

Abstract
Worldwide, 10% of babies are born preterm, defined as birth before 37 weeks’ gestation. We have had little success in developing strategies to prevent preterm births, the majority of which are due to infection or are idiopathic. An emerging hypothesis is that the maternal microbiome – the bacteria that inhabit the mother’s body and play vital functions in normal health – contributes to the etiology of preterm birth. Here, we highlight the latest data revealing correlations between preterm birth and maternal intestinal, vaginal, cervical, and placental microbiomes. Additionally, we describe the most commonly used comparative microbiome analysis methods and highlight important issues to consider when conducting such studies.
Keywords: vagina, placenta, 16S rRNA profiling, cervical microbiome
Introduction
The World Health Organization estimates that each year, 15 million infants are born preterm, putting them at increased risk of morbidity and mortality1. Simplistically, preterm birth (PTB) occurs when normal term labor events – uterine contractions and cervical remodeling – occur early2. However, PTB is challenging to explain, predict, and prevent because up to 40 to 45% of cases are idiopathic (spontaneous), and numerous risk factors are known, including maternal history, demographics, nutritional status, stress, and infection2.
Approximately 30% of PTB cases are caused by infection and inflammation2. Traditionally, infection-related PTB was thought to ensue from foreign microbes reaching the uterus via ascending infection or hematogenous transfer. During ascending infections, microbes from the vagina travel upward through the cervix to reach the fetal membranes. For example, the presence of Mycoplasma spp. (Ureaplasma nucleatum and Ureaplasma parvum) and Candida spp., in the vagina is associated with PTB3. Hematogenous infection occurs when bacteria travel through the blood stream from another site in the body and then traverse the placenta at the maternal-fetal interface2.
Although foreign bacteria are important causes of PTB, current research in this area is building on the observation that, far from being sterile, the human body is home to millions of microorganisms4. Collectively, all of the bacterial genomes present in or on our body surfaces are known as the human microbiome, and each body niche has its own resident microbes. These bacteria contribute to human health in many ways, such as providing resistance to pathologic infection, breaking down nutrients, and educating the immune system4,5. In addition to contributing to physiology, microbiome communities respond to physiology. Pregnancy is a period of major physiological changes6, such as immunological shifts and the vascular remodeling and metabolic changes needed to promote exchange of nutrients, gases, and wastes with the developing fetus7. Thus, the microbial community structure in various maternal niches has the potential to shift during pregnancy6. Although many of these changes may benefit or cause no harm to the mother and fetus, we are beginning to learn that dysbiosis of the maternal microbiomes is associated with adverse pregnancy outcomes such as PTB.
Here, we review human microbiome studies that define the microbiomes in key maternal niches and identify associations with both term and preterm birth. Additionally, we describe common technical, analytical, and statistical approaches used to conduct maternal microbiome studies. Rigorous studies together with curated microbiome data will provide an in-depth understanding of the maternal microbiomes and their impact on pregnancy and, hopefully, identify new therapeutic strategies to decrease the incidence and burden of PTB.
Maternal Intestinal Microbiome Changes During Pregnancy
A healthy gastrointestinal tract, dominated by Bacteriodetes and Firmicutes, contains numerous beneficial microbes that generate vitamins, break down complex foods, and synthesize products that can keep potentially harmful microbes at bay 8 (Figure 1). Because the intestinal microbiome regulates critical metabolic process, diseases, such as obesity and allergy, may arise when the community structure is abnormal 9.
Figure 1: Schematic depicting the intestinal and reproductive tract microbial communities during pregnancy.
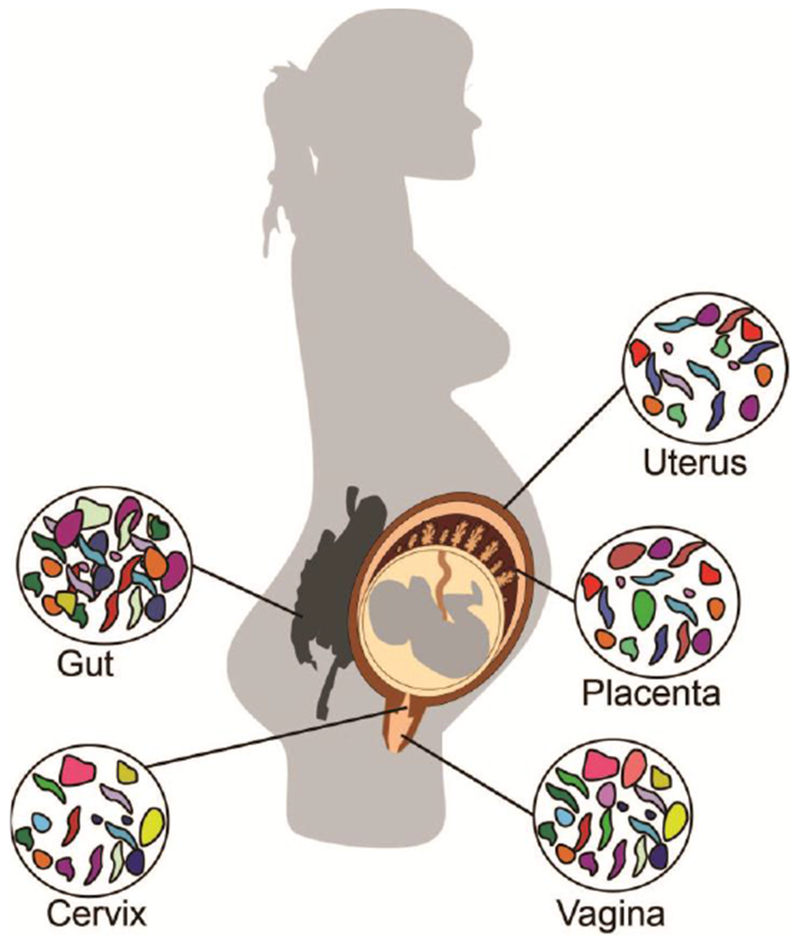
Adverse pregnancy outcomes may be associated with microbes that originate from reproductive tract (vaginal and cervical) and ascend to colonize the gestational compartment. However, mounting evidence suggests that other microbial communities including those in the gut and placenta may also impact pregnancy health.
