biol Chem 283: 25988–25999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wiche G, Osmanagic‐Myers S, Castanon MJ (2015) Networking and anchoring through plectin: a key to IF functionality and mechanotransduction. Curr Opin Cell Biol 32: 21–29 [DOI] [PubMed] [Google Scholar]
- 20. Klaus A, Saga Y, Taketo MM, Tzahor E, Birchmeier W (2007) Distinct roles of Wnt/beta‐catenin and Bmp signaling during early cardiogenesis. Proc Natl Acad Sci USA 104: 18531–18536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Desai R, Sarpal R, Ishiyama N, Pellikka M, Ikura M, Tepass U (2013) Monomeric alpha‐catenin links cadherin to the actin cytoskeleton. Nat Cell Biol 15: 261–273 [DOI] [PubMed] [Google Scholar]
- 22. Giles RH, van Es JH, Clevers H (2003) Caught up in a Wnt storm: Wnt signaling in cancer. Biochem Biophys Acta 1653: 1–24 [DOI] [PubMed] [Google Scholar]
- 23. Rothbauer U, Zolghadr K, Muyldermans S, Schepers A, Cardoso MC, Leonhardt H (2008) A versatile nanotrap for biochemical and functional studies with fluorescent fusion proteins. Mol Cell Proteomics 7: 282–289 [DOI] [PubMed] [Google Scholar]
- 24. Asakura T, Sasaki T, Nagano F, Satoh A, Obaishi H, Nishioka H, Imamura H, Hotta K, Tanaka K, Nakanishi H et al (1998) Isolation and characterization of a novel actin filament‐binding protein from Saccharomyces cerevisiae . Oncogene 16: 121–130 [DOI] [PubMed] [Google Scholar]
- 25. Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M, Bradke F, Jenne D, Holak TA, Werb Z et al (2008) Lifeact: a versatile marker to visualize F‐actin. Nat Methods 5: 605–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Daigle N, Beaudouin J, Hartnell L, Imreh G, Hallberg E, Lippincott‐Schwartz J, Ellenberg J (2001) Nuclear pore complexes form immobile networks and have a very low turnover in live mammalian cells. J Cell Biol 154: 71–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Marcotrigiano J, Gingras A‐C, Sonenberg N, Burley SK (1997) Cocrystal structure of the messenger RNA 5′ cap‐binding protein (eIF4E) bound to 7‐methyl‐GDP. Cell 89: 951–961 [DOI] [PubMed] [Google Scholar]
- 28. Napoli I, Mercaldo V, Boyl PP, Eleuteri B, Zalfa F, De Rubeis S, Di Marino D, Mohr E, Massimi M, Falconi M et al (2008) The fragile X syndrome protein represses activity‐dependent translation through CYFIP1, a new 4E‐BP. Cell 134: 1042–1054 [DOI] [PubMed] [Google Scholar]
- 29. Schmidt EK, Clavarino G, Ceppi M, Pierre P (2009) SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods 6: 275 [DOI] [PubMed] [Google Scholar]
- 30. Dai M‐S, Zeng SX, Jin Y, Sun X‐X, David L, Lu H (2004) Ribosomal protein L23 activates p53 by inhibiting MDM2 function in response to ribosomal perturbation but not to translation inhibition. Mol Cell Biol 24: 7654–7668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen C‐S, Ho D‐R, Chen F‐Y, Chen C‐R, Ke Y‐D, Su J‐GJ (2014) AKT mediates actinomycin D‐induced p53 expression. Oncotarget 5: 693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Poetsch‐Schneider T, Ju J, Eyler DE, Dang Y, Bhat S, Merrick WC, Green R, Shen B, Liu JO (2010) Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat Chem Biol 6: 209–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jefferies HB, Fumagalli S, Dennis PB, Reinhard C, Pearson RB, Thomas G (1997) Rapamycin suppresses 5′ TOP mRNA translation through inhibition of p70s6k. EMBO J 16: 3693–3704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sharma A, Hoeffer CA, Takayasu Y, Miyawaki T, McBride SM, Klann E, Zukin RS (2010) Dysregulation of mTOR signaling in fragile X syndrome. J Neurosci 30: 694–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Haegel H, Larue L, Ohsugi M, Fedorov L, Herrenknecht K, Kemler R (1995) Lack of beta‐catenin affects mouse development at gastrulation. Development 121: 3529–3537 [DOI] [PubMed] [Google Scholar]
