

Abstract
Cells of hematopoietic origin express high levels of the immunoproteasome, a cytokine‐inducible proteasome variant comprising the proteolytic subunits LMP2 (β1i), MECL‐1 (β2i), and LMP7 (β5i). Targeting the immunoproteasome in pre‐clinical models of autoimmune diseases with the epoxyketone inhibitor ONX 0914 has proven to be effective. ONX 0914 was previously described as a selective LMP7 inhibitor. Here, we show that PRN1126, developed as an exclusively LMP7‐specific inhibitor, has limited effects on IL‐6 secretion, experimental colitis, and experimental autoimmune encephalomyelitis (EAE). We demonstrate that prolonged exposure of cells with ONX 0914 leads to inhibition of both LMP7 and LMP2. Co‐inhibition of LMP7 and LMP2 with PRN1126 and LMP2 inhibitors LU‐001i or ML604440 impairs MHC class I cell surface expression, IL‐6 secretion, and differentiation of naïve T helper cells to T helper 17 cells, and strongly ameliorates disease in experimental colitis and EAE. Hence, co‐inhibition of LMP2 and LMP7 appears to be synergistic and advantageous for the treatment of autoimmune diseases.
Keywords: autoimmune disease, immunoproteasome, immunoproteasome inhibitor design, proteasome
Subject Categories: Immunology
Introduction
The ubiquitin–proteasome system is expressed in all eukaryotic cells and exerts numerous essential regulatory functions in nearly all cell biological pathways. The 26S proteasome degrades polyubiquitylated protein substrates and consists of a 19S regulator bearing ubiquitin receptors and an ATPase ring in charge of protein unfolding as well as a 20S proteolytic core complex. The 20S proteasome has a barrel‐shaped structure consisting of four rings with seven subunits each. In the inner two rings, made of β‐subunits, the catalytically active β1c, β2c, and β5c subunits are located 1. In hematopoietic cells and in cells stimulated with interferon (IFN)‐γ or tumor necrosis factor (TNF)‐α, these proteolytically active subunits are replaced by β1i [low molecular mass polypeptide (LMP)2], β2i [multicatalytic endopeptidase complex‐like (MECL)‐1], and β5i (LMP7) forming the immunoproteasome. Proteasome inhibitors are used in anti‐cancer therapy to treat multiple myeloma and relapsed mantle cell lymphoma 2. To improve pharmacological and toxicological profiles, new proteasome inhibitors with immunoproteasome selectivity have been developed. In 2009, the epoxyketone inhibitor PR‐957 (later renamed ONX 0914) was described and characterized to be selective for LMP7 in a concentration‐dependent manner 3. The molecular basis for this selectivity was explained after solving crystal structures of ONX 0914‐soaked immunoproteasomes, showing that the LMP2 substrate pocket could theoretically favor ONX 0914 binding, but steric hindrance by Phe31 of LMP2 counteracts this 1. Hence, ONX 0914 was used as the prototype LMP7‐selective inhibitor in many studies. LMP7 inhibition with ONX 0914, for example, protected from immunopathological damage in the brain after virus infection 4, exacerbated the pathogenesis of experimental systemic Candida albicans infection 5, protected from colitis‐associated cancer formation 6, 7, and prevented several autoimmune diseases in pre‐clinical mouse models 3, 8, 9, 10, 11, 12, 13, 14 (summarized in Ref. 13). At the cellular level, these effects were shown to involve two major pathways of disease development, namely cytokine secretion and T helper cell differentiation 15. The secretion of different proinflammatory cytokines from LPS‐stimulated human PBMCs or mouse splenocytes as well as TCR‐activated T cells was strongly suppressed by LMP7 inhibition with ONX 0914 3, 8, 10, 12, 16, 17. Additionally, ONX 0914 treatment prevented the differentiation of naïve T helper cells to polarized Th17 cells in vitro 3, 7, 14, 18. Hence, these findings propose the immunoproteasome to be an interesting therapeutic target for the treatment of inflammatory diseases.
Therefore, the novel highly selective LMP7‐specific inhibitor PRN1126 was developed. However, PRN1126, in contrast to ONX 0914, had no effect on cytokine secretion, Th17 differentiation, and autoimmune diseases. We demonstrated that prolonged exposure of cells with ONX 0914 led to inhibition of both LMP7 and LMP2. Combining the strictly LMP7‐selective inhibitor PRN1126 with LMP2‐specific inhibitors, we found that co‐inhibition of both LMP2 and LMP7 is required to reduce IL‐6 secretion and to impair Th17 differentiation. In vivo, only combined LMP2 and LMP7 inhibition could significantly ameliorate experimental colitis and inhibit EAE disease induction under dosing conditions where PRN1126 treatment alone was not efficacious.
Results
PRN1126 is an LMP7‐specific inhibitor
PRN1126 was discovered at Principia Biopharma during a medicinal chemistry effort focused on the discovery of inhibitors with selectivity for the immunoproteasome LMP7 subunit. PRN1126 is a reversible covalent inhibitor of LMP7, covalently anchoring via reversible interactions to Cys48 on LMP7 to enhance potency, selectivity, and prolonged target inhibition in immune cells (Figs 1A and EV1A).
Figure 1. PRN1126 is selective for the chymotrypsin‐like activity of the immunoproteasome.
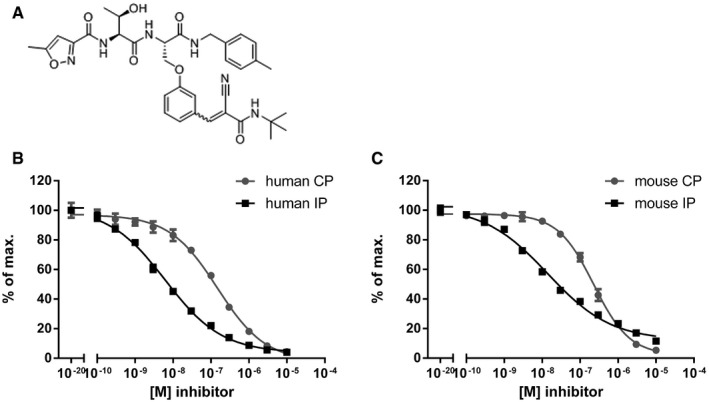
-
AStructure of PRN1126, a reversible covalent selective LMP7 inhibitor.
-
B, CHydrolysis of fluorogenic substrates (Suc‐LLVY‐AMC) for the chymotrypsin‐like activity of human (B) or mouse (C) 20S constitutive proteasome (CP) or 20S immunoproteasome (IP) at various PRN1126 concentrations. Data are presented as the means ± s.d. from quadruplicate assays. The highest fluorescence value was set to 100%. The experiments were repeated twice with similar results.
Figure EV1. Extended LMP7 inhibition in human PBMC .
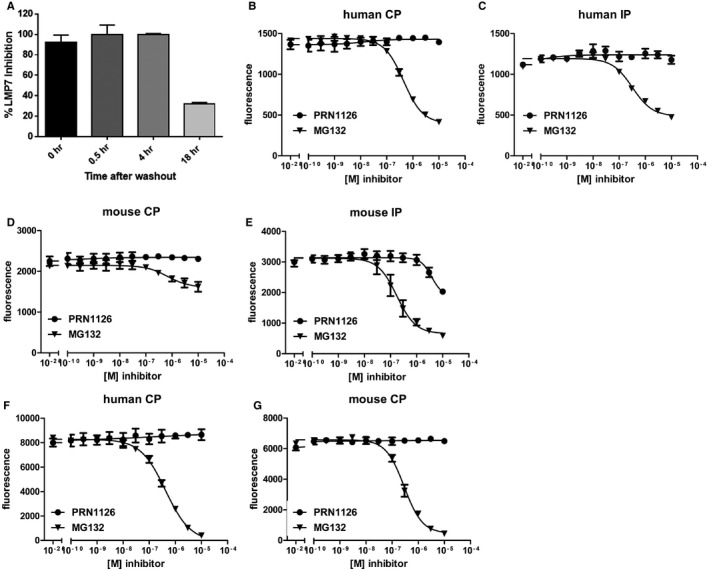
-
APBMCs were treated with PRN1126 at 1 μM for 1 h, washed to remove compound, and incubated in media for the time indicated. LMP7 inhibition was determined by hydrolysis of fluorogenic substrate (Suc‐LLVY‐AMC). Data points represent the mean ± s.d. (n = 3).
-
B–GHydrolysis of fluorogenic substrates Bz‐VGR‐AMC for trypsin‐like activity (B–E) or z‐LLE‐βNA for caspase‐like activity (F, G) of human (B, C, F) or mouse (D, E, G) 20S constitutive proteasome (B, D, F, G) or immunoproteasome (C, E) at various concentrations of PRN1126 and MG132. Data are presented as the means of fluorescence ± s.d. from quadruplicate assays. The experiments were repeated three times with similar results.
To further characterize the selectivity of PRN1126, fluorogenic peptide assays with purified proteasomes were performed. To this aim, human constitutive proteasomes (CPs) and human immunoproteasomes (IPs) were respectively purified from LCL721.174 (source of CP) or LCL721.145 (source of IP) lymphoblastoid cell lines 19. CP and IP were incubated with different concentrations of PRN1126 and assayed with a fluorogenic substrate specific for the chymotrypsin‐like activity of the proteasome (Suc‐LLVY‐AMC; Fig 1B). PRN1126 showed IC50 values for the IP of 7.2 ± 1.3 × 10−9 M and 2.1 ± 0.8 × 10−7 M for the CP, and is therefore approximately 30 times more selective for the IP than for the CP. A similar experiment was performed with mouse proteasome. Immunoproteasomes were isolated from livers of BALB/c mice 8 days after infection with lymphocytic choriomeningitis virus (LCMV), and constitutive proteasomes were purified from livers of uninfected LMP7−/−/MECL‐1−/− gene‐targeted mice 1, 20. PRN1126 showed IC50 values for the mouse IP of 1.6 ± 0.3 × 10−8 M and 2.1 ± 0.3 × 10−7 M for the mouse CP, and is therefore at least 13 times more selective for the mouse IP than for the mouse CP (Fig 1C). Next, mouse and human immunoproteasome were incubated with PRN1126, and the trypsin‐like activity was assessed with the fluorogenic substrate Bz‐VGR‐AMC (Fig EV1B–E). PRN1126 did not affect the trypsin‐like activity below 10−6 M, indicating that PRN1126 does not inhibit β2c in the CP and MECL‐1 in the IP, the proteolytically active subunits in the proteasome responsible for the trypsin‐like activity. To investigate the impact of PRN1126 on the caspase‐like activity, which is evoked by β1c, human and mouse CPs were incubated with different inhibitor concentrations and assayed with the fluorogenic substrate z‐LLE‐βNA (Fig EV1F and G). PRN1126 did not reduce the caspase‐like activity. Taken together, PRN1126 is a potent and LMP7‐selective inhibitor of the mouse and human immunoproteasome.
