

Abstract
Chemical toxicity can arise from disruption of specific biomolecular functions or through more generalized cell stress and cytotoxicity-mediated processes. Here, responses of 1060 chemicals including pharmaceuticals, natural products, pesticidals, consumer, and industrial chemicals across a battery of 815 in vitro assay endpoints from 7 high-throughput assay technology platforms were analyzed in order to distinguish between these types of activities. Both cell-based and cell-free assays showed a rapid increase in the frequency of responses at concentrations where cell stress/cytotoxicity responses were observed in cell-based assays. Chemicals that were positive on at least 2 viability/cytotoxicity assays within the concentration range tested (typically up to 100 μM) activated a median of 12% of assay endpoints whereas those that were not cytotoxic in this concentration range activated 1.3% of the assays endpoints. The results suggest that activity can be broadly divided into: (1) specific biomolecular interactions against one or more targets (eg, receptors or enzymes) at concentrations below which overt cytotoxicity-associated activity is observed; and (2) activity associated with cell stress or cytotoxicity, which may result from triggering specific cell stress pathways, chemical reactivity, physico-chemical disruption of proteins or membranes, or broad low-affinity non-covalent interactions. Chemicals showing a greater number of specific biomolecular interactions are generally designed to be bioactive (pharmaceuticals or pesticidal active ingredients), whereas intentional food-use chemicals tended to show the fewest specific interactions. The analyses presented here provide context for use of these data in ongoing studies to predict in vivo toxicity from chemicals lacking extensive hazard assessment.
Keywords: In vitro, high-throughput screening, oxidative stress, cytotoxicity, cell stress
Toxicology research is making increasing use of in vitro assay data to help elucidate potential adverse outcome pathways (AOP) (Ankley et al., 2010) or modes of action (MOA) (Boobis et al., 2008; Meek et al., 2003; Seed et al., 2005) through which chemicals may cause adverse effects in vivo. In vitro assay results from human cellular and molecular targets can aid in elucidating the underlying mechanism(s) of toxicity and, hence, assessing the human relevance of any adverse findings in animals. These issues were extensively discussed in the NRC Toxicity Testing in the 21st Century report (NRC, 2007). The U.S. EPA ToxCast program (Dix et al., 2007; Judson et al., 2010; Kavlock et al., 2012) and the U.S. federal cross-agency Tox21 program (Attene-Ramos et al., 2013; Collins et al., 2008; Shukla et al., 2010; Tice et al., 2013) are building large collections of in vitro assay data on diverse sets of environmentally-relevant chemicals to which humans are potentially exposed, including pesticides, food, cosmetics and personal care ingredients, pharmaceuticals, and industrial chemicals. Chemicals are being tested for bioactivity at various levels of biological organization in a broad battery of in vitro assays using multiple technologies. These include cell-free systems, cell lines, primary cells from multiple tissue types, complex culture systems, and alternative small model organisms (eg, zebrafish). The data from this program can enable a better understanding of the advantages and drawbacks of different in vitro technologies and analysis approaches.
Here, we report some key findings regarding assay performance and behavior based on analyses of a large data set from the EPA ToxCast program. The data set considered here was generated on 1060 unique chemicals in up to 815 distinct assay endpoints (Houck et al., 2009; Knight et al., 2009; Martin et al., 2010; Rotroff et al., 2010 , 2013; Sipes et al., 2013). The Tox21 program has run these same chemicals as part of a much larger set of more than 8300 chemicals, but in a smaller number of assays (Attene-Ramos et al., 2015; Hsu et al., 2014; Huang et al., 2011, 2014). The current assay portfolio covers 7 distinct technology platforms (see Table 1 in “Materials and Methods” section).
TABLE 1.
Summary of Assay Sets.
| Source | Assay endpoints | Note | References |
|---|---|---|---|
ACEA
|
2 | Real-time cell electronic sensing | (Abassi et al., 2009; Rotroff et al., 2013) |
Apredica
|
58 | High-content cell imaging; Duplicate in up and down direction | (Giuliano et al., 2005, 2006, 2010; Taylor and Giuliano 2005) |
Attagene
|
82 | Multiplex transcription reporter | (Martin et al., 2010; Romanov et al., 2008) |
BioSeek
|
174 | Biologically multiplexed activity profiling (BioMAP); Duplicate in up and down direction | (Berg et al., 2005, 2006, 2010; Houck et al., 2009; Kleinstreuer et al., 2014) |
Tox21
|
55 | Cell-based qHTS | (Attene-Ramos et al., 2015; Hsu et al., 2014; Huang et al., 2011, 2014; Xia et al., 2008) |
NovaScreen ADME
|
30 | Cell-free HTS Cytochrome P-450 activity assays; Duplicate in activator direction | (Knudsen et al., 2011; Sipes et al., 2013) |
NovaScreen Enzyme
|
115 | Cell-free HTS other enzyme activity assays; Duplicate in activator direction | (Knudsen et al., 2011; Sipes et al., 2013) |
NovaScreen GPCR
|
77 | Cell-free HTS G-protein coupled receptor assays | (Knudsen et al., 2011; Sipes et al., 2013) |
NovaScreen Ion Channel
|
22 | Cell-free HTS ion channel assays, including ligand-gated ion channels | (Knudsen et al., 2011; Sipes et al., 2013) |
NovaScreen Nuclear Receptor
|
20 | Cell-free HTS radioligand binding nuclear receptor assays | (Knudsen et al., 2011; Sipes et al., 2013) |
NovaScreen Transporter
|
9 | Cell-free HTS transporter activity assays | (Knudsen et al., 2011; Sipes et al., 2013) |
NovaScreen Other
|
2 | Cell-free HTS activity assays | (Knudsen et al., 2011; Sipes et al., 2013) |
Odyssey Thera
|
17 | Protein complementation | (Bolt et al., 2015; Stossi et al., 2014) |
The total number of assay endpoints is 815. The bulleted items in the first column are the suffixes used in the different assay sets. Information on individual assays is available from http://actor.epa.gov/dashboard, last accessed May 19, 2016.
The capacity of in vitro data and in silico models to reliably predict in vivo toxicity is a major consideration for the application and translation of ToxCast and Tox21 methods. One critical issue is the ability to infer molecular initiating events (MIEs) or key events (KEs) from the assay data for a wide range of MOAs and AOPs. Chemical toxicity can occur in many ways, but we hypothesize that it can be broadly classified into 2 major categories: disruption of specific biomolecular targets or pathways (eg, receptor agonist/antagonist effects, enzyme activation/inhibition), or generalized disruption of cellular machinery that can lead to cell stress and cytotoxicity. Cell-disruptive processes include protein, DNA or lipid reactivity; physico-chemical disruption of proteins or membranes (eg, by surfactants); or processes such as apoptosis, oxidative stress response, mitochondrial disruption, endoplasmic reticulum (ER) stress, microtubule disruption, or heat shock response.
Profiling the concentration-dependent responses of large chemical inventories across a battery of in vitro assays provides a new approach to enable a more comprehensive, systematic, and quantitative analysis of chemical activity across multiple, broad biological targets at concentrations relative to those that broadly invoke cell stress responses or cytotoxicity. First, we explore the issue of target selectivity across concentrations. Many chemicals show activation of large numbers of assays over a narrow range of concentrations in which cell stress and cytotoxicity are also seen. We term this phenomenon the cytotoxicity-associated “burst”. Whereas some of the assay activity in this concentration range may represent chemical effects on the intended target of the assay, some of it is not. In such situations, activity represents a false positive response that can be ascribed to assay interference processes (Baell and Holloway, 2010; Bruns and Watson, 2012; Thorne et al., 2010). This phenomenon raises the need to establish a concentration threshold at which each chemical begins to drive activity across multiple and diverse cell stress and cytotoxicity assays by initiating this cytotoxicity-associated burst of activity. Second, we test the degree to which the burst activity is associated with cell stress processes such as oxidative stress or mitochondrial disruption, uniquely or in addition to cytotoxicity. The large number of chemicals and assays in this dataset enable an assessment of this burst phenomenon across multiple technologies and in both cell-based and cell-free assays. (Note that it can be confusing to associate activity in cell-free assays to concentrations where cytotoxicity, an inherently cell-based phenomenon, occurs. We believe that this is because some mechanisms leading to cytotoxicity are acting through disruption at the molecular/physical level, which also operates in the cell-free assays.) Third, we describe an analysis strategy to separate the burst activity from what is more likely to be specific biomolecular interactions against one or more targets. We then present a promiscuity metric to quantify the number of off-target interactions below the concentration at which the cytotoxicity-associated burst occurs for pharmaceuticals, pesticidal active ingredients, or natural products for which there is an intended molecular target. These findings aim to better characterize the performance of the large collection of in vitro data generated by ToxCast, and to provide the background needed for follow-on analyses exploring the use of these data in systems toxicology (Sturla et al., 2014; Thomas et al., 2013).
MATERIALS AND METHODS
Chemicals and chemical annotation
The analyses presented here focused on 1060 unique chemicals which were run in at least 90% of the assays described below. These form a subset of the larger ToxCast chemical library that consisted of 1860 unique chemicals for which data were generated in at least some of the assays. Further information on the complete chemical library can be found at http://www.epa.gov/chemical-research/toxicity-forecasting (last accessed May 19, 2016), release date December 2014. The majority of chemicals were prepared and shipped as nominal 20 mM stock plates in DMSO. A few chemicals insoluble at 20 mM were provided at 2–15 mM concentrations. More details can be found in earlier papers on the ToxCast program (Dix et al., 2007; Judson et al., 2010; Kavlock et al., 2012).