Koren et al., used stool samples to characterize the maternal gut microbiome during the 1st and 3rd trimesters of pregnancy and post-partum10. They found that Proteobacteria was more abundant and Faecalibacterium was less abundant in the 3rd trimester compared to the other two stages of pregnancy. Although the underlying mechanisms and implications of this shift are not fully understood, the authors suggested that types of taxa present during these stages may be related to both anti-inflammatory (Faecalibacterium) and pro-inflammatory diseases (Proteobacteria).10 The maternal gut microbiota may change as a result of shifts in the environment during pregnancy as a result the demand for the transfer of nutrients to the fetus11. Future work will hopefully reveal the role of the gut microbiome during pregnancy and neonatal outcomes, which impact neonatal birth weight and infant gut colonization12,13.
Vaginal Microbiome and Preterm Birth:
The hallmark of a healthy vaginal microbiome in non-pregnant, reproductive-age (12 to 45 years old) women of multiple ethnic groups is a preponderance of the Lactobacillus genera14. These bacteria thrive in anaerobic niches and contribute to the acidic vaginal environment by fermenting sugars and producing lactic acid14–16. Lactic acid-producing species, like Lactobacillus, are thought to help protect the uterus from ascending infections such as sexually transmitted infection and urinary tract infections17,18. For example, diminished lactic acid-producing bacteria is associated with greater susceptibility to human immunodeficiency virus15,19.
The normal vaginal microbiome differs between pregnant and non-pregnant women20. Romero et al. found that whereas the community structure of the vaginal microbiota remained consistent across multiple gestational ages (and included species such as L. vaginalis, L. crlspatus, L. gasserl, and L. jensenii), the vaginal microbiota of non-pregnant women was more variable across different time points, with differences in the dominating Lactobacillus species. Similarly, another study revealed that the vaginal microbiome six weeks post-delivery was more diverse than the vaginal microbiome during pregnancy, regardless of ethnic background21. However, the dominating Lactobacillus species in the vagina differed depending on the region from which the women originated21, suggesting that region-specific factors such as diet affect the community structure of the vaginal microbiome. One possibility to explain the shifts before, during, and after pregnancy is that hormonal, nutritional, and immunological pressures alter the vaginal microbiota during pregnancy. Alternatively, these shifts in the vaginal microbiome may help maintain maternal and fetal health during pregnancy20.
Thus far, evidence that the community structure of the vaginal microbiota plays a role in PTB is limited. Nonetheless, a number of studies found that PTB correlates with bacterial vaginosis, a dysbiosis of the vaginal microbiome characterized by the emergence of pathogens such as F. nucleatum, Mycoplasma hominis, and Bacteriodetes urealyticus and the loss of Lactobacillus22, particularly during the 2nd trimester of pregnancy23. Prematurity in association with BV is also accompanied by low neonatal birth weight23. However, whether there is a casual relationship between BV and preterm birth has not been shown.
Cervical and uterine microbiomes
Vaginal microbes associated with PTB most likely reach the uterus by traveling through the cervix (Figure 1). Thus, more and more studies are beginning to address how microbes inhabiting this particular niche contribute to the PTB outcome. During the 2nd trimester of pregnancy, in women with a short cervix the preponderance of specific Lactobacillus species correlates with the gestational age at delivery—where increased vaginal L. iners is associated with preterm birth and L. crispatus is associated with term birth24. Further, the administration of vaginal progesterone, which normally used as a therapeutic for delaying early delivery, does not alter the vaginal microbiota24. Recently, Ghartey et al. analyzed metabolites in the cervicovaginal fluid at two time points in women who went on to deliver either preterm or at term and identified several that differed between the two groups25. Although these authors did not assess the microbiome in these women, these alterations in the metabolic environment could make the cervix and vagina more or less hospitable to PTB-related microbes26. Additionally, metabolites produced by the cervical microbiome could induce cervical remodeling and thereby promote labor, but there is currently no evidence to support this hypothesis25. Further, the microbiota within the cervical fluid of women with premature rupture of membranes (PPROM) exhibited variations in inflammatory markers in association with specific bacterial community state types (CST)—namely cervical fluid with increased non-Lactobacillus CSTs had increased IL-627.
Less studied is the uterine microbiome in association with PTB outcomes because little is known about its origins. Uterine biopsies sampled from non-pregnant women exhibited the presence of L. crispatus, L. iners, Prevotella spp., and other bacterial phylotypes28. Further, oral microbes, like Fusobacterium nucleatum, are thought to travel to the uterine cavity and contribute to PTB, as shown using a murine model29,30. Thus, many more studies must be conducted in order to understand the relevance of both the cervical and uterine microbiome in the etiology of PTB.
