

Abstract
Genome-wide association studies have established BIN1 (Bridging Integrator 1) as the most significant late-onset Alzheimer disease (AD) susceptibility locus after APOE . We analyzed BIN1 protein expression using automated immunohistochemistry on the hippocampal CA1 region in 19 patients with either no, mild, or moderate-to-marked AD pathology, who had been assessed by Clinical Dementia Rating and CERAD scores. We also examined the amygdala, prefrontal, temporal, and occipital regions in a subset of these patients. In non-demented controls without AD pathology, BIN1 protein was expressed in white matter, glia, particularly oligodendrocytes, and in the neuropil in which the BIN1 signal decorated axons. With increasing severity of AD, BIN1 in the CA1 region showed: 1) sustained expression in glial cells, 2) decreased areas of neuropil expression, and 3) increased cytoplasmic neuronal expression that did not correlate with neurofibrillary tangle load. In patients with AD, both the prefrontal cortex and CA1 showed a decrease in BIN1-immunoreactive (BIN1-ir) neuropil areas and increases in numbers of BIN1-ir neurons. The numbers of CA1 BIN1-ir pyramidal neurons correlated with hippocampal CERAD neuritic plaque scores; BIN1 neuropil signal was absent in neuritic plaques. Our data provide novel insight into the relationship between BIN1 protein expression and the progression of AD-associated pathology and its diagnostic hallmarks.
Keywords: Alzheimer disease, BIN1, Glia, Hippocampus, Neuropil, Pyramidal neurons, White matter
INTRODUCTION
Because late-onset Alzheimer disease (LOAD) has a largely unknown genetic component, genome-wide association studies have investigated thousands of human genomes to identify LOAD-associated risk loci ( 1–5 ). The 2 most significant LOAD-associated risk loci that have repeatedly been confirmed by genome-wide association studies are APOE followed by Bridging Integrator 1 ( BIN1) ( 1–3 , 5 ). The BIN1 risk locus remains significant after controlling for APOE genotype, suggesting that BIN1 is an independent risk allele associated with LOAD ( 4 ). BIN1 risk alleles are associated with earlier age of onset and faster global cognitive decline in LOAD ( 6 , 7 ). As a potential target for future therapies, an understanding of the cellular distribution of the BIN1 encoded protein during LOAD progression is needed to elucidate its relationship with Alzheimer disease (AD)-associated pathological changes.
BIN1 is a nucleocytoskeletal adaptor protein with ubiquitous expression and multiple tissue-specific isoforms, including the brain-specific and largest isoform (iso1), and a smaller ubiquitous isoform (iso9) ( 8–10 ). Western blot analyses of AD brains found that levels of the iso1 were significantly reduced whereas levels of iso9 were significantly increased compared to controls ( 11 ). The known cellular functions of neuronal BIN1 include regulation of membrane curvature, clathrin-mediated endocytosis, and vesicular transport ( 9 , 12–18 ). All isoforms of BIN1 bind the plasma membrane through an evolutionarily conserved Bin1/Amphiphysin/RVS167 (BAR) domain that is involved in sensing and generating membrane curvature; brain-specific isoforms contain a clathrin-AP2 binding region (CLAP) domain, which interacts with endocytosis mediators clathrin and AP-2 (9, 12, 13, 16, 17, 19). These biological functions may link BIN1 to β-amyloid (Aβ) and tau pathology because both amyloid precursor protein (APP) and soluble tau are subjected to clathrin-mediated endocytosis, reportedly facilitating Aβ production ( 20–22 ) and trans-neuronal tau spreading ( 23–26 ), respectively . BIN1 is also suggested to act as a modulator of synaptic repair via lipoprotein ApoE ( 27 ). Astrocytes and activated microglia produce ApoE ( 28–31 ), which is internalized by neurons via BIN1-dependent receptor-mediated endocytosis ( 32 , 33 ).
In a multidisciplinary approach, Chapuis et al studied the expression of a BIN1 risk variant associated with increased mRNA expression and tau load, and concluded that BIN1 mediates AD risk by modulating tau pathology ( 34 ). However, while critically important for understanding the relationship between BIN1 and AD-associated histopathology, the patterns of subcellular and cell-type specific distribution of BIN1 in healthy cortex and white matter versus that in progressive stages of AD remain unknown.
In this study, we examined BIN1 protein expression primarily in the CA1 region because it is a site of the earliest AD-associated pathology and undergoes incremental neurodegeneration through advancing Braak and Braak stages ( 35–37 ). To evaluate the consistency of AD-related changes in BIN1 subcellular distribution, we examined BIN1 immunoreactivity in additional regions that are differentially affected during AD progression. Our results provide the groundwork for novel hypotheses regarding mechanisms by which BIN1 may affect the onset and advancement of AD pathology.
MATERIALS AND METHODS
Human Postmortem Brain Region Selection
Human formalin-fixed, paraffin-embedded (FFPE) blocks of 19 hippocampi, 4 amygdalae, 8 superior temporal gyri ([STG], Brodmann area 22, [BA22]), 8 middle frontal gyri (prefrontal cortex [PFC], BA9), and 8 medial occipital cortices ([OCC], BA17) acquired through the Framingham Heart Study Brain Donation Program, Framingham, Massachusetts, the Netherlands Brain Bank, Amsterdam, The Netherlands, and Boston Medical Center, were used in this study (Table). The brains were stratified into 3 groups based on the Clinical Dementia Rating (CDR) score and Braak and Braak (BB) stage (Table). All patients were de-identified and the authors were blinded to the CDR scores and BB stage during data acquisition. We performed anti-tau immunohistochemistry (IHC) on all analyzed hippocampi to corroborate reported BB stages by the brain banks and to perform quantitative analysis of neurofibrillary tangle (NFT) load in the CA1 region. In cognitively healthy control subjects (CDR0), we performed anti-tau IHC to confirm the complete absence of AD-associated NFT pathology, and categorized these subjects as Braak and Braak stage 0 (BB0). We omitted the ultimate degenerative stage, BBVI, from our analysis because the pervasive neuronal loss in the CA1 region at this stage precludes consistent analysis.
Table 1.
Hippocampal CA1 Regions Analyzed in Cases Grouped According to Clinical Dementia Rating Score and Braak and Braak Stage
| Subject | Braak & Braak stage |
CERAD plaque density/presence of D and N plaques
* |
Cognitive status | Age (years) | Sex | PMI (h) | |||
|---|---|---|---|---|---|---|---|---|---|
| Hippocampus | Prefrontal cortex $ | ||||||||
| CDR 0, BB 0 (Group 1) | |||||||||
| NBB 2012 052 | 0 | 0 | No neurological history; no evidence of dementia | 64 | F | 5.7 | |||
| NBB 2010 115 $,† | 0 | 0 | 1 + (D) | No neurological history; no evidence of dementia | 70 | M | 3.6 | ||
| NBB 2011 091 | 0 | 1 + (D) | No neurological history; no evidence of dementia | 76 | M | 6.8 | |||
| BM 23 | 0 | 0 | No neurological history; no evidence of dementia | 66 | F | 24 | |||
| BM 29 | 0 | 0 | No neurological history; no evidence of dementia | 61 | M | 24 | |||
| CDR 0-0.5, BB II-III (Group 2) | |||||||||
| NBB 2012 059 $ | II | 1 + (D) | 2 + (D) | 0 | 98 | F | 4.6 | ||
| NBB 2012 070 | II | 3 + (D), 1 + (N) | 0 | 79 | M | 5.8 | |||
| NBB 2012 092 | II | 0 | 0 | 90 | M | 5.8 | |||
| NBB 2013 011 † | III | 2 + (D) | 0 | 92 | F | 4.4 | |||
| NBB 2012 001 $ | II | 3 + (D) | 3 + (D), 1 + (N) | 0.5 | 79 | M | 5.7 | ||
| NBB 2013 013 | II | 0 | 0.5 | 89 | M | 6.8 | |||
| NBB 2013 010 | III | 0 | 0.5 | 89 | F | 6.6 | |||
| NBB 2012 067 $ | III | 2 + (D) | 0.5 | 102 | M | 5.0 | |||
| CDR 1-2, BB IV-V (Group 3) | |||||||||
| NBB 2013 009 $ | IV | 1 + (D), 3 + (N) | 2 + (D), 1 + (N) | 1 | 92 | F | 7.4 | ||
| NBB 2008 075 † | IV | 3 + (D), 1 + (N) | Overt dementia; CDR not available | 88 | M | 5.0 | |||
| BVAX 100 $ | IV | 1 + (D), 2 + (N) | 3 + (D), 2 + (N) | 1 | 89 | M | 3.0 | ||
| BVAX 205 $ | IV | 2 + (N) | 2 + (D), 2 + (N) | 1 | 96 | F | 3.5 | ||
| BVAX 170 $ | V | 1 + (D), 1 + (N) | 4 + (D), 3 + (N) | 2 | 90 | F | 3.5 | ||
| BVAX 253 $ | V | 1 + (D), 1 + (N) | 4 + (D), 2 + (N) | 2 | 82 | M | 24 | ||
Group 1: cognitively intact subjects without neurofibrillary tangles (NFTs) in the hippocampus (CDR0, BB0), N = 5; Group 2: cognitively normal or mildly impaired subjects with or without AD-associated NFT pathology (CDR0-0.5, BBII-III), N = 8; and Group 3: patients with AD with AD-associated NFT pathology (CDR1-2, BBIV-V), N = 6.