Chemicals were annotated with a primary use category and a chemical structural class. The structural classification started by clustering chemicals using a set of structure fingerprints. Chemicals with available structures were fingerprinted using information from publicly available SMARTS sets FP3, FP4, MACCS (O'Boyle et al., 2011), PADEL (Yap, 2011), and PubChem (NCBI, 2008). OpenBabel was used to perform fingerprinting, after which chemicals were subjected to hierarchical clustering (Ward’s method, Tanimoto similarity, implemented in R package stats). This was followed by manual labeling of 665 clusters with short descriptive names (eg, primary alcohol, phthalate, and conazoles). In many cases, these clusters are one-to-one with common chemical groups such as phthalates, conazoles, etc. In all files, the chemicals were tracked by a unique code, which is usually the Chemical Abstracts Services Registry Number (CASRN) with dashes removed and a “C” prepended (eg, 80-05-7 = C80057). By convention, chemicals without a CASRN are given a value in the CASRN field of “NOCAS_xxxxx” where xxxxx is the DSSTox generic substance ID (GSID) (http://www.epa.gov/chemical-research/distributed-structure-searchable-toxicity-dsstox-database) (last accessed May 19, 2016). The chemical code for these chemicals is then of the form “CNOCASxxxxx”. All chemicals were assigned a primary use (eg, fungicide, food additive, and solvent) based on a survey of information from the internet carried out for this effort. Although a chemical could fall into multiple use categories, labeling with a primary use category, where possible, helps to highlight trends in the data. Based on the primary use categories, chemicals were then mapped to a smaller set of 8 aggregated-use categories including: FoodFlavorFragrance (later usually shortened to “Food”), Herbicide, Microbicide, Pesticide, Pharmaceutical, Solvent, Surfactant, and Other. The “Other” category contains a large number of structurally diverse industrial chemicals, with no specifically known intended consumer or industrial use. Note that some chemicals have many uses across these categories (Dionisio et al., 2013; Wambaugh et al., 2014), so the analyses here show the main trends but are not meant to be definitive. Summary chemical information is contained in a Supplementary Data with the code, CASRN, DSSTox GSID, chemical names, chemical structure (SMILES notation), structure and use classes, and mapping to a variety of chemical use lists. This file also contains a URL pointing to information used to assign the primary use.
In vitro assay data
The ToxCast in vitro data was generated by 7 commercial and governmental laboratories each using a different suite of technologies and assays. Most of these technologies and the associated ToxCast library assay results have been the subject of primary data publications (references provided in Table 1), so the assay types are only briefly summarized here. More detailed descriptions are given in Supplementary Data and in the primary publications. The majority of these assays were run on the original ToxCast Phase I (v1) library (Judson et al., 2010). Table 1 summarizes the assay sets. Not all chemicals were run in all assays. With the exception of the cell-free biochemical assays (NovaScreen), all assays were run in concentration-response format. For NovaScreen, there was an initial single-point screen (10 μM for ADME/CYP450 assays, 25 μM for remaining assays), and chemicals showing activity above a threshold value [3 median absolute deviations (MAD) above the median for all samples in the assay or an absolute response of 30% of positive control] were then run in concentration-response mode. This 2-stage process was driven by the prohibitive cost of running each of the several hundred NovaScreen assays in concentration-response mode for all chemicals.
All of the concentration-response data were analyzed using a standardized data analysis pipeline that automates the processes of baseline correction, normalization, curve-fitting, hit-calling, and detection of a variety of potential confounders. This pipeline, along with all of the raw and processed data, and annotations are publicly available (http://www.epa.gov/chemical-research/toxicity-forecasting and http://actor.epa.gov/dashboard, last accessed May 19, 2016). The data from each chemical-assay pair was fit to 3 models: a constant model, a Hill model, and a Gain-Loss model. The last allows the curve to rise from zero to a plateau, and then fall off again. This curve shape allowed us to account for non-specific assay interference, such as cytotoxicity occurring at high concentrations. Activity (hit) calls were determined based on a chemical-assay pair reaching a set of significance thresholds:
median of normalized response values at a single concentration above the established response cutoff;
modeled top (T) of the curve above the established response cutoff; and
Hill or Gain-Loss model was the selected model over the constant model.
In order to establish the response cutoff, the baseline median absolute deviation (BMAD) was calculated per assay using the distribution of the normalized response values for the lowest 2 concentrations for all chemicals run in the in vitro assay. The response cutoff was then selected per assay as being the maximum of 3-BMAD, 20% above baseline, or an assay-specific cutoff, eg, 6-BMAD or 10-BMAD. The Akaike Information Criterion (AIC) (Akaike, 1998) was then calculated for each model, and the model with the lowest AIC was selected. For each model, the output included parameters as well as a number of diagnostics. The diagnostics were assigned to specific chemical sample-assay pairs and indicate the presence of potential confounding factors such as curves that are marginally active and hence could be the result of non-normally distributed background noise instead of true activity. An AC50 (activity concentration at half-maximal response), Hill-slope, and maximum activity (T or Top value) were extracted.
All assay data are publicly available as both a series of flat files and a MySQL database dump at http://www.epa.gov/chemical-research/toxicity-forecasting, last accessed May 19, 2016. For all of the analyses reported here, the assay data were formatted into a series of files. Each of the files was organized as 1860 chemical rows by 815 assay endpoint columns. As stated above, for the following analyses, only the subset of 1060 chemicals with >90% of the assays run is used. There are 5 assay files, with 1 column per assay containing the following values: AC50 (concentration at which the activity reaches 50% of its maximal values for a particular assay-chemical pair, units of μM), AC50 modifier (eg, ==, <, >), Hill model parameters W and T, and the maximum observed efficacy (Emax). Inactive assay-chemicals pairs are assigned an AC50 of 1000000 μM for computational purposes. Attagene and BioSeek assay results are provided on a log-10 fold-change scale. We report logAC50 values [ie, −log10(AC50/1000000)]. This process sets inactive assay-chemical pairs to a value of 0 and 1 μM hits to a value of 6. For some comparisons in the current analyses, it was useful to scale all assays so that the T/top parameter was on a 0%–100% scale. For these cases, a scaled T-value was derived by using a linear scaling, setting the 95th percentile of the T-value (in log fold change) to 100%. This scaling was done on a per-assay basis. The raw data files contain the original log fold-change T-values.
Assay genes and categories
All assays were manually mapped to a high level biological process, usually based on their associated gene target and the gene to Gene Ontology mapping (Ashburner et al., 2000). These include processes such as inflammation (up/down), hypoxia, heat shock, and ion channel activity. The mappings are not unique, but serve to illustrate some useful trends in the data. The complete mapping between assays, genes, biological process, etc. is given in Supplementary Data.
Cytotoxicity assays
A total of 33 cytotoxicity-related assays are included in the assay set, and are used for much of the subsequent analysis. The assay battery is listed in Table 2, and indicated in Supplementary Data with “biological_process” of “cytotoxicity BLA,” “cytotoxicity SRB” or “proliferation decrease”. BLA indicates assays run alongside beta-lactamase readout assays, and SRB indicates cytotoxicity assays using the SulfoRhodamine B colorimetric technology (Vichai and Kirtikara, 2006). Note that “proliferation decrease” assays were sometimes active alone, with no frank cytotoxicity being present (ie, cytostatic effects without obvious cell loss). Cytotoxicity was then defined by a minimum threshold of activity; here, taken to be a positive call in 2 or more assays in the battery of 33 cytotoxicity-related or reduced proliferation assays. The threshold of 2 was selected based on our observation that most chemicals with 2 or more of these assays active displayed the burst.
TABLE 2.
34 Cytotoxicity-Related Assays.
| Assays | Biological process | Organism | Tissue | Cell type/Cell line |
|---|---|---|---|---|
| Tox21_AR_BLA_Antagonist_viability | Cytotoxicity BLA | Human | Kidney | HEK293T |
| Tox21_ERa_BLA_Antagonist_viability | Cytotoxicity BLA | Human | Kidney | HEK293T |
| Tox21_ESRE_BLA_viability | Cytotoxicity BLA | Human | Cervix | HeLa |
| Tox21_FXR_BLA_antagonist_viability | Cytotoxicity BLA | Human | Kidney | HEK293T |
| Tox21_GR_BLA_Antagonist_viability | Cytotoxicity BLA | Human | Cervix | HeLa |
| Tox21_HSE_BLA_agonist_viability | Cytotoxicity BLA | Human | Cervix | HeLa |
| Tox21_MMP_viability | Cytotoxicity BLA | Human | Liver | HepG2 |
| Tox21_NFkB_BLA_agonist_viability | Cytotoxicity BLA | Human | Cervix | ME-180 |
| Tox21_p53_BLA_p1_viability [1] | Cytotoxicity BLA | Human | Intestinal | HCT116 |
| Tox21_p53_BLA_p2_viability [1] | Cytotoxicity BLA | Human | Intestinal | HCT116 |
| Tox21_p53_BLA_p3_viability [1] | Cytotoxicity BLA | Human | Intestinal | HCT116 |
| Tox21_p53_BLA_p4_viability [1] | Cytotoxicity BLA | Human | Intestinal | HCT116 |
| Tox21_p53_BLA_p5_viability [1] | Cytotoxicity BLA | Human | Intestinal | HCT116 |
| Tox21_PPARg_BLA_antagonist_viability | Cytotoxicity BLA | Human | Kidney | HEK293 |
| Tox21_VDR_BLA_Agonist_viability | Cytotoxicity BLA | Human | Kidney | HEK293T |
| Tox21_VDR_BLA_antagonist_viability | Cytotoxicity BLA | Human | Kidney | HEK293T |
| BSK_3C_SRB_down | Cytotoxicity SRB | Human | Vascular | Umbilical vein endothelium |
| BSK_4H_SRB_down | Cytotoxicity SRB | Human | Vascular | Umbilical vein endothelium |
| BSK_BE3C_SRB_down | Cytotoxicity SRB | Human | Lung | Bronchial epithelial cell |
| BSK_CASM3C_SRB_down | Cytotoxicity SRB | Human | Vascular | Umbilical vein endothelium and coronary artery smooth muscle cells |
| BSK_hDFCGF_SRB_down | Cytotoxicity SRB | Human | Skin | Foreskin fibroblast |
| BSK_KF3CT_SRB_down | Cytotoxicity SRB | Human | Skin | Keratinocytes and foreskin fibroblasts |
| BSK_LPS_SRB_down | Cytotoxicity SRB | Human | Vascular | Umbilical vein endothelium and peripheral blood mononuclear cells |
| BSK_SAg_PBMCCytotoxicity_down | Cytotoxicity SRB | Human | Vascular | Umbilical vein endothelium and peripheral blood mononuclear cells |
| BSK_SAg_SRB_down | Cytotoxicity SRB | Human | Vascular | Umbilical vein endothelium and peripheral blood mononuclear cells |
| ACEA_T47D_80hr_Negative | Proliferation decrease | Human | Breast | T47D |
| APR_HepG2_CellLoss_24h_dn | Proliferation decrease | Human | Liver | HepG2 |
| APR_HepG2_CellLoss_72h_dn | Proliferation decrease | Human | Liver | HepG2 |
| BSK_3C_Proliferation_down | Proliferation decrease | Human | Vascular | Umbilical vein endothelium |
| BSK_3C_Vis_down | Proliferation decrease | Human | Vascular | Umbilical vein endothelium |
| BSK_CASM3C_Proliferation_down | Proliferation decrease | Human | Vascular | Umbilical vein endothelium and coronary artery smooth muscle cells |
| BSK_hDFCGF_Proliferation_down | Proliferation decrease | Human | Skin | Foreskin fibroblast |
| BSK_SAg_Proliferation_down | Proliferation decrease | Human | Vascular | Umbilical vein endothelium and peripheral blood mononuclear cells |
[1] These assays all use the same protocol but were performed as part of a chemical stability study, so were run on the same batch of chemicals that had been in solution at room temperature for increasing lengths of time, up to 6 months.
