

Abstract
Sudden infant death syndrome (SIDS) and sudden unexplained death in childhood (SUDC) are defined as sudden death in a child remaining unexplained despite autopsy and death scene investigation. They are distinguished from each other by age criteria, i.e. with SIDS under 1 year and SUDC over 1 year. Our separate studies of SIDS and SUDC provide evidence of shared hippocampal abnormalities, specifically focal dentate bilamination, a lesion classically associated with temporal lobe epilepsy, across the 2 groups. In this study, we characterized the clinicopathologic features in a retrospective case series of 32 children with sudden death and hippocampal formation (HF) maldevelopment. The greatest frequency of deaths was between 3 weeks and 3 years (81%, 26/32). Dentate anomalies were found across the pediatric age spectrum, supporting a common vulnerability that defies the 1-year age cutoff between SIDS and SUDC. Twelve cases (38%) had seizures, including 7 only with febrile seizures. Subicular anomalies were found in cases over 1 year of age and were associated with increased risk of febrile seizures. Sudden death associated with HF maldevelopment reflects a complex interaction of intrinsic and extrinsic factors that lead to death at different pediatric ages, and may be analogous to sudden unexplained death in epilepsy.
Keywords: Dentate gyrus, Febrile seizures, Granule cell dispersion, Sudden unexplained death in childhood (SUDC), Sudden infant death syndrome (SIDS), Sudden unexplained death in epilepsy (SUDEP), Sudden unexpected death in pediatrics with hippocampal formation maldevelopment (SUDP-HFM)
INTRODUCTION
Contemporary clinical definitions for sudden unexplained death in young children had their origins in 1969 at the Second International Conference on the Causes of Sudden Death in Infants ( 1 ). Beckwith and colleagues advanced the term “sudden infant death syndrome” (SIDS), defining it as the sudden and unexpected death in “any infant or young child” that was unexplained by a thorough postmortem examination ( 1 ). Two decades later, a consensus panel convened by the National Institutes of Health restricted SIDS to infants under 1 year of age in its operational definition ( 2 ). In 2005, Krous and colleagues introduced the term sudden unexplained death in childhood (SUDC) for children over 1 year of age (with no defined upper age limit) for death unexplained by autopsy ( 3 ). The age distinction is rooted in epidemiology indicating that the incidence of sudden unexplained death peaks in the first 6 months of life ( 4 ), declines, rises to another smaller peak in the first 3 years of life, and then declines throughout the remainder of childhood ( 3 ). This age distribution informs current research, which separately considers potential processes underlying sudden death before and after 1 year. Our group has reported developmental abnormalities in the dentate gyrus of the hippocampus in independent subsets of both SIDS ( 5 , 6 ) and SUDC ( 7–10 ). The key feature shared in these groups is focal double layering in the otherwise single layer of granule cells (GCs) in the dentate gyrus, called focal dentate bilamination (DB). Detectable with the standard light microscope, focal DB is a morphologic variant of GC dispersion in the dentate gyrus ( 11–14 ). Focal DB and the associated GC abnormalities have been classically, and almost exclusively, associated with temporal lobe epilepsy (TLE) ( 11–14 ). Its presence in SIDS and SUDC cases, with death typically occurring during a sleep period, raises the possibility that sudden death in affected children may be the consequence of a seizure or seizure-related, paroxysmal event that is generated from the abnormal dentate gyrus, triggered by stress (e.g. asphyxia or fever), and results in autonomic and/or cardiorespiratory instability. In the vulnerable infant or young child, this instability during sleep putatively cannot be compensated for by homeostatic mechanisms in processes analogous to those hypothesized in sudden unexpected death in epilepsy (SUDEP), in which sudden death occurs in patients with epilepsy without a fully understood mechanism of death ( 15–20 ).
In this report, we provide evidence supporting the hypothesis that developmental abnormalities of the hippocampal formation (HF), defined by the dentate gyrus, Ammon’s horn, subiculum, and entorhinal cortex ( 21 ), are in some cases associated with sudden death in children without regard to age. We characterized the clinical, general autopsy, and neuropathologic features in a retrospective case series of 32 children defined by HF maldevelopment and sudden, unexpected death. We delineate the topography and histopathology of brain abnormalities of the cohort using neuronal, glial, and myelin staining, and identify an entity we term, “sudden unexpected death in pediatrics—hippocampal formation maldevelopment” (SUDP-HFM). The techniques used are basic to neuropathology practice and establish a foundation for future studies.
MATERIALS AND METHODS
Experimental Design
The cases in this series were referred to Robert’s Program on Sudden Unexpected Death in Pediatrics (SUDP) at Boston Children’s Hospital (BCH) ( 22 ) by pediatricians, pediatric neurologists, medical examiners, pediatric pathologists, and families for analysis into the causes of sudden, unexpected/unexplained death. The Institutional Review Board of BCH approved the study; parental consent was obtained for each case. The study’s pediatrician (R.D.G.) and pediatric neurologist (A.H.P.) reviewed obstetrical, perinatal, and postnatal case records, as well as the circumstances of death. Data were recorded concerning gestational age, newborn metabolic screening, postnatal age at death, gender, race, and variables from the pre-, peri-, and postnatal period, seizure onset and type if applicable, circumstances of death, and family history. The autopsies in all cases were performed under the auspices of the medical examiner system in the jurisdiction of death, as mandated by law for sudden and unexpected death. The study’s pediatric pathologist (L.T.) reviewed the general autopsy reports and ancillary studies, and evaluated available somatic microscopic slides.