*0 = none; 1+ = mild; 2+ = moderate; 3+ = severe; 4+ = very severe. (D) = diffuse plaques; (N) = neuritic plaques. NBB = Netherlands Brain Bank. BM = Boston Medical Center. BVAX = Brain Bank of the Boston University Alzheimer Disease Center; CERAD = The Consortium to Establish a Registry for Alzheimer’s Disease; F = female; M = male; PMI (h) = post mortem interval (hours).
$ indicates prefrontal cortex (BA9) was analyzed qualitatively and quantitatively, and superior temporal (BA22) and occipital (BA17) cortices qualitatively.
† indicates amygdala was analyzed qualitatively.
Quantitative and qualitative analysis of BIN1 signal was first performed in the CA1 region (n = 19), followed by PFC (BA9) analysis in a subset of subjects (n = 8) (Table). For further qualitative neocortical analysis, we examined BIN1 immunoreactivity in the superior temporal gyrus cortex (STG, BA22) and primary visual cortex (OCC, BA17) (Table). We chose these regions because they exhibited differential NFT load between subject groups and, in the case of Group 3 (AD patients), within individual brains. The superior temporal and prefrontal cortices were moderately burdened with NFTs in Group 3 subjects, but contained few or no NFTs in Group 2 subjects. Conversely, primary visual cortex was spared of NFTs in all subjects. Finally, to assess BIN1 immunoreactivity in a subcortical region that undergoes early AD-associated changes and atrophy correlating with CDR score ( 38 , 39 ), we qualitatively examined BIN1 signal in the amygdala in 4 subjects (Group 1, n = 1; Group 2, n = 2, and Group 3, n = 1; Table).
Boston University Medical Center’s Institutional Review Board approved this study; the authors state adherence to the requisite standards.
Immunohistochemistry
FFPE blocks were sectioned at 5-µm thickness, dried at room temperature for 24 hours, and heated at 80 °C for 24 hours before IHC processing. Deparaffinization, antigen retrieval, and subsequent staining was performed with Boston Medical Center Pathology Department’s Ventana Benchmark Ultra automated IHC instrument using commercially available primary antibodies and Ventana Medical System reagents including ultraView Universal DAB Detection Kit (Cat#760-500), ultraView Universal AP Red Detection Kit (Cat#760-501), Hematoxylin II (Cat#790-2208), and Bluing Reagent (Cat#760-2037) (Ventana Medical Systems, Inc., Roche Diagnostics Ltd., West Sussex, UK). The primary BIN1 antibody used was a mouse anti-BIN1 [2F11] monoclonal antibody, a pan-BIN1 marker ( 8 ), with specificity to exons 7 and 8 of the conserved N-terminal BAR domain common to all BIN1 isoforms (1:160, (Cat#ab84974) Abcam, Cambridge, MA) ( 40 ). Monoclonal anti-BIN1 [2F11] immunohistochemical specificity in FFPE tissues has been previously characterized ( 8 ), and we replicated this specificity (Supplemental Digital Content 1). Additionally, we compared our anti-BIN1 2F11 clone to the anti-BIN1 99D clone recently used by others ( 11 , 34 , 41 , 42 ). The 99D antibody is specific for exon 13 of the C-terminal Myc domain ( 9 , 10 , 40 ), common to most BIN1 isoforms (1:3200, (Cat#05-449) Millipore, Billerica, MA) and yields identical signal to 2F11 antibody as described in the Results section. Phosphorylation-independent tau polyclonal antibody (K9JA; 1:3200, (Cat#A0024) Dako, Glostrup, Denmark), approved for clinical in vitro diagnostic use, was used to label NFTs ( 43 , 44 ). This antibody labels all 6 adult brain tau isoforms that in hyperphosphorylated state give rise to paired helical filaments (PHFs) which form NFTs ( 45 ). Phospho-PHF-tau [AT8] monoclonal antibody (1:2000, (Cat#MN1020) Pierce, Rockford, IL) was used to confirm specificity of K9JA antibody in labeling NFTs (Supplemental Digital Content 2). Other primary antibodies included mouse anti-human β-amyloid [6F/3D] monoclonal antibody (1:50, (Cat#M0872) Dako), mouse anti-neurofilament [SMI-312] monoclonal antibody (1:4000, (Cat#ab24574) (Abcam), and rabbit anti-GFAP (EP672Y) monoclonal antibody (pre-diluted, (Cat#760-4345) Ventana Medical Systems, Inc.).
For quantitative analysis, BIN1 protein expression was assessed in 3 independent IHC experiments for each FFPE block, and all samples were processed collectively. Experiments performed in triplicate yielded 3 independently stained sections per subject. Automated IHC with the Ventana Benchmark Ultra allowed for maximally reproducible conditions in IHC experiments, eliminating variability in reagent composition, quantity, incubation time, and human error. Internal control sections from established subjects were stained collectively with any newly added subjects to ensure reproducibility of staining. Quantitative analysis of FFPE tissue samples utilized 3 stepwise sections separated by at least 10 μm; quantitative data from imaged sections were averaged to obtain representative values for each subject.
Image Analysis
Slides were imaged using an Olympus BX60 light microscope, QImaging Retiga 2000R camera, and QCapture Suite and Suite-PLUS software. Three 20x field images encompassing the majority of the CA1 region were analyzed from each hippocampal section (Supplemental Digital Content 3A, B). Similarly, in tandem PFC analysis, 3 20x fields from PFC pyramidal layer III and 3 20x fields from PFC pyramidal layers V-VI were used for quantitative analysis. For tangled neuron evaluation of CA1 region, 10x field images from 3 independent tau IHC sections from all subjects were analyzed. All cell count data are presented as a percentage of total cells to control for neuronal loss over the course of AD. To examine changes in BIN1 immunoreactivity in control, early, and moderately advanced AD-affected CA1 region and PFC pyramidal layers, 20x field images were used to quantify BIN1-immunoreactive (BIN1-ir) cell type (neurons or glia) and area of BIN1-ir neuropil. All field images were analyzed with ImageJ, version 1.48, Bethesda, MD: National Institutes of Health. Mean data values from 3 20x field images encompassing CA1 region, PFC layer III, and PFC layers V-VI comprise representative values of BIN1 immunoreactivity for each examined subject.
Specifically, BIN1-immunostained images were processed in ImageJ software to quantify BIN1-ir neuropil in each 20x field. The threshold value for BIN1 signal in neuropil was determined empirically and size restriction criteria were used to minimize non-neuropil signal from cell bodies. The percent area of above-threshold and size-restricted BIN1 neuropil signal was determined (Supplemental Digital Content 3C). The percentages of BIN1-ir neurons and glial cells were obtained manually using the ImageJ CellCounter plugin. Cell types were determined by morphological criteria including cell size, cell shape, and nuclear characteristics, which differentiate neurons from glia. BIN1 immunoreactivity in CA1 glia was binned collectively, regardless of glial cell subtype. ImageJ Stitching plugin was used to compose high-resolution and low-magnification representative images ( 46 ).