Modeling software and data availability
All software, except where noted, is written in R (version 3.1.0). This includes all statistical analyses. All input data and code is available from the U.S. EPA website [http://epa.gov/comptox/toxcast/data.html, last accessed May 19, 2016].
RESULTS
In Vitro Cytotoxicity
For many chemicals, we observed a disproportionately large number of positive assay responses (hits) in the concentration range where cytotoxicity was observed. This phenomenon is termed here as the cell stress or cytotoxicity-associated “burst”, which refers to the observed steep increase in number of assays showing activity upon reaching a concentration threshold. This phenomenon is illustrated in Figure 1. (Note that this phenomenon is also observed in cell-free biochemical assays, so it is not necessarily driven by cytotoxicity; however, it does occur at the concentrations at which cytotoxicity is seen in cell-based assays. This issue is discussed further below.) The burst region was delineated as follows. For chemicals with 2 or more hits in cytotoxicity assays, we calculated the median logAC50(cytotox) and the MAD of the logAC50(cytotox) for the hits. Next, we calculated the median of the MAD of the logAC50(cytotox) distributions across all chemicals to define the global cytotoxicity MAD. A new value (the Z-score) was then assigned to each chemical-assay hit combination:
| (1) |
FIG. 1.

Illustration of the high incidence of assay hits overlapping the cytotoxicity concentration region for 3 hypothetical chemicals (left panel), compared with the same distribution normalized to the Z-score space (right panel) to allow cross-chemical cytotoxicity region comparisons. For approximately 50% of chemicals tested, we observe cytotoxicity in the range of concentrations tested (up to ∼100 μM). For most of these chemicals, we also observe a large number of assay hits with AC50 values within the cytotoxicity region, represented by gray shading. The Z-score is a measure of how many MAD units below the median cytotoxicity value we observe a specific AC50, and allows for alignment of cytotoxicity regions across chemicals, independent of specific AC50s. The red “T” indicates the median and width (3 MAD) of the cytotoxicity AC50s. The activities of the third (bottom) chemical are inferred, because they occur at concentrations above those tested.
If fewer than 2 cytotoxicity-related assays were active, the median cytotoxicity concentration was arbitrarily set to 1000 μM for computational purposes. A chemical-assay hit with a large Z value indicates that activity occurred at concentrations far below the cytotoxicity threshold, as defined by equation 1. By definition, high-Z chemical-assay hits occurred in concentration regions where there was no evidence of cytotoxicity (and usually no evidence of cell stress). We hypothesize that these hits are then more likely to be associated with specific biomolecular interactions with the intended biological process or target that the assays are designed to measure. The global cytotoxicity MAD = 0.293 log units. The choice of 1000 μM value for the default median cytotoxicity concentration yields Z-score of >3 for all chemical-assay hits for these chemicals not observed to be cytotoxic, based on the upper testing concentration of 100 μM. (The lower bound of the cytotoxicity region for non-cytotoxic chemicals is 132 μM, equal to 3 global MAD below 1000 μM.) Because further analyses largely divide hits into bins with Z < 3 and Z > 3, any choice of the default >1000 will yield the same results. Figure 1 illustrates the concept of the burst in relation to the Z-score for 3 hypothetical chemicals.
The cutoff of Z = 3 is a somewhat arbitrary value used for illustration purposes, and other values would yield quantitatively, but not qualitatively different results. An important point about this the cutoff is that in practice, one would often use the Z-score more as a flag than an absolute cutoff. (Note that in our data analysis pipeline, we produce other “flags” such as to indicate noisy data, or hits due to a single point crossing the statistical threshold. These do not change the hit call, but provide the user a set of cautions or warnings to use when looking at data for a particular chemical-assay pair.) Hits with low Z-scores (deeper in the cytotoxicity region) are more likely to be associated with an interference process than will hits with high Z-scores (active well below where cytotoxicity is seen). However, given quantitative uncertainties in AC50 values (see the discussion), and our incomplete understanding of the biological linkages between cell-stress, cytotoxicity, and specific activity, a hard filter will not always be appropriate.
The selection of the use of 2 cytotoxicity assays as the cutoff for setting the cytotoxicity threshold below the default value of 1000 μM is also somewhat arbitrary. The total number of assays active as a function of the number of cytotoxicity hits is approximately linear (see Supplementary Data, first plot). One hypothesis is that chemicals that are cytotoxic at lower concentrations will show more cytotoxicity assays as being active, and hence more overall hits. However, with the exception of the chemicals with 1 assay active and with 33 active, there is at most a weak trend in the median cytotoxicity concentration vs. the number of cytotoxicity assays that are active (see Supplementary Data, second plot). From this plot, the 1-cytotoxity assay hit chemicals are somewhat anomalous, which is one reason for requiring 2 positive assays in the cytotoxicity set. There are numerous reasons that cytotoxicity values do not agree across all assays. Multiple cell types are used including cell lines and primary cells. Growth rates differ, and thus sensitivities to various toxicity mechanisms. Adaptive responses to various types of insults differ in each cell type. Assay technologies differ and respond to different cytotoxicity mechanisms with different kinetics. Levels of xenobiotic metabolism will also differ in each cell type.
Note that this default value for the median cytotoxicity concentration of 1000 μM is another example of an arbitrary hard cutoff. Other choices would yield quantitatively different results in the subsequent analyses, but the overall trends will hold. In particular for this default choice, we are biasing our result towards not “filtering” hits without strong evidence that they are in the cytotoxicity region. This minimizes false negatives when detecting specific activity, which is a conservative choice appropriate in a toxicity screening context. An alternative, and equally legitimate choice, would be to recognize that, historically, many hits in the 10–100 μM range are false positives. One could then bias the result to filter these hits even without direct evidence of cytotoxicity using a default closer to 100 μM. This choice would tend to minimize false positives for detecting specific activity, which would be more appropriate in a pharmaceutical efficacy screening context.
Figure 2 shows the activity profiles and structures for 4 exemplar chemicals (bisphenol A and 3 of its structural analogs) illustrating the presence of a peak of assay activity in the cytotoxicity region. For each of these chemicals, only a small fraction of hits fell outside of the cytotoxicity range. Bisphenol A, bisphenol B and bisphenol AF are estrogenic in vitro, whereas the tetrabromo analogue is not (Shen et al., 2013). The estrogen receptor assay hits, marked by triangles in Figure 2, occurred at concentrations below the cytotoxicity region in 3 out of 4 bisphenol compounds. The bisphenol compounds bind the ER through hydrogen-bond interactions with the hydroxyl groups. The tetrabromo variant is, at most, a weak binder because the bulky bromines sterically hinder the required ligand-protein interactions (Kitamura et al., 2005). Plots for all 1060 chemicals are provided in Supplementary Data.
FIG. 2.