The study’s neuropathologists (H.C.K., J.B.C., D.D.A.) systematically reviewed the neuropathologic materials, including uncut whole brains when submitted, neuropathology reports, brain photographs, and microscopic sections of the brain and spinal cord. Consensus was reached between the 2 senior neuropathologists (H.C.K. and D.D.A.) after independent assessments. We received the whole brain for neuropathologic examination in 8/32 (25%) of cases. The hippocampus was examined in step sections in the anterior (pes), body (level of the lateral geniculate nucleus) and posterior level (posterior to the lateral geniculate nucleus) in these cases. Two young child brains were part of the San Diego SUDC Research Project, and were included in less detail in a previous case series published by us ( 10 ); they are included in the present cohort because DNA for genetic testing has become available, and their DNA results will be reported in the follow-up genetic study of this pathologic study (work in progress) for consideration of brain phenotypic-genotypic relationships in those rare cases with both DNA and histopathologic data available. The brains were fixed in formalin; microscopic sections from regions from the entire brain were surveyed. The hippocampal sections, 4–6–µm thick, were stained with hematoxylin and eosin (H&E) in all cases. Sections were stained with H&E with Luxol fast blue (LFB) (H&E/LFB) in 6 infants and 9 children over 1 year. Microscopic sections were examined with a standard light microscope. Immunostaining for glial fibrillary acidic protein and/or microglial activation (CD68) was performed in selected cases according to standard methods. Because of variation in the cellular architecture of the human hippocampus throughout its structure and variations in sectioning by different forensic groups, inclusion in the study required that the sections be oriented properly in the coronal plane, as identified by adjacent anatomic landmarks and in comparison to normative human atlases of the hippocampus ( 21 , 23 ). Cases that were technically unsatisfactory, and were in equivocal planes due to uneven cuts were excluded.
Microscopic sections were available for assessment from the following brain regions in the total cohort: HF proper, 100% (32/32), temporal lobe cortex, 100% (32/32), subiculum, 97% (31/32), cerebellum, 94% (30/32), non-temporal cerebral cortex, 91% (29/32), medulla, 88% (28/32), caudate, 78% (25/32), putamen, 72% (23/32), midbrain, 66% (21/32), thalamus, 66% (21/32), pons, 66% (21/32), globus pallidus, 59% (19/32), hypothalamus, 31% (11/32), amygdala, 22% (7/32), and spinal cord, 25% (8/32). The differences in frequency of regions sectioned reflect the lack of standardized neuropathologic practices across the different medical examiner systems from which the cases originated. In 47% (15/32) of cases, the right and left hippocampus were available for comparison. Multiple (serial or step [i.e. every 10–20 µm]) sections were examined in blocks of the hippocampus available to us for further analysis in 47% (15/32) of cases. In the hippocampal sections, we scored the presence of 44 key standardized microscopic features, which are defined in detail in our previous publications ( 6 , 10 ). Normative references were controls analyzed in previous studies ( 6 , 10 ), and other publications of the normal human hippocampus ( 21 , 23 ).
Statistical Analysis
Four morphological patterns (A–D) of developmental hippocampal pathology were identified in the cohort. Because cases with Pattern C appeared to have different characteristics from those with Patterns A, B, and D, we compared patterns using the Student t-test for symmetrically distributed variables, the Wilcoxon rank-sum test for variables with skewed distributions, and the Fisher exact test for categorical variables. A post hoc analysis was performed to determine the prevalence of Pattern C by age group, with a pairwise comparison of age groups performed due to an overall age effect at the 0.05 significance level. Logistic regression was used to identify independent correlates of the presence vs absence of Pattern C. Head circumference percentiles were calculable only for age 0 to 5 years using normative data from the World Health Organization. Wilcoxon signed rank test with p value < 0.10 was used for comparison to a percentile of 50 (median for healthy children). Otherwise, a p value of 0.05 was considered statistically significant. All analyses were performed in SAS version 9.4 (Statistical Analysis System, Cary, NC).
RESULTS
Clinical Information
Fifty-seven cases of sudden and unexpected death in pediatrics were enrolled for research by the Robert’s Program between 2012 and 2015; 81% (46/57) had at least 1 hippocampal section available for examination. Of these 46 cases, 32 (70%) had an abnormal HF and constitute the retrospective cohort of this study. Eleven of the 46 cases (24%) had no hippocampal anomalies; the hippocampal sections of another 3 (6%) were technically unsatisfactory for review.
Demographic and clinical features of the cohort of SUDP-HFM are summarized in Table 1 . The cases ranged in age from 3 weeks to 16 years with a mean of 25.7 months: 13 were infants, and 19 were over 1 year of age at the time of death. Four of the 32 cases (13%) were born pre-term, their gestational ages ranging from 33 to 35 6/7 weeks ( Table 1 ). Thirteen were ages 1 to <3 years; 4 were ages 3 to <6 years; and 2 were over 6 years. (We use the term “young child” in this report for children between 1 and 6 years.) Of the 32 cases, 28 were Caucasian, 1 African-American, and 3 siblings were of mixed ancestry; 53% were male. The cause of death as listed on the death certificate was SIDS or sudden unexpected infant death (SUID) for 6 cases, SUDC for 6 cases, SUDEP for 2 cases, seizure-related for 4 cases, positional asphyxia for 1 case, unascertained/undetermined for 5 cases, and specific causes for 6 cases, with 1 case “pending” at the time of this report ( Table 1 ). The manner of death was listed as natural in 19 cases, undetermined in 4, accidental in 1, pending in 1, and not listed for the remaining 8 cases per local practice ( Table 1 ). The 1 death unrelated to a sleep period was in a 4-year-old who died in status epilepticus after a witnessed in-hospital cardiac arrest during an admission for febrile seizures and dehydration. Seven children had a febrile illness in the 48 h prior to death. Among the 27 for whom the position was recorded in the forensic records, 16 were found prone ( Table 1 ). Maternal alcohol during pregnancy was acknowledged by history in 8 of the 32 cases, self-reported as minimal, occasional, and possible in 6 cases and not qualified in the other 2. Two cases, including 1 reporting alcohol use, had a positive history of tobacco use in pregnancy. The only medical condition for which children in this series were under medical supervision was seizures. Twelve cases in our series (38%) had a history of seizures, including 7 with febrile seizures only (1 to <6 years), 4 with febrile and afebrile seizures (1–11 years), and 1 infant with afebrile seizures only. All 11 children diagnosed with febrile seizures were presented with sudden death at ≥1 year of age; none of the infants had a history of personal febrile seizures. Febrile seizures began within the typical age range for febrile seizures, i.e. from 7.5 months to 3 years. Five children in the cohort had a history of afebrile seizures (epilepsy) (4 additionally having febrile seizures), and 1 with afebrile seizures only.
TABLE 1.