Statistical Analyses
Quantitative data among the subject groups were analyzed by two-tailed one-way ANOVA (alpha = 0.05) and post-hoc Tukey multiple comparison test in CA1 region analysis of 19 subjects. All correlation analyses were performed by Pearson r correlation. Duplicate quantitative evaluation of BIN1 immunoreactivity in neurons and neuropil in pyramidal layers of PFC in 8 representative subjects was analyzed by non-repeated measures two-way ANOVA with Bonferroni post-test of subject group (non-AD, BB0-III; patients with AD, BBIV-V) and anatomical region (CA1; PFC Layer III; PFC Layer V-VI). Anatomical region and subject group are 2 factors that we used to independently predict a continuous, dependent variable, i.e. BIN1-ir status. Student t-test (unpaired, two-tailed) was used to confirm neuronal loss in patients with AD versus subjects with non-AD. Statistical methods were performed with GraphPad Prism, version 5.0b, (GraphPad Software Inc., San Diego, CA).
RESULTS
BIN1 Protein Expression in CA1 Region of Cognitively Intact Subjects
We defined control subjects as neurologically healthy individuals with no NFT in CA1 region (CDR0, BB0) according to tau IHC (Table, Group 1; Supplemental Digital Content 2). These healthy controls and cognitively intact subjects with early AD-associated NFT pathology (CDR0, BBII-III; Table, Group 2) displayed similar BIN1 expression pattern, with strong BIN1 immunoreactivity (2F11 antibody) in the white matter (alveus) and in small, non-neuronal cortical cells ( Fig. 1A ). We identified BIN1 immunoreactivity in oligodendrocytes within the white matter of the alveus ( Fig. 1B ), as well as in CA1 cortex. In addition, there was a punctate BIN1 signal in the neuropil ( Fig. 1C ). This pattern of BIN1 immunoreactivity was also highlighted by the anti-BIN1 99D antibody (Supplemental Digital Content 4). Cells with typical astrocyte morphology, often adjacent to neuronal somata, were also labeled by BIN1 IHC ( Fig. 1D ). However, in a CDR0, BBII subject, CA1 comparison of the expression of BIN1 ( Fig. 1E ) and GFAP ( Fig. 1F ), an astrocyte marker, in CA1 adjacent sections suggested a minor astrocytic contribution to the overall BIN1 signal in the gray and white matter.
FIGURE 1.
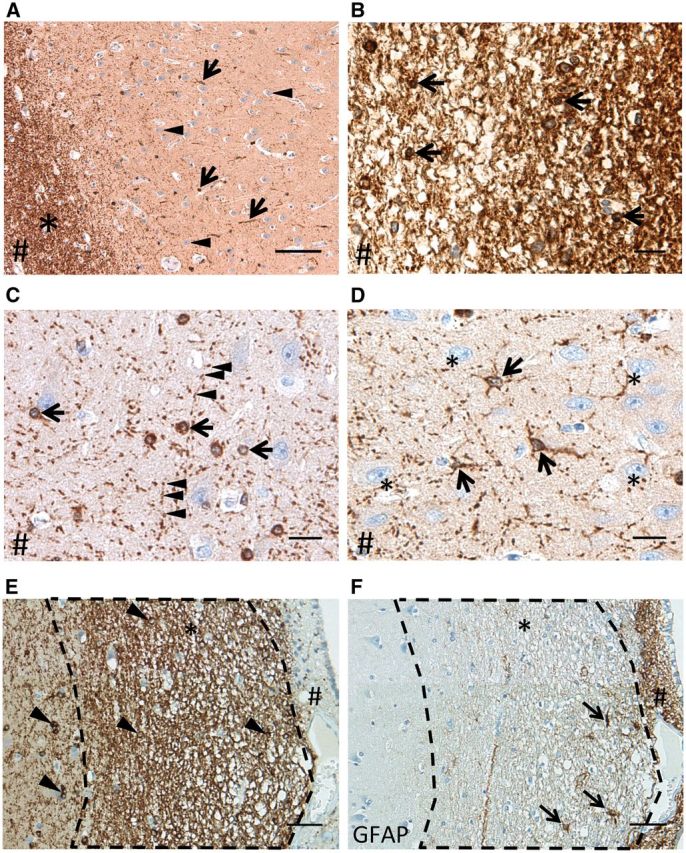
BIN1 immunohistochemistry in the CA1 region of cognitively intact subjects without or with early AD-associated NFT pathology highlights white matter, glia, and neuropil. (A) BIN1 immunoreactivity in CA1 region of a healthy control subject (CDR0, BB0) in alveus (white matter, asterisk) and in non-neuronal CA1 cells (arrows). Note the absence of BIN1-immunoreactive (-ir) signal in neurons (arrowheads). (B) White matter of alveus strongly expresses BIN1 and harbors BIN1-ir oligodendrocytes (arrows). (C) BIN1-ir oligodendrocytes (arrows) and a punctate BIN1 signal in the neuropil (arrowheads). (D) BIN1-ir astrocytes (arrows) among BIN1-non-ir pyramidal neurons (asterisks) in a CDR0, BBII subject. In A-D, # indicates the direction of the ependymal surface. (E, F) CA1 region of a CDR0, BBII hippocampus (asterisk indicates alveus). BIN1 IHC shows BIN1-ir oligodendrocytes (arrowheads) in the hippocampal CA1 cortex and in alveus (asterisk) (E) . There is minimal BIN1 immunoreactivity in area of subependymal astrocytosis (#). This area is highlighted by GFAP IHC in the adjacent section (F) , which highlights GFAP-ir astrocytes (arrows) and subependymal astrocytosis (#). Scale bars: A = 100 μm, B-D = 20 μm; E, F = 50 μm.
BIN1 Protein Expression in CA1 and Select Neocortical Regions during Progression of AD-Associated NFT Pathology and Concomitant Cognitive Impairment
Healthy control subjects (Group 1) and cognitively intact or minimally impaired subjects in early Braak and Braak stages (Group 2) exhibited strong BIN1-immunoreactive (-ir)-ir neuropil in the CA1 region whereas patients with AD (CDR1-2, BBIV-V; Table, Group 3) showed a weaker BIN1 signal ( Fig. 2A ). Patients with AD had significantly decreased BIN1-ir neuropil area compared to other groups (p = 0.002, Fig. 2B; Supplemental Digital Content 5, table 1.1). Pearson analysis revealed that BIN1-ir neuropil area correlated positively with BIN1-ir glial population, but negatively with BIN1-ir neuronal somata (which were rare); there was a negative correlation with rare BIN1-ir neuronal somata mostly encountered in AD cases, as described below (p = 0.004; Fig. 2C; Supplemental Digital Content 5, table 1.2-3). However, BIN1-ir neuropil area also correlated positively with the total number of CA1 pyramidal neurons (p = 0.009, Fig. 3A; Supplemental Digital Content 5, table 1.4). Since the percentage of BIN1-ir glial cells in CA1 remained consistent across all the examined subjects (Supplemental Digital Content 5, table 1.5), and the number of CA1 neurons became significantly reduced in Group 3 (Supplemental Digital Content 5, table 1.6), the decrease in BIN1-ir neuropil area in patients with AD likely reflects diminished numbers of axons. Consistently, dual IHC for BIN1 (using either 2F11 or 99D antibody) and the axon marker neurofilament confirmed that BIN1-ir neuropil punctate signal decorates axons ( Fig. 3B–D; Supplemental Digital Content 6). In Groups 1 and 2, very few CA1 pyramidal neurons yielded cytoplasmic BIN1 (2F11 ab) signal (mean = 3.34% and 3.16%, respectively), whereas in patients with AD (Group 3), BIN1 cytoplasmic immunoreactivity became apparent in a variable but significant percentage of CA1 pyramidal neurons (mean = 32.21%, p < 0.001) ( Fig. 4 ; Supplemental Digital Content 5, table 1.7). The BIN1 presence in CA1 pyramidal neurons of patients with AD was highlighted with BIN1 99D antibody as well (Supplemental Digital Content 7). As indicated above, more frequent BIN1-ir neurons found in AD cases did not contribute to BIN1-ir neuropil area, which was actually diminished in patients with AD as shown in Figure 2B . Because the BIN1-ir neuropil area correlated with BIN1-ir glial numbers and the total number of CA1 neurons, oligodendrocyte-derived myelin sheaths on preserved axons may be responsible for the BIN1-ir neuropil signal.