Next, we tested the cell permeability of PRN1126. LCL721.174 (contain CP) or LCL721.145 (contain IP) cells were incubated with different concentrations of PRN1126 and assayed with the cell‐permeable substrate MeO‐Suc‐GLF‐AMC 21 specific for the chymotrypsin‐like activity (Fig EV2A). PRN1126 inhibited the cleavage of this substrate in cells containing immunoproteasomes, demonstrating that PRN1126 is cell‐permeable.
Figure EV2. PRN1126 cell permeability and target occupancy of LMP7 or LMP2 in mice.
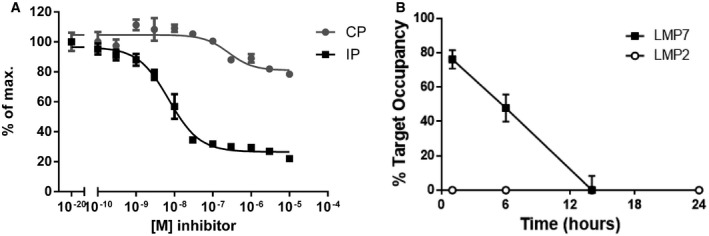
- LCL721.174 cells expressing constitutive proteasomes (CP) or LCL721.145 cells expressing immunoproteasomes (IP) were treated with the indicated concentrations of the LMP7 inhibitor PRN1126. The chymotrypsin‐like activity in the cells was determined by the hydrolysis of the cell‐permeable fluorogenic proteasome substrate Meo‐Suc‐GLF‐AMC. Depicted is the mean ± s.d. % of maximal hydrolytic activity of triplicate cultures. The highest fluorescence value was set to 100%.
- Mice were treated with a single dose of PRN1126 (40 mg/kg, s.c.) or vehicle. Spleens were harvested at the time points indicated, and the drug occupancy of LMP7 or LMP2 subunits was assessed in splenocytes with the ProCISE assay. Data points represent the means ± s.d. of three mice.
PRN1126 affects the presentation of an LMP7‐dependent epitope
Since LMP7 plays an important role in major histocompatibility complex (MHC) class I‐restricted antigen presentation, we investigated whether PRN1126 can alter the presentation of an LMP7‐dependent MHC‐I T‐cell epitope. Therefore, the CTL epitope UTY246–254 (ubiquitously transcribed tetratricopeptide repeat gene, Y‐linked), derived from the endogenously expressed Y chromosome‐encoded HY‐Ag, was chosen. It has been shown that the generation of the UTY246–254 epitope is strictly LMP7‐dependent 22, 23. Furthermore, the presentation of the UTY246–254 epitope can be reduced by an LMP7‐selective inhibitor (ONX 0914), but not an LMP2‐selective inhibitor 3, 23. Male derived mouse splenocytes were incubated with PRN1126, and the UTY246–254 presentation on the H‐2Db class I molecule was determined with an UTY246–254‐specific T‐cell hybridoma in lacZ assays (Fig 2A). The presentation of UTY246–254 was reduced in a dose‐dependent manner by PRN1126, confirming previous results obtained with different inhibitors targeting LMP7 3, 19 and corroborating that PRN1126 is an LMP7‐specific inhibitor.
Figure 2. PRN1126 does not affect dextran sulfate sodium (DSS)‐induced colitis or experimental autoimmune encephalomyelitis (EAE).
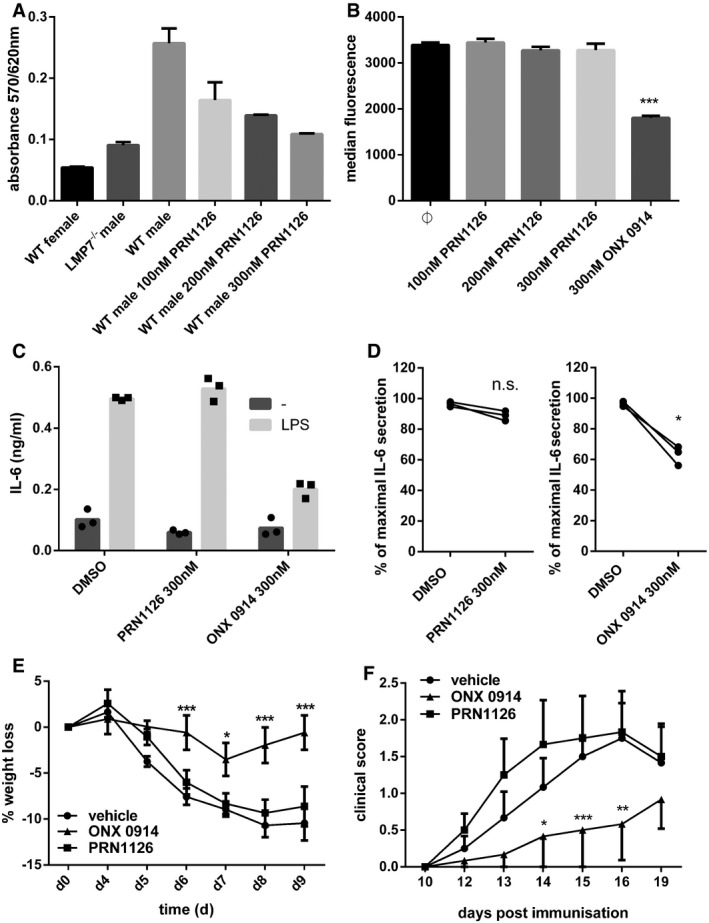
-
APresentation of UTY246–254 on splenocytes after exposure to indicated concentrations of PRN1126. Data are presented as the mean absorbance ± s.d. of three replicate cultures. The experiment has been performed twice, yielding similar results.
-
BFlow cytometry analysis of H‐2Kb surface expression on splenocytes derived from C57BL/6 mice treated with the indicated concentrations of PRN1126 overnight. Pooled data from three independent experiments (n = 9) are shown as the means of median fluorescence intensity ± s.e.m. All data were statistically compared to the DMSO‐treated group. ***P < 0.001. One‐way ANOVA.
-
C, DSplenocytes from C57BL/6 mice (C) or human PBMCs (D) were exposed (continuous treatment) to 300 nM PRN1126, or vehicle (DMSO), or 300 nM ONX 0914 and stimulated with LPS overnight. IL‐6 concentrations in the supernatant were analyzed by ELISA. (C) IL‐6 concentrations are presented as mean and individual data points from triplicate wells. The experiment has been performed twice, yielding similar results. (D) Data are presented as single dots from three independent donors. The highest cytokine concentration was set to 100%. *P < 0.05. Unpaired Student's t‐test.
-
EColitis was induced by oral administration of 3% DSS for 5 days. Mice were treated daily (s.c.) with either PRN1126 (40 mg/kg), or ONX 0914 (10 mg/kg), or vehicle. Data points represent mean ± s.e.m. of 15 mice pooled from three independent experiments. All data were statistically compared to the vehicle‐treated group. *P < 0.05, ***P < 0.001. Two‐way ANOVA.
-
FMice were immunized with MOG35–55 peptide and were monitored daily for clinical symptoms of EAE. Mice were treated three times a week (s.c.) with either PRN1126 (40 mg/kg), or ONX 0914 (10 mg/kg), or vehicle. All data were statistically compared to the vehicle‐treated group. *P < 0.05, **P < 0.01, ***P < 0.001. Two‐way ANOVA. Shown are the means of the clinical scores ± s.e.m. (n = 6 per group). The experiments have been performed twice, yielding similar results.
PRN1126 does not reduce MHC class I cell surface expression and cytokine secretion
LMP7‐deficient mice display reduced levels of MHC‐I surface expression 24, and MHC‐I surface expression on splenocytes can be reduced with LMP7‐selective inhibitors 3, 19. Therefore, splenocytes from wild‐type C57BL/6 mice were incubated with 300 nM PRN1126 or with the previously described LMP7‐selective inhibitor ONX 0914 3. Whereas the expression of the MHC‐I molecule H‐2Kb on splenocytes from wild‐type mice was reduced with ONX 0914, no effect could be observed with PRN1126 (Fig 2B).
Next, we investigated whether PRN1126 can alter cytokine secretion as previously reported for different LMP7‐selective inhibitors 3, 12, 16, 19. Mouse splenocytes or human PBMCs were exposed to PRN1126 or ONX 0914, stimulated with LPS and IL‐6 secretion into the supernatant was determined (Fig 2C and D). In contrast to ONX 0914, no influence on IL‐6 secretion could be observed with PRN1126 up to a concentration of 300 nM.
PRN1126 does not ameliorate DSS‐induced colitis and EAE
Targeting the immunoproteasome has been shown to protect mice from DSS‐induced colitis 8 and to ameliorate experimental autoimmune encephalomyelitis (EAE) 12. DSS‐induced colitis was elicited by oral administration of 3% DSS in the drinking water for 5 days. Mice were treated daily with 40 mg/kg PRN1126, a dose which inhibits LMP7 in vivo (as shown in naïve mice achieving 76, 48, and 0% LMP7 occupancy at 1, 6, and 14 h, respectively post‐dose; Fig EV2B). Compared to vehicle‐treated mice, a minor reduction in colitis‐associated weight loss could be observed in mice treated with PRN1126 as expected based on intermittent target inhibition (Fig 2E). In contrast, ONX 0914 protected mice from colitis as previously observed 8. Similar to colitis, in EAE, a mouse model for multiples sclerosis, PRN1126 had no significant impact on the clinical manifestation of EAE when dosed intermittently (3×/week), while ONX 0914 delayed the onset and reduced the severity of disease symptoms (Fig 2F) with the same dosing schedule.