Clinical Features of Total Cohort of Patients with SUDP-HFM
| 0 to < 1 year | 1 to < 6 years | ≥6 years | |
|---|---|---|---|
| Total n in each age group | 13 | 17 | 2 |
| Postnatal age (weeks) at death (mean, SD) | 59.7 ± 14.4 | 149.2 ± 46.1 | 751.4 ± 170.2 |
| Term pregnancy | 10 | 16 | 2 |
| Female | 7 | 7 | 1 |
| Male | 6 | 10 | 1 |
| Caucasian race | 9 | 17 | 2 |
| Manner of death per death certificate | |||
| Natural | 8 | 10 | 1 |
| Undetermined | 1 | 3 | 0 |
| Accidental | 1 | 0 | 0 |
| Pending | 1 | 0 | 0 |
| Not listed | 2 | 4 | 1 |
| Cause of death per death certificate | |||
| SIDS | 3 | Not relevant | Not relevant |
| SUID | 3 | Not relevant | Not relevant |
| SUDC | Not relevant | 5 | 1 |
| Undetermined | 2 | 3 | 0 |
| Positional Asphyxia | 1 | 0 | 0 |
| Specific Total | 3 | 7 | 1 |
| Specific: infection | 1 | 3 | 0 |
| Specific: long QT Syndrome | 1 | 0 | 0 |
| Specific: primary pulmonary hypertension | 1 | 0 | 0 |
| Seizure-related | 1 | 4 | 0 |
| Pending | 1 | 0 | 0 |
| SUDEP | 0 | 2 | 0 |
| Seizure mentioned in death definition | 0 | 8 | 0 |
| Sleep-related death | |||
| Yes | 13 | 16 | 2 |
| No | 0 | 1 | 0 |
| Found Position | |||
| Prone | 3 | 12 | 1 |
| Not Prone | 9 | 2 | 0 |
| Unknown | 3 | 3 | 1 |
We reviewed 31 of the cases for a family history of febrile seizures and/or epilepsy (non-febrile seizures) in first- or second-degree relatives, which was positive in 68% (22/31), including 8/13 (62%) under 1-year old and 13/18 (72%) of those older than 1 year. Family history was positive for only febrile seizures in 9/31 (19%), only epilepsy in 10% (3/31), and both febrile seizures and epilepsy in the family in 9/31 (19%), including 2 families with at least 1 relative with both febrile seizures and epilepsy. We also ascertained family history of sudden death. Three cases of SIDS in 1 family were full siblings, and 1 SIDS and 1 SUDC in a second family were maternal half-siblings. Including these familial cases, family history was positive for SIDS in 7/31 (23%), and SUDC in 1/31 (3%). No case had a family history of SUDEP.
General Autopsy Findings
We reviewed the autopsy reports in all 32 cases. In 21, microscopic slides of somatic organs were evaluated, with evidence of antigenic stimulation, defined as mucosal or submucosal inflammation and/or increased numbers and prominence of lymphoid follicles with active germinal centers in upper respiratory tract, gastrointestinal tract, splenic white pulp, and/or enlarged lymph nodes in twenty. Focal, lymphocytic inflammation was noted in the leptomeninges in 13 of 32 cases, suggesting mild (non-lethal) aseptic meningitis. Microbial cultures were reported as positive for pathogens in 3 of the 20 cases with antigenic stimulation. There was no definitive evidence of terminal/acute or gastric aspiration in the 21 cases. Esophagitis with eosinophils was present in 4 cases. Pulmonary edema was present in 10/21 (48%), and pulmonary hemorrhage in 14/21 (67%), analogous to autopsy features reported in, but not specific to, SUDEP, and considered related to a neurogenic pathogenesis ( 18 , 24 ). Of 9 young child cases with age-appropriate dentition in the autopsy reports, bite marks on the tongue were reported as present in 3 cases and equivocally in 1, consistent with an acute, terminal seizure.
Dentate/Hippocampal/Subicular Findings
The most frequent abnormality of the HF was disorganization of the dentate gyrus defined by DB, which occurred in 31/32 (97%) of the cohort ( Table 2 ; Fig. 1 ), present in 100% (13/13) of infants, and in 18/19 (95%) of children ≥1 year. The 1 case without DB was a young child with abnormal folding of the subiculum. DB, a readily recognized variant of GC dispersion seen in TLE ( 11–14 ), is characterized by 2 partial or complete layers of GCs, the upper layer displaced in the molecular layer of the dentate gyrus ( Fig. 1F, G ). In contrast, the normal dentate gyrus forms a single layer of GCs ( Fig. 1A–E ). Focal bilamination was observed in the upper and lower blades of the dentate gyrus, as well as at the bend. In series of published controls, we found a frequency of 7.7% (3/39) of focal DB in infants dying of known (non-seizure related) diseases ( 6 ) and in 0/20 in controls 1 to <6 years, in a second, age-related archive ( 10 ) ( Table 2 ).
Table 2.
Frequency of Features in the Dentate Gyrus by Age and Compared with Archived Controls in 30 Cases Between 0 and 6 Years
| Feature | Cohort 0 to < 1 year n = 13 | Control a 0 to < 1 year n = 39 | Cohort 1 to < 6 years n = 17 | Control b 1 to < 6 years (n = 20) |
|---|---|---|---|---|
| DB | 100% (13/13) | 8% (3/39) | 94% (16/17) | 0% (0/20) |
| Single or clusters of ectopic GCs in ML | 100% (13/13) | 33% (13/39) | 100% (17/17) | 50% (10/20) |
| Subgranular clusters of immature cells | 100% (13/13) | 11% (4/39) | 29% (5/17) | 5% (1/20) |
| Focal lack of GCs in DG | 69% (9/13) | 13% (5/39) | 94% (16/17) | 25% (5/20) |
| Irregularity with caterpillar formation | 77% (10/13) | 10% (4/39) | 59% (10/17) | 10% (2/20) |
FIGURE 1.