Placental Microbiome
Although the placenta was long thought to be germ-free, multiple histological and high-throughput sequencing studies have suggested that the placenta harbors its own microbiome31 (Figure 1). For example, our group used a variety of staining methods to demonstrate the presence of intracellular microbes in the basal plate (the maternal-fetal interface) of term and preterm placentas32. Aagaard et al. used 16S sequencing and whole-genome shotgun sequencing to profile bacteria in term and preterm-cross placental biopsies. These authors identified E. coli, Prevotella species, Bacteriodetes species, and Fusobacterium species33. Notably, preterm placentas had a preponderance of Burkholderia, Actinomycetales, and Alphaproteobacteria. In a later study, the same group profiled the fetal membrane microbiome in patients with chorioamnionitis (infection of the fetal membrane) and spontaneous PTB and found that it contained species previously identified in PTB. These included Ureaplasma and species associated with the oral microbiome, such as Fusobacteria. Microbiome changes in the fetal membranes from PTB subjects with chorioamnionitis was accompanied by shifts related to glucose metabolism, inflammatory markers and the siderophore biosynthesis34. 16S rRNA sequencing has also been used to determine that there is spatial organization to the placental microbial communities with distinct differences noted in the basal plate, fetal membranes (which encapsulate the amniotic cavity), and placental villi which are bathed in maternal blood35. The study noted that the fetal membranes were dominated by Firmicutes including Lactobacillus crispatus and Lactobacillus iners, which have previously been detected in the human placenta36. Since these species are prevalent in the intestinal and vaginal flora, this study suggests that the fetal membranes may be exposed to microbial species ascending from the vagina. In contrast, the basal plate was dominated by Proteobacteria, including Bacteriodetes, which have been found in the non-pregnant uterus28. Given the location of the basal plate adjacent to the uterine epithelium, the uterus may be a site from which the microbes in the basal plate originate. These findings provide new avenues for investigation into sites of origin of placental microbiota. Whether the spatial differences in microbial community membership impacts pathogenesis of PTB remains unknown.
The mechanisms by which microbes inhabit the placenta is far from understood. Recently, our lab reported that the cellular recycling pathway autophagy is an important mechanism for limiting infection in placental trophoblasts37,38, as preterm placentas showed lower levels of autophagy and higher levels of infection-associated markers than term placentas37. Collectively, these data indicate that, in addition to the placental microbiome, dysfunction in placental levels of autophagy activity could contribute to PTB38. Further characterizing maternal microbiomes in association with PTB using clinical specimens is currently underway. These efforts, along with the advancement of technology with greater sensitivity for detecting microbes in low biomass niches, greater understanding of geographical ecology of the placenta, and the development of suitable models for testing the function of these communities in association with PTB may be available in the near future.
Methodology of Pregnancy Microbiome Research
The human maternal microbiome studies using high-throughput sequencing have revealed promising associations between microbial composition and health and PTB and have provided a wealth of sequencing datasets, some of which has been made publically available. The majority of associations noted thus far between maternal microbiomes and PTB are qualitative, which is a critical first step. A second priority is to develop the most complete and concise methodology for performing high-throughput maternal microbiome analysis in order to ensure both accuracy and validity of the datasets derived from clinical specimens. A standard pipeline for conducting human microbiome research is well-described by the Human Microbiome Project Consortium39 and in a recent review by Goodrich et al.40.
Here, for a clinical perinatology research audience, we present the key steps for conducting a high-throughput microbiome analysis of pregnant subjects including: 1) designing the study, 2) sampling the subjects, 3) extracting the DNA, generation 16S amplicons, sequencing on a high-throughput platform, 4) performing community analysis, and reporting findings. We have also addressed ways to control for contamination and have presented analytic limitations when using the generated sequencing dataset. Thus, fine-tuning high-throughput approaches to address the link between maternal microbiomes and PTB will strengthen the foundational evidence, lead to future studies that can address the function of the microbiota, and pave the way for the development of the microbiome-related diagnostics and therapeutics.
1. Designing the study
The study design is the most critical aspect of conducting a microbiome-based study. In a case-control study of pregnant subjects, the microbiota from the control group (e.g., women who went on to deliver spontaneously at term, >37 weeks) is compared to the microbiota from the cases (e.g., women who went on to deliver preterm). The inclusion criteria for the control cohort most often include uncomplicated pregnancies and no evidence of maternal viral or bacterial maternal infection. Additionally, one should consider maternal factors that can affect the community structure of the microbiota. For example, Aagaard et al. found that chorioamnionitis is associated with a modified fetal membrane community34. Aagaard et al. also showed that the community structure of the placental microbiome correlates with weight gain during pregnancy41. Thus, one should record and consider as possible confounders, factors such as maternal infection history, weight, race/ethnicity, and even the hospitals from which samples are collected.
2. Sampling the subjects
To avoid contaminating samples, it is critical to use an appropriate sampling technique, which will vary depending on the niche of interest. The vagina, or a particular region in the vagina, is sampled by circumferential swabbing using sterile technique. The swabs, which are also used in the microbiology clinic for aerobic and anaerobic cultures, can either be stored long term at −80 °C or kept on ice for direct DNA extraction42,43. During sampling, most investigators also measure the vaginal pH42. In placental sampling, one can either take samples representing the entire placenta or target particular compartments (e.g., amniotic fluid, amniotic membrane, or basal plate). In one study, tissue was washed in sterile phosphate buffer saline 44 to remove blood contamination. Once sampled, tissue can be frozen in liquid-phase nitrogen40 and then frozen at −80 °C or used immediately for DNA extraction. To sample the intestinal microbiome, investigators use stool, which can be stored between −80 °C and −20 °C without significantly altering the microbial abundance or diversity within samples40.