In contrast to ONX 0914, PRN1126 is selective for LMP7 and does not inhibit LMP2
It has been reported previously that LMP7 inhibition alters MHC‐I surface expression 3, reduces cytokine secretion 3, 12, 16, protects from experimental colitis 8, and ameliorates EAE 12 using the inhibitor ONX 0914, and this was further confirmed in this study (Fig 2). Nevertheless, PRN1126 alone had limited effect in these experiments, although PRN1126 was LMP7‐selective as shown in Fig 1. These discrepant results posed the question how two LMP7‐selective inhibitors can have such different effects in biological assays in vitro and in vivo? Therefore, we had a closer look at the activity profile of the two immunoproteasome inhibitors PRN1126 and ONX 0914. Ac‐PAL‐AMC is a fluorogenic peptide substrate exclusively cleaved by LMP2 23, 25. Mouse and human immunoproteasomes were incubated with different concentrations of PRN1126 and assayed with the fluorogenic substrate specific for LMP2 activity (Fig 3A). Whereas PRN1126 did not alter Ac‐PAL‐AMC cleavage up to 10 μM, ONX 0914 partially inhibited LMP2 starting at concentrations above 10 nM [human: IC50 1.1 ± 0.5 × 10−7 M (Fig 3A upper panel); mouse IC50 6.5 ± 0.5 × 10−8 M (Fig 3A lower panel)]. We have observed that binding of ONX 0914 to LMP7 and LMP2 leads to a shift of LMP7 and LMP2 bands to higher apparent molecular weights in Western blot. Incubation of enriched lymphocytes from mouse spleens with 300 nM ONX 0914 for 2 h led to an irreversible modification of almost all LMP2 molecules with ONX 0914 (Fig 3B). These results indicate that ONX 0914 is inhibiting both LMP7 and LMP2 at 300 nM in cells if they are incubated with ONX 0914 for extended periods of time. Therefore, we hypothesized that inhibition of both LMP7 and LMP2 might be required to induce broad anti‐inflammatory effects in vitro and in vivo. To investigate this hypothesis, the LMP7‐specific inhibitor PRN1126 was combined with the well‐characterized LMP2‐specific inhibitors LU‐001i 19, 26 or ML604440 23, and inhibition of IL‐6, T‐cell differentiation, and therapeutic efficacy in mouse models of inflammatory bowel disease and multiple sclerosis was assessed. LU‐001i is a pure LMP2‐specific inhibitor not affecting other subunits of the CP or the IP 19, 26 and leads to a shift of LMP2 but not LMP7 bands to higher apparent molecular weights in Western blot (Fig EV3A). In contrast to the epoxyketone LU‐001i, the dipeptide boronate ML604440 does not lead to a shift of immunoproteasome subunits bands to higher apparent molecular weights in Western blot (Fig EV3A). However, it has been demonstrated that ML604440 does not affect the chymotrypsin‐like 23 and the trypsin‐like activity (Fig EV3B–E) and is therefore an LMP2‐specific inhibitor.
Figure 3. ONX 0914 but not PRN1126 inhibits LMP2.
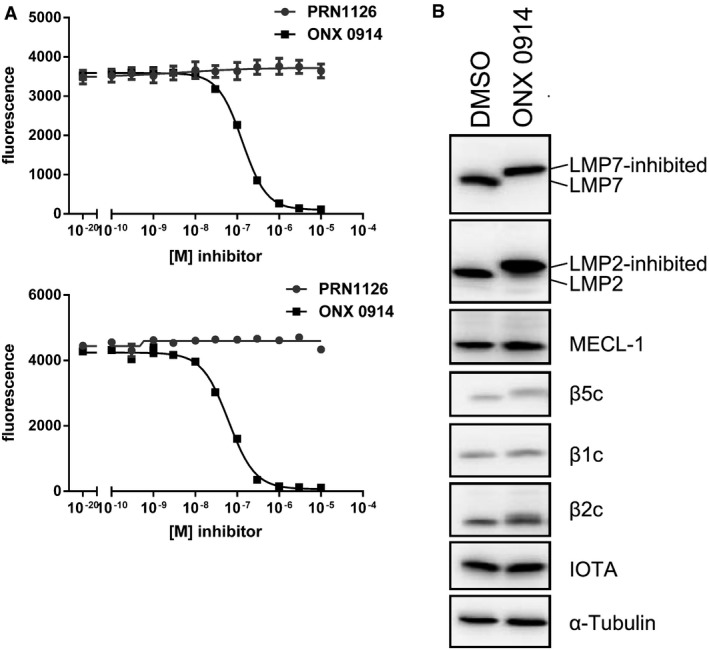
- Hydrolysis of fluorogenic substrates (Ac‐PAL‐AMC) for LMP2 activity of human (upper panel) or mouse (lower panel) 20S immunoproteasome at various concentrations of PRN1126 or ONX 0914. Data are presented as the means of fluorescence ± s.d. from quadruplicate assays. The experiments were repeated three times with similar results.
- Altered electrophoretic mobility of IP subunits by covalent modification with ONX 0914. Ficoll‐enriched lymphocytes from C57BL/6 mice were treated with DMSO or 300 nM ONX 0914 for 2 h in vitro. SDS–PAGE and immunoblotting against indicated proteins were performed. Shown are representative Western blots out of three independent experiments with similar outcome.
Source data are available online for this figure.
Figure EV3. Electrophoretic mobility shifts of IP subunits with different inhibitors.
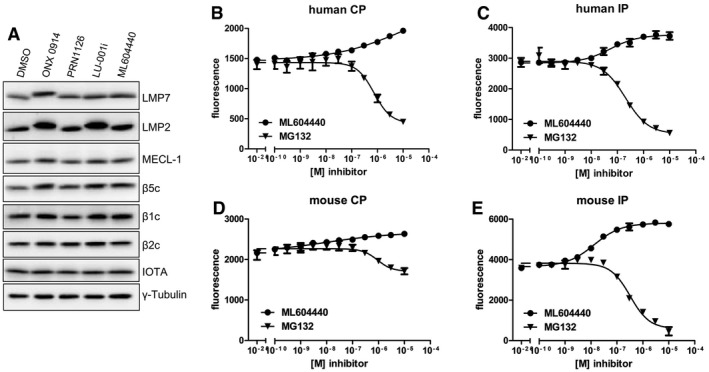
-
AFicoll‐enriched lymphocytes from C57BL/6 mice were treated with DMSO, 300 nM ONX 0914, 300 nM PRN1126, 300 nM LU‐001i, or 300 nM ML604440 for 2 h in vitro. SDS–PAGE and immunoblotting against indicated proteins were performed. Shown are representative Western blots out of two independent experiments with similar outcome.
-
B–EHydrolysis of the fluorogenic substrate Bz‐VGR‐AMC for trypsin‐like activity of human (B, C) or mouse (D, E) 20S constitutive proteasome (B, D) or immunoproteasome (C, E) at various concentrations of ML604440 and MG132. Data are presented as the means of fluorescence ± s.d. from quadruplicate assays. The experiments were repeated three times with similar results.
Source data are available online for this figure.
A reduction of MHC class I surface expression requires the co‐inhibition of LMP7 and LMP2
Splenocytes derived from wild‐type or LMP7‐deficient mice were incubated with 300 nM ONX 0914, 300 nM PRN1126, 300 nM ML604440, 300 nM LU‐001i, 300 nM PRN1126 + 300 nM ML604440, or 300 nM PRN1126 + 300 nM LU‐001i overnight. On the next day, the H‐2Kb surface expression on wild‐type or LMP7‐deficient splenocytes was analyzed by flow cytometry (Fig 4). Whereas LMP2 inhibition with ML604440 or LU‐001i alone or LMP7 inhibition with PRN1126 alone had no influence on the surface expression of H‐2Kb, combined LMP2 and LMP7 inhibition (PRN1126 + ML604440 or PRN1126 + LU‐001i) reduced H‐2Kb expression to an extent, which was similar to that achieved with ONX 0914. Basal H‐2Kb expression on LMP7−/− splenocytes, which is approximately 50% lower relative to wild‐type levels, was not affected when LMP2 and LMP7 inhibitors were combined, thus confirming the specificity of the inhibitors.
Figure 4. Co‐inhibition of LMP7 and LMP2 reduces H‐2Kb surface expression on splenocytes.
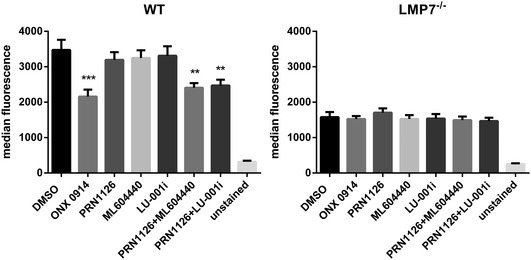
Flow cytometry analysis of H‐2Kb surface expression on splenocytes derived from C57BL/6 wild‐type (left side) or LMP7−/− (right side) mice treated with vehicle (DMSO), ONX 0914 (300 nM), PRN1126 (300 nM), ML604440 (300 nM), LU‐001i (300 nM), PRN1126 + ML604440 (300 nM each), or PRN1126 + LU‐001i (300 nM each) overnight. Unstained splenocytes were used as negative control. Pooled data from three independent experiments (n = 9) are shown as the means of median fluorescence intensity ± s.e.m. All data were statistically compared to the DMSO‐treated group. **P < 0.01, ***P < 0.001. One‐way ANOVA.
The reduction in IL‐6 production depends on combined inhibition of LMP7 and LMP2
Next, we tested whether co‐inhibition of LMP2 and LMP7 is also needed to alter cytokine secretion. Mouse splenocytes or human PBMCs from different healthy volunteers were incubated with 300 nM ONX 0914, 300 nM PRN1126, 300 nM ML604440, 300 nM LU‐001i, 300 nM PRN1126 + 300 nM ML604440, or 300 nM PRN1126 + 300 nM LU‐001i. After stimulation with LPS overnight, the concentration of IL‐6 in the supernatant was determined by ELISA (Fig 5A and B). No significant inhibition of IL‐6 secretion by mouse splenocytes (Fig 5A) or human PBMCs (Fig 5B) was observed when LMP7 (PRN1126) or LMP2 (ML604440, LU‐001i) alone were targeted. In contrast, co‐inhibition of LMP2 and LMP7 reduced IL‐6 secretion to a similar degree as ONX 0914. This reduction in IL‐6 secretion is not due to increased cell death, since splenocytes incubated overnight with LMP7 (PRN1126) and LMP2 (ML604440, LU‐001i) inhibitors did not result in increased propidium iodide‐positive cells (Fig EV4). Hence, these data indicate that the previously reported reduction in cytokine secretion of cells exposed to ONX 0914 was due to the co‐inhibition of LMP2 and LMP7.