The normal dentate gyrus compared with the abnormal focal DB in sudden unexpected/unexplained death in children and TLE. (A) Low-power view of the normal hippocampus of an infant demonstrating the interlocking relationship of the dentate gyrus and Ammon’s horn of the hippocampus proper, and the subiculum, the major outflow site, adjacent to CA1. (B) Diagram of the GC layer of the dentate gyrus showing the site of genesis of GCs in the subgranular layer from neuronal precursors (mitotic zone), and transition to differentiated GCs (postmitotic zone), with upward migration and elaboration of their dendritic arbor in the molecular layer of the dentate gyrus (from reference 60 Copyright (2006) National Academy of Sciences, U.S.A). Normal dentate gyrus of an infant (C) compared with that of a young child (D) and adolescent (E) . H&E-LFB, ×20. There is normally a single layer of packed GCs between the molecular layer and pleomorphic layer of the hilus, the 3 components of the dentate gyrus. (F) Focal DB of the GC layer in a SIDS infant with a row of GCs in 1 layer (asterisks) separated by a cell free zone from the main body of GCs. There are immature cells in the subgranular layer (arrows). Focal DB is seen in Patterns A–D. H&E-LFB, ×20. The displaced isolated layer, 1- to multiple-cells-thick, occurs at different points and with different lengths along the upper and/or lower blade of the dentate gyrus; the acellular layer of neuropil separates the 2 GC layers, and gliosis is not present. There is no gliosis or neuronal loss in Ammon’s horn, supporting the concept of a developmental lesion without major acquired changes. (G) GC dispersion in TLE, as first defined by Houser ( 13 ), with a widened GC layer and GCs dispersed in the molecular layer. Abbreviations: DG, dentate gyrus; GC, granule cell; ML, molecular layer; TLE, temporal lobe epilepsy.
We observed a spectrum of anomalies in the formation of the hippocampus and/or subiculum, all associated with DB, which we have categorized into 4 morphological patterns (Patterns A–D; Fig. 2 ). These patterns were assessed at the mid-body of the hippocampus, at or near the lateral geniculate body, and rostral to the pulvinar. Two of these patterns were not previously identified (Patterns C and D). Pattern A was defined as focal DB with single or ectopic clusters of GCs in the molecular layer and/or hilus of the dentate gyrus, irregular configuration of the dentate gyrus, and no asymmetry or malrotation of the hippocampus proper (i.e. dentate gyrus and Ammon’s horn) or abnormal folding of the subiculum ( Fig. 2 ). Pattern B was defined as DB with single or clustered ectopic GCs in the molecular layer or hilus, irregular configuration of the dentate gyrus, and, in contradistinction to Pattern A, asymmetry and/or malrotation of the hippocampus proper ( Figs. 2, 3A–C ). Pattern C had all the anomalous features of the dentate gyrus in Patterns A and B, with the distinguishing feature of abnormal folding of the subiculum ( Figs. 3, 4A, B ), with or without asymmetry or malrotation of the hippocampus proper. Pattern D was defined as hippocampal dysplasia in which there was a displacement of elements of the GC and molecular layer and blood vessels undulating towards and away from the center of the hilus ( Figs. 2, 5A–C ) at the mid-body level, e.g. the level of the lateral geniculate nucleus. Small arteries and veins, side by side in the hilus, were partially surrounded by a collar of GCs ( Fig. 5D ), which at some point connected with the main dentate gyrus ( Fig. 5C ). Closed loops of the GC layer were often situated outside the hilus on the medial side of the hippocampus; with serial sectioning, these loops connected with the main GC layer ( Fig. 5C ). In the infants, the leading pattern was Pattern A (46%, 6/13), followed by Pattern D (38%, 5/13). Pattern C (abnormal subicular folding) was not seen in the infants (0%, 0/13). Patterns A, B, and D, which are all characterized by DB without subicular folding, were present in 24/32 (75%) of the cohort, compared to Pattern C in 25% (8/32) of the cohort, the latter all ≥1 year of age. Thus, the common entity across the arbitrary divide of 1 year involved dentate gyral abnormalities without subicular folding defects.
FIGURE 2.

Schematic drawings of 4 morphological patterns of developmental pathology in the hippocampus and subiculum of the cohort of 32 infants and children with sudden unexpected death.
FIGURE 3.

Hippocampal asymmetry and/or malrotation in coronal slices of the brains (mid-thalamic level) of an infant (A) , young child (B) , and adolescent (C) . Hippocampal asymmetry is defined as a difference in the size and shape of the right and left hippocampi compared to each other, as noted in each of the 3 cases at different ages. Malrotation of the hippocampus proper is notable for a rounded and upright shape, exemplified in the brain of the infant bilaterally (A) , but more pronounced on the encircled hippocampus, and in the young child (B) , also distinct in the encircled hippocampus. The young child also has abnormal folding of the subiculum (Pattern C) (arrow), whereas the subiculum is not abnormal in the infant (A) or adolescent (C) . In the young child, the right hippocampus is abnormally formed as well as the left, and the right is smaller than the left (encircled), both without distinct landmarks. In this case (B) , the insular cortex is also asymmetric (asterisk on each side of insular cortex). In the adolescent (C) , the left and right superior temporal gyri are asymmetric (asterisk). The hippocampus on the left side (encircled) is larger than on the right side, and slanted downward and to the right. Abbreviations: CC, corpus callosum; In, insula; LGN, lateral geniculate nucleus; STG, superior temporal gyrus; Th, thalamus.
FIGURE 4.

Abnormal folding of the subiculm (Pattern C) in the brains of 2 different young children of the cohort in whole mount sections of the HF stained with H& E/LFB. (A) Duplication of the subiculum is characterized by the “splitting” of CA1 of Ammon’s horn into 2 thick “branches” of subiculum (arrows, enclosed in dashed circles). Whole mount. (B) Abnormal subicular folding is characterized by an uneven thickness in its shape. Whole mount. Of note, the degree of myelination is age-appropriate in the intrinsic and extrinsic temporal lobe pathways. Abbreviations: CP, choroid plexus; DG, dentate gyrus; LGN, lateral geniculate nucleus; OT, optic tract.
FIGURE 5.

Hippocampal dysplasia (Pattern D) demonstrated in the brains of 1 infant and 1 young child in the cohort. (A) Hippocampal dysplasia (Pattern D) in a SIDS infant with excessive convolutions of the GC layer of the dentate gyrus at the medial surface (arrows). A GC heterotopia is in the molecular layer (asterisk). H&E, Whole mount. (B) In a young child with SUDC, there is displacement of elements of the GC and molecular layer and blood vessels undulating towards and away from the center of the hilus (all arrows). In the molecular layer, there is a central blood vessel (long arrow) (Pattern D); there is also focal DB is present (short arrows). There is a blood vessel in the hilus with a partial collar of GCs towards the body of the DG (asterisk). H&E-LFB, ×40. (C) In a second SIDS infant, at 1 level there is a closed loop of GCs separate from the main body of the granular cell layer that becomes attached to the main body (asterisk) on deeper cuts, H&E, ×10. (D) A small artery and vein, side by side in the hilus, are partially surrounded by a collar of GCs (arrows) on the side of the main body of the DG, H&E, ×20. BVs, blood vessels; DG, dentate gyrus; GC, granule cell; ML, molecular layer.