FIGURE 2.

BIN1 immunoreactivity in CA1 neuropil declines in patients with AD. (A) Representative images of BIN1 signal in neuropil of a subject with early NFT accumulation but cognitively intact (CDR0, BBIII), and an AD patient (CDR2, BBV). Note the prominent punctate BIN1-immunoreactive (-ir) neuropil as well as BIN1-ir astrocytes (arrows) and oligodendrocyte (arrowheads) (left panel). In an AD patient, the BIN1 signal is largely reduced in the neuropil (right panel). #Indicates direction of ependymal surface. (B) Area of BIN1-ir neuropil is significantly decreased in patients with AD (CDR1-2, BBIV-V) vs. control and cognitively intact or mildly impaired subjects (CDR0-0.5, BBII-III) (p = 0.002). Mean subject group values marked by horizontal bar; error bars represent SEM. (C) Pearson analysis showing that BIN1-ir neuropil area positively correlates with BIN1-ir glia (p = 0.004), but negatively correlates with BIN1-ir neurons (p = 0.019). Symbols indicate subject group: (•) CDR0, BB0; (▪) CDR0-0.5, BBII-III; (▲) CDR1-2, BBIV-V. Scale bars = 20 μm.
FIGURE 3.

BIN1-immunoreactive (-ir) neuropil signal in CA1 reflects axonal density. (A) Area of BIN1-ir neuropil positively correlates with total number of pyramidal neurons in CA1 (p = 0.009). Symbols indicate subject group: (•) CDR0, BB0; (▪) CDR0-0.5, BBII-III; (▲) CDR1-2, BBIV-V. (B) BIN1 (brown) and neurofilament (red) dual IHC in CA1 with insets c, d. *Indicates alveus. (C) Inset (from area ‘c’ in B ) highlights BIN1-ir neuropil puncta (brown, arrows) along a neurofilament-ir process (red, arrowheads) in CA1. (D) Inset (from area ‘d’ in B ) highlights a similar staining pattern at the junction with alveus. Scale bars: B , 100 μm; C, D , 20 μm.
FIGURE 4.
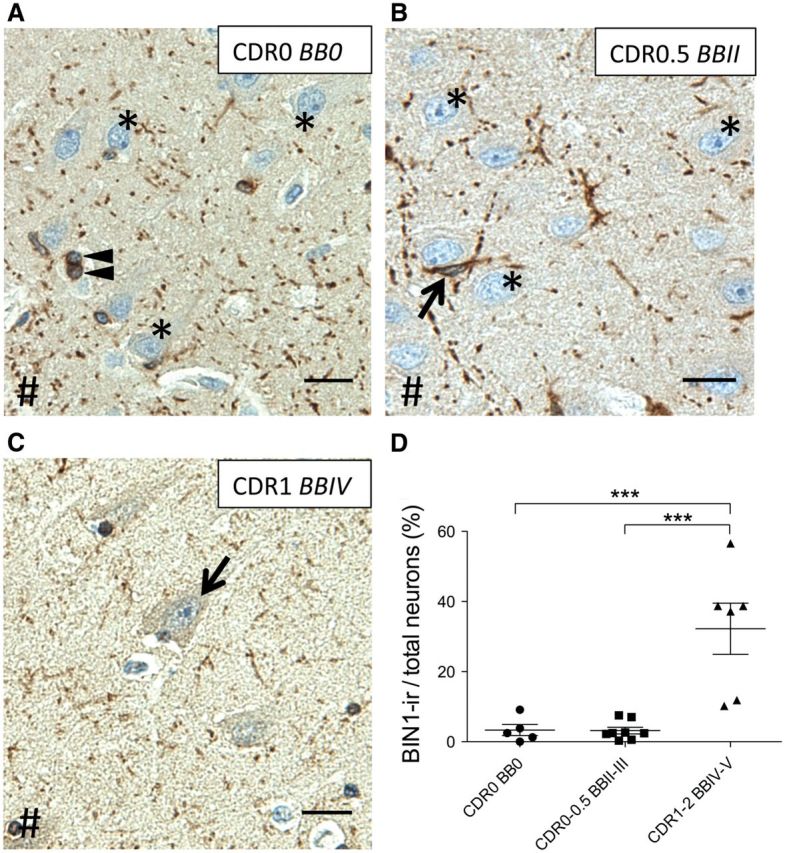
BIN1 immunoreactivity of CA1 pyramidal neurons increases in patients with AD. (A, B) Representative images of BIN1 immunoreactivity show many CA1 pyramidal neurons are BIN1-non-immunoreactive (asterisks) in healthy controls (A) and in subjects with early cognitive decline (B) . (C) CA1 BIN1-ir neurons (arrow) in a patient with AD. The BIN1 signal in the neuropil and glial cells, i.e. oligodendrocytes ( A , arrowheads) and astrocytes ( B , arrow) are less prominent or absent. #Indicates direction of ependymal surface. (D) In patients with AD (CDR1-2, BBIV-V), BIN1 expression in pyramidal neurons increases in variable but significant fractions (***p < 0.001). Scale bars = 20 μm.
To evaluate the consistency of AD-related changes in BIN1 expression, we examined BIN1 immunoreactivity in the superior temporal gyrus cortex (STG, BA22), prefrontal cortex (PFC, BA9), and primary visual cortex (OCC, BA17). We chose these neocortical regions because they exhibited differential NFT load across the Groups (see Human Postmortem Brain Region Selection in Materials and Methods). Because Group 1 and Group 2 subjects exhibited significantly different CA1 BIN1-ir properties compared to the subjects with AD (Group 3) (Supplemental Digital Content 5, table 1.1, 1.6, 1.7), we used 3 subjects with non-AD (Group 1, n = 1; Group 2, n = 2), and almost all subjects with AD (Group 3, n = 5), with values closest to the group mean of CA1 BIN1-ir data as representative subjects. PFC samples were examined because there was considerable variability in the CA1 data. The experimental design of PFC BIN1 IHC analysis replicated the CA1 analysis. As in CA1, increased BIN1-ir in the cytoplasm of several pyramidal neurons was found in the temporal, prefrontal, and primary visual cortices in patients with AD ( Fig. 5 ), but not in healthy controls (CDR0, BB0) or subjects with early BB stages without or with early cognitive decline (CDR0-0.5, BBII-III) (Supplemental Digital Content 8). We performed a quantitative analysis of BIN1 immunoreactivity in PFC of 3 representative subjects with non-AD (BB0-III) and 5 subjects with AD (BBIV-V) from Groups 1–3. Two-way ANOVA of subject group (non-AD, BB0-III; AD, BBIV-V) and anatomical region (CA1, PFC Layer III, PFC Layer V-VI) revealed a significant overall effect of subject group (non-AD vs. AD) in BIN1-ir neuropil area and BIN1-ir pyramidal neurons (p < 0.0001 and p < 0.0001, respectively) ( Fig. 6A, B; Supplemental Digital Content 5, table 2.1-2). Anatomical region showed a significant overall effect in BIN1-ir neuropil area (p < 0.0001), likely due to anatomical variation in myelinated axon density in each analyzed layer. Additionally, BIN1-ir neuropil area showed a significant interaction between subject group and anatomical area (p = 0.019), as anatomical region influenced how well the subject group predicted BIN1-ir measures. Overall, the subject group had the strongest overall effect; therefore, the decrease in BIN1-ir neuropil area and increase in BIN1-ir pyramidal neurons were consistent in both CA1 and PFC of patients with AD. The BIN1 neuronal cytoplasmic signal was not specific to cortical (pyramidal) neurons in AD because subcortical neurons of amygdala displayed consistent findings (Supplemental Digital Content 9).
FIGURE 5.