Figure 5. The reduction in IL‐6 production and the suppression of Th17 differentiation rely on the co‐inhibition of LMP7 and LMP2.
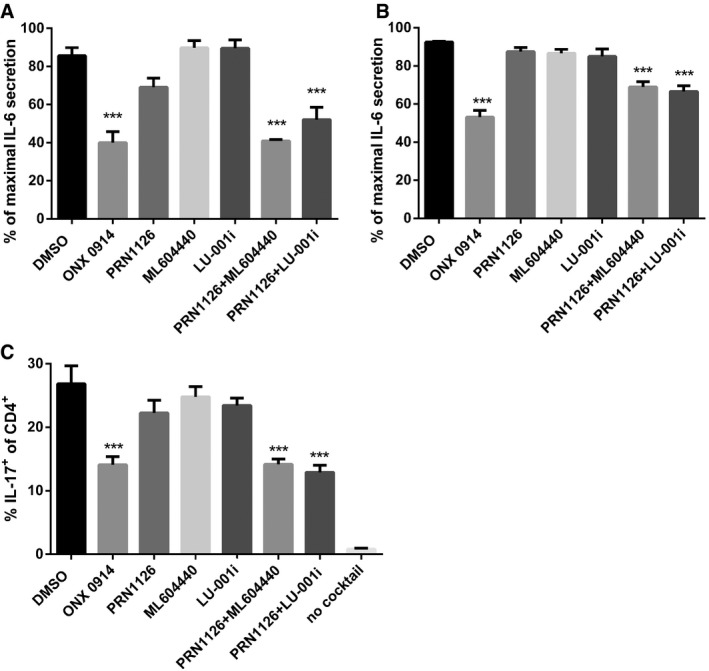
-
A, BSplenocytes from C57BL/6 mice (A) or human PBMCs (B) were incubated (continuous treatment) with DMSO, ONX 0914 (300 nM), PRN1126 (300 nM), ML604440 (300 nM), LU‐001i (300 nM), PRN1126 + ML604440 (300 nM each), or PRN1126 + LU‐001i (300 nM each) and stimulated with LPS overnight. The IL‐6 concentration in the supernatant was analyzed by ELISA. The highest IL‐6 concentration from each experiment was set to 100%. (A) Data are presented as the mean ± s.e.m. of four independent experiments each measured in triplicates. (B) Data are presented as the mean ± s.e.m. of six different healthy donors. All data were statistically compared to the DMSO‐treated group. ***P < 0.001. One‐way ANOVA.
-
CThe differentiation of CD4+ T cells isolated from the spleens of C57BL/6 mice that were exposed (continuous treatment) to DMSO, ONX 0914 (300 nM), PRN1126 (300 nM), ML604440 (300 nM), LU‐001i (300 nM), PRN1126 + ML604440 (300 nM each), or PRN1126 + LU‐001i (300 nM each) and that were stimulated with plate‐bound antibodies to CD3/CD28 in the presence of TGF‐β and IL‐6 and scavenging antibodies neutralizing IL‐4 and IFN‐γ was measured in 3‐day cultures. IL‐17 expression was detected by intracellular cytokine staining after a short restimulation with PMA/ionomycin. IL‐17 expression in activated T cells cultured in the absence of Th17‐polarizing conditions is shown as a comparison (no cocktail). Values reflect the percentage of CD4+ cells that are IL‐17A+. Data are presented as the means ± s.e.m. of three independent experiments each measured in triplicates. All data were statistically compared to the DMSO‐treated group. ***P < 0.001. One‐way ANOVA.
Figure EV4. Effect of different inhibitors on cell viability.
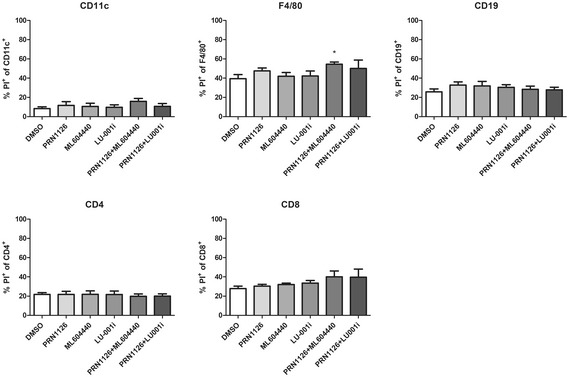
Splenocytes derived from C57BL/6 mice were treated with DMSO, 300 nM PRN1126, 300 nM ML604440, 300 nM LU‐001i, 300 nM PRN1126 + 300 nM ML604440, and 300 nM PRN1126 + 300 nM LU‐001i for 24 h. Cell death (PI+) of CD11c+, F4/80+, CD19+, CD4+, and CD8+ was analyzed by flow cytometry. Shown are the means of propidium iodide‐positive (PI+) populations ± s.e.m. from splenocytes derived from three different mice (n = 3) measured in triplicates. All data were statistically compared to the DMSO‐treated group. *P < 0.05. One‐way ANOVA.
The suppression of Th17 differentiation relies on the co‐inhibition of LMP7 and LMP2
Th17 cells play a crucial role in the pathogenesis of autoimmunity 27. It has previously been shown that LMP7 inhibition strongly diminishes the differentiation of naïve T helper cells to polarized Th1 or Th17 cells in vitro 3, 18. Therefore, the effect of LMP2 and LMP7 co‐inhibition was investigated. Mouse splenic CD4+ T cells were magnetically sorted and cultured in vitro under Th17‐polarizing conditions in the presence of 300 nM ONX 0914, 300 nM PRN1126, 300 nM ML604440, 300 nM LU‐001i, 300 nM PRN1126 + 300 nM ML604440, or 300 nM PRN1126 + 300 nM LU‐001i. After 3 days of culture, the number of Th17 cells was determined by intracellular IL‐17A staining of CD4+ T cells (Fig 5C). The percentage of IL‐17A‐producing CD4+ T cells cultured in the presence of PRN1126, ML604440, or LU‐001i alone was similar to that in T‐cell cultures treated with the vehicle DMSO. On the contrary, CD4+ T cells simultaneously exposed to an LMP7‐ and an LMP2‐specific inhibitor significantly reduced Th17 polarization when applied at the same concentration. These data suggest that LMP7 together with LMP2 controls the differentiation into inflammatory effector cells in the presence of polarizing cytokines.
The amelioration of DSS‐induced colitis and EAE relies on the combined inhibition of LMP7 and LMP2
Based on the beneficial impact of combined LMP7 and LMP2 inhibition on IL‐6 production and Th17 differentiation in vitro, we then investigated whether the administration of the LMP2‐specific inhibitor LU‐001i together with PRN1126 might be effective in DSS colitis and EAE disease models.
LU‐001i has been shown to inhibit LMP2 activity in mice, but, nevertheless, mice were not protected from DSS‐induced colitis when treated with LU‐001i 19. DSS‐induced colitis was induced by oral administration of 3% DSS in the drinking water. Mice were daily treated with LU‐001i (15 mg/kg), PRN1126 (40 mg/kg), LU‐001i + PRN1126 (15 + 40 mg/kg), or vehicle, and the body weight of the mice was recorded for 9 days (Fig 6A). In mice treated with LU‐001i or PRN1126 alone, the weight loss was similar to vehicle‐treated mice, confirming previous observations (Fig 2E). However, when mice were simultaneously treated with LU‐001i and PRN1126 the weight loss was significantly reduced. Induction of colitis is associated with the shrinkage of the colon in diseased mice. In agreement with the body weight data (Fig 6A), only a minor colon length shortening in mice simultaneously treated with PRN1126 and LU‐001i could be observed (Fig 6B).
Figure 6. The amelioration of DSS‐induced colitis and EAE relies on the joint inhibition of LMP7 and LMP2.
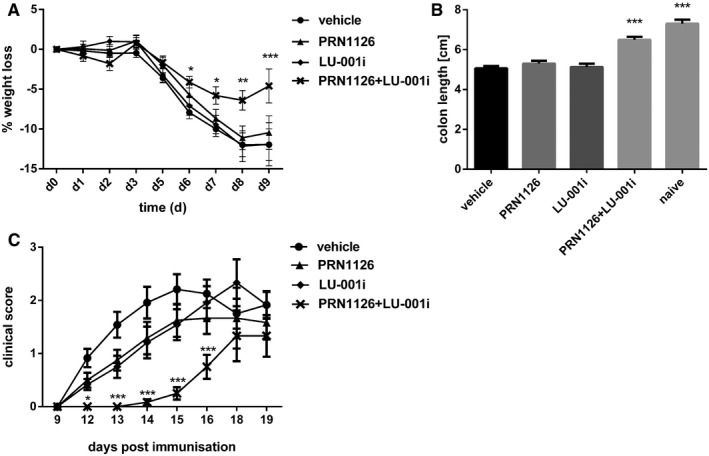
-
A, BColitis was induced by oral administration of 3% DSS. Mice were treated daily (s.c.) with LU‐001i (15 mg/kg), PRN1126 (40 mg/kg), PRN1126 + LU‐001i (40 + 15 mg/kg), or vehicle starting from the begin of the experiment. Data points represent means ± s.e.m. of 15 mice pooled from three independent experiments. (A) The body weight of individual mice was monitored daily, and the percent weight loss (y‐axis) was plotted versus time (x‐axis). All data were statistically compared to the vehicle‐treated group. *P < 0.05, **P < 0.01, ***P < 0.001. Two‐way ANOVA. (B) On day 9 after initiation of DSS treatment, colon lengths were measured (n = 15). Naïve mice (n = 5) were used as healthy controls. All data were statistically compared to the vehicle‐treated group. ***P < 0.001. One‐way ANOVA.