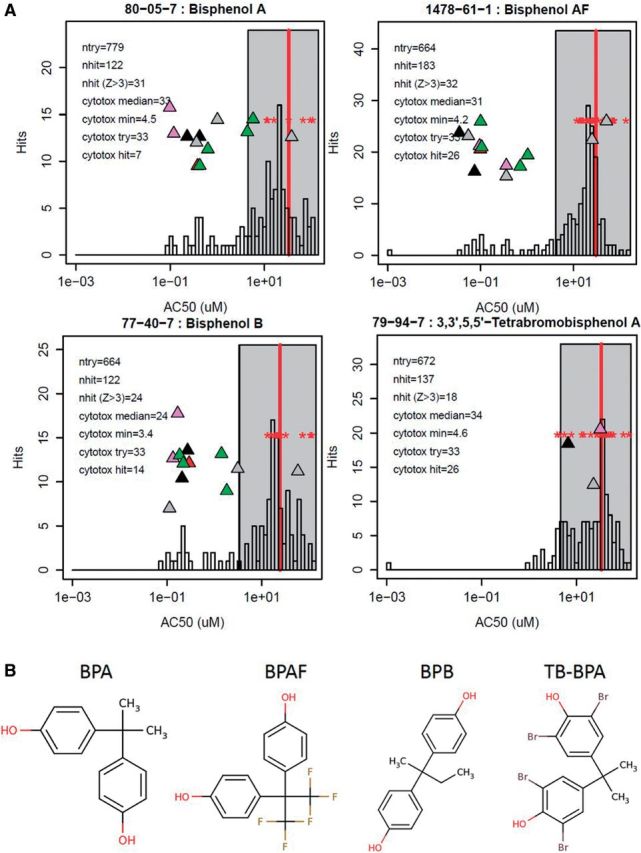
A, Examples showing assay activities both within and outside of the region of cytotoxicity for 4 bisphenol compounds. The histogram shows the distribution of AC50 values across the full assay set for each chemical. The vertical red bar shows the median of the log(AC50) of the cytotoxicity assays, and the gray box indicates the cytotoxicity region, starting 3 cytotoxicity-MAD (0.293 log units) below the median. The red stars indicate the AC50 values for the cytotoxicity assays activated by these chemicals (8 or more cytotoxity-related assays were active in each case). The x-axis (concentration) positions of the triangles indicate the AC50 values for estrogen receptor pathway assays. The y-axis positions for the triangles are randomly generated to prevent overlap of the symbols. Different colors indicate different technologies (black = NovaScreen, pink = Attagene, green = Tox21, gray = Odyssey Thera, red = ACEA). Up-arrows are agonist assays and down-arrows are antagonist assays. In the legend, ntry is the number of assay endpoints tested, excluding cytotoxicity endpoints, nhit is the number active, and nhit (Z > 3) is the number active with Z > 3. The cytotoxicity median and min are in units of μM and give the position of the red vertical line and the left end of the gray cytotoxicity region box (Z = 3). B, Structures of the example chemicals. BPA = bisphenol A, BPAF = bisphenol AF, BPB = bisphenol B, TB-BPA = 3,3’,5,5’-tetrabromo bisphenol A.
The distribution of Z-scores for almost all assays and technologies considered in this study had a strong peak around Z = 0, with a long tail towards higher Z-scores. Due to inherent differences in sensitivity between assay technologies, these distributions were shifted so that the peak occurred somewhat below zero (most common in cell-based assays, which tend to require relatively higher concentrations than cell-free assays to trigger activity), or somewhat above zero (most common for cell-free assays, which tend to require lower concentrations to trigger activity). To compensate for these technology-specific shifts, the Z-scores for all assays in a technology group were shifted to place the peak of the distribution at zero. The technology groups are: ACEA, Attagene, Apredica (positive and negative responses were run separately—“up” and “down” are respective suffixes on the assay names), BioSeek (positive and negative as with Apredica), Tox21, Odyssey Thera, and NovaScreen. For NovaScreen, the different assay technologies were further separated out according to the molecular functions grouped as listed in Table 1. The initial positions of the lower Z-score peak by assay technology (ie, before shifting, by subtracting the median log(AC50) to set the first peak at Z = 0) are given in Table 3. There is a general trend that the activity in cell-free assays is centered in the positive region before shifting, whereas the activity in the cell-based assays is centered in the negative region. A general trend of increased sensitivity in the cell-free assays is not surprising because chemicals have direct access to their potential targets without complications of subcellular transport, xenobiotic metabolism, or compartmentation. The NovaScreen ADME assays are the most right-shifted on the Z-score plot (shifted to higher positive values). These are predominantly cell-free cytochrome P450 activity assays. Perhaps this sensitivity reflects the native role of CYP P450 systems in xenobiotic metabolism or lower susceptibility to generalized interference.
TABLE 3.
Center of first peaks in Z-score (lower values of Z) by technology prior to normalization, in units of standard deviations.
| Source | Z Center |
|---|---|
| APR_dn | −1.64 |
| Tox21_BLA_Agonist | −1.53 |
| APR_up | −1.46 |
| Tox21_LUC_Antagonist | −1.27 |
| APR_Cytotoxicity | −1.16 |
| Tox21_Cytotoxicity | −1.15 |
| Tox21_BLA_Antagonist | −1.09 |
| ATG_CIS | −0.92 |
| ATG_TRANS | −0.82 |
| OT | −0.70 |
| NVS_ENZ | −0.17 |
| NVS_IC | 0.00 |
| BSK_down | 0.05 |
| BSK_Cytotoxicity | 0.15 |
| NVS_NR | 0.17 |
| NVS_GPCR | 0.20 |
| NVS_MP | 0.21 |
| NVS_TR | 0.53 |
| BSK_up | 0.73 |
| NVS_ADME | 1.60 |
| Tox21_LUC_Agonist | 1.76 |
The cell-free technologies are bolded.
Figure 3 illustrates the distributions of Z-scores for all chemicals for 4 representative technologies, 2 cell-free and 2 cell-based. We have separated out the contribution to the distribution from chemicals showing cytotoxicity (left column) from that due to chemicals not showing cytotoxicity (right column). Recall that the actual Z-scores for the non-cytotoxic chemicals are somewhat arbitrary due to the selection of 1000 μM as the default cytotoxicity center for these chemicals. The total area under the histogram for the cytotoxic chemicals is significantly greater than that for the non-cytotoxic chemicals, despite the fact that the 2 groups contain about the same number of chemicals (529 vs 531). The complete set of technology-wise summary plots are in Supplementary Data, and for all assays in Supplementary Data.
FIG. 3.
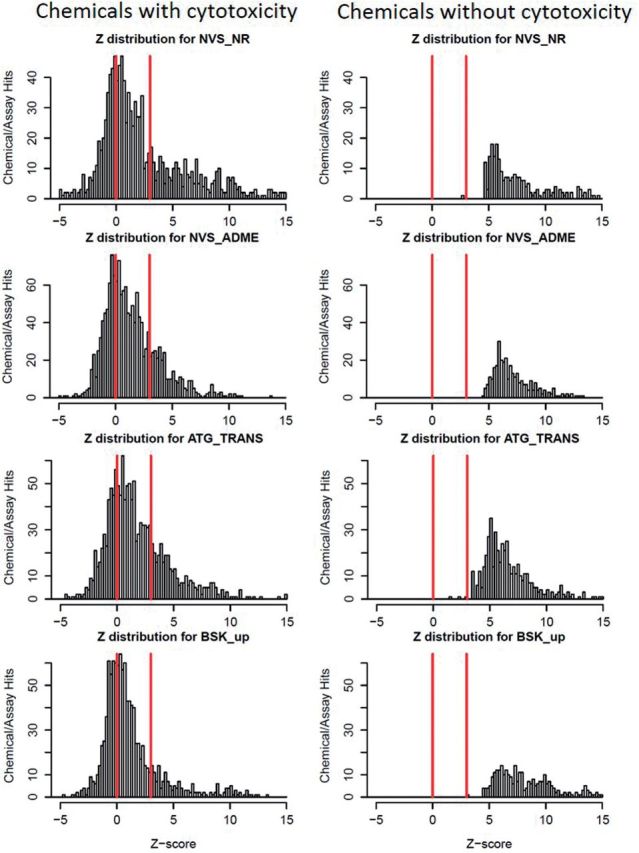
Distributions of Z-scores for all assays from 4 representative technologies, 2 cell-free (NVS_NR, NVS_ADME) and 2 cell-based (ATG_TRANS and BSK_up). Each histogram bar represents the number of assay-active chemicals in a particular Z-score range (AC50s transformed to Z-score based on cytotoxicity threshold for a particular chemical, equation 1), and the y-axis is scaled to the maximum number of hits in each case. The left hand column contains data for the cytotoxic chemicals (n = 529, ≥2 cytotoxicity assays hit), whereas the right hand contains data for the non-cytotoxic chemicals (n = 531, <2 cytotoxicity assays hit). The lines are placed at Z = 0 and 3. Plots for all technologies are given in Supplementary Data.
One test of the relative specificity indicated by the Z-score is a comparison of chemicals with their design targets versus others. (The design target is the intended molecular target of a pharmaceutical or pesticidal compound.) We designated chemicals with a design target that is probed by at least 1 assay as our in-group or “Target Class”. These chemicals are mostly either pharmaceuticals or pesticidal active ingredients, in addition to a small number of natural products or industrial chemicals that are used as reference compounds (eg, bisphenol A for ESR1). Some pharmaceuticals have multiple design targets (eg, different isoforms of the estrogen receptor). There were a total of 495 such chemical-assay pairs in the data set. The target genes in this list then determined the set of assays to be used for the comparison. We then calculated the hit rate and distribution of Z-scores for the Target Class chemicals with only their intended targets. The out-group (Others) was the balance of the 1060 chemicals, tested against all of the assays in the targeted gene set. The Target Class had a hit rate of 78% versus 6.7% for the out-group. The median values of Z for the 2 groups were 7.8 and 1.1 respectively, which are significantly different (P = 3.7E−65). The Z-score separation indicates that “true” hits tend to occur well outside of the cytotoxicity region, so are mostly not confounded with cytotoxicity. The fact that the overall hit (or confirmation) rate for these reference-like chemicals is well below 100% requires further explanation. The confirmation rate varies significantly by assay, indicating that some assays are under-sensitive (ie, they detect potent compounds but not weak ones). In the situation of the estrogen receptor assays, we have analyzed this extensively elsewhere (Judson et al., 2015). Another notable instance arises with the acetylcholinesterase (ACHE) assay, which only detects 30% of ACHE-targeting pesticides. This is because many of these compounds require metabolic activation. Because there are a large number of these compounds (56), the poor performance of this assay significantly brings down the total confirmation rate. The hit rates for all assays for these reference compounds are given in Supplementary Data.