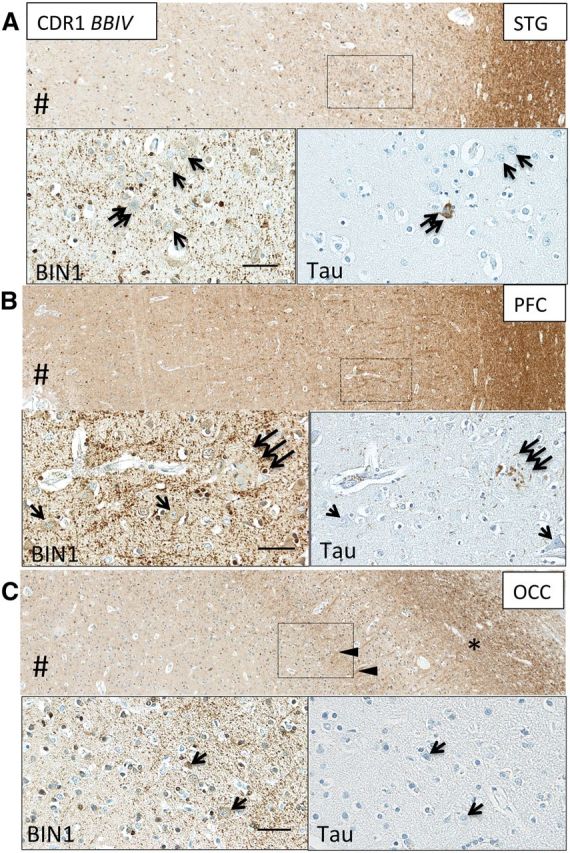
Neocortical BIN1 immunoreactivity in neurons, neuropil and white matter axons in AD. BIN1-immunoreactive (-ir) cortical neurons without NFTs (single arrows, insets) were found in STG (A) , PFC (B) , and OCC (C) in patients with AD. (A) Neuronal NFT, highlighted by tau IHC in STG, obscured BIN1 cytoplasmic signal (double arrows). (B) Neuritic plaque, highlighted by tau IHC in PFC, disrupted BIN1 signal in the neuropil (triple arrows). (C) BIN1-ir optic radiation axons (arrowheads) surround non-tangled BIN1-ir neurons in layer IV of OCC. *Indicates white matter. #Indicates direction of pial surface. Scale bars = 50 μm.
FIGURE 6.

Quantification of BIN1 immunoreactivity yields similar results in PFC and CA1. (A) Subject group (non-AD/BB0-III; AD/BBIV-V) and anatomical region (CA1; PFC Layer III; PFC Layer V-VI) show significant overall effect in BIN1-immunoreactive (-ir) neuropil area (p < 0.0001 for both) by two-way ANOVA. Significant interaction between subject group and anatomical area also exists (p = 0.019). (B) BIN1-ir pyramidal neurons show a significant subject group overall effect (p < 0.0001).
BIN1 Protein Expression and AD Pathological Hallmarks in CA1 and Neocortex
Some BIN1-ir neurons contained pretangles or tangles, as observed in CA1 and PFC of patients with AD ( Fig. 7A, B ). Neuritic plaques colocalized with interrupted neuropil BIN1 immunoreactivity ( Fig. 7C ). Quantitative analysis of the CA1 NFT burden revealed, as expected, negative correlation with total CA1 pyramidal neurons (p = 0.016, Fig. 8A; Supplemental Digital Content 5, table 3.1) and also with BIN1-ir neuropil area (p = 0.017, Supplemental Digital Content 5, table 3.2). This is consistent with the earlier observed positive correlation between total CA1 pyramidal neurons and BIN1-ir neuropil area, reflecting the density of axons as shown in Figure 3A-C . However, there was no significant correlation between BIN1-ir neurons and NFT-containing neurons in CA1 ( Fig. 8A; Supplemental Digital Content 5, table 3.1). As expected, hippocampal CERAD scores of diffuse and neuritic plaque load revealed that neuritic plaques negatively correlated with total neurons and total glia in CA1 (p = 0.030 and p < 0.010, respectively, Supplemental Digital Content 5, table 3.3). However, the percentage of BIN1-ir CA1 neurons positively correlated with hippocampal CERAD neuritic plaque scores (p < 0.0001) ( Fig. 8B; Supplemental Digital Content 5, table 3.4). Additionally, in PFC Layers V-VI, BIN1-ir pyramidal neurons also positively correlated with the PFC neuritic plaque score (Supplemental Digital Content 5, table 2.3).
FIGURE 7.
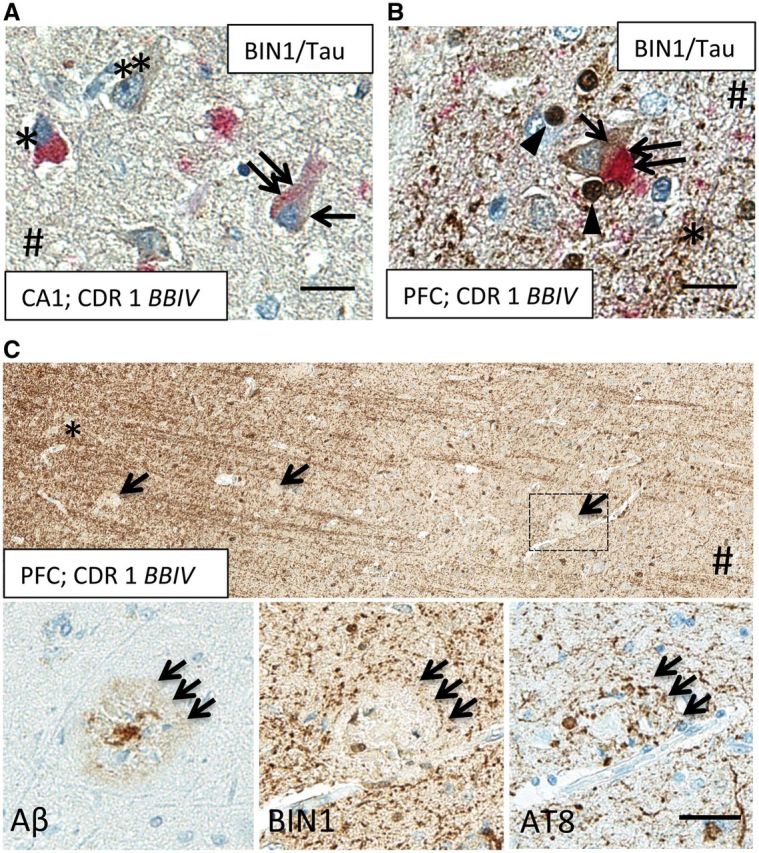
BIN1 immunoreactivity coexists with NFTs but not with neuritic plaques in AD cortex. (A) BIN1 (brown) and tau (red) dual IHC shows BIN1 signal (arrow) in pre-tangled CA1 neuron (double arrow) in an AD patient. Note a nearby tangled neuron (asterisk) and an exclusively BIN1-immunoreactive (-ir) neuron (double asterisks). # Indicates direction of ependymal surface. (B) A pyramidal neuron at the gray/white matter junction in PFC of an AD patient shows colocalization of BIN1 signal (brown, arrow) with tau-ir NFT (red, double arrow). Note BIN1-ir axons (asterisk) and BIN1-ir oligodendrocytes (arrowheads). Scale bars A, B = 20 μm. (C) BIN1-ir neuropil signal is disrupted by the presence of neuritic plaque (middle inset), highlighted by AT8 (right inset, triple arrows) and β-amyloid IHC (left inset, triple arrows), in adjacent PFC sections of an AD patient. *Indicates white matter; # indicates direction of pial surface. Scale bar: C = 40 μm.
FIGURE 8.

The percentage of BIN1-immunoreactive (-ir) CA1 pyramidal neurons correlates positively with CERAD hippocampal neuritic plaque score but not with tangled neuron fraction. (A) The percentage of BIN1-ir neurons does not correlate with the percentage of tangled neurons in CA1 region. As expected, the fraction of tangled neurons negatively correlates with the total number of pyramidal neurons in CA1. (B) Hippocampal CERAD neuritic (N) plaque scores correlates positively with the percentage of BIN1-ir neurons in CA1. Diffuse (D) plaque score does not correlate. Symbols indicate subject group: (•) CDR0, BB0; (▪) CDR0-0.5, BBII-III; (▲) CDR1-2, BBIV-V.