In a post hoc analysis there was a significant association of age group and the presence vs absence of Pattern C (p = 0.013), with a significant difference in a pairwise comparison of age groups between 1 and <6 years and <1 year of age (p = 0.01). In additional post hoc analyses, we attempted to define a clinical subphenotype distinguishing the morphological pattern C from Patterns A, B, and D combined ( Table 3 ). The 2 significant distinguishing features were age at death and personal history of febrile seizures ( Table 3 ). The median age for Pattern C was 2.32 years compared to 0.83 year for Patterns A, B, and D combined (p = 0.007). The frequency of Pattern C in children <1 year of age was 0% (0/13) compared with 88% (7/8) in children 1 to <6 years (p = 0.013). Using a multivariable logistic regression model, a personal history of febrile seizures was the only independent predictor of Pattern C. The odds ratio (95%, confidence interval) for a personal history of febrile seizures with Pattern C was 11.4 (1.7, 74.7) (p = 0.011). Age at death was highly correlated with the case having a personal history of febrile seizure (p < 0.001), which explains why age was not significant in a model that accounts for a personal history of febrile seizures. The mean head circumference for age was significantly higher in cases with Pattern C than those without Pattern C (mean 87th vs 57th percentile, p = 0.030) ( Table 3 ). The percentage with head circumference above the 90th percentile was also higher in cases with Pattern C (60%) than those without Pattern C (60% vs 5%, p = 0.018). Mean brain weights for age were in the normative age range compared with historical controls (data not shown) ( 25 ), although the brain weight in Group C tended to be higher than that of Patterns A, B, and D combined ( Table 3 ). Pattern C was not significantly associated with prenatal adverse exposures to alcohol and tobacco ( Table 3 ).
TABLE 3.
Case Characteristics by Morphological Pattern C vs A, B, and D Combined
|
Pathology pattern
|
|||
|---|---|---|---|
| Variable | C | A,B,D | p Value * |
| Number of cases | 8 | 24 | |
| Age, years [Mean ± SD] | 3.65 ± 3.42 | 1.66 ± 3.18 | 0.144 |
| Age, years [Median, range] | 2.32 (1.13, 11.3) | 0.83 (0.07, 16.0) | 0.007 |
| Infant (<1 year) | 0 (0%) | 13 (54%) | 0.010 |
| Age group, year | 0.013 | ||
| <1 | 0 (0%) | 13 (54%) | |
| 1 to < 6 | 7 (88%) | 10 (42%) | |
| ≥6 | 1 (13%) | 1 (4%) | |
| Maternal exposure during pregnancy | |||
| Alcohol | 0 (0%) | 8 (33%) | 0.082 |
| Tobacco | 0 (0%) | 2 (8%) | 1.00 |
| Seizure History | |||
| Febrile, personal | 6 (75%) | 5 (21%) | 0.010 |
| Febrile, family ** | 6 (86%) | 13 (54%) | 0.202 |
| Non-febrile, personal | 2 (25%) | 3 (13%) | 0.578 |
| Non-febrile, family ** | 2 (29%) | 10 (42%) | 0.676 |
| Language delay *** | 1 (13%) | 1 (4%) | 0.456 |
| Motor delay *** | 1 (13%) | 0 (0%) | 0.258 |
| Family History | |||
| SIDS | 0 (0%) | 7 (29%) | 0.161 |
| SUDC | 0 (0%) | 1 (4%) | 1.00 |
| Brain weight at death, grams (restricted to ≥ 1 and <6 years, n = 7 and 9) | 1300 ± 194 | 1173 ± 165 | 0.178 |
| Mean control (historical) for 1–6 years ( 25 ), grams | 1168 (range: 938–1266) | ||
| Anthropometric assessment at death a | |||
| Head circumference for age percentile (n = 5 and 19) | 87 ± 14 b | 57 ± 30 | 0.030 |
| Head circumference for age percentile >90% (n = 5 and 19) | 3 (60%) | 1 (5%) | 0.018 |
| Head circumference for age percentile <10% (n = 5 and 19) | 0 (0%) | 2 (11%) | 1.000 |
| Weight for age percentile (n = 6 and 20) | 76 ± 30 | 65 ± 26 b | 0.201 |
| Height for age percentile (n = 7 and 23) | 51 ± 42 | 54 ± 30 | 0.980 |
*p Value from Student t-test for comparison of means; Wilcoxon rank-sum test for comparison of medians and growth percentiles; Fisher exact test for comparison of percentages; **n = 7 in Group C; ***n = 23 in Group A, B, and D. a World Health Organization standard. Head circumference percentiles are calculable only for age 0 to 5 years. b Wilcoxon signed rank test p value <0.10 for comparison to a percentile of 50 (median for healthy children).
Other abnormalities identified in association with DB included: (i) thick walled blood vessels in the hilus, GC layer, and/or molecular layer of the dentate gyrus ( Fig. 6A ); (ii) ectopic GCs (isolated or clustered) in the molecular layer and/or hilus ( Fig. 6B, C ); (iii) heterotopia of GCs in the molecular layer or hilus ( Fig. 6D, E ); (iv) focal lack of GCs in the dentate gyrus, non-adjacent to blood vessels ( Fig. 6F ); (v) GCs bridging between dentate convolutions ( Fig. 6H ); and (vi) alternating thinness and thickness of the dentate gyrus, appearing like a caterpillar’s undulating shape in movement ( Fig. 6I ; Table 2 ). There were also clusters of immature neurons in the subgranular dentate layer ( Fig. 6G ) previously reported by us to express Tuj1, a marker of neuronal precursors ( 6 ) in 10.3% (4/39) in control infants ( 6 ), and in 5% (1/30) of controls between 1 and 6 years ( 10 ) ( Table 2 ). Clusters of immature neurons decreased in frequency with increasing age in this cohort; they were identified in 100% (13/13) of the infants and 29% (5/17) of the young children 1 to <6 years ( Table 2 ).
FIGURE 6.