- 36. Nelson WJ, Nusse R (2004) Convergence of Wnt, β‐catenin, and cadherin pathways. Science 303: 1483–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fagotto F, Jho E, Zeng L, Kurth T, Joos T, Kaufmann C, Costantini F (1999) Domains of Axin involved in protein–protein interactions, Wnt pathway inhibition, and intracellular localization. J Cell Biol 145: 741–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cong F, Varmus H (2004) Nuclear‐cytoplasmic shuttling of Axin regulates subcellular localization of β‐catenin. Proc Natl Acad Sci USA 101: 2882–2887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. He T‐C, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW (1998) Identification of c‐MYC as a target of the APC pathway. Science 281: 1509–1512 [DOI] [PubMed] [Google Scholar]
- 40. Tetsu O, McCormick F (1999) β‐Catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 398: 422–426 [DOI] [PubMed] [Google Scholar]
- 41. Mann B, Gelos M, Siedow A, Hanski ML, Gratchev A, Ilyas M, Bodmer WF, Moyer MP, Riecken EO, Buhr HJ et al (1999) Target genes of β‐catenin–T cell‐factor/lymphoid‐enhancer‐factor signaling in human colorectal carcinomas. Proc Natl Acad Sci USA 96: 1603–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yuskaitis CJ, Mines MA, King MK, Sweatt JD, Miller CA, Jope RS (2010) Lithium ameliorates altered glycogen synthase kinase‐3 and behavior in a mouse model of fragile X syndrome. Biochem Pharmacol 79: 632–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bechara EG, Didiot MC, Melko M, Davidovic L, Bensaid M, Martin P, Castets M, Pognonec P, Khandjian EW, Moine H (2009) A novel function for fragile X mental retardation protein in translational activation. PLoS Biol 7: e1000016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Miyashiro KY, Beckel‐Mitchener A, Purk TP, Becker KG, Barret T, Liu L, Carbonetto S, Weiler IJ, Greenough WT, Eberwine J (2003) RNA cargoes associating with FMRP reveal deficits in cellular functioning in Fmr1 null mice. Neuron 37: 417–431 [DOI] [PubMed] [Google Scholar]
- 45. Matic K, Eninger T, Bardoni B, Davidovic L, Macek B (2014) Quantitative phosphoproteomics of murine Fmr1‐KO cell lines provides new insights into FMRP‐dependent signal transduction mechanisms. J Proteome Res 13: 4388–4397 [DOI] [PubMed] [Google Scholar]
- 46. Santoro MR, Bray SM, Warren ST (2012) Molecular mechanisms of fragile X syndrome: a twenty‐year perspective. Annu Rev Pathol 7: 219–245 [DOI] [PubMed] [Google Scholar]
- 47. Darnell JC, Van Driesche SJ, Zhang C, Hung KYS, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW (2011) FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146: 247–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bhatia VN, Perlman DH, Costello CE, McComb ME (2009) Software tool for researching annotations of proteins: open‐source protein annotation software with data visualization. Anal Chem 81: 9819–9823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman WH, Pages F, Trajanoski Z, Galon J (2009) ClueGO: a cytoscape plug‐in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25: 1091–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bindea G, Galon J, Mlecnik B (2013) CluePedia cytoscape plugin: pathway insights using integrated experimental and in silico data. Bioinformatics 29: 661–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ray JL, Leach R, Herbert JM, Benson M (2002) Isolation of vascular smooth muscle cells from a single murine aorta. Methods Cell Sci 23: 185–188 [DOI] [PubMed] [Google Scholar]
- 53. Vizcaíno JA, Csordas A, Del‐Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez‐Riverol Y, Reisinger F, Ternent T (2015) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res 44: D447–D456 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 5A
Source Data for Figure 6A
Source Data for Figure 6B
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE 53 partner repository with the dataset identifier PXD010421.