DISCUSSION
Our study elucidates several new aspects of BIN1 protein expression in human brains that may relate to the progression of AD-associated pathological changes.
First, 2 BIN1 antibodies raised against sequences within distinct BIN1 domains (2F11 and 99D clones) highlighted the presence of BIN1 in white matter, neuropil, and glial cells in human (and rat) brains ( Fig. 1 ; Supplemental Digital Content 4, 6). BIN1 protein is associated with myelinated axons and oligodendrocytes in the white and gray matter of CA1 and neocortex. This finding is consistent with previous reports of strongest BIN1 mRNA expression in the cerebral white matter of healthy subjects ( 47 ). Moreover, BIN1 punctate signal was observed in the rat brain white matter ( 48 ) and colocalization of BIN1 signal and axonal neurofilaments was previously visualized in AD-affected human hippocampus ( 34 ). In addition, Holler et al found increased BIN1 protein expression in the frontal cortex in myotonic dystrophy ( 11 ), reportedly a white matter disease ( 49 ), as well as a neurodegenerative tauopathy ( 50 ). They also reported a positive correlation between smaller BIN1 isoform (iso9) level and NFT load in AD brains ( 11 ). However, a later study showed that BIN1 iso9 is predominately expressed in astrocytes and not in neurons ( 42 ); therefore, this correlation may have reflected reactive astrocytosis. Considering the strong BIN1 expression in oligodendrocytes ( Figs. 1B, C, 7B ) and BIN1 punctate immunoreactivity along axons ( Fig. 3C, D ), the BIN1-ir neuropil signal likely highlights oligodendrocyte-derived myelin sheaths. Consistent with a potential role in myelin ultrastructure, BIN1 has been implicated in inducing and stabilizing membrane curvature ( 12 , 18 , 19 , 51 ).
In addition to oligodendrocytes, we observed BIN1 immunoreactivity in astrocytes ( Figs. 1D, 2A, 4B ), and, as reported by others ( 34 ), in microglia (not shown). Glial cell subtypes may each modulate AD pathogenesis. For example, astrocytes become activated by inflammatory stimuli such as Aβ, and secrete inflammatory mediators, leading to reactive astrocytosis around Aβ plaques ( 52 , 53 ). Microglia, the innate immune cells of the CNS, similarly undergo activation, phagocytose Aβ ( 54 , 55 ), and release inflammatory mediators ( 56 , 57 ). Inflammatory byproducts damage surrounding structures, including myelinated axons, and may therefore impair neurotransmission. Oligodendrocytes maintain axonal integrity and neuronal health, structurally via myelin sheaths and metabolically by trophic factor release ( 58–60 ). We demonstrated that the percentages of BIN1-ir glia in CA1 across our subject groups were not significantly changed, whereas BIN1-ir neuropil area decreased significantly in patients with AD in which there is neuronal loss ( Fig. 2B; Supplemental Digital Content 5, Table 1.5-6). These findings suggest that the BIN1-signal in neuropil may reflect the density of myelinated axons.
Second, we found a minimal BIN1 signal in CA1 pyramidal neurons of healthy controls and subjects with early NFT accumulation that were either cognitively intact or mildly impaired (CDR0-0.5). However, in more advanced Braak and Braak stages (BBIV-V) accompanied by overt dementia (CDR1-2), we observed a variable but significant increase in the percentage of pyramidal neurons with BIN1 cytoplasmic immunoreactivity ( Fig. 4D ). The increase in BIN1-ir pyramidal neurons in patients with AD was also observed in associative and sensory neocortical areas (BA22 and BA17, respectively), and quantitatively confirmed in prefrontal cortex (BA9); the latter, like CA1, showed a concomitant decrease in BIN1-ir neuropil area ( Figs. 5, 6 ). We found a negative correlation between BIN1-ir neuropil area and NFT burden (Supplemental Digital Content 5, table 3.2). Considering the positive correlation between BIN1-ir neuropil area and number of neurons ( Fig. 3A ), our finding is expected as NFT accumulation contributes to neuronal loss. Reduction in the numbers of neurons results in diminished numbers of myelinated axons and, therefore, a decreased size of the BIN1-ir neuropil area.
Third, BIN1-ir pyramidal neurons in CA1 positively correlated with hippocampal CERAD neuritic plaque score in patients with AD ( Fig. 8B ), whereas the neuritic plaque score expectedly negatively correlated with total number of neurons (Supplemental Digital Content 5, table 3.3). BIN1 has previously been shown to correlate with NFT load in AD brain ( 11 , 34 ). Chapuis et al reported increased BIN1 mRNA in AD brain, identified a non-coding BIN1 AD risk variant that increased mRNA levels in a reporter assay, and found an association between this BIN1 risk variant and tau load in patients with AD ( 34 ). Using a pan-BIN1 antibody with specificity to the conserved BAR domain present in all BIN1 isoforms ( 8 ), we found a nearly significant positive correlation between BIN1-ir neuronal population and tangled neurons (p = 0.0505) (Supplemental Digital Content 5, table 3.1).
The positive correlation between neuritic plaque load and BIN1-ir neurons ( Fig. 8B ) may suggest alternative pathophysiological functions of BIN1 in the presence of classic amyloid pathology. Furthermore, the interruption in BIN1-ir neuropil signal at the sites of neuritic plaques ( Fig. 7C ) likely reflects axonal defects. APP aggregates have negative effects on both axonal integrity ( 61 ) and myelin repair ( 62 ). In addition, transcriptome analysis of human brain in trisomy 21 (Down syndrome), a disorder the pathogenesis of which includes 3 copies of APP that causes the early appearance of AD-like pathology ( 63 ), revealed possible defects in oligodendrocyte maturation and myelination ( 64 ). Thus, human brain, genetic, and animal model studies suggest a link between oligodendrocyte function and amyloid pathology.
Our findings, together with reported functions of BIN1 in vitro, further support the role of BIN1 in Alzheimer disease. β-Amyloid has been reported to inhibit receptor-mediated endocytosis in rat hippocampal neurons, and, consequently, causes the localization of endocytic machinery at the plasma membrane ( 65 ), of which BIN1 is a constituent ( 12 , 16 , 17 ). The BIN1-ir signal that we observed in pyramidal neurons of subjects with AD may reflect altered receptor-mediated endocytosis activity at the neuronal plasma membrane. This possibility is supported by the increase in cytoplasmic BIN1 signal in pyramidal neurons, which positively correlated with neuritic plaque load. Additionally, BIN1 and tau can form complexes that are regulated by tau-phosphorylation and partly colocalize with the actin cytoskeleton in primary rat neurons ( 41 ). Increased BIN1 expression in neurons of patients with AD may occur together with changes in actin cytoskeleton as pathological tau species accumulate ( 66 ). Indeed, BIN1 signal colocalizes with pretangles and tangles in some neurons ( Fig. 7A, B ). BIN1-ir neurons are found not only in CA1 and prefrontal cortex of patients with AD, but also in frontotemporal dementia, another tauopathy accompanied by cognitive decline (Supplemental Digital Content 10). However, neuronal cytoplasmic BIN1-immunoreactivity does not occur exclusively in the vicinity of NFTs because primary visual cortex (OCC, BA17) of subjects with AD (BBIV-V) did not contain NFTs but did harbor BIN1-ir neurons ( Fig. 5C ). Thus, the alterations in BIN1 signal in AD cortex appear to be global and not exclusive to NFT-burdened regions.
The limitations of our study include a relatively modest sample size and inherent demographic constraint in CDR0, BB0 controls, which tend to belong to the young subjects. However, none of the findings presented here correlated with subject age, sex, or the postmortem interval (Supplemental Digital Content 11). These limitations did not preclude our study from revealing the patterns of BIN1-ir that may be summarized as follows: 1) the percentage of BIN1-ir CA1 glia remains constant; 2) the percentage of BIN1-ir pyramidal neurons increases significantly in CA1 and neocortex in patients with AD of Braak and Braak stages IV-V; and 3) BIN1-ir CA1 neuropil signal declines significantly in BBIV-V hippocampi and is disrupted in the presence of neuritic plaques.