Additional features in the abnormal dentate gyrus associated with DB in representative infants and young children with SUDP-HFM. A: Blood vessels with thick walls, shown here in the hilus and molecular layer (arrows), H&E, ×40. (B) Ectopic GCs in a row in the hilus below the subgranular layer (arrow), H&E, ×20. (C) Clusters (asterisk) and single (arrow) ectopic GCs in the molecular layer of the DG, H&E, ×40. (D) GC heterotopia in the molecular layer (asterisk), H&E, ×10. (E) GC heterotopia in the hilus (asterisk), H&E, ×10. (F) Focal lack of GCs (thick arrow), suggesting abnormal cell migration to this position, in an uneven GC layer with alternating areas of cell thickness of different degrees. H&E, ×10. (G) Immature cells in the subgranular layer of the dentate gyrus (arrows), characterized by dark nuclei and scant or rod shaped cytoplasm in clusters, H&E, ×20. (H) Marked GC dispersion with a “smear” of GCs bridging 2 convolutions (asterisk) of the abnormal GC layer, H&E ×10. (I) Marked undulation of the GC layer of the dentate gyrus mimicking the shape of a caterpillar in movement, H&E, ×2. DB, dentate bilamination; DG, dentate gyrus; ML, molecular layer; GC, granule cell.
Anomalous Features Outside of the Temporal Lobes
Multiple microdysgenetic features, defined as microscopic findings of abnormal cellular development predominantly in neurons ( 11 , 12 ) were noted in different frequencies in the extra-temporal regions of the SUDP-HFM cohort ( Figs. 7, 8 ; Table 4 ). These features have been reported in association with temporal and extra-TLE in autopsy and surgically resected specimens ( 11–14 , 26 , 27 ). In the present cohort, the most common of these features, present in 18/28 (64%) of all cases, was focal cortical dysplasia (FCD), Type I (with vertical layering of neurons in columns) or unclassified type ( Fig. 7 ; Table 4 ). In the brain of 1 young child with a history of febrile and non-febrile seizures, developmental delay, and a family history of febrile seizures and epilepsy, there was severe macroscopic asymmetry of the hippocampus associated with asymmetry of the insular cortex ( Fig. 3B ). In the case of a 16-year-old boy with no history of febrile and/or non-febrile seizures and sudden unexpected death there was asymmetry of the superior temporal gyri, in addition to asymmetry of the hippocampus ( Fig. 3C ).
FIGURE 7.

Anomalies of development in the temporal and extra-temporal cerebral cortex in infants and children of the SUDP-HFM cohort. (A) Normal cerebral cortex of an infant for comparison, ×10. (B) FCD in a SIDS infant with vertical layering of neurons in rows of at least eight cells (arrows), H&E, ×10. (C) Higher power of vertical neuronal layering in FCD, Type I, ×20. (D) Disorganized cortex in a SIDS infant with an undulating molecular layer and absence of an organized laminar cortex directly underneath the molecular layer, ×10. (E) Fused gyrus between 2 cortical gyri (arrows), ×10. All are H&E.
FIGURE 8.

Microdysgenetic features in extra-temporal lobe sites in infants and children of the SUDP-HM cohort. (A) Hamartia (asterisk) in the putamen of the basal ganglia, H&E, ×10. (B) Hamartia (asterisk) in the periventricular region of the lateral ventricle of the temporal horn. H&E, ×20. (C) Microdysgenetic focus with clusters of immature neurons (thin arrows) in the molecular layer of the cerebral cortex around thickened leptomeningeal vessels between 2 gyri, with a single mature neuron (thick arrow), H&E, ×20. (D) A heterotopia in the subcortical white matter of the frontal lobe. Whole mount, H&E-LFB. Put, putamen.
TABLE 4.
Extra-temporal Lobe Microdysgenetic Features in Cohort of 32 Cases, Including in the Cerebral Cortex and Rhombic-Lip-Derived Structures
| Feature | Cohort 0 to < 1 year n = 13 | Cohort 1 to < 6 years n = 17 | Cohort >`6 years n = 2 |
|---|---|---|---|
| FCD, unclassified | 75% (9/12) | 56% (9/16) | 0/2 |
| Hamartia-TC | 31% (4/13) | 18% (3/17) | 1/2 |
| Hamartia-extra TC | 8% (1/13) | 6% (1/17) | 0/2 |
| Heterotopia-extra TC | 23% (3/13) | 29% (8/17) | 0/2 |
| Anomalies of the lateral temporal lobe | 0% (0/13) | 8% (2/17) | 1/2 |
| Asymmetry of the insula | 0% (0/13) | 6% (1/17) | 0/2 |
| Olivary Heterotopia | 40% (4/10) | 44% (7/16) | 1/2 |
| Olivary Dysplasia | 20% (2/10) | 19% (3/16) | 1/2 |
| Arcuate hypoplasia | 0% (0/10) | 18% (3/16) | 1/2 |
| Arcuate hyperplasia | 10% (1/10) | 13% (2/16) | 0/2 |
| Fused Pyramids | 10% (1/10) | 18% (3/16) | 1/2 |
| GCs in ML of cerebellum | 25% (3/12) | 18% (3/16) | 2/2 |
| Cerebellar heterotopia | 25% (3/12) | 0% (0/16) | 0/2 |
Not all cases had all sections for assessment (see text).
FCD, focal cortical dysplasia; GCs, granular cells; ML, molecular layer; TC, temporal cortex.
Hippocampal Findings in Families
In the cohort there were 2 families in which hippocampal anomalies were found in deceased siblings upon neuropathologic examination. In 1 family, a 1-month-old boy, 3-month-old girl, and 3-week old girl all died suddenly during a sleep period. In all 3 cases, focal DB was found; this was characterized by Pattern D in the 2 oldest siblings and Pattern A in the youngest ( Fig. 9 ). In the second family, maternal half-siblings, a boy who died at 2.5 months and a girl at 15 months, occurred 2 years apart. These half-siblings both had Pattern A (data not shown). Two fathers of young children in the cohort, 1 young child with Pattern A and the other with Pattern D, had TLE that developed in adolescence and had temporal lobe resections for refractory epilepsy as young adults. In both resections, Ammon’s horn sclerosis (AHS) was diagnosed, as demonstrated in available microscopic sections from 1 father’s surgical material ( Fig. 10 ).
FIGURE 9.

Familial DB. In 1 family, a 1-month-old boy (A) , 3-month-old girl (B) , and 3-week old girl (C) died suddenly and unexpectedly during a sleep period. In all 3 cases, DB (arrows) and GC dispersion were found upon microscopic examination in sibling A (A) , sibling B (B) , and sibling C (C) , H&E, ×40. In sibling C, there is a single layer of GCs deep in the hilus (arrow) underneath the subgranular gyrus (D) . DG, dentate gyrus; GC, granule cell; ML, molecular layer.