-
CMice were immunized with MOG35–55 peptide and were monitored daily for clinical symptoms of EAE. Mice were treated intermittently with three times a week (s.c.) schedule with LU‐001i (15 mg/kg), PRN1126 (40 mg/kg), PRN1126 + LU‐001i (40 + 15 mg/kg), or vehicle starting from the beginning of the experiment. Data points represent the means of the clinical scores ± s.e.m. of 12 mice pooled from two independent experiments. All data were statistically compared to the vehicle‐treated group. *P < 0.05, ***P < 0.001. Two‐way ANOVA.
Encephalomyelitis, a disease model for multiple sclerosis (MS) in rodents, shares clinical and pathological features with similarity to MS in humans. To test whether co‐inhibition of LMP2 and LMP7 is effective in EAE, mice were immunized with the MOG35–55 peptide derived from the MS antigen myelin oligodendrocyte glycoprotein. Mice were treated intermittently three times a week with LU‐001i (15 mg/kg), PRN1126 (40 mg/kg), LU‐001i + PRN1126 (15 + 40 mg/kg), or vehicle beginning on the day of immunization with MOG35–55 (Fig 6C) using a suboptimal individual dosing schedule to enable evaluation of the combined pharmacological effects on clinical scores. First, clinical symptoms were observed on day 12 in LU‐001i, PRN1126, or vehicle‐treated mice, whereas simultaneous PRN1126 and LU‐001i treatment resulted in a significantly later onset of the disease (vehicle d 12.3 ± 0.3; PRN1126 + LU‐001i d 16.8 ± 0.6) and lower disease incidence [vehicle 12/12 (100%); PRN1126 + LU‐001i 7/12 (58%)]. Together, these results indicate that combined targeting of LMP7 and LMP2 achieved significant and broad therapeutic efficacy in the treatment of autoimmune inflammation in two distinct pre‐clinical models.
Discussion
Immunoproteasome inhibitors have been shown to be effective in pre‐clinical animal models for autoimmune diseases 13, 14, in tumor models 6, 7, 28, 29, and in organ transplantation 17. Broad‐spectrum proteasome inhibitors, such as bortezomib or carfilzomib, which are used to treat patients with multiple myeloma, have numerous adverse side effects and dose‐limiting toxicities since the proteasome is constitutively expressed in all types of eukaryotic cells, controlling cell cycle, cell signaling, transcription, and apoptosis 2. In contrast, immunoproteasomes are expressed in immune cells and in an inflammatory environment, and therefore, selective inhibitors of the immunoproteasome can be applied below their maximally tolerated dose still retaining their therapeutic efficacy but lacking untoward toxicity 3. The number of immunoproteasome inhibitors tested in pre‐clinical animal models for autoimmune diseases is limited. Therefore, the novel LMP7‐selective inhibitor PRN1126 was developed, a novel covalent inhibitor anchoring via reversible interaction with Cys48 on LMP7 to enhance potency, selectivity, and prolonged target inhibition (Figs 1 and EV1). Although PRN1126 potently and selectively inhibited LMP7, previous observations made with immunoproteasome inhibitors reportedly selective for LMP7 could not be confirmed 3, 8, 12. Our studies demonstrated that PRN1126 could not reduce MHC‐I surface expression on splenocytes, had no effect on IL‐6 secretion, and could not ameliorate DSS‐induced colitis with daily dosing or EAE with infrequent dosing (Fig 2). A comparison of the specificity of the previously used ONX 0914 with PRN1126 revealed that ONX 0914 significantly inhibited both LMP2 and LMP7 at concentrations above 100 nM when cells were incubated with this irreversible epoxyketone inhibitor for longer time periods, whereas PRN1126 was highly selective for LMP7 and did not inhibit LMP2 (Fig 3). In most in vitro studies, ONX 0914 was used at 300 nM, a concentration almost completely blocking LMP2 (Fig 3B). Why has it previously remained unrecognized that ONX 0914 inhibits LMP2 to such an extent? Using an active‐site ELISA, Muchamuel et al 3 showed an LMP2 inhibition by ONX 0914 of approximately 35% at 200 nM and 75% at 500 nM. In this study, we used the hydrolysis of a peptide substrate (Ac‐PAL‐AMC) exclusively cleaved by LMP2 23, 25 to determine the LMP2 specificity. This sensitive method, not dependent on specific antibodies, indicates that ONX 0914 binding to LMP2 has previously been underestimated. Additionally, the crystal structure of ONX 0914‐soaked immunoproteasomes supported the view of LMP7 selectivity 1. Furthermore, to demonstrate the specificity of ONX 0914, cells derived from LMP7‐deficient mice were used previously 3. Therefore, no effect on MHC‐I surface expression and cytokine secretion could be observed with LMP7‐deficient splenocytes, concluding that ONX 0914 is LMP7‐specific. Nevertheless, it has been shown that LMP7‐deficient mice have a strongly reduced LMP2 incorporation into immunoproteasomes 30. Hence, the inhibition of LMP2 by ONX 0914 remained unrecognized in cells lacking LMP7.
To investigate whether co‐inhibition of both LMP2 and LMP7 is required to affect cytokine release and inflammatory diseases, LMP2 and LMP7 inhibitors were combined. ML604440 23 and LU‐001i 26 are LMP2‐specific inhibitors. It has been shown that LMP2 inhibition alone does not affect MHC‐I surface expression, cytokine release, Th17 differentiation, and DSS‐induced colitis 19. Co‐inhibition of both LMP2 and LMP7, in contrast, reduced MHC‐I surface expression on splenocytes (Fig 4). These results indicate that the lack of both LMP2 and LMP7 contributes to the reduced MHC‐I surface expression observed for LMP7‐deficient cells and mice, which has not been realized in the past 24. The secretion of IL‐6, i.e., a cytokine centrally involved in inflammation and autoimmunity, from endotoxin‐stimulated mouse splenocytes or human PBMCs was not reduced by PRN1126, but was significantly lowered when both LMP2 and LMP7 were co‐inhibited with LMP2‐ and LMP7‐specific inhibitors (Fig 5A and B). Notably, this result was obtained with the peptide boronate inhibitor ML604440 and the peptide epoxyketone inhibitor LU‐001i, which are both selective for LMP2 but carry different warheads. Th17 cells have a pivotal role in the pathogenesis of multiple autoimmune‐mediated inflammatory diseases 31. It has been shown that LMP7 inhibition with ONX 0914 biases T helper cell differentiation against the development of proinflammatory Th17 cells 3, 18. Whereas PRN1126 could not alter Th17 differentiation at the concentration of 300 nM tested, addition of LMP2 inhibitor to the culture reduced Th17 differentiation similar to ONX 0914 (Fig 5C). Inflammatory cytokines and Th17 cells play crucial roles in the development of inflammatory diseases. Since co‐inhibition of LMP2 and LMP7 altered both pathways, we investigated whether co‐inhibition of LMP2 and LMP7 is also required in vivo in pre‐clinical animal models. Previously, ONX 0914 was frequently used at a concentration of 10 mg/kg in mice 8, 12. At this concentration, LMP2 is inhibited up to app. 75% in blood and kidney 1 h post‐intravenous ONX 0914 application 3. Hence, the previously observed beneficial effects observed with ONX 0914 in pre‐clinical models might well be due to inhibition of both LMP7 and LMP2. Indeed, only co‐inhibition of both LMP7 and LMP2 could ameliorate DSS‐induced colitis and EAE (Fig 6).
Our results indicate that targeting LMP7 and LMP2 can significantly interfere with the development of inflammatory diseases. Whether blocking of LMP7 and MECL‐1 or LMP2 and MECL‐1 would have similar effects as observed with LMP7 and LMP2 co‐inhibition remains to be determined. Blocking LMP2 and LMP7 does not simply kill immune cells, since repeated application of ONX 0914 to mice did not lower the numbers of CD8+ or CD4+ T cells, CD19+ B cells, and CD11c+ dendritic cells 8. The mechanism how immunoproteasome inhibition can prevent autoimmune diseases still remains elusive. It has been suggested that the immunoproteasome might selectively processes a factor that is required for regulating cytokine production and T helper cell differentiation 15. However, since two different subunits with two different proteolytic activities have to be targeted, this notion seems less likely unless a short bioactive peptide jointly generated by LMP2 and LMP7 is involved. An involvement of the immunoproteasome in NF‐κB activation has remained controversial 32, 33, 34, 35. A recent study using IFN‐γ‐stimulated peritoneal macrophages and mouse embryonic fibroblasts derived from mice deficient for the immunoproteasome showed that the immunoproteasome has no specialized function for canonical NF‐κB activation 36. Moreover, Muchamuel et al 3 showed no difference in NF‐κB activation in an ONX 0914‐inhibited reporter cell line. Recently, it has been reported that the mTORC1 immunoproteasome pathway is important for cell survival against stress 37. Whether this pathway is affected in immunoproteasome‐targeted mice in autoimmune diseases remains to be determined.
In a recent study, Sula Karreci et al 17 described a novel highly selective LMP7 inhibitor (named DPLG3), which did not detectably inhibit LMP2 in vitro. This non‐covalent inhibitor promoted long‐term cardiac allograft acceptance in mice across a major histocompatibility barrier. This is in apparent contrast to our data, which strongly suggest that co‐inhibition of LMP7 and LMP2 is required to ameliorate autoimmune diseases. It seems that in this transplantation model the inhibition of LMP7 alone is sufficient to prevent rejection; however, the selectivity of DPLG3 in vivo has not yet been disclosed. Additionally, whether DPLG3 has off‐target effects not related to proteasome binding remains elusive as experiments in LMP2−/− and LMP7−/− mice were not reported.
Taken together, our study confirms that the immunoproteasome is indeed a valid target for the treatment of inflammatory diseases. Our data strongly suggest that combined LMP7 and LMP2 inhibition is synergistic, and both subunits have to be jointly targeted for the treatment of IL‐6‐mediated inflammatory disorders. These new insights will promote the design of new generations of immunoproteasome inhibitors simultaneously targeting LMP2 and LMP7 in immunoproteasomes with optimal therapeutic efficacy for clinical use in the treatment of autoimmunity and cancer.
Materials and Methods
Chemical synthesis of PRN1126
PRN1126 was prepared as described according to procedures described in patent WO2015195950 A1, “Preparation of isoxazolidines and thiazolidinones as Lmp7 inhibitors”. A detailed description of the synthesis and characterization of the compound is shown below. Unless otherwise noted, all chemicals were obtained from commercial sources and used without further purification. Analytical thin layer chromatography (TLC) was performed on EM Reagent 0.25 mm silica gel 60 F254 plates with visualization by ultraviolet (UV) irradiation at 254 nm and/or staining with potassium permanganate. Purifications by flash chromatography were performed using EM silica gel 60 (230–400 mesh). The eluting system for each purification was determined by TLC analysis.