3. DNA Extraction, Amplicon Generation, and Sequencing
In most cases, DNA is extracted directly from samples by using commercially available kits as shown in Table 1. Kits such as the QIAamp DNA Stool Mini Kit (QIAgen)14,21,24,45, are used to extract microbial DNA from primarily from stool, but have been used to isolate microbial DNA from other human niches. However, since DNA extraction kits that target low abundance microbial communities like the placenta are not yet available, investigators have used kits such as the DNeasy PowerSoil Kit (MOBIO)42,44, which can capture DNA from a variety of biological sources. Given samples with low microbial biomass, it is recommended that multiple kits be used along with extraction blanks and water, for microbiome analysis to minimize the possibility of contaminants and false positives44.
Table 1:
Methods for maternal 16S sequencing studies. Universal primers F (F, forward primer) R (R, reverse primer); Ribosomal Database Project (RDP); Quantitative Insights into Microbial Ecology (QIIME); Linear discriminate analysis effect size (LEfSe).
| Location | Sequencing Platform | Sampling | DNA Isolation Kit (Manufacturer) | Variable Regions (Primers) | rRNA reference database | Microbiome Analysis Program | Ref. |
|---|---|---|---|---|---|---|---|
| Placental | 454-FLX Titanium Sequencer (Roche). | 1-cm × 1-cm × 1-cm biopsy | PowerSoil DNA Isolation Kits (MOBIO) | V1-V3 (27F, 534R) | RDP | QIIME LefSe | 33 |
| Placental | Miseq (Illumina) | 0.11 g – 0.66 g biopsy | SP Stool DNA Plus kit (STRATEC
Biomedical) PowerSoil DNA Isolation Kits (MOBIO) |
V1V2 (27F, 338R) | Greengenes v. 13_8 | QIIME | 44 |
| Placental | Genome Sequencer FLX Titanium Series (454 Life Science) | 10 mg biopsy | QIAamp DNA Stool Mini Kit (QIAgen) | V1-V3 (27F, 533R) | GreenGenes v 13_8 | QIIME LefSe | 45 |
| Placental | Miseq (Illumina) | 1 cm × 1 cm × 1 cm biopsy | E.Z.N.A. DNA Kit (Omega
Bio-tek) AxyPrep DNA Gel Extraction Kit (Axygen Biosciences) |
V3-V4 (338F, 806R) | RDP Classifier and SILVA (SSU115) | QIIME | 67 |
| Vaginal | 454 FLX Titanium pyrosequencing | Swab | ZR Fecal DNA extraction kit (ZYMO Research, | V1-V2 27F, 338R | Version 0.2 of the vaginal community 16S rRNA gene reference tree (from pplacer) | pplacer version v1.1.alpha 08 | 20 |
| Vaginal (introitus, posterior fornix, midvagina) | Genome Sequencer GS-FLX Titanium platform (Roche) | Swab | PowerSoil DNA Isolation Kit (MoBio) | V3V5 (354F, 926R) |
RDP classifier v2.2 | QIIME R packages (BiodiversityR; vegan) LefSe |
42 |
| Vagina | 454 FLX pyrosequencing | Swab | QIAamp DNA Mini Kit (Qiagen) | V1-V2 (27F and 338R) | RDP | Ribosomal Database Project (RDP) Naïve Bayesian Classifier | 14 |
| Vaginal (posterior fornix) | Miseq (Illumina) | Swab | QIAamp DNA Mini kit (Qiagen) | V1-V3 (28F-519R) | Silva | Mothur | 24 |
| Vaginal (posterior fornix) | Miseq (Illumina) | Swab | QIAamp DNA Mini kit (Qiagen) | V1-V2 (28F-388R) |
Silva | Mothur | 21 |
| Vagina | 454 pyrosequencing using 454 Life Sciences (Roche/454 Life Sciences) | Swab | CellFree500 kit | V1-V3 (27 F and 534R) |
Pplacer speciateIT | QIIME | 26 |
| Stool | Illumina (HiSeq) | - | - | V1V2 | Greengenes/LCA | QIIME | 10 |
Because DNA extracted directly from human samples will contain both bacterial and human genomes, one must specifically amplify the bacterial DNA. This is achieved by PCR amplifying regions of the small subunit ribosomal RNA gene sequence (16S)46. In addition to conserved regions, this gene contains nine variable regions that can be used to determine the individual species from which the sequence derived.47 For many studies, primers are designed to amplify a sequence that spans one or multiple variable regions. Alternatively one can amplify and sequence random regions of the genome in a process called whole-genome shotgun sequencing48. In both cases, the most commonly used high-throughput amplicon sequencing platforms are Illumina Miseq and 454/Roche49 (Table 1).
4. Performing community analysis: Analytic Considerations and Pitfalls
Based on a similarity threshold, quality filtered sequences are given taxonomic assignments or are ascribed to an operational taxonomic unit (OTU), a group of similar sequences50, by using a reference database48. However, one must be cautious of comparing the taxonomic assignments derived from amplification of different rRNA regions because their relative abundances will not necessarily be correlated based on factors that are not fully understood, but may be related to the structure or function of the different regions of the ribosomal structure51. Assignments are commonly done using the software Quantitative Insights into Microbial Ecology (QIIME)49, which requires basic programming experience. Other programs that can be used for taxonomic assignments and microbial community analysis are shown in Table 2.
Table 2: Common analysis tools used for microbiome analysis.