The onset of neuronal BIN1 immunoreactivity separates overt dementia (CDR1-2, BBIV-V) from early AD-associated pathological changes in subjects without or with mild cognitive impairment (CDR0-0.5, BBII-III), and correlates with neuritic plaque load in hippocampal CA1 region. The role of BIN1 in oligodendrocytes and the ubiquitous processes that utilize membrane dynamics indicate that this LOAD-associated risk allele may affect axonal integrity and AD pathogenesis in a multifaceted manner. Future studies are needed to elucidate how BIN1 risk variants affect the myelin-axon unit, amyloid and tau accumulation, and glial homeostasis.
ACKNOWLEDGMENTS
We thank Terri Lima for providing expert immunohistochemistry advice, Dr. Joel Henderson and Emily Aniskovich for the use of imaging equipment, Dr. Jayandra Jung Himali for helping acquire subject clinical information, Drs. Z. Xie and J. K. Blusztajn for careful reading of the manuscript, and the Framingham Heart Study Brain Donation Program and Netherlands Brain Bank participants for their contributions to science.
REFERENCES
- 1. Seshadri S, Fitzpatrick AL, Ikram MA , et al. . Genome-wide analysis of genetic loci associated with Alzheimer disease . JAMA 2010. ; 303 : 1832 – 40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lambert JC, Ibrahim-Verbaas CA, Harold D , et al. . Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease . Nature Genetics 2013. ; 45 : 1452 U206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Carrasquillo MM, Belbin O, Hunter TA , et al. . Replication of BIN1 association with Alzheimer's disease and evaluation of genetic interactions . J Alzheimers Dis 2011. ; 24 : 751 – 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wijsman EM, Pankratz ND, Choi Y , et al. . Genome-wide association of familial late-onset Alzheimer's disease replicates BIN1 and CLU and nominates CUGBP2 in interaction with APOE . PLoS Genet 2011. ; 7 : e1001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhu R, Liu X, He Z. The bridging integrator 1 gene polymorphism rs744373 and the risk of Alzheimer's disease in caucasian and asian populations: an updated meta-analysis . Mol Neurobiol 2016. Feb 4 [ePub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 6. Naj AC, Jun G, Reitz C , et al. . Effects of multiple genetic loci on age at onset in late-onset Alzheimer disease a genome-wide association study . JAMA Neurol 2014. ; 71 : 1394 – 404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vivot A, Glymour MM, Tzourio C , et al. . Association of Alzheimer's related genotypes with cognitive decline in multiple domains: results from the three-city Dijon study . Mol Psych 2015. ; 20 : 1173 – 8 [DOI] [PubMed] [Google Scholar]
- 8. DuHadaway JB, Lynch FJ, Brisbay S , et al. . Immunohistochemical analysis of Bin1/Amphiphysin II in human tissues: diverse sites of nuclear expression and losses in prostate cancer . J Cell Biochem 2003. ; 88 : 635 – 42 [DOI] [PubMed] [Google Scholar]
- 9. Tan MS, Yu JT, Tan L. Bridging integrator 1 (BIN1): Form, function, and Alzheimer's disease . Trends Mol Med 2013. ; 19 : 594 – 603 [DOI] [PubMed] [Google Scholar]
- 10. Wechsler-Reya R, Sakamuro D, Zhang J , et al. . Structural analysis of the human BIN1 gene - Evidence for tissue-specific transcriptional regulation and alternate RNA splicing . J Biol Chem 1997. ; 272 : 31453 – 8 [DOI] [PubMed] [Google Scholar]
- 11. Holler CJ, Davis PR, Beckett TL , et al. . Bridging integrator 1 (BIN1) protein expression increases in the Alzheimer's disease brain and correlates with neurofibrillary tangle pathology . J Alzheimers Dis 2014. ; 42 : 1221 – 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Prokic I, Cowling BS, Laporte J. Amphiphysin 2 (BIN1) in physiology and diseases . J Mol Med (Berl) 2014. ; 92 : 453 – 63 [DOI] [PubMed] [Google Scholar]
- 13. Ren G, Vajjhala P, Lee JS , et al. . The BAR domain proteins: molding membranes in fission, fusion, and phagy . Microbiol Mol Biol Rev 2006. ; 70 : 37 – 120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Leprince C, Le Scolan E, Meunier B , et al. . Sorting nexin 4 and amphiphysin 2, a new partnership between endocytosis and intracellular trafficking . J Cell Sci 2003. ; 116 : 1937 – 48 [DOI] [PubMed] [Google Scholar]
- 15. Kajiho H, Saito K, Tsujita K , et al. . RIN3: a novel Rab5 GEF interacting with amphiphysin II involved in the early endocytic pathway . J Cell Sci 2003. ; 116 : 4159 – 68 [DOI] [PubMed] [Google Scholar]
- 16. Ramjaun AR, McPherson PS. Multiple amphiphysin II splice variants display differential clathrin binding: identification of two distinct clathrin-binding sites . J Neurochem 1998. ; 70 : 2369 – 76 [DOI] [PubMed] [Google Scholar]
- 17. Ramjaun AR, Micheva KD, Bouchelet I , et al. . Identification and characterization of a nerve terminal-enriched amphiphysin isoform . J Biol Chem 1997. ; 272 : 16700 – 6 [DOI] [PubMed] [Google Scholar]
- 18. Adam J, Basnet N, Mizuno N. Structural insights into the cooperative remodeling of membranes by amphiphysin/BIN1 . Sci Rep 2015. ; 5 : 15452 doi:10.1038/srep15452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Itoh T, De Camilli P. BAR, F-BAR (EFC) and ENTH/ANTH domains in the regulation of membrane-cytosol interfaces and membrane curvature . Biochim Biophys Acta 2006. ; 1761 : 897 – 912 [DOI] [PubMed] [Google Scholar]
- 20. Bohm C, Chen F, Sevalle J , et al. . Current and future implications of basic and translational research on amyloid-β peptide production and removal pathways . Mol Cell Neurosci 2015. ; 66 : 3 – 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chyung JH, Selkoe DJ. Inhibition of receptor-mediated endocytosis demonstrates generation of amyloid β-protein at the cell surface . J Biol Chem 2003. ; 278 : 51035 – 43 [DOI] [PubMed] [Google Scholar]
- 22. Zhang YW, Thompson R, Zhang H , et al. . APP processing in Alzheimer's disease . Mol Brain 2011. ; 4 : 3 doi:10.1186/1756-6606-4-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Avila J, Gomez-Ramos A, Bolos M. AD genetic risk factors and tau spreading . Front Aging Neurosci 2015. ; 7 : 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. de Calignon A, Polydoro M, Suarez-Calvet M , et al. . Propagation of tau pathology in a model of early Alzheimer's disease . Neuron 2012. ; 73 : 685 – 97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu L, Drouet V, Wu JW , et al. . Trans-synaptic spread of tau pathology in vivo . Plos One 2012. ; 7 : e31302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pooler AM, Phillips EC, Lau DH , et al. . Physiological release of endogenous tau is stimulated by neuronal activity . EMBO Rep 2013. ; 14 : 389 – 94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ueno S, Ito J, Nagayasu Y , et al. . An acidic fibroblast growth factor-like factor secreted into the brain cell culture medium upregulates apoE synthesis, HDL secretion and cholesterol metabolism in rat astrocytes . Biochim Biophys Acta 2002. ; 1589 : 261 – 72 [DOI] [PubMed] [Google Scholar]
- 28. Ito J, Nagayasu Y, Lu R , et al. . Astrocytes produce and secrete FGF-1, which promotes the production of apoE-HDL in a manner of autocrine action . J Lipid Res 2005. ; 46 : 679 – 86 [DOI] [PubMed] [Google Scholar]
- 29. Ito J, Nagayasu Y, Miura Y , et al. . Astrocytes endogenous apoE generates HDL-like lipoproteins using previously synthesized cholesterol through interaction with ABCA1 . Brain Res 2014. ; 1570 : 1 – 12 [DOI] [PubMed] [Google Scholar]