FIGURE 10.

Temporal lobe resection for TLE in a young adult father of a young child with SUDP-HFM showed AHS. (A) Irregular dentate gyrus with focal depopulation of GCs (arrow), ×10. (B) Focal bilamination of the dentate gyrus with GCs in a separate layer from the main body (arrows). H&E, ×20. (C) Neuronal loss and gliosis in CA1; insert, reactive astrocyte with hypertrophic, eosinophilic cytoplasm, ×20. All are H&E. DG, dentate gyrus; ML, molecular layer.
Rhombic Lip-Derived Anomalies
In addition to hippocampal and cerebral cortical microdysgenetic features, we found anomalies that involved derivatives of the rhombic lip, the germinal zone at the junction of the pons and medulla in the human embryo ( Table 4 ). The anomalies included hypoplasia of the arcuate nucleus, with the caveat that in all cases with medullary sections, only 1 section was available for examination rather than the preferable multiple sections to assess the heterogeneous distribution of arcuate neurons in the rostrocaudal plane. Nevertheless, no arcuate neurons were noted in the single sections ( Fig. 11B ). Arcuate hyperplasia was noted ( Fig. 11C ), as were olivary heterotopia, dysplasia, and hypoplasia ( Fig. 12A–D ). Misplaced (dispersed) GCs of the internal granular cell layer of the cerebellar cortex were observed above the Purkinje cell layer ( Fig. 12E ).
FIGURE 11.

Arcuate nucleus anomalies associated with SUDP-HFM. (A) Normal arcuate nucleus in clusters (long arrows) for comparison at ventral surface of the medulla within the rim of the pyramid, H&E./LFB, ×2. (B) Absence of clusters of arcuate nucleus along the ventral rim of the medullary surface. H&E, ×2. (C) Hyperplasia of the arcuate nucleus (arrows) extending along the entire ventral rim of the medullary surface associated with fusion of the pyramids. There is a perivascular space entrapped between the 2 fused pyramids (arrowhead). PIO, principal inferior olive; PYR, pyramid.
FIGURE 12.
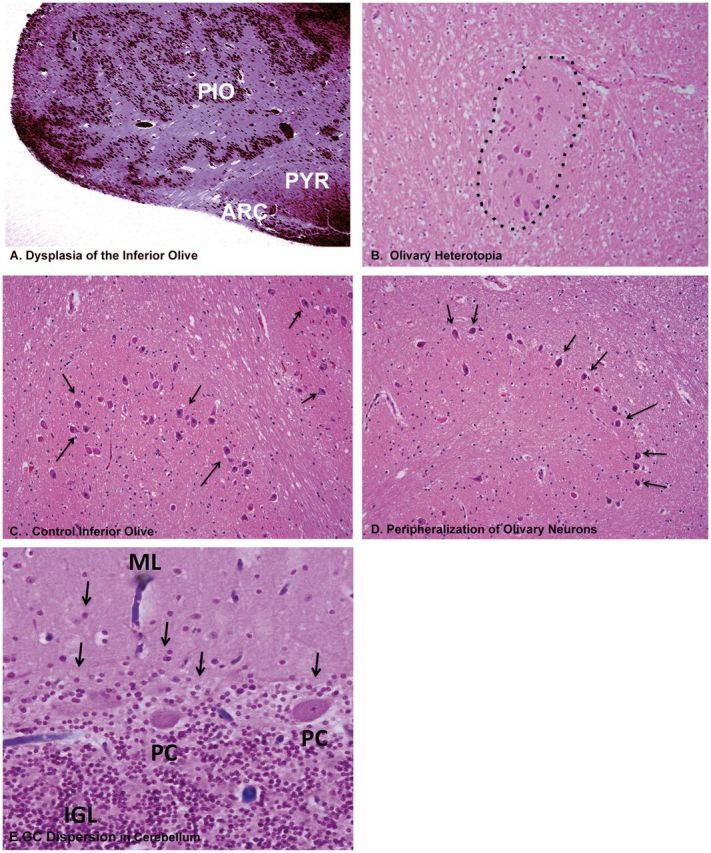
Abnormal derivatives of the rhombic lip in the infant and children cases of the SUDP-HFM cohort. (A) Olivary dysplasia, exhibiting abnormal configuration of a hyperconvoluted principal inferior olive, with closely packed convolutions H&E, ×2. (B) Olivary heterotopia (misplaced clusters of olivary neurons) (dotted line) in the inferior cerebellar peduncle, H&E, ×20. (C) High-power view of normal inferior olive for comparison showing loosely packed separated neurons (arrows). H&E, ×20. (D) High-power view of olivary neurons (arrows) that are marginalized to the periphery of the convolutions of the nucleus (arrows). H&E, ×20. (E) A putative defect in migration of GCs from the external to internal granular cell layer in the cerebellum in infants and children in the SUDP-HFM cohort. Illustrated is dispersion of GCs (arrows) in the molecular layer of the cerebellar cortex, presumably having not reached the internal granular layer from its site of migration from the external granular cell layer, H&E, ×20. ARC, arcuate nucleus; GC, granule cell; IGL, internal granular layer; ML, molecular layer. PC, Purkinje cell. PIO, principal inferior olive; PYR, pyramid.
Acquired Lesions
Of the cases under 1 year of age, 10/13 (77%) had diffuse cerebral white matter gliosis ( Supplementary Data, Fig. 1A ), with a decline thereafter to 6% (1/17) in young children under 6 years, and none of the 2 cases over 6 years ( Table 5 ). The majority of cases in the first year of life had brainstem tegmental gliosis, including the dorsal raphe and olivary gliosis ( Supplementary Data, Fig. S1 ); both of these features were reduced by half in early childhood ( Table 5 ). There was sparing of gliosis and neuronal loss in the thalamus, putamen, caudate, globus pallidus, and amygdala, and relative sparing of the hypothalamus ( Table 5 ). In the hippocampus there was hilar gliosis in both the infants (0 to <1 year, 46% [6/13]) and young children (1 to <6 years old, 47% [8/17]), without obvious neuronal loss. There was neither gliosis nor neuronal loss in CA1-3 in any age group ( Table 4 ). The degree of myelination was age-appropriate in all forebrain and brainstem regions in which myelin counter-staining with LFB was available (6 infants and 9 children over 1 year).