CloVR (Cloud Virtual Resource); LEfSe (Linear discriminate analysis effect size); MEGAN (MEtaGenome Analyzer); MG-RAST (MetaGenomic - Rapid Annotation using Subsystem Technology); PICRUSt (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States); QIIME (Quantitative Insights into Microbial Ecology); STAMP (Statistical Analysis of Metagenomic Profiles)
| Analysis Tools | Access | Purpose | Ref. |
|---|---|---|---|
| CloVR | Web (can be downloaded onto Desktop) | Software used to analyze microbial communities from high-throughput sequencing outputs | 68 |
| Galaxy | Web-based | Software that uses various tools for metagenomic comparisons between communities. | 45 |
| LEfSe | Web-based | Online tool that incorporates statistical approaches to identify differential features between microbial communities. | 34,42,45,69 |
| MEGAN | Software | Downloadable bioinformatics software that constructs taxonomic hierarchical trees. | 45 |
| MG-RAST | Web-based | Performs microbial metagenomics analysis–both functional and phylogenetic. | 10,33,34,70 |
| Mothur | Command-line interface | Software used to analyze microbial communities from high-throughput sequencing outputs. | 21,71 |
| PICRUSt | Web-based | Online tool that uses 16S data to make metagenomic predictions about microbial communities. | 45,72 |
| QIIME | Command-line interface | Software used to analyze microbial communities. | 33,34,44,52 |
| R | Software | Programming software for statistical and computational analysis (commonly used packages include Phyloseq and Vegan). | 21,73 |
| STAMP | Software | Uses the output files from QIIME, PICRUST, MG-RAST, etc. to generate graphics and perform statistical tests and confidence intervals in order to test the null hypothesis. | 24,45,74 |
An important step in analyzing maternal microbiomes is to determine the types and relative abundances of bacteria within a sample. This is usually done with two measures: first, alpha diversity, defines the diversity within each sample49. The most commonly used measure is the Shannon Diversity index; Chao1 and Simpson are also popular choices, and many publications report more than one measure. Alpha diversity of each sample can be computed by using a number of different programs and software including QIIME 49,52, R VEGAN package53, and others listed in Table 2. Although some publications compare the diversity measures between groups with a t-test, this test requires that values be normally distributed, which is difficult with a small number of study subjects. Thus, Kruskal-Wallis and Mann-Whitney tests are usually more appropriate because they do not require normality and can be used to determine if at least one group out of many is different (Kruskal-Wallis) or if two groups differ from each other (Mann-Whitney). These tests can be performed in almost any programming environment (Table 3).
Table 3: Common statistical methods for microbiome analyses.
ANOVA (Analysis of variance); PERMANOVA (Permutational multivariate analysis of variance); PERMDISP (Permutational analysis of multivariate dispersion).
| Statistical Methods | Purpose | Ref. |
|---|---|---|
| ANOVA | Compares the differences in the means of multiple groups to determine whether they are statistically significant. | 45 |
| Jensen-Shannon divergence | Used to predict the similarity between two groups of probability distributions | 20 |
| Kruskall-Wallis | Non-parametric; evaluates the statistical significance of two or more groups that do not meet the normality assumption | 42,44 |
| Mann-Whitney U-test | Non-parametric; evaluates the statistical significance of two groups that do not meet the normality assumption. | 34 |
| PERMANOVA | Statistical significance of beta diversity | 34 |
| PERMDISP | Statistical significance of beta diversity | 34 |
| Spearman’s Correlation | Generates a coefficient that describes the degree to which two variables are negatively or positively correlated. | 10,14,67 |
| Student t-test | Parametric; evaluates the statistical significance of two groups that meet the normality assumption; can be either paired or unpaired. | 67 |
The second measure is beta diversity, which indicates how the microbial diversity differs between individuals or between cohorts49. Beta diversity calculations can be completed in the same programs used to calculate alpha diversity, but the outcome variable is more complicated. The QIIME output of beta diversity is a matrix comparing every sample to every other sample49. For example, the output of a beta diversity calculation for five samples will be a five-by-five matrix (or just the upper diagonal) of phylogenetic dissimilarity between species or OTUs. Several formulas can be used to calculate each measurement in the matrix, but publications usually only report one. Bray-Curtis and unweighted UniFrac are commonly used metrics. Each method has its own formula and calculation that must be considered when deciding which one to use for analysis. For example, Bray Curtis accounts for both the abundance of microbiota54. In contrast, unweighted UniFrac uses only the phylogenetic distance between the different taxa55. Each measure has its own set of assumptions that must be met before the results can be properly interpreted.
To test the null hypothesis that microbiomes from clinical groups are identical to one another, the current analytic convention is to run the statistical test, PERMANOVA56, which can be used for any of the phylogenetic dissimilarity measures used for microbiome analysis. Flowever, for this test to be appropriate, the dispersions, or relative phylogenetic distances, of samples in one group to others in the same group must be statistically similar across groups.
Another method for analyzing beta diversity is to create a multi-dimensional scaling or principle components analysis plot57. This is a graphical representation of the similarity of individual subjects’ microbiomes. Although there is no specific test to complete on the plot itself, the plot is useful for a few reasons. First, one can use different colored dots to distinguish subjects by a categorical or dichotomous variable (preterm premature rupture of membrane status, age group, pregnancy term, etc.) and then visualize where the different groups cluster, if at all. Second, most programs indicate the percentage of the difference between samples that is shown by each axis. The higher the percentage, the more one can conclude that samples cluster as a result of a handful of specific taxa within the microbiomes. Conversely, low percentages imply that many taxa are responsible for the differences. Such a finding indicates that no specific microbiota correlates with outcome of interest, such as PTB.
Notably, programs that use sequencing outputs, such as QIIME, will only provide OTU abundance measures relative to other taxa within a sample, not absolute abundance58. To compare the relative abundance microbial communities at the same number of reads, investigators often use rarefaction and normalize their samples to a single read depth59.