An application of the Z-score approach described here is equivalent to use of in vitro assays to predict differences in concentrations required to engage therapeutic targets versus those that cause intrinsic or specific toxicities, ie, the therapeutic index (TI). Such methods are commonly used in early stage drug development to be followed eventually by a similar approach comparing efficacy versus toxicity using animal testing (Muller and Milton 2012). For non-pharmaceuticals, ie, for compounds without desired human efficacy, chemicals at concentrations corresponding to high Z-score would suggest potential activation of molecular initiating events leading to specific toxicity pathways. Achieving concentrations equivalent to the low-Z assay endpoints would likely induce additional, systemic toxicities. With respect to a TI approach for non-pharmaceuticals and other chemicals not intended for human efficacy, the concentration range used in place of the drug target efficacy range could be the known or predicted exposure ranges, which then would define the safety index. This is related to the in vitro-derived lower bound of the biological-pathway altering dose (BPAD) (Judson et al., 2011). Overall, the vast majority of assay hits are seen for chemicals with cytotoxicity observed in the concentration range tested, and seen with Z-scores <3. There are a total of 785 694 tested assay-chemical combinations, and 52 428 (6.7%) active combinations. Of the active combinations, 41 220 (79%) are in the cytotoxicity region (Z < 3), whereas 11 208 (21%) occur at lower concentrations, outside of this cytotoxicity region, ie, Z > 3. This latter group of 21% of hits is likely to be useful for elucidating molecular targets in pathway-based (AOP-based) analyses of chemical toxicity. There is a near-even split between chemicals that show cytotoxicity in the tested concentration range (<100 μM; 529 or 50%) and those that do not (531 or 50%). Chemicals showing cytotoxicity in the tested concentration range were active in an average of 12% of the tested assays (SD = 7.6%), whereas chemicals not showing cytotoxicity were active in an average of 1.3% of the tested assays (SD = 1.4%). The 2 groups do not differ significantly in the fraction of high-Z hits.
One major predictor of the overall activity (ie, number of assay hits) of a chemical across all assays was its aggregated use category (see “Materials and Methods” section, and Supplementary Data). Figure 4 shows boxplots for the fractions of assays hit for chemicals in the aggregated use groups, separately for hits at Z > 3 (high-Z) or Z < 3 (low-Z). From this, one can see that the trend for chemicals to be more active in the low-Z region extends across chemical use classes, although most Food and Solvent category chemicals are relatively inactive overall, and are much less likely to display cytotoxicity below 100 μM than are other categories of chemicals. The Food group was in general the least active set of chemicals and thus was used as a baseline for comparisons of overall activity. A t-test was used to compare the fraction of active assays between Food chemicals and chemicals in the other groups. The numbers in the plot are the number of chemicals in each group and the t-test P-values. We see that in both the high-Z (lower AC50) and low-Z (cytotoxicity) regions, most aggregated use groups are more active than Food chemicals, with the exception of Solvents. Several points are worth noting. First, within the biocide group (Herbicide, Microbicide, and Pesticide), the Herbicide chemicals were the least active, which may reflect the fact that the targets for which they were designed are phylogenetically distant from humans (the source species for most assays used here) relative to the targets for the Microbicide and Pesticide chemicals. Pharmaceutical compounds, many of which are “failed” drugs or drug candidates, are among the most active chemicals, but most of that activity is in the low-Z region. As discussed above, the intended targets of these chemicals also tend to occur with high Z-scores. Surfactant chemicals are very active in the low-Z region, which is likely to reflect large-scale disruption of the cells through processes such as disruption of protein–protein interactions, lipid disruption, or denaturation of cell membranes. Interestingly, Solvent chemicals are largely inactive. This implies a lack of membrane disruption leading to overall cell stress and cytotoxicity at the concentrations tested. Many commonly used solvents for compound dissolution, eg, DMSO, DMF, and EtOH, are used in cellular assays at concentrations of 0.1% and higher without cytotoxic effects. Even the low end of this range is equivalent to tens of millimolar which suggests the physicochemical disruptions of solvent effects may occur chiefly at concentrations above our testing range. Volatility may contribute to lack of significant effects. Finally, the “Other” category, which is made up mainly of industrial chemicals, has an almost bimodal distribution, with most chemicals largely inactive, but with a small subset that is much more active. The Food group also shows this trend. The subsets of active chemicals in these classes are further investigated below.
FIG. 4.

Boxplots showing the fraction of assays per chemical with Z > 3 (High-Z, left) and Z < 3 (Low-Z, right) in the aggregated use categories. The number at the top of each column is the number of chemicals in each group. Below that is the t-test P-value comparing the distribution of hit-fractions for each group versus the Food category. In all cases except for Solvents in the High-Z plot, all groups were more active than the Food group. NS indicates that there was no statistically significant difference. In each boxplot, the median is indicated by the dark horizontal line; the second and third quartiles by the box; the 95th percentile by the whiskers; and outliers by the circles.
Table 4 lists the most active (in terms of number of active assays) of the Food class chemicals. The chemicals with the greatest number of assays with high Z-values (ie, those active in 2% or more of the tested assays) are mostly natural products (genistein, eugenol, methyl salicylate, folic acid, and thymol). Table 5 lists the “Other” chemicals that are active in 10% or more of the tested assays at low Z-values. Note that the majority of these are phenols. Similar data is provided for all chemicals and structure and use categories (as described in Methods) in Supplementary Data.
TABLE 4.
Most active food-use chemicals (those active in 2% or more of the assays tested).
| Name | Structure category | Use category | Z-class | Fraction active |
|---|---|---|---|---|
| Tannic acid | Phenol benzoic acid | Natural product | Low-Z | 0.14 |
| Quercetin | Genistein-like | Natural product | Low-Z | 0.12 |
| Genistein | Genistein-like | Natural product | Low-Z | 0.12 |
| 1-Dodecanol | Alcohol primary | Flavor agent | Low-Z | 0.10 |
| 2-Benzylideneoctanal | Phenyl aldehyde | Food and flavor agent (natural product) | Low-Z | 0.09 |
| Dicyclohexyl disulfide | Alkane sulfide | Flavor agent | Low-Z | 0.08 |
| 1-Undecanol | Alcohol primary | Flavor and fragrance agent | Low-Z | 0.06 |
| 1-Pentadecanol | Alcohol primary | Natural product | Low-Z | 0.06 |
| Daidzein | Genistein-like | Flavone | Low-Z | 0.05 |
| Clove leaf oil | Oil | Natural product | Low-Z | 0.05 |
| 4-Cyclohexylcyclohexanone | Ketone cycloalkyl | Flavor and fragrance agent | Low-Z | 0.04 |
| Isoeugenol | Phenol alkoxy | Natural product | High-Z | 0.04 |
| 3,7-Dimethyl-2,6-octadienal | Aldehyde ene | Flavor and fragrance agent | High-Z | 0.04 |
| Acetyl tributyl citrate | Carboxylate multi | Flavor agent | Low-Z | 0.03 |
| Octadecanoic acid | Carboxylic acid | Natural product | Low-Z | 0.03 |
| Genistein | Genistein-like | Natural product | High-Z | 0.03 |
| Methyl salicylate | Salicylate | Flavor agent | High-Z | 0.02 |
| Eugenol | Phenol alkoxy | Natural product | High-Z | 0.02 |
| Benzophenone | Phenyl-phenyl [co] | Flavor agent | High-Z | 0.02 |
| 4-(2-Methylbutan-2-yl)cyclohexanol | Alcohol sec alkane cyclo | Fragrance agent | Low-Z | 0.02 |
| 2,4-Dimethylphenol | Phenol alkyl | Flavor agent | High-Z | 0.02 |
| Thymol | Phenol alkyl | Flavor agent | High-Z | 0.02 |
Z-class is High-Z: Z > 3 and Low-Z: Z < 3. The table separately lists the fraction of assays active in the Low-Z and High-Z regions, so chemicals can be listed twice. The rows corresponding to High-Z activity are bolded. The table is ordered by fraction active, regardless of the Z-class. Data on all chemicals and Use-category subtypes is given in Supplementary Data.
TABLE 5.
Chemicals in the “Other” class that are active in 10% or more of the tested assays at low Z-values.
| Name | structure_category | use_category | Fraction active |
|---|---|---|---|
| 1-Tridecanol | alcohol pri | Lubricant | 0.12 |
| 1-Tetradecanol | alcohol pri | Chemical intermediate | 0.10 |
| Hexachlorocyclopentadiene | alkane cyclo chloro | Chemical intermediate | 0.16 |
| N-Phenyl-1,4-benzenediamine | aniline-phenyl [N] | Chemical intermediate | 0.12 |
| 2-Amino-5-azotoluene | aniline-phenyl [NN] | Chemical intermediate (dyes) | 0.15 |
| 3,3'-Dimethoxybenzidine dihydrochloride | bisanliline alkoxy | Chemical intermediate (dyes) | 0.13 |
| Octyl gallate | gallate | Antioxidant | 0.22 |
| 7,12-Dimethylbenz(a)anthracene | PAH | Research chemical | 0.15 |
| 1-Hydroxypyrene | PAH-ol | Metabolite of Pyrene | 0.15 |
| 3-Hydroxyfluorene | PAH-ol | Metabolite of Fluorene | 0.11 |
| tert-Butylhydroquinone | phenol | Antioxidant | 0.14 |
| 2,4-Bis(2-methylbutan-2-yl)phenol | phenol alkyl | Chemical intermediate | 0.20 |
| 4-(1,1,3,3-Tetramethylbutyl)phenol | phenol alkyl | Chemical intermediate | 0.19 |
| 2,4-Di-tert-butylphenol | phenol alkyl | Antioxidant | 0.17 |
| 4-Octylphenol | phenol alkyl | Plastics | 0.15 |
| 2,5-Di-tert-butylbenzene-1,4-diol | phenol alkyl | Antioxidant | 0.14 |
| Hydroquinone | phenol-2 | Chemical intermediate | 0.10 |
| Bisphenol B | phenol-phenol [C] | Plastics | 0.14 |
| Bisphenol A | phenol-phenol [C] | Plastics | 0.13 |
| Bisphenol AF | phenol-phenol [C] halide | Plastics | 0.21 |
| 3,3',5,5'-Tetrabromobisphenol A | phenol-phenol [C] halide | Flame retardant | 0.18 |
| Phenolphthalein | phenol-phenol [Cn] carboxylate | Dye | 0.14 |
| 4,4'-Sulfonylbis[2-(prop-2-en-1-yl)phenol] | phenol-phenol [SO2] | Thermal recording material | 0.22 |
| 4-Cumylphenol | phenol-phenyl [C] | Chemical intermediate | 0.20 |
| 2,4-Bis(1-methyl-1-phenylethyl)phenol | phenol-phenyl [C] phenyl | Chemical intermediate | 0.18 |
| 2-Chloroacetophenone | phenyl ketone halide | Irritant e.g. tear gas | 0.14 |
| Triphenyl phosphate | phenyl phosphate | Flame retardant | 0.12 |
| Dicumyl peroxide | phenyl-phenyl [COOC] | Chemical intermediate | 0.13 |
| TDCPP | phosphate alkyl halide | Flame retardant | 0.11 |
| Triglycidyl isocyanurate | triazinone epoxide | Chemical intermediate (resins) | 0.11 |
| 4,4',4-Ethane-1,1,1-triyltriphenol | triphenol [C] | Plastics | 0.15 |
Data on chemicals, structure and use categories are given in Supplementary Data. These categories are described in the “Materials and Methods” section. Chemicals are ordered by structure category.