- 30. Beffert U, Danik M, Krzywkowski P , et al. . The neurobiology of apolipoproteins and their receptors in the CNS and Alzheimer's disease . Brain Res Brain Res Rev 1998. ; 27 : 119 – 42 [DOI] [PubMed] [Google Scholar]
- 31. Xu Q, Bernardo A, Walker D , et al. . Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus . J Neurosci 2006. ; 26 : 4985 – 94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McMahon HT, Boucrot E. Molecular mechanism and physiological functions of clathrin-mediated endocytosis . Nat Rev Mol Cell Biol 2011. ; 12 : 517 – 33 [DOI] [PubMed] [Google Scholar]
- 33. Poirier J, Miron J, Picard C , et al. . Apolipoprotein E and lipid homeostasis in the etiology and treatment of sporadic Alzheimer's disease . Neurobiol Aging 2014. ; 35 Suppl 2 : S3 – 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chapuis J, Hansmannel F, Gistelinck M , et al. . Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology . Mol Psychiatry 2013. ; 18 : 1225 – 34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. West MJ, Kawas CH, Martin LJ , et al. . The CA1 region of the human hippocampus is a hot spot in Alzheimer's disease . Ann Ny Acad Sci 2000. ; 908 : 255 – 9 [DOI] [PubMed] [Google Scholar]
- 36. Padurariu M, Ciobica A, Mavroudis I , et al. . Hippocampal neuronal loss in the CA1 and CA3 areas of Alzheimer's disease patients . Psychiatr Danub 2012. ; 24 : 152 – 8 [PubMed] [Google Scholar]
- 37. Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes . Acta Neuropathol 1991. ; 82 : 239 – 59 [DOI] [PubMed] [Google Scholar]
- 38. Poulin SP, Dautoff R, Morris JC , et al. . Amygdala atrophy is prominent in early Alzheimer's disease and relates to symptom severity . Psychiatry Res 2011. ; 194 : 7 – 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Unger JW, Lapham LW, McNeill TH , et al. . The amygdala in Alzheimer's disease: neuropathology and Alz 50 immunoreactivity . Neurobiol Aging 1991. ; 12 : 389 – 99 [DOI] [PubMed] [Google Scholar]
- 40. Thomas S, Mercado JM, DuHadaway J , et al. . Novel colitis immunotherapy targets Bin1 and improves colon cell barrier function . Dig Dis Sci 2016. ; 61 : 423 – 32 [DOI] [PubMed] [Google Scholar]
- 41. Sottejeau Y, Bretteville A, Cantrelle FX , et al. . Tau phosphorylation regulates the interaction between BIN1's SH3 domain and Tau's proline-rich domain . Acta Neuropathol Commun 2015. ; 3 : 58 doi: 10.1186/s40478-015-0237-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhou Y, Hayashi I, Wong J , et al. . Intracellular clusterin interacts with brain isoforms of the bridging integrator 1 and with the microtubule-associated protein Tau in Alzheimer's disease . Plos One 2014. ; 9 : e103187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Trojanowski JQ, Lee VM. The role of tau in Alzheimer's disease . Med Clin North Am 2002. ; 86 : 615 – 27 [DOI] [PubMed] [Google Scholar]
- 44. Kosik KS, Orecchio LD, Binder L , et al. . Epitopes that span the tau molecule are shared with paired helical filaments . Neuron 1988. ; 1 : 817 – 25 [DOI] [PubMed] [Google Scholar]
- 45. Goedert M, Jakes R, Spillantini MG , et al. . Tau protein in Alzheimer's disease . Biochem Soc Trans 1995. ; 23 : 80 – 5 [DOI] [PubMed] [Google Scholar]
- 46. Preibisch S, Saalfeld S, Tomancak P. Globally optimal stitching of tiled 3D microscopic image acquisitions . Bioinformatics 2009. ; 25 : 1463 – 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Holton P, Ryten M, Nalls M , et al. . Initial assessment of the pathogenic mechanisms of the recently identified Alzheimer risk loci . Ann Hum Genet 2013. ; 77 : 85 – 105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Butler MH, David C, Ochoa GC , et al. . Amphiphysin II (SH3P9; BIN1), a member of the amphiphysin/Rvs family, is concentrated in the cortical cytomatrix of axon initial segments and nodes of ranvier in brain and around T tubules in skeletal muscle . J Cell Biol 1997. ; 137 : 1355 – 67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Minnerop M, Weber B, Schoene-Bake JC , et al. . The brain in myotonic dystrophy 1 and 2: evidence for a predominant white matter disease . Brain 2011. ; 134 : 3530 – 46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies . Annu Rev Neurosci 2001. ; 24 : 1121 – 59 [DOI] [PubMed] [Google Scholar]
- 51. Frost A, Unger VM, De Camilli P. The BAR domain superfamily: membrane-molding macromolecules . Cell 2009. ; 137 : 191 – 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Avila-Munoz E, Arias C. When astrocytes become harmful: functional and inflammatory responses that contribute to Alzheimer's disease . Ageing Res Rev 2014. ; 18 : 29 – 40 [DOI] [PubMed] [Google Scholar]
- 53. Phillips EC, Croft CL, Kurbatskaya K , et al. . Astrocytes and neuroinflammation in Alzheimer's disease . Biochem Soc Trans 2014. ; 42 : 1321 – 5 [DOI] [PubMed] [Google Scholar]
- 54. Wilcock DM, DiCarlo G, Henderson D , et al. . Intracranially administered anti-Aβ antibodies reduce β-amyloid deposition by mechanisms both independent of and associated with microglial activation . J Neurosci 2003. ; 23 : 3745 – 51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wilcock DM, Rojiani A, Rosenthal A , et al. . Passive immunotherapy against Aβ in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage . J Neuroinflammation 2004. ; 1 : 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Doens D, Fernandez PL. Microglia receptors and their implications in the response to amyloid β for Alzheimer's disease pathogenesis . J Neuroinflammation 2014. ; 11 : 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Roth AD, Ramirez G, Alarcon R , et al. . Oligodendrocytes damage in Alzheimer's disease: β-amyloid toxicity and inflammation . Biol Res 2005. ; 38 : 381 – 7 [DOI] [PubMed] [Google Scholar]
- 58. Court FA, Alvarez J. Local regulation of the axonal phenotype, a case of merotrophism . Biol Res 2005. ; 38 : 365 – 74 [PubMed] [Google Scholar]
- 59. Du Y, Dreyfus CF. Oligodendrocytes as providers of growth factors . J Neurosci Res 2002. ; 68 : 647 – 54 [DOI] [PubMed] [Google Scholar]
- 60. Fruhbeis C, Frohlich D, Kuo WP , et al. . Neurotransmitter-triggered transfer of exosomes mediates oligodendrocyte-neuron communication . PLoS Biol 2013. ; 11 : e1001604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhan X, Jickling GC, Ander BP , et al. . Myelin basic protein associates with AβPP, Aβ1-42, and amyloid plaques in cortex of Alzheimer's disease brain . J Alzheimers Dis 2015. ; 44 : 1213 – 29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bartzokis G. Alzheimer's disease as homeostatic responses to age-related myelin breakdown . Neurobiol Aging 2011. ; 32 : 1341 – 71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lemere CA, Blusztajn JK, Yamaguchi H , et al. . Sequence of deposition of heterogeneous amyloid β-peptides and APO E in Down syndrome: implications for initial events in amyloid plaque formation . Neurobiol Dis 1996. ; 3 : 16 – 32 [DOI] [PubMed] [Google Scholar]
- 64. Olmos-Serrano JL, Kang HJ, Tyler WA , et al. . Down syndrome developmental brain transcriptome reveals defective oligodendrocyte differentiation and myelination . Neuron 2016. ; 89 : 1208 – 22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kelly BL, Ferreira A. Beta-amyloid disrupted synaptic vesicle endocytosis in cultured hippocampal neurons . Neuroscience 2007. ; 147 : 60 – 70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bramblett GT, Goedert M, Jakes R , et al. . Abnormal tau phosphorylation at Ser396 in Alzheimer's disease recapitulates development and contributes to reduced microtubule binding . Neuron 1993. ; 10 : 1089 – 99 [DOI] [PubMed] [Google Scholar]