Another approach to determining the absolute abundance of species detected within a sample is to perform quantitative PCR with species-specific primers targeting the 16S gene of the microbe of interest60. In this approach, one must generate a standard curve of the quantification cycle 61 values of a known quantity of serially diluted genomic DNA. Cq values derived from samples can then be used to calculate the 16S copy number per quantity of DNA. It is important to note that quantitative PCR will not distinguish between 16S rRNA from viable bacteria or from free nucleic acid within the tissue62.
Reporting findings
One should always report the distance, measure, and index used for analysis, and explain the rationale behind the choices as there is no simple answer for choosing one method over the other. Each available method must be considered deliberately as reporting only the method that results in the “most significant findings” in a post hoc pick-and-choose process undermines the necessity for multiple analysis methods and formulae and, more alarmingly, raises ethical concerns about future clinical applications of the data.
Prospectus
The majority of associations noted thus far between microbiomes and PTB are qualitative. Long term, the goal of this field will be to identify unique bacteria or bacterial communities that can be targeted to prevent PTB. In such work, it will be important to consider the ecological dynamics of bacterial species and their interactions with the host environment. Currently, infection- and inflammation-related therapies to reduce PTB focus on either targeting the causal microbes, which requires timely and accurate diagnostic measures, or targeting cytokines and inflammatory pathways that promote labor63. Given that shifts in the maternal microbiota might contribute to PTB, we must consider how such therapeutics may alter the normal microbiota in various niches64,65. A leader in the field of gut microbiome research, Dr. Jeffrey Gordon, and colleagues have recently called for the establishment of human-associated microbial communities, termed ‘human microbial observatories’66. Development of a broad view of human development, health, physiology, and disease as being a composite result of the genome, epigenome, and the microbiome, is needed to truly understand etiologies of conditions such as PTB. We hope that studies such as those described above will use appropriate analytical methods and thereby elucidate the role of the maternal microbiome in PTB, leading to development of microbiome-based therapeutic strategies to prevent this outcome. Long term, this understanding can lead to refining our definitions of a healthy microbiome during pregnancy and may aid in development of strategies to restore the normal microbiota during pregnancy
Acknowledgements
We thank Dr. Deborah Frank for editing. This work was supported by a Preventing Prematurity Initiative grant from the Burroughs Wellcome Fund, a Prematurity Research Initiative Investigator award 21-FY13-28 from the March of Dimes, NIH grant R01HD091218 (to IUM), and NIH grant 2T32GM007067-42 (supporting LAP).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.March of Dimes, P., Save the Children, WHO. Born Too Soon: The Global Action Report on Preterm Birth. (World Health Organization, Geneva, 2012). [Google Scholar]
- 2.Goldenberg RL, Culhane JF, Iams JD & Romero R Epidemiology and causes of preterm birth. Lancet 371, 75–84, doi:10.1016/S0140-6736(08)60074-4 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Payne MS et al. Ureaplasma parvum genotype, combined vaginal colonization with Candida albicans, and spontaneous preterm birth in an Australian cohort of pregnant women. BMC Pregnancy Childbirth 16, 312, doi:10.1186/s12884-016-1110-x (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Turnbaugh PJ et al. The human microbiome project. Nature 449, 804–810, doi:10.1038/nature06244 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grice EA & Segre JA The human microbiome: our second genome. Annu Rev Genomics Hum Genet 13, 151–170, doi:10.1146/annurev-genom-090711-163814 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nuriel-Ohayon M, Neuman H & Koren O Microbial Changes during Pregnancy, Birth, and Infancy. Front Microbiol 7, 1031, doi:10.3389/fmicb.2016.01031 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soma-Pillay P, Nelson-Piercy C, Tolppanen H & Mebazaa A Physiological changes in pregnancy. Cardiovasc J Afr 27, 89–94, doi:10.5830/CVJA-2016-021 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shreiner AB, Kao JY & Young VB The gut microbiome in health and in disease. Curr Opin Gastroenterol 31, 69–75, doi:10.1097/MOG.0000000000000139 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carding S, Verbeke K, Vipond DT, Corfe BM & Owen LJ Dysbiosis of the gut microbiota in disease. Microb Ecol Health Dis 26, 26191, doi:10.3402/mehd.v26.26191 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koren O et al. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell 150, 470–480, doi:10.1016/j.cell.2012.07.008 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.King JC Physiology of pregnancy and nutrient metabolism. Am J Clin Nutr 71, 1218S–1225S (2000). [DOI] [PubMed] [Google Scholar]
- 12.Chernikova DA et al. Fetal exposures and perinatal influences on the stool microbiota of premature infants. J Matern Fetal Neonatal Med 29, 99–105, doi:10.3109/14767058.2014.987748 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stinson LF, Payne MS & Keelan JA Planting the seed: Origins, composition, and postnatal health significance of the fetal gastrointestinal microbiota. Crit Rev Microbiol 43, 352–369, doi:10.1080/1040841X.2016.1211088 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Ravel J et al. Vaginal microbiome of reproductive-age women. Proceedings of the National Academy of Sciences of the United States of America 108 Suppl 1, 4680–4687, doi:10.1073/pnas.1002611107 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ravel J & Brotman RM Translating the vaginal microbiome: gaps and challenges. Genome Med 8, 35, doi:10.1186/s13073-016-0291-2 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klebanoff SJ, Hillier SL, Eschenbach DA & Waltersdorph AM Control of the microbial flora of the vagina by H2O2-generating lactobacilli. J Infect Dis 164, 94–100 (1991). [DOI] [PubMed] [Google Scholar]