1H and 13C spectra were measured with a JEOL ECZ 400 (400 MHz) spectrometer. 1H NMR chemical shifts are reported as δ in units of parts per million (ppm) relative to DMSO‐d 6 (δ 2.50, septet) or chloroform‐d (δ 7.26, singlet). Multiplicities are reported as follows: s (singlet), d (doublet), t (triplet), q (quartet), dd (doublet of doublets), dt (doublet of triplets), or m (multiplet). Coupling constants are reported as a J value in Hertz (Hz). The number of protons (n) for a given resonance is indicated as nH and is based on spectral integration values. 13C NMR chemical shifts are reported as δ in units of parts per million (ppm) relative to DMSO‐d 6 (δ 39.5, septet) or chloroform‐d (δ 77.2, triplet).
Synthesis of N‐((2S,3R)‐1‐(((S)‐3‐(3‐(3‐(tert‐butylamino)‐2‐cyano‐3‐oxoprop‐1‐en‐1‐yl)phenoxy)‐1‐((4‐methylbenzyl)amino)‐1‐oxopropan‐2‐yl)amino)‐3‐hydroxy‐1‐oxobutan‐2‐yl)‐5‐methylisoxazole‐3‐carboxamide (PRN1126) (Fig 1A):
Step 1
To a 500‐ml four‐neck round‐bottomed flask, tert‐butyl (S)‐(3‐(3‐formylphenoxy)‐1‐((4‐methylbenzyl)amino)‐1‐oxopropan‐2‐yl)carbamate (10 g, 24.2 mmol) and N‐(tert‐butyl)‐2‐cyanoacetamide (3.38 g, 24.2 mmol) were dissolved in CH2Cl2 (200 ml). The reaction mixture was cooled to 10°C. To the reaction mixture, pyrrolidine (7.04 g, 99.2 mmol) was added dropwise and stirred for 10 min. TMS‐Cl (7.84 g, 72.6 mmol) was added dropwise to the reaction mixture and stirred at room temperature for 2 h. A solution of NaHCO3 (500 ml) was added to the reaction mixture and extracted with CH2Cl2. The combined organic layer was washed with water (200 ml), dried, and concentrated, and then was dissolved in dioxane (40 ml) and water (5 ml). Sulfamic acid (1.41 g, 14.5 mmol) was added, and the reaction mixture was cooled to 10°C. To the above reaction mixture, the aqueous solution of NaClO2 (0.43 g, 4.84 mmol) in water (10 ml) followed by aqueous solution of KH2PO4 (0.39 g, 29 mmol) in water (15 ml) was added at 10°C. After completion of addition, the reaction mixture was warmed up to room temperature and stirred for 3 h. A saturated solution of NaHCO3 was added to the reaction mixture and extracted with ethyl acetate. The combined organics were washed with brine and dried over Na2SO4. Following filtration, the solution was concentrated to yield 7.5 g of tert‐butyl (S)‐(3‐(3‐(3‐(tert‐butylamino)‐2‐cyano‐3‐oxoprop‐1‐en‐1‐yl)phenoxy)‐1‐((4‐methylbenzyl)amino)‐1‐oxopropan‐2‐yl)carbamate, which was used in the next step without further purification.
Step 2
In a 250‐ml single‐neck round‐bottomed flask, tert‐butyl (S)‐(3‐(3‐(3‐(tert‐butylamino)‐2‐cyano‐3‐oxoprop‐1‐en‐1‐yl)phenoxy)‐1‐((4‐methylbenzyl)amino)‐1‐oxopropan‐2‐yl)carbamate (7.5 g, 14 mmol) was dissolved in 1,4‐dioxane (100 ml). To the reaction mixture, 5 N HCl in dioxane (40 ml) was added dropwise and stirred at 50°C for 1 h. The reaction mixture was concentrated, and traces of HCl was removed by azeotrope with toluene to yield 6.5 g of (S)‐3‐(3‐(2‐amino‐3‐((4‐methylbenzyl)amino)‐3‐oxopropoxy)phenyl)‐N‐(tert‐butyl)‐2‐cyanoacrylamide hydrochloride, which was used in the next step without further purification.
Step 3
In a 250‐ml single‐neck round‐bottomed flask under nitrogen atmosphere, (5‐methylisoxazole‐3‐carbonyl)‐L‐threonine (3.46 g, 15.2 mmol) was dissolved in DMF (40 ml) and cooled to 0°C. To this reaction mixture, HATU (7.86 g, 20.7 mmol) was added and stirred for 30 min. A solution of (S)‐3‐(3‐(2‐amino‐3‐((4‐methylbenzyl)amino)‐3‐oxopropoxy)phenyl)‐N‐(tert‐butyl)‐2‐cyanoacrylamide hydrochloride (6.5 g, 13.8 mmol) in DMF (25 ml) and DIPEA (7.1 ml, 41 mmol) was added dropwise at 0°C. The reaction mixture was further stirred for 1 h. The reaction mixture was diluted with cold water and extracted with ethyl acetate. The combined organic layer was washed with water, dried, and concentrated to get the crude, which was purified using flash column purification in 60–70% ethyl acetate in hexanes to yield 3.1 g of the title compound.
1HNMR (400 MHz, DMSO‐d6), 8.60–8.57 (m, 2H), 8.06 (d, J = 8.8, 1H), 8.01 (s, 1H), 7.83 (s, 1H), 7.55–7.44 (m, 3H), 7.15–7.05 (m, 5H), 6.56 (d, J = 0.8, 1H), 5.17 (d, J = 6.4, 1H), 4.79–4.73 (m, 1H), 4.52 (dd, J = 4.4, 8.8, 1H), 4.30–4.22 (m, 4H), 4.16–4.08 (m, 1H), 2.47 (bs, 3H), 2.25 (bs, 3H), 1.35 (s, 9H), 1.09 (d, J = 6.4, 1H). 13C NMR (100 MHz, DMSO‐d6: δ 172.1, 170.1, 168.9, 161.3, 159.0, 158.9, 158.8, 149.8, 136.3, 136.2, 133.8, 130.8, 129.2, 127.4, 122.8, 118.9, 117.0, 116.1, 108.7, 101.8, 68.2, 67.3, 58.8, 52.9, 52.0, 42.3, 28.6, 21.1, 20.3, 12.3. LC‐MS (ES, m/z): 645 [M + H].
Mice, cell lines, and media
C57BL/6 mice (H‐2b) were originally purchased from Charles River, Germany. LMP7 24 gene‐targeted mice were kindly provided by Dr. John J. Monaco (Department of Molecular Genetics, Cincinnati Medical Center, Cincinnati, OH, USA). Male or female mice were kept in a specific pathogen‐free facility and used at 8–10 weeks of age. All the animals were housed in groups of three to five animals in Eurotype II long clear‐transparent plastic cages with autoclaved dust‐free sawdust bedding. They were fed a pelleted and extruded mouse diet ad libitum and had unrestricted access to drinking water. The light/dark cycle in the room consisted of 12/12 h with artificial light. Animal experiments were approved by the Review Board of Regierungspräsidium Freiburg. LCL721.174 cells (contain CP) and LCL721.145 cells (contain IP) 38 were kindly provided by Hansjörg Schild (Mainz University). All media were purchased from Invitrogen (Life Technologies, Karlsruhe, Germany) and contained GlutaMAX, 10% FCS, and 100 U/ml penicillin/streptomycin.
Proteasome inhibitors
PRN1126 that was provided by Principia Biopharma (for synthesis, see above), ONX 0914 3 (formerly called PR‐957; Onyx Pharmaceuticals), LU‐001i 19, 26, and ML604440 23 were previously described and dissolved at a concentration of 10 mM in DMSO and stored at −80°C. For all in vitro experiments, a final DMSO concentration of 0.3% was used. For proteasome inhibition in mice, LU‐001i was dissolved in 5% ethanol, 10% PEG300 (Sigma) in an aqueous solution of 20% (w/v) sulfobutylether‐β‐cyclodextrin and 10 mM sodium citrate (pH 6), and administered to mice s.c. (15 mg/kg), and PRN1126 was dissolved in 5% ethanol, 45% PEG300 (Sigma) in an aqueous solution of 20% (w/v) sulfobutylether‐β‐cyclodextrin, and administered to mice s.c. (40 mg/kg). ONX 0914 was formulated in an aqueous solution of 10% (w/v) sulfobutylether‐β‐cyclodextrin and 10 mM sodium citrate (pH 6) and administered to mice as an s.c. bolus dose of 10 mg/kg.
Purification of 20S proteasome from mouse organs and fluorogenic assays
The purification of 20S proteasomes from the liver of MECL‐1−/−/LMP7−/− mice 39 (a source of mouse constitutive proteasome), the liver of LCMV‐infected BALB/c mice (8 days post‐infection with 200 pfu LCMV‐WE i.v.; source of mouse immunoproteasome), LCL721.174 cells (source of human constitutive proteasome), or LCL 721.145 cells (source of human immunoproteasome) as well as the analysis of their subunit composition on two‐dimensional non‐equilibrium pH gradient electrophoresis (NEPHGE)/SDS–PAGE was performed as described previously 40, 41.
Hydrolytic assays for proteasome activity using fluorogenic substrates were performed, as detailed previously 41. Substrates were dissolved in DMSO (10 mM) and used at final concentrations of 12.5 μM (Ac‐PAL‐AMC; Boston Biochem) or of 100 μM (Suc‐LLVY‐AMC; Bachem). IC50 values were determined using GraphPad Prism Software (version 6.07).
To investigate the cell permeability of PRN1126, LCL721.174 cells (contain CP) and LCL721.145 cells (contain IP) were treated with different inhibitor concentrations in PBS + 25 mM HEPES for 30 min at 37°C. The cell‐permeable substrate MeoSuc‐GLF‐AMC (Bachem; in DMSO 10 mM) was added at 40 μM to the cells and incubated at 37°C. The fluorescence intensity in the wells containing the cells was measured at an excitation wavelength of 360 nm and emission wavelength of 465 nm (Infinite M200 PRO; Tecan).