The data set includes a number of assays that probe for mechanisms of cell stress response or other disruptive cellular phenotypes, including apoptosis, oxidative stress, endoplasmic reticulum (ER) stress, heat shock, microtubule disorganization, and mitochondrial disruption. Most of the activity in these assays occurs in the cytotoxicity region, which makes it difficult to de-convolute direct versus indirect modes of cellular disruption/stress. Understanding specific mechanisms may be useful in identifying potential target tissues in vivo because cell types have different capacities to handle different types of stressors. Our current assay suite has only very limited temporal data collections; one potential future approach to identify the direct stress response leading to generalized cytotoxicity is to run assays in both concentration-response and time-response mode (Shah et al. (forthcoming)).
In lieu of existing temporal data, we attempted to better elucidate direct chemical effects by mapping cell stress assays to 6 classes: apoptosis, oxidative stress, endoplasmic reticulum stress, heat shock, microtubule disruption, and mitochondrial disruption. Chemicals were clustered based on activity against the related assays, in addition to the 3 classes of cytotoxicity assays, shown in Figure 5. The assay order was fixed, but chemicals were hierarchically clustered using Ward’s method. We see clustering with distinct patterns of cell stress response, decreased proliferation and cytotoxicity. Cluster 1 had almost no activity in any of these assays, and correspondingly little activity in the total set of assays (see the color bar at the top of the figure and its meaning in the figure legend). Clusters 2, 3, and 6 showed decreased proliferation but little overt cytotoxicity, and mainly differed in the types of cell stress observed (eg, clusters 3 and 6 had more evidence of oxidative stress than cluster 2). Cluster 6 is interesting in that many of the chemicals triggered oxidative stress and showed activity in mitochondrial disruption assays, but with little effect on overall cell health. Cluster 4 showed decreased cell proliferation and cytotoxicity in the sensitive primary cell assays, cluster 7 shows more evidence of cytotoxicity in the primary cells and some in the cell lines, and cluster 5 is the most toxic set of chemicals, with significant toxicity in all cell types. Supplementary Data is a spreadsheet containing the data in Figure 5.
FIG. 5.

Heatmap of activity in the 64 cytotoxicity and cell stress response assays. The ordering of assays was fixed as shown, but chemicals were hierarchically clustered. The top color bar indicates the total number of assays hit by a chemical: blue (<10), green (<50), yellow (<100), orange (<150), and red (>150). The color code in the heatmap is: no activity (white), activity with Z < 3 (light red), and activity with Z > 3 (dark red/brown).
Trying to understand the linkages between different types of cell stress and cytotoxicity is a difficult task, and it will require more research to fully understand the patterns seen in Figure 5. For instance, oxidative stress is a complex process occurring through a variety of mechanisms. Cells have varying degrees of adapting to such stress and the outcome is modulated by concentration, duration and type of reactive oxygen species. Thus, it is not surprising that oxidative stress can be detected in cells that have not undergone cell death. Indeed, Shah et al. (forthcoming) examine what is involved in reaching a tipping point where cells can no longer recover from such stress. An important issue that the current set of assays and experiments do not allow us to sort out is what is the initial cause of cell stress and cytotoxicity? For instance, one could initially disrupt the mitochondria, which would then lead to the production of free radicals, leading to oxidative stress and finally cell death. Alternatively, some other process might have caused oxidative stress, and then the mitochondrial disruption occurred later. One of our areas of research focus is trying to sort out the initial event that leads to cytotoxicity, and involves running selected time series experiments and chemical structure/reactivity modeling.
The analysis so far has used the Z-score as a metric which is based on the concentration range where cytotoxicity was observed. However, we found an increased number of hits in cell-free assays as well as in cell-based ones in the cytotoxicity range. As such, cell death/cytotoxicity cannot be the sole causal driver of this phenomenon. Some cytotoxicity may be driven by physico-chemical factors, such as protein denaturation or reactivity, which would affect both the cell-free and cell-based assays. Another possibility is very low-affinity non-covalent binding to receptors, enzymes, etc. that only occurs at very high concentrations. Figure 6 illustrates the relationship between the fraction of cell-free and cell-based hits in the low-Z region. Most chemicals activated a higher fraction of cell-based assays than cell-free ones, with some notable exceptions. There are a group of microbicides (named chemicals, blue dots) that affected a larger portion of cell-free assays than average. With the exception of myclobutanil, all of these are organometallics which might be expected to disrupt protein structure directly or through a reactive mechanism. Similarly, a set of surfactants (red dots) also affect a larger portion of cell-free assays, which could be due to their ability to denature proteins. Finally, there are several pharmaceuticals (white dots) that are active against specific targets that are related to, but not identical with, their intended design targets. This is what we call promiscuity rather than assay interference. “Dirty” psychoactive compounds such as methadone hydrochloride are typical examples.
FIG. 6.

Analysis of hit-frequency in cell-based versus cell-free assays: Each circle show the information for a single chemical where the x and y values are the fraction of assays in the cell-free and cell-based classes, respectively. The diagonal line shows a 1:1 ratio. Points are colored as indicated in the legend. The text discusses the named chemicals.
Several papers have been published on structure-based rules to detect chemicals that are potentially reactive or may cause assay interference (Baell and Holloway 2010; Bruns and Watson 2012; Thorne et al., 2010). We ran the current set of chemicals through the Bruns and Watson structure-based prediction model (Bruns and Watson 2012) (https://tripod.nih.gov/?p=418, last accessed May 19, 2016). These authors place chemicals into 3 categories: “clean”, “dirty”, and “fail”, where the latter 2 classes are expected to have increasingly larger numbers of hits (and by definition most of these are off-target). We compared the distribution of hits between chemicals placed into these 3 classes, for hits in the low and high Z-value regions. At low Z-values (in the cytotoxicity region), the chemicals classified as “fail” had significantly more hits than those classified as “clean” (P = .003), but there was no significant difference in hit frequency between the “clean” and the intermediate-class “dirty” chemicals. Therefore, the Bruns–Watson “fail” category captures many chemicals that hit many assays, but our analysis would label most of these hits as being cytotoxicity/cell stress-related. For activity in the high Z-value region (at lower concentrations than the cytotoxicity region), there was no significant difference in activity between any of the 3 Bruns–Watson categories.
DISCUSSION
We have provided a summary of analyses of trends in in vitro activity and the effects of cell stress and cytotoxicity in a data set covering 1060 chemicals screened in a battery of 815 high-throughput in vitro assay endpoints, covering 7 different technologies and multiple biological/cellular backgrounds and targets. A first key finding is that for many chemicals (50% of the 1060), we observed a large number of assay hits (cell-based, cell-free, and all assay technologies) in a narrow concentration range corresponding to where cell stress and cytotoxicity were observed. For chemicals that show cytotoxicity in the tested concentration range (typically up to 100 μM), the median percentage of assays that were active in the cytotoxic concentration range is 12%. For these same assays, the average number of active chemicals having AC50s with Z > 3 (well below cytotoxic range) is only 1.3%. The distribution for chemicals that do not show cytotoxicity in the tested concentration range closely mimics the Z > 3 distribution for cytotoxic chemicals. This suggests that even the activity in the cell stress/cytotoxicity region is meaningful in that it reflects true dysregulation of cellular machinery at higher exposure concentrations. However, this cytotoxicity-related phenomenon is not evident for all chemicals. Perhaps some chemicals were simply not tested to high enough concentrations (see the third chemical in Figure 1). Our test systems are mostly limited to ∼100 μM; therefore, if cytotoxicity occurs at concentrations well above this, then we will only pick up the tail of the distribution, if at all. Some chemicals may have low affinity for biological molecules, as well as lacking reactivity, so that no bioactivity would be expected up to solubility-limiting concentrations.
We distinguish the cell stress/cytotoxicity associated activity from what is often called “assay interference” (Baell and Holloway, 2010; Bruns and Watson, 2012; Harding et al., 2003; Thorne et al., 2010). The latter is assumed to arise from interference of chemicals with the physics or chemistry of the technology platform, such as fluorescence emission or quenching, colorimetric interference, or large pH changes. Here instead, we hypothesize that the large numbers of cytotoxicity-related hits observed mostly reflect cellular or biochemical activity that may also be seen in vivo. For instance, chemicals that cause lipid peroxidation, sulfhydryl group modification, protein acylation, redox cycling resulting in oxidative stress, DNA damage, or chelation of required metal ions will cause significant effects that will be observed as increased assay responses affecting groups of assays that have biological features in common, regardless of the assay technology. In cell-based systems, they can lead to cytotoxicity, whereas in cell-free assays, the molecular assay machinery could be disrupted through, for example, protein reactivity or denaturation. Alternatively, some of this activity could be due to very low affinity non-covalent binding to receptors and enzymes that coincidentally occurs at cytotoxic concentrations. Concentrations affecting the cell-free assays are often equivalent to concentrations inducing cytotoxicity. Nevertheless, chemicals such as maneb, mancozeb, and titanium(4+) 2-[bis(2-hydroxyethyl)amino]ethanolate propan-2-olate are active in a number of cell-free enzyme assays at concentrations below where cytotoxicity occurs. Cells may have varying levels of protective or adaptive mechanisms that buffer the effects near the concentrations where cytotoxicity is observed.