- 17.Dunlop AL et al. Maternal Microbiome and Pregnancy Outcomes That Impact Infant Health: A Review. Adv Neonatal Care 15, 377–385, doi:10.1097/ANC.0000000000000218 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cao B, Stout MJ, Lee I & Mysorekar IU Placental Microbiome and Its Role in Preterm Birth. Neoreviews 15, e537–e545, doi:10.1542/neo.15-12-e537 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brotman RM Vaginal microbiome and sexually transmitted infections: an epidemiologic perspective. J Clin Invest 121, 4610–4617, doi:10.1172/JCI57172 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Romero R et al. The composition and stability of the vaginal microbiota of normal pregnant women is different from that of non-pregnant women. Microbiome 2, 4, doi:10.1186/2049-2618-2-4 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.MacIntyre DA et al. The vaginal microbiome during pregnancy and the postpartum period in a European population. Scientific reports 5, 8988, doi:10.1038/srep08988 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chaim W, Mazor M & Leiberman JR The relationship between bacterial vaginosis and preterm birth. A review. Arch Gynecol Obstet 259, 51–58 (1997). [DOI] [PubMed] [Google Scholar]
- 23.Hillier SL et al. Association between bacterial vaginosis and preterm delivery of a low-birth-weight infant. The Vaginal Infections and Prematurity Study Group. The New England journal of medicine 333, 1737–1742, doi:10.1056/NEJM199512283332604 (1995). [DOI] [PubMed] [Google Scholar]
- 24.Kindinger LM et al. The interaction between vaginal microbiota, cervical length, and vaginal progesterone treatment for preterm birth risk. Microbiome 5, 6, doi:10.1186/s40168-016-0223-9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghartey J, Bastek JA, Brown AG, Anglim L & Elovitz MA Women with preterm birth have a distinct cervicovaginal metabolome. American journal of obstetrics and gynecology 212, 776 e771–776 e712, doi:10.1016/j.ajog.2015.03.052 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Romero R et al. The vaginal microbiota of pregnant women who subsequently have spontaneous preterm labor and delivery and those with a normal delivery at term. Microbiome 2, 18, doi:10.1186/2049-2618-2-18 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kacerovsky M et al. Cervical microbiota in women with preterm prelabor rupture of membranes. PloS one 10, e0126884, doi:10.1371/journal.pone.0126884 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Verstraelen H et al. Characterisation of the human uterine microbiome in non-pregnant women through deep sequencing of the V1–2 region of the 16S rRNA gene. PeerJ 4, e1602, doi:10.7717/peerj.1602 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Payne MS & Bayatibojakhi S Exploring preterm birth as a polymicrobial disease: an overview of the uterine microbiome. Front Immunol 5, 595, doi:10.3389/fimmu.2014.00595 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Han YW et al. Fusobacterium nucleatum induces premature and term stillbirths in pregnant mice: implication of oral bacteria in preterm birth. Infect Immun 72, 2272–2279 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pelzer E, Gomez-Arango LF, Barrett HL & Nitert MD Review: Maternal health and the placental microbiome. Placenta, doi:10.1016/j.placenta.2016.12.003 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Stout MJ et al. Identification of intracellular bacteria in the basal plate of the human placenta in term and preterm gestations. Am J Obstet Gynecol 208, 226 e221–227, doi:10.1016/j.ajog.2013.01.018 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aagaard K et al. The placenta harbors a unique microbiome. Sci Transl Med 6, 237ra265, doi:10.1126/scitranslmed.3008599 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prince AL et al. The placental membrane microbiome is altered among subjects with spontaneous preterm birth with and without chorioamnionitis. American journal of obstetrics and gynecology 214, 627 e621–627 e616, doi:10.1016/j.ajog.2016.01.193 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parnell LA; Briggs CM;B; Delannoy-Bruno O; Schrieffer AE; Mysorekar IU. Geographical influences on the placental microbiome: spatial variation in microbial communities at the maternal fetal interface. Scientific reports (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Satokari R, Gronroos T, Laitinen K, Salminen S & Isolauri E Bifidobacterium and Lactobacillus DNA in the human placenta. Lett Appl Microbiol 48, 8–12, doi:10.1111/j.1472-765X.2008.02475.x (2009). [DOI] [PubMed] [Google Scholar]
- 37.Cao B, Macones C & Mysorekar IU ATG16L1 governs placental infection risk and preterm birth in mice and women. JCI Insight 1, e86654, doi:10.1172/jci.insight.86654 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cao B, Camden AJ, Parnell LA & Mysorekar IU Autophagy regulation of physiological and pathological processes in the female reproductive tract. Am J Reprod Immunol, doi:10.1111/aji.12650 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Human Microbiome Project C A framework for human microbiome research. Nature 486, 215–221, doi:10.1038/nature11209 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goodrich JK et al. Conducting a microbiome study. Cell 158, 250–262, doi:10.1016/j.cell.2014.06.037 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Antony KM et al. The preterm placental microbiome varies in association with excess maternal gestational weight gain. American journal of obstetrics and gynecology 212, 653 e651–616, doi:10.1016/j.ajog.2014.12.041 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aagaard K et al. A metagenomic approach to characterization of the vaginal microbiome signature in pregnancy. PloS one 7, e36466, doi:10.1371/journal.pone.0036466 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bai G et al. Comparison of storage conditions for human vaginal microbiome studies. PloS one 7, e36934, doi:10.1371/journal.pone.0036934 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lauder AP et al. Comparison of placenta samples with contamination controls does not provide evidence for a distinct placenta microbiota. Microbiome 4, 29, doi:10.1186/s40168-016-0172-3 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Collado MC, Rautava S, Aakko J, Isolauri E & Salminen S Human gut colonisation may be initiated in utero by distinct microbial communities in the placenta and amniotic fluid. Scientific reports 6, 23129, doi:10.1038/srep23129 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clarridge JE 3rd. Impact of 16S rRNA gene sequence analysis for identification of bacteria on clinical microbiology and infectious diseases. Clin Microbiol Rev 17, 840–862, table of contents, doi:10.1128/CMR.17.4.840-862.2004 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chakravorty S, Helb D, Burday M, Connell N & Alland D A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria. J Microbiol Methods 69, 330–339, doi:10.1016/j.mimet.2007.02.005 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ranjan R, Rani A, Metwally A, McGee HS & Perkins DL Analysis of the microbiome: Advantages of whole genome shotgun versus 16S amplicon sequencing. Biochem Biophys Res Commun 469, 967–977, doi:10.1016/j.bbrc.2015.12.083 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuczynski J et al. Using QIIME to analyze 16S rRNA gene sequences from microbial communities. Current protocols in microbiology Chapter 1, Unit 1E 5, doi:10.1002/9780471729259.mc01e05s27 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schloss PD & Westcott SL Assessing and improving methods used in operational taxonomic unit-based approaches for 16S rRNA gene sequence analysis. Applied and environmental microbiology 77, 3219–3226, doi:10.1128/AEM.02810-10 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang B, Wang Y & Qian PY Sensitivity and correlation of hypervariable regions in 16S rRNA genes in phylogenetic analysis. BMC Bioinformatics 17, 135, doi:10.1186/s12859-016-0992-y (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Caporaso JG et al. QIIME allows analysis of high-throughput community sequencing data. Nature methods 7, 335–336, doi:10.1038/nmeth.f.303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dixon P VEGAN, a package of R functions for community ecology. Journal of Vegetation Science 14, 927–930 (2003). [Google Scholar]
- 54.Birtel J, Walser JC, Pichon S, Burgmann H & Matthews B Estimating bacterial diversity for ecological studies: methods, metrics, and assumptions. PloS one 10, e0125356, doi:10.1371/journal.pone.0125356 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lozupone CA, Hamady M, Kelley ST & Knight R Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Applied and environmental microbiology 73, 1576–1585, doi:10.1128/AEM.01996-06 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Anderson MJ PERMANOVA: A new method for non-parametric multivariate analysis of variance. Austral Ecology 26, 32–46 (2001). [Google Scholar]
- 57.Navas-Molina JA et al. Advancing our understanding of the human microbiome using QIIME. Methods Enzymol 531, 371–444, doi:10.1016/B978-0-12-407863-5.00019-8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stammler F et al. Adjusting microbiome profiles for differences in microbial load by spike-in bacteria. Microbiome 4, 28, doi:10.1186/s40168-016-0175-0 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Carcer DA, Denman SE, McSweeney C & Morrison M Evaluation of subsampling-based normalization strategies for tagged high-throughput sequencing data sets from gut microbiomes. Applied and environmental microbiology 77, 8795–8798, doi:10.1128/AEM.05491-11 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ling Z et al. Molecular analysis of the diversity of vaginal microbiota associated with bacterial vaginosis. BMC Genomics 11, 488, doi:10.1186/1471-2164-11-488 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Borbely AU et al. The term basal plate of the human placenta as a source of functional extravillous trophoblast cells. Reprod Biol Endocrinol 12, 7, doi:10.1186/1477-7827-12-7 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nocker AR-H,T; Montijn R; Schuren F; Kort R Discrimination between live and dead cells in bacterial communities from environmental water samples analyzed by 454 pyrosequencing. Int Microbiol 13, 59–65 (2010). [DOI] [PubMed] [Google Scholar]
- 63.Taguchi A et al. Recent Progress in Therapeutics for Inflammation-Associated Preterm Birth: A Review. Reprod Sci, doi:10.1177/1933719115618282 (2015). [DOI] [PubMed] [Google Scholar]
- 64.Nishijima K, Shukunami K & Kotsuji F Probiotics affects vaginal flora in pregnant women, suggesting the possibility of preventing preterm labor. J Clin Gastroenterol 39, 447–448 (2005). [DOI] [PubMed] [Google Scholar]
- 65.Yang S et al. Is there a role for probiotics in the prevention of preterm birth? Front Immunol 6, 62, doi:10.3389/fimmu.2015.00062 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Charbonneau MR et al. A microbial perspective of human developmental biology. Nature 535, 48–55, doi:10.1038/nature18845 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zheng J et al. The Placental Microbiome Varies in Association with Low Birth Weight in Full-Term Neonates. Nutrients 7, 6924–6937, doi:10.3390/nu7085315 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Angiuoli SV et al. CloVR: a virtual machine for automated and portable sequence analysis from the desktop using cloud computing. BMC Bioinformatics 12, 356, doi:10.1186/1471-2105-12-356 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Segata N et al. Metagenomic biomarker discovery and explanation. Genome Biol 12, R60, doi:10.1186/gb-2011-12-6-r60 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Meyer F et al. The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9, 386, doi:10.1186/1471-2105-9-386 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schloss PD et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and environmental microbiology 75, 7537–7541, doi:10.1128/AEM.01541-09 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Langille MG et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31, 814–821, doi:10.1038/nbt.2676 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McMurdie PJ & Holmes S phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PloS one 8, e61217, doi:10.1371/journal.pone.0061217 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Parks DH, Tyson GW, Hugenholtz P & Beiko RG STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 30, 3123–3124, doi:10.1093/bioinformatics/btu494 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]