To investigate the durability of LMP7 inhibition, human PBMCs from healthy volunteers were incubated with 1 μM PRN1126 at 37°C for 1 h. PBMC wells were then washed three times with media to remove unbound compound and incubated for either 0, 0.5, 4 or 18 h in media. Hydrolytic assays for proteasome activity using fluorogenic substrates were performed, as detailed previously 41 at each time point to assess the durability of inhibition.
Immunoblotting
Equal numbers of Ficoll‐enriched lymphocytes were lysed in whole cell lysis buffer [1% NP‐40, 137 mM NaCl, 1 mM EDTA, 20 mM Tris–HCl pH 7.2 at 25°C, 2 mM Na3VO4, 0.15% SDS 1× PhosSTOP (Roche), 1× protease inhibitors (Roche), 0.1% sodium deoxycholate] on ice for 20–30 min and centrifuged for 15 min at 20,000 g. Protein extracts were separated by SDS–PAGE and transferred (wet blot) onto nitrocellulose membrane. The expression levels of LMP7 39, LMP2 39, MECL‐1 39, β5c (D1H68; Cell Signaling), β1c (clone E1K90; Cell Signaling), β2c (E1L5H; Cell Signaling), iota [clone IB5, recognizes the α1‐subunit iota of the proteasome and was generously contributed by Klaus Scherrer (Institut Jacques Monod, Paris, France)], and α‐tubulin (clone B‐5‐1‐2; Sigma) or γ‐tubulin (clone GTU‐88; Sigma) were analyzed by immunoblotting with specific antibodies combined with a secondary polyclonal goat anti‐mouse IgG antibody or swine anti‐rabbit IgG antibody conjugated to horseradish peroxidase (HRP; Dako, Hamburg, Germany).
LacZ assay
The LacZ assay was exactly performed as previously described 42. Shortly, 105 cells of the UTY246–254‐specific T‐cell hybridoma (kindly contributed by Nilabh Shastri, University of California, Berkeley, CA) were co‐cultured with 1.5 × 106 stimulator cells in 96‐well plates overnight. The lacZ‐based color reaction was conducted and measured, as detailed elsewhere 43.
Cytokine secretion assay
Splenocytes (106/well) of C57BL/6 mice were incubated with proteasome inhibitors or DMSO and stimulated with plate‐bound anti‐CD3 (5 μg/ml, clone 145‐2C11; eBioscience) and anti‐CD28 (5 μg/ml, clone 37.51; eBioscience) antibodies or stimulated with 3 μg/ml LPS (from Escherichia coli 0111:B4; Sigma). After 20 h, IL‐6 in the supernatant was determined by ELISA, according to the manufacturer's protocol (eBioscience).
Human PBMCs from healthy volunteers were incubated with proteasome inhibitors or DMSO at 37°C and stimulated with 3 μg/ml LPS. After 20 h, IL‐6 in the supernatant was determined by ELISA, according to the manufacturer's protocol (eBioscience).
MHC‐I cell surface expression
Splenocytes from C57BL/6 or LMP7−/− mice were incubated overnight with various concentrations of PRN1126, ML604440, LU‐001i, or DMSO as control. H‐2Kb surface staining was performed as previously described 44. Shortly, splenocytes were incubated with anti‐mouse CD16/32 (clone 2.4G2) to block Fc receptors and then stained for H‐2Kb‐PE (clone AF6‐88.5; BD Biosciences) for 30 min. After two washes, cells were acquired with the use of the Accuri C6 flow cytometer system (BD Biosciences).
Propidium iodide staining
Splenocytes from C57BL/6 mice were incubated with different proteasome inhibitors or DMSO control for 24 h. Cells were stained with antibodies for CD11c‐BV421 (clone N418; BD Biosciences), F4/80‐APC (clone BM8; eBioscience), CD4‐FITC (clone H1 29.19; BD Biosciences), CD8‐BV421 (clone 56‐6.7; BioLegend), and CD19‐FITC (clone eBio1D3; eBioscience). The viability of these cells was assessed with propidium iodide (PI; eBioscience) using the BD FACSVerse™ flow cytometer (BD Biosciences).
Th17 cell differentiation
Magnetically purified murine CD4+ T cells (MACS; Miltenyi Biotec) were stimulated (8 × 104 cells per well) with plate‐bound antibodies to CD3 (5 μg/ml, clone 145‐2C11; eBioscience) and CD28 (5 μg/ml, clone 37.51; eBioscience) in the presence of 2.5 ng/ml TGF‐β (PeproTech), 30 ng/ml IL‐6 (eBioscience) and antibodies to IL‐4 (10 μg/ml; eBioscience) and IFN‐γ (10 μg/ml; eBioscience) for 3 days. Intracellular IL‐17A (anti IL‐17‐APC, clone eBio17B7; eBioscience) expression in CD4+ cells (CD4‐FITC, clone H1 29.19; BD Biosciences) was measured 4 h after exposure to 50 ng/ml phorbol 12‐myristate 13‐acetate (PMA; Sigma) and 500 ng/ml ionomycin (Sigma) in the presence of 10 μg/ml brefeldin A (Sigma) by flow cytometry (Accuri C6; BD Biosciences).
Mouse LMP7 and LMP2 occupancy
Mice were treated with a single dose of PRN1126 (40 mg/kg, s.c.) or vehicle. Spleens were harvested at 1, 6, 14, and 24 h after dosing, and the occupancy of LMP7 or LMP2 subunits was assessed in splenocytes using the subunit‐specific ProCISE (proteasome constitutive/immunoproteasome subunit enzyme‐linked immunosorbent) assay.
Induction of colitis
Colitis was induced in 8–10‐week‐old mice by adding 3% dextran sulfate sodium (DSS; m.w. 36,000–50,000; MP Biomedicals, Solon, OH) to the drinking water, beginning on day 0 for 5 to 7 days; thereafter, mice were given regular drinking water. The body weight was measured daily throughout the experiment.
Induction of EAE
C57BL/6 mice were immunized subcutaneously in the lateral abdomen with 300 μg MOG35–55 peptide in CFA, and 200 ng pertussis toxin in PBS was administered on day 0 (i.p.) and day 2 (i.v.). Clinical disease was scored as follows: 0, no detectable signs of EAE; 0.5, distal limp tail; 1, complete limp tail; 1.5, limp tail and hind limb weakness; 2, unilateral partial hind limb paralysis; 2.5, bilateral partial hind limb paralysis; 3, complete bilateral hind limb paralysis; 3.5, complete hind limb paralysis and unilateral forelimb paralysis; and > 3, to be killed. Each time point shown is the average disease score of each group ±s.e.m.
Statistical analysis
The statistical significance of the differences was determined using the Student's t‐test, one‐way ANOVA with Dunnett's multiple comparison test, or two‐way ANOVA. Dunnett's multiple comparison test was run only if F achieved P < 0.05, and there was no significant variance inhomogeneity as determined with Bartlett's test. All statistical analyses were performed using GraphPad Prism Software (version 6.07; GraphPad, San Diego, CA). Statistical significance was achieved when P < 0.05. *P < 0.05; **P < 0.01; ***P < 0.001.
Author contributions
MB designed and performed experiments, wrote the manuscript, and supervised the project. CS, MML, JJLS, and JMB performed experiments. CJK, CT, EM, and HSO provided inhibitors. TDO designed PRN1126. CLL supervised the collaboration, provided inhibitors, and interpreted data. MG supervised the project and refined the manuscript.
Conflict of interest
M.B., C.S., E.M., H.S.O., and M.G. have no financial conflict of interest. M.M.L., J.J.LS., J.M.B., T.D.O., and C.L.L. are current or former employees of Principia Biopharma, Inc. C.T. is an employee of Takeda Pharmaceuticals International Co. C.J.K is President and Chief Scientific Officer of Kezar Life Sciences.
Supporting information
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 3
Acknowledgements
Ulrike Beck and Heike Goebel are acknowledged for excellent technical assistance. This work was funded by the German Research Foundation grant BA 4199/2‐1 to M.B. and GR 1517/2.4, GR 1517/10‐2, and SFB969 project C01 to M.G., the SwissLife Jubiläumsstiftung to M.B., and Swiss Cancer Research grant KFS‐3687‐08‐2015 to M.G. H.S.O. and E.M. acknowledge funding (TOP‐PUNT grant) from the Netherlands Organization of Scientific Research. Some of the flow cytometry experiments were performed at the flow cytometry facility FlowKon of the University of Konstanz.
EMBO Reports (2018) 19: e46512
This manuscript is dedicated to the late Jens Oliver Funk, to acknowledge his contributions to the initial design of studies and supervision of the collaboration.
Contributor Information
Michael Basler, Email: michael.basler@uni-konstanz.de.
Marcus Groettrup, Email: marcus.groettrup@uni-konstanz.de.