A major goal of the ToxCast and Tox21 programs is to provide biological activity data for large numbers of chemicals to which humans and ecological species are potentially exposed, and to provide inputs for predictive models of toxicity. The current analysis provides important context around the data. The assay activity profiles observed for many chemicals can be subdivided into specific molecular target-based activity on one hand, and general cell stress or cytotoxicity-related activity on the other. The former set potentially can be used to understand or model target-mediated activities in relation to toxicity, for instance through the adverse outcome pathway/molecular initiating event approach. Knowledge of the specific molecular targets of chemicals provides an ability to understand specific modes of toxicity, better extrapolate across species, determine relevance, and design targeted in vivo experiments. The more general cell stress response/cytotoxicity effects should help inform non-specific or common mechanisms such as necrosis, regenerative proliferation, or secondary inflammation. Such activity may suggest a focus on understanding internal dosimetry and toxicokinetics, which may govern target organ toxicity. This may underlie liver toxicity frequently encountered with high dose toxicity testing, in particular for chemicals showing enhanced toxicity with biotransformation.
An overall trend that we document is that chemical classes that are designed to be active against particular molecular targets (pharmaceuticals and pesticidal active ingredients) were also among the most broadly active. Whereas pharmaceuticals and pesticidal active ingredients showed specific, high affinity interaction for their intended target (activity occurs well below cytotoxicity), they also showed the most promiscuous activity in the sub-cytotoxic region. In contrast, chemically simple molecular classes (ie, aliphatic chains, low molecular weight chemicals, and chemicals with few or no reactive functional groups) are relatively inactive, with the potential exception of broad cell stress and cytotoxicity.
Many of these findings were not readily apparent in the preliminary ToxCast Phase I data set (Dix et al., 2007; Houck et al., 2009; Huang et al., 2014; Judson et al., 2010; Kavlock et al., 2012; Knight et al., 2009; Knudsen et al., 2011; Rotroff et al., 2010, 2013) largely because it tested almost exclusively pesticidal actives, which have a limited spectrum of target selectivity, but are mostly highly active and cytotoxic. With the inclusion of, at one end of the spectrum, a large number of pharmaceuticals with many target classes, and, at the other end, many industrial and food-use chemicals showing little to no activity, the broad trends across the chemical landscape have become clearer.
We finally note that the most important of the finding here are the broad trends, which can provide “rules of thumb” for interpreting in vitro data. Specific details would change if we used different values for the default we have used (default median cytotoxicity potency for non-cytotoxic compounds, the use of 2 active cytotoxicity assays to label a chemical cytotoxic, the use of Z = 3 as hard cutoff for label hits in or out of the cytotoxicity regions). These are actually a subset of several “default” choices that we have made, and which are inherent in any screening program. Others include the selection of experimental factors such as the specific set of assays to be run (including here the selection of cytotoxicity assays), the set of tested concentrations, (including the upper testing concentration), and the number of replicates. On the computational side, there is a selection of the functional form to which the data is fit (a Hill curve or some more general set of functions), the numerical algorithm used for curve fitting (which determines for instance how outliers are treated), the method for determining whether a chemical-assay pair is a hit (which depends on the model of the underlying noise process, and an arbitrary selection of a statistical cutoff).
We need to emphasize though that there is uncertainty in all of the quantities that we present, and not just in the bright lines of the defaults. This issue is driving a significant next step in our analyses of the ToxCast data, which is the quantification of uncertainties. The first phase of this is the development of a variety of bootstrapping and Bayesian methods to derive confidence intervals around AC50s and other fitting parameters, and around the hit call itself (instead of an assay being a hit or no-hit, the hit probability ranges from zero to one). This uncertainty can be propagated through any model based on the concentration-response data, for instance providing a confidence interval around the Z-score for any hit. This should further emphasize the need to look at the full context around the data for any given chemical when using it in a particular decision-making context.
Supplementary Material
ACKNOWLEDGMENTS
We would like to acknowledge the detailed and very helpful comments from the anonymous reviewers of this manuscript, which served to greatly improve the final version.
FUNDING
All funding provided by the U.S. EPA.
REFERENCES
- Abassi Y. A., Xi B., Zhang W., Ye P., Kirstein S. L., Gaylord M. R., Feinstein S. C., Wang X., Xu X. (2009). Kinetic cell-based morphological screening: prediction of mechanism of compound action and off-target effects. Chem. Biol. 16, 712–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akaike H. (1998). Information Theory and an Extension of the Maximum Likelihood Principle. Springer, New York. [Google Scholar]
- Ankley G. T., Bennett R. S., Erickson R. J., Hoff D. J., Hornung M. W., Johnson R. D., Mount D. R., Nichols J. W., Russom C. L., Schmieder P. K., et al. (2010). Adverse outcome pathways: a conceptual framework to support ecotoxicology research and risk assessment. Environ. Toxicol. Chem. 29, 730–741. [DOI] [PubMed] [Google Scholar]
- Ashburner M., Ball C. A., Blake J. A., Botstein D., Butler H., Cherry J. M., Davis A. P., Dolinski K., Dwight S. S., Eppig J. T., et al. (2000). Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attene-Ramos M. S., Huang R., Michael S., Witt K. L., Richard A., Tice R. R., Simeonov A., Austin C. P., Xia M. (2015). Profiling of the Tox21 chemical collection for mitochondrial function to identify compounds that acutely decrease mitochondrial membrane potential. Environ. Health Perspect. 123, 49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attene-Ramos M. S., Miller N., Huang R., Michael S., Itkin M., Kavlock R. J., Austin C. P., Shinn P., Simeonov A., Tice R. R., and., et al. (2013). The Tox21 robotic platform for the assessment of environmental chemicals—From vision to reality. Drug Discov. Today 18, 716–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell J. B., Holloway G. A. (2010). New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 53, 2719–2740. [DOI] [PubMed] [Google Scholar]
- Berg E. L., Kunkel E. J., Hytopoulos E. (2005). Biological complexity and drug discovery: A practical systems biology approach. Syst. Biol. (Stevenage) 152, 201–206. [DOI] [PubMed] [Google Scholar]
- Berg E. L., Kunkel E. J., Hytopoulos E., Plavec I. (2006). Characterization of compound mechanisms and secondary activities by BioMAP analysis. J. Pharmacol. Toxicol. Methods 53, 67–74. [DOI] [PubMed] [Google Scholar]
- Berg E. L., Yang J., Melrose J., Nguyen D., Privat S., Rosler E., Kunkel E. J., Ekins S. (2010). Chemical target and pathway toxicity mechanisms defined in primary human cell systems. J. Pharmacol. Toxicol. Methods 61, 3–15. [DOI] [PubMed] [Google Scholar]
- Bolt M. J., Stossi F., Callison A. M., Mancini M. G., Dandekar R., Mancini M. A. (2015). Systems level-based RNAi screening by high content analysis identifies UBR5 as a regulator of estrogen receptor-alpha protein levels and activity. Oncogene 34, 154–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boobis A. R., Doe J. E., Heinrich-Hirsch B., Meek M. E., Munn S., Ruchirawat M., Schlatter J., Seed J., Vickers C. (2008). IPCS framework for analyzing the relevance of a noncancer mode of action for humans. Crit. Rev. Toxicol. 38, 87–96. [DOI] [PubMed] [Google Scholar]
- Bruns R. F., Watson I. A. (2012). Rules for identifying potentially reactive or promiscuous compounds. J. Med. Chem. 55, 9763–9772. [DOI] [PubMed] [Google Scholar]
- Collins F. S., Gray G. M., Bucher J. R. (2008). Toxicology. Transforming environmental health protection. Science 319, 906–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dionisio K. L., Frame A., Goldsmith M. R., Wambaugh J., Liddell A., Cathey T., Smith D., Vail J., Ernstoff A. S., et al. (2013). Exploring consumer exposure pathways and patterns of use for chemicals in the environment. Toxicol. Rep. 2, 228–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dix D. J., Houck K. A., Martin M. T., Richard A. M., Setzer R. W, Kavlock R. J. (2007). The ToxCast program for prioritizing toxicity testing of environmental chemicals. Toxicol. Sci. 95, 5–12. [DOI] [PubMed] [Google Scholar]
- Giuliano K. A., Cheung W. S., Curran D. P., Day B. W., Kassick A. J., Lazo J. S., Nelson S. G., Shin Y, Taylor D. L. (2005). Systems cell biology knowledge created from high content screening. Assay Drug Dev. Technol. 3, 501–514. [DOI] [PubMed] [Google Scholar]
- Giuliano K. A., Gough A. H., Taylor D. L., Vernetti L. A, Johnston P. A. (2010). Early safety assessment using cellular systems biology yields insights into mechanisms of action. J. Biomol. Screen. 15, 783–797. [DOI] [PubMed] [Google Scholar]
- Giuliano K. A., Johnston P. A., Gough A, Taylor D. L. (2006). Systems cell biology based on high-content screening. Methods Enzymol. 414, 601–619. [DOI] [PubMed] [Google Scholar]