References
- 1. Huber EM, Basler M, Schwab R, Heinemeyer W, Kirk CJ, Groettrup M, Groll M (2012) Immuno‐ and constitutive proteasome crystal structures reveal differences in substrate and inhibitor specificity. Cell 148: 727–738 [DOI] [PubMed] [Google Scholar]
- 2. Manasanch EE, Orlowski RZ (2017) Proteasome inhibitors in cancer therapy. Nat Rev Clin Oncol 14: 417–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Muchamuel T, Basler M, Aujay MA, Suzuki E, Kalim KW, Lauer C, Sylvain C, Ring ER, Shields J, Jiang J et al (2009) A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat Med 15: 781–787 [DOI] [PubMed] [Google Scholar]
- 4. Mundt S, Engelhardt B, Kirk CJ, Groettrup M, Basler M (2016) Inhibition and deficiency of the immunoproteasome subunit LMP7 attenuates LCMV‐induced meningitis. Eur J Immunol 46: 104–113 [DOI] [PubMed] [Google Scholar]
- 5. Mundt S, Basler M, Buerger S, Engler H, Groettrup M (2016) Inhibiting the immunoproteasome exacerbates the pathogenesis of systemic Candida albicans infection in mice. Sci Rep 6: 19434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Koerner J, Brunner T, Groettrup M (2017) Inhibition and deficiency of the immunoproteasome subunit LMP7 suppress the development and progression of colorectal carcinoma in mice. Oncotarget 8: 50873–50888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vachharajani N, Joeris T, Luu M, Hartmann S, Pautz S, Jenike E, Pantazis G, Prinz I, Hofer MJ, Steinhoff U et al (2017) Prevention of colitis‐associated cancer by selective targeting of immunoproteasome subunit LMP7. Oncotarget 8: 50447–50459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Basler M, Dajee M, Moll C, Groettrup M, Kirk CJ (2010) Prevention of experimental colitis by a selective inhibitor of the immunoproteasome. J Immunol 185: 634–641 [DOI] [PubMed] [Google Scholar]
- 9. Zaiss DM, Bekker CP, Grone A, Lie BA, Sijts AJ (2011) Proteasome immunosubunits protect against the development of CD8 T cell‐mediated autoimmune diseases. J Immunol 187: 2302–2309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ichikawa HT, Conley T, Muchamuel T, Jiang J, Lee S, Owen T, Barnard J, Nevarez S, Goldman BI, Kirk CJ et al (2012) Novel proteasome inhibitors have a beneficial effect in murine lupus via the dual inhibition of type i interferon and autoantibody secreting cells. Arthritis Rheum 64: 493–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nagayama Y, Nakahara M, Shimamura M, Horie I, Arima K, Abiru N (2012) Prophylactic and therapeutic efficacies of a selective inhibitor of the immunoproteasome for Hashimoto's thyroiditis, but not for Graves’ hyperthyroidism, in mice. Clin Exp Immunol 168: 268–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Basler M, Mundt S, Muchamuel T, Moll C, Jiang J, Groettrup M, Kirk CJ (2014) Inhibition of the immunoproteasome ameliorates experimental autoimmune encephalomyelitis. EMBO Mol Med 6: 226–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Basler M, Mundt S, Bitzer A, Schmidt C, Groettrup M (2015) The immunoproteasome: a novel drug target for autoimmune diseases. Clin Exp Rheumatol 33: 74–79 [PubMed] [Google Scholar]
- 14. Liu H, Wan C, Ding Y, Han R, He Y, Xiao J, Hao J (2017) PR‐957, a selective inhibitor of immunoproteasome subunit low‐MW polypeptide 7, attenuates experimental autoimmune neuritis by suppressing Th17 cell differentiation and regulating cytokine production. FASEB J 31: 1756–1766 [DOI] [PubMed] [Google Scholar]
- 15. Basler M, Kirk CJ, Groettrup M (2013) The immunoproteasome in antigen processing and other immunological functions. Curr Opin Immunol 25: 74–80 [DOI] [PubMed] [Google Scholar]
- 16. Basler M, Beck U, Kirk CJ, Groettrup M (2011) The antiviral immune response in mice devoid of immunoproteasome activity. J Immunol 187: 5548–5557 [DOI] [PubMed] [Google Scholar]
- 17. Sula Karreci E, Fan H, Uehara M, Mihali AB, Singh PK, Kurdi AT, Solhjou Z, Riella LV, Ghobrial I, Laragione T et al (2016) Brief treatment with a highly selective immunoproteasome inhibitor promotes long‐term cardiac allograft acceptance in mice. Proc Natl Acad Sci USA 113: E8425–E8432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kalim KW, Basler M, Kirk CJ, Groettrup M (2012) Immunoproteasome subunit LMP7 deficiency and inhibition suppresses Th1 and Th17 but enhances regulatory T cell differentiation. J Immunol 189: 4182–4193 [DOI] [PubMed] [Google Scholar]
- 19. Basler M, Maurits E, de Bruin G, Koerner J, Overkleeft HS, Groettrup M (2018) Amelioration of autoimmunity with an inhibitor selectively targeting all active centres of the immunoproteasome. Br J Pharmacol 175: 38–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Khan S, van den Broek M, Schwarz K, de Giuli R, Diener PA, Groettrup M (2001) Immunoproteasomes largely replace constitutive proteasomes during an antiviral and antibacterial immune response in the liver. J Immunol 167: 6859–6868 [DOI] [PubMed] [Google Scholar]
- 21. Harding CV, France J, Song R, Farah JM, Chatterjee S, Iqbal M, Siman R (1995) Novel dipeptide aldehydes are proteasome inhibitors and block the MHC‐I antigen‐processing pathway. J Immunol 155: 1767–1775 [PubMed] [Google Scholar]
- 22. Palmowski MJ, Gileadi U, Salio M, Gallimore A, Millrain M, James E, Addey C, Scott D, Dyson J, Simpson E et al (2006) Role of immunoproteasomes in cross‐presentation. J Immunol 177: 983–990 [DOI] [PubMed] [Google Scholar]
- 23. Basler M, Lauer C, Moebius J, Weber R, Przybylski M, Kisselev AF, Tsu C, Groettrup M (2012) Why the structure but not the activity of the immunoproteasome subunit low molecular mass polypeptide 2 rescues antigen presentation. J Immunol 189: 1868–1877 [DOI] [PubMed] [Google Scholar]
- 24. Fehling HJ, Swat W, Laplace C, Kuehn R, Rajewsky K, Mueller U, von Boehmer H (1994) MHC class I expression in mice lacking proteasome subunit LMP‐7. Science 265: 1234–1237 [DOI] [PubMed] [Google Scholar]
- 25. Blackburn C, Gigstad KM, Hales P, Garcia K, Jones M, Bruzzese FJ, Barrett C, Liu JX, Soucy TA, Sappal DS et al (2010) Characterization of a new series of non‐covalent proteasome inhibitors with exquisite potency and selectivity for the 20S beta5‐subunit. Biochem J 430: 461–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. de Bruin G, Huber EM, Xin BT, van Rooden EJ, Al‐Ayed K, Kim KB, Kisselev AF, Driessen C, van der Stelt M, van der Marel GA et al (2014) Structure‐based design of beta1i or beta5i specific inhibitors of human immunoproteasomes. J Med Chem 57: 6197–6209 [DOI] [PubMed] [Google Scholar]
- 27. Gaffen SL, Jain R, Garg AV, Cua DJ (2014) The IL‐23‐IL‐17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol 14: 585–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Singh AV, Bandi M, Aujay MA, Kirk CJ, Hark DE, Raje N, Chauhan D, Anderson KC (2011) PR‐924, a selective inhibitor of the immunoproteasome subunit LMP‐7, blocks multiple myeloma cell growth both in vitro and in vivo . Br J Haematol 152: 155–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wehenkel M, Ban JO, Ho YK, Carmony KC, Hong JT, Kim KB (2012) A selective inhibitor of the immunoproteasome subunit LMP2 induces apoptosis in PC‐3 cells and suppresses tumour growth in nude mice. Br J Cancer 107: 53–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. De M, Jayarapu K, Elenich L, Monaco JJ, Colbert RA, Griffin TA (2003) Beta 2 subunit propeptides influence cooperative proteasome assembly. J Biol Chem 278: 6153–6159 [DOI] [PubMed] [Google Scholar]
- 31. Patel DD, Kuchroo VK (2015) Th17 cell pathway in human immunity: lessons from genetics and therapeutic interventions. Immunity 43: 1040–1051 [DOI] [PubMed] [Google Scholar]
- 32. Hayashi T, Faustman D (1999) NOD mice are defective in proteasome production and activation of NF‐ kappa B. Mol Cell Biol 19: 8646–8659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hayashi T, Faustman D (2000) Essential role of human leukocyte antigen‐encoded proteasome subunits in NF‐kappa B activation and prevention of tumor necrosis factor‐ alpha‐induced apoptosis. J Biol Chem 275: 5238–5247 [DOI] [PubMed] [Google Scholar]
- 34. Runnels HA, Watkins WA, Monaco JJ (2000) LMP2 expression and proteasome activity in NOD mice. Nat Med 6: 1064–1065; author reply 1065–1066 [DOI] [PubMed] [Google Scholar]
- 35. Kessler BM, Lennon‐Dumenil AM, Shinohara ML, Lipes MA, Ploegh HL (2000) LMP2 expression and proteasome activity in NOD mice. Nat Med 6: 1064; author reply 1065–1066 [DOI] [PubMed] [Google Scholar]
- 36. Bitzer A, Basler M, Krappmann D, Groettrup M (2017) Immunoproteasome subunit deficiency has no influence on the canonical pathway of NF‐kappaB activation. Mol Immunol 83: 147–153 [DOI] [PubMed] [Google Scholar]
- 37. Yun YS, Kim KH, Tschida B, Sachs Z, Noble‐Orcutt KE, Moriarity BS, Ai T, Ding R, Williams J, Chen L et al (2016) mTORC1 coordinates protein synthesis and immunoproteasome formation via PRAS40 to prevent accumulation of protein stress. Mol Cell 61: 625–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Salter RD, Cresswell P (1986) Impaired assembly and transport of HLA‐A and ‐B antigens in a mutant TxB cell hybrid. EMBO J 5: 943–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kremer M, Henn A, Kolb C, Basler M, Moebius J, Guillaume B, Leist M, Van den Eynde BJ, Groettrup M (2010) Reduced immunoproteasome formation and accumulation of immunoproteasomal precursors in the brains of lymphocytic choriomeningitis virus‐infected mice. J Immunol 185: 5549–5560 [DOI] [PubMed] [Google Scholar]
- 40. Basler M, Moebius J, Elenich L, Groettrup M, Monaco JJ (2006) An altered T cell repertoire in MECL‐1‐deficient mice. J Immunol 176: 6665–6672 [DOI] [PubMed] [Google Scholar]
- 41. Basler M, Groettrup M (2012) Immunoproteasome‐specific inhibitors and their application. Methods Mol Biol 832: 391–401 [DOI] [PubMed] [Google Scholar]
- 42. Basler M, Groettrup M (2013) Using protease inhibitors in antigen presentation assays. Methods Mol Biol 960: 31–39 [DOI] [PubMed] [Google Scholar]
- 43. Basler M, Youhnovski N, Van Den Broek M, Przybylski M, Groettrup M (2004) Immunoproteasomes down‐regulate presentation of a subdominant T cell epitope from lymphocytic choriomeningitis virus. J Immunol 173: 3925–3934 [DOI] [PubMed] [Google Scholar]
- 44. Basler M, Groettrup M (2007) No essential role for tripeptidyl peptidase II for the processing of LCMV‐derived T cell epitopes. Eur J Immunol 37: 896–904 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 3