- Harding H. P., Zhang Y., Zeng H., Novoa I., Lu P. D., Calfon M., Sadri N., Yun C., Popko B., Paules R., et al. (2003). An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11, 619–633. [DOI] [PubMed] [Google Scholar]
- Houck K. A., Dix D. J., Judson R. S., Kavlock R. J., Yang J, Berg E. L. (2009). Profiling bioactivity of the ToxCast chemical library using BioMAP primary human cell systems. J. Biomol. Screen. 14, 1054–1066. [DOI] [PubMed] [Google Scholar]
- Hsu C. W., Zhao J., Huang R., Hsieh J. H., Hamm J., Chang X., Houck K., Xia M. (2014). Quantitative high-throughput profiling of environmental chemicals and drugs that modulate farnesoid X receptor. Sci. Rep. 4, 6437.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R., Sakamuru S., Martin M. T., Reif D. M., Judson R. S., Houck K. A., Casey W., Hsieh J. H., Shockley K. R., Ceger P., et al. (2014). Profiling of the Tox21 10K compound library for agonists and antagonists of the estrogen receptor alpha signaling pathway. Sci. Rep. 4, 5664.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R., Xia M., Cho M. H., Sakamuru S., Shinn P., Houck K. A., Dix D. J., Judson R. S., Witt K. L., Kavlock R. J., et al. (2011). Chemical genomics profiling of environmental chemical modulation of human nuclear receptors. Environ. Health Perspect. 119, 1142–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imran Shah, Woodrow Setzer, John Jack, Keith Houck, Thomas Knudsen, Matt Martin, David Reif, Ann M. Richard, David J. Dix, Robert J. Kavlock. Elucidating dynamic modulation of cellular state function during chemical perturbation. Environ. Health Perspect. Forthcoming. [Google Scholar]
- Judson R. S., Houck K. A., Kavlock R. J., Knudsen T. B., Martin M. T., Mortensen H. M., Reif D. M., Rotroff D. M., Shah I., Richard A. M, et al. (2010). In vitro screening of environmental chemicals for targeted testing prioritization: the ToxCast project. Environ. Health Perspect. 118, 485–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judson R. S., Kavlock R. J., Setzer R. W., Cohen Hubal E. A., Martin M. T., Knudsen T. B., Houck K. A., Thomas R. S., Wetmore B. A, Dix D. J. (2011). Estimating toxicity-related biological pathway altering doses for high-throughput chemical risk assessment. Chem. Res. Toxicol. 24, 451–462. [DOI] [PubMed] [Google Scholar]
- Kavlock R., Chandler K., Houck K., Hunter S., Judson R., Kleinstreuer N., Knudsen T., Martin M., Padilla S., Reif D., et al. (2012). Update on EPA's ToxCast program: Providing high throughput decision support tools for chemical risk management. Chem. Res. Toxicol. 25, 1287–1302. [DOI] [PubMed] [Google Scholar]
- Kitamura S., Suzuki T., Sanoh S., Kohta R., Jinno N., Sugihara K., Yoshihara S., Fujimoto N., Watanabe H., Ohta S. (2005). Comparative study of the endocrine-disrupting activity of bisphenol A and 19 related compounds. Toxicol. Sci. 84, 249–259. [DOI] [PubMed] [Google Scholar]
- Kleinstreuer N. C., Yang J., Berg E. L., Knudsen T. B., Richard A. M., Martin M. T., Reif D. M., Judson R. S., Polokoff M., Dix D. J., et al. (2014). Phenotypic screening of the ToxCast chemical library to classify toxic and therapeutic mechanisms. Nat. Biotechnol. 32, 583–591. [DOI] [PubMed] [Google Scholar]
- Knight A. W., Little S., Houck K., Dix D., Judson R., Richard A., McCarroll N., Akerman G., Yang C., Birrell L, et al. (2009). Evaluation of high-throughput genotoxicity assays used in profiling the US EPA ToxCast chemicals. Regul. Toxicol. Pharmacol. 55, 188–199. [DOI] [PubMed] [Google Scholar]
- Knudsen T. B., Houck K. A., Sipes N. S., Singh A. V., Judson R. S., Martin M. T., Weissman A., Kleinstreuer N. C., Mortensen H. M., Reif D. M., J. R., et al. (2011). Activity profiles of 309 ToxCast chemicals evaluated across 292 biochemical targets. Toxicology 282, 1–15. [DOI] [PubMed] [Google Scholar]
- Martin M. T., Dix D. J., Judson R. S., Kavlock R. J., Reif D. M., Richard A. M., Rotroff D. M., Romanov S., Medvedev A., Poltoratskaya N., et al. (2010). Impact of environmental chemicals on key transcription regulators and correlation to toxicity end points within EPA's ToxCast program. Chem. Res. Toxicol. 23, 578–590. [DOI] [PubMed] [Google Scholar]
- Meek M. E., Bucher J. R., Cohen S. M., Dellarco V., Hill R. N., Lehman-McKeeman L. D., Longfellow D. G., Pastoor T., Seed J., Patton D. E. (2003). A framework for human relevance analysis of information on carcinogenic modes of action. Crit. Rev. Toxicol. 33, 591–653. [DOI] [PubMed] [Google Scholar]
- Muller P. Y., Milton M. N. (2012). The determination and interpretation of the therapeutic index in drug development. Nat. Rev. Drug Discov. 11, 751–761. [DOI] [PubMed] [Google Scholar]
- NCBI. (2008). PubChem. Available at http://pubchem.ncbi.nlm.nih.gov/. Accessed August 8, 2008.
- NRC. (2007). Toxicity Testing in the 21st Century: A Vision and a Strategy. National Academies Press, Washington, DC. [Google Scholar]
- O'Boyle N. M., Banck M., James C. A., Morley C., Vandermeersch T, Hutchison G. R. (2011).“Open Babel: An open chemical toolbox. J. Cheminform. 3, 33.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanov S., Medvedev A., Gambarian M., Poltoratskaya N., Moeser M., Medvedeva L., Diatchenko L., Makarov S. (2008). Homogeneous reporter system enables quantitative functional assessment of multiple transcription factors. Nat. Methods 5, 253–260. [DOI] [PubMed] [Google Scholar]
- Rotroff D. M., Beam A. L., Dix D. J., Farmer A., Freeman K. M., Houck K. A., Judson R. S., LeCluyse E. L., Martin M. T., Reif D. M, et al. (2010). Xenobiotic-metabolizing enzyme and transporter gene expression in primary cultures of human hepatocytes modulated by ToxCast chemicals. J. Toxicol. Environ. Health B Crit. Rev. 13, 329–346. [DOI] [PubMed] [Google Scholar]
- Rotroff D. M., Dix D. J., Houck K. A., Kavlock R. J., Knudsen T. B., Martin M. T., Reif D. M., Richard A. M., Sipes N. S., Abassi Y. A., et al. (2013). Real-time growth kinetics measuring hormone mimicry for Toxcast chemicals in T-47D human ductal carcinoma cells. Chem. Res. Toxicol. 26, 1097–1107. [DOI] [PubMed] [Google Scholar]
- Seed J., Carney E. W., Corley R. A., Crofton K. M., DeSesso J. M., Foster P. M., Kavlock R., Kimmel G., Klaunig J., Meek M. E., et al. (2005). Overview: Using mode of action and life stage information to evaluate the human relevance of animal toxicity data. Crit. Rev. Toxicol. 35, 664–672. [DOI] [PubMed] [Google Scholar]
- Shen J., Xu L., Fang H., Richard A. M., Bray J. D., Judson R. S., Zhou G., Colatsky T. J., Aungst J. L., Teng C., et al. (2013). EADB: An estrogenic activity database for assessing potential endocrine activity. Toxicol. Sci. 135, 277–291. [DOI] [PubMed] [Google Scholar]
- Shukla S. J., Huang R., Austin C. P., Xia M. (2010). The future of toxicity testing: A focus on in vitro methods using a quantitative high-throughput screening platform. Drug Discov. Today 15, 997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipes N. S., Martin M. T., Kothiya P., Reif D. M., Judson R. S., Richard A. M., Houck K. A., Dix D. J., Kavlock R. J, Knudsen T. B. (2013). Profiling 976 ToxCast chemicals across 331 enzymatic and receptor signaling assays. Chem. Res. Toxicol. 26, 878–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stossi F., Bolt M. J., Ashcroft F. J., Lamerdin J. E., Melnick J. S., Powell R. T., Dandekar R. D., Mancini M. G., Walker C. L., Westwick J. K, et al. (2014). Defining estrogenic mechanisms of bisphenol A analogs through high throughput microscopy-based contextual assays. Chem. Biol. 21, 743–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturla S. J., Boobis A. R., FitzGerald R. E., Hoeng J., Kavlock R. J., Schirmer K., Whelan M., Wilks M. F, Peitsch M. C. (2014). Systems toxicology: from basic research to risk assessment. Chem. Res. Toxicol. 27, 314–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor D. L., Giuliano K. A. (2005). Multiplexed high content screening assays create a systems cell biology approach to drug discovery. Drug Discov. Today Suppl, 13–18. [PubMed] [Google Scholar]
- Thomas R. S., Philbert M. A., Auerbach S. S., Wetmore B. A., Devito M. J., Cote I., Rowlands J. C., Whelan M. P., Hays S. M., Andersen M. E., M. E., et al. , (2013). Incorporating new technologies into toxicity testing and risk assessment: Moving from 21st century vision to a data-driven framework. Toxicol. Sci. 136, 4–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorne N., Auld D. S., Inglese J. (2010). Apparent activity in high-throughput screening: origins of compound-dependent assay interference. Curr. Opin. Chem. Biol. 14, 315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tice R. R., Austin C. P., Kavlock R. J., Bucher J. R. (2013). Improving the human hazard characterization of chemicals: A tox21 update. Environ. Health Perspect. 121, 756–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vichai V., Kirtikara K. (2006). Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 1, 1112–1116. [DOI] [PubMed] [Google Scholar]
- Wambaugh J. F., Wang A., Dionisio K. L., Frame A., Egeghy P., Judson R, Setzer R. W. (2014). High throughput heuristics for prioritizing human exposure to environmental chemicals. Environ. Sci. Technol. 48, 12760–12767. [DOI] [PubMed] [Google Scholar]
- Xia M., Huang R., Witt K. L., Southall N., Fostel J., Cho M. H., Jadhav A., Smith C. S., Inglese J., Portier C. J., et al. (2008). Compound cytotoxicity profiling using quantitative high-throughput screening. Environ. Health Perspect. 116, 284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap C. W. (2011). PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 32, 1466–1474. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.