TABLE 5.
Acquired Injury (Gliosis) in Cohort of 32 Cases with SUDP-HM
| Feature | Cohort 0 to < 1 year n = 13 | Cohort 1 to < 6 years n = 17 | Cohort >6 years n = 2 |
|---|---|---|---|
| Cerebral WM gliosis | 77% (10/13) | 6% (1/17) | 0% (0/2) |
| Thalamus gliosis | 9% (1/11) | 0% (0/8) | 0% (0/2) |
| Caudate gliosis | 0% (0/12) | 0% (0/11) | 0% (0/2) |
| Putamen gliosis | 0% (0/11) | 10% (1/10) | 0% (0/2) |
| Globus pallidus gliosis | 9% (1/11) | 17% (1/6) | 0% (0/2) |
| Hypothalamus gliosis | 25% (1/4) | 29% (2/7) | 50% (1/2) |
| Amygdala gliosis | 0% (0/1) | 20% (1/5) | 0% (0/1) |
| Bergmann gliosis | 8% (1/12) | 6% (1/16) | 0% (0/2) |
| CA1-3 gliosis | 8% (1/13) | 6% (1/17) | 0% (0/2) |
| Hilar gliosis | 46% (6/13) | 47% (8/17) | 0% (0/2) |
| Dorsal raphe gliosis | 67% (4/6) | 42% (5/12) | 0% (0/2) |
| MB tegmentum gliosis | 29% (3/7) | 17% (2/12) | 50% (1/2) |
| Pons tegmentum gliosis | 43% (3/7) | 8% (1/13) | 0% (0/2) |
| Medulla tegmentum gliosis | 50% (5/10) | 13% (2/15) | 0% (0/2) |
| Olivary gliosis | 70% (7/10) | 13% (2/176) | 0% (0/2) |
Not all cases had all sections for assessment (see text).
BG, basal ganglia; DG, dentate gyrus; MB, midbrain; WM, white matter.
DISCUSSION
We report abnormalities of the HF associated with sudden unexpected death in children across the operational 1-year division between SIDS and SUDC in which there is a bimodal distribution of sudden death. We name this abnormality SUDP-HFM. Because we recognized the HF abnormalities in a child dying in late childhood and 1 in adolescence, we emphasize that they can be detected across the age spectrum from early infancy through adolescence. We hypothesize that cases with SUDP-HFM, currently categorized under the diagnoses of SIDS, SUDC, and SUDEP, represent a distinct clinicopathologic entity. In some instances, SUDP-HFM is associated with non-neural processes that medical examiners may consider the cause of death, e.g. infection, but may in fact represent precipitating disease processes; we speculate that these non-neural processes may not have been lethal except in the context of SUDP-HFM.
We identified 4 morphological patterns of anomalous development in different substructures of the HF (Patterns A–D) within SUDP-HFM. These patterns may prove to be related to specific clinical subtypes, and/or variable genetic and environmental factors involved in pathogenesis. All patterns display focal DB, with or without asymmetry/malrotation of the hippocampus proper (dentate gyrus and Ammon’s horn). The major distinction among them is the presence of abnormal subicular folding (Pattern C) vs the absence of such folding (Patterns A, B, and D). While Patterns A, B, and D were present in all age groups, Pattern C was only observed in children ≥1 year of age. Pattern C also carried an 11-fold increase in risk of personal febrile seizures compared to Patterns A, B, and D combined, and a tendency toward larger head/brain size compared to Patterns A, B, and D combined, the latter observation needing confirmation in larger samples and comparison with parental head circumference data.
The neuropathologic findings in these children provide a plausible mechanism for sudden and unexpected death via an epilepsy-like mechanism. Seizures, known to arise in all HF substructures ( 28–32 ), may be generated in the abnormal HF in SUDP-HFM, triggered by stress (e.g. asphyxia or fever). The dentate gyrus is a well-recognized site of epileptogenesis ( 31 , 33 ). Mouse models indicate that dentate morphological disorganization is responsible for the origin of seizures, and not secondary to them ( 29 , 33 ). In epileptic brains, dentate GC dispersion is thought to result in aberrant synaptic connectivity increasing susceptibility to seizures through hyperexcitability ( 31 ). We speculate that abnormal electrical discharges in the disorganized HF are propagated to regions of the brainstem involved in breathing and/or autonomic function during sleep, leading to lethal disruption of vital functions and sudden death ( 34 ) during sleep, when the threshold for epileptogenesis is lowered ( 28 , 35 ). We agree with Noebels who recently coined the term “epilepsy in situ ” in SUDC cases with hippocampal maldevelopment ( 9 , 10 ) as an apt “new term that may be usefully applied to a microscopic epileptiform lesion with or without evidence of actual seizures ( 36 ).”
Similar mechanisms have been postulated in SUDEP complicating TLE ( 15–20 ). Similarities with SUDEP are further supported by pulmonary edema in the majority of the SUDP-HFM cohort at autopsy, consistent with neurogenic edema described in SUDEP ( 18 , 24 ), and tongue bites in several children ( 37 ). Our group has previously reported abnormalities in serotonergic (5-HT) parameters in the brainstem in SIDS cases ( 38–41 ). Because the dentate gyrus receives 5-HT innervation from the 5-HT domains of the brainstem ( 42 ), and its development is orchestrated in part by the neurotropic influences of 5-HT, research is needed to determine if dentate abnormalities in SUDP-HFM are secondary to primary defects in the brainstem 5-HT cell domains. Importantly, recent experimental evidence suggests that brainstem 5-HT systems are critical in cardiorespiratory changes during and after seizures ( 43 ), with implications for seizure-related sudden death.
The operational definition of epilepsy proposed by the International League Against Epilepsy contends that, in contrast to the prior requirement of more than 1 unprovoked seizure, a child with a provoked (e.g. fever-associated) seizure and a high likelihood of recurrence is considered to have epilepsy ( 44 ). Although in principle the knowledge of a hippocampal malformation might establish risk of recurrence, particularly in the setting of afebrile seizures or complex febrile seizures, the determination of the hippocampal lesion was made only at autopsy in our cohort. Today, the unexpected death of a child with epilepsy, even if not obviously related to a witnessed seizure, would likely be classified as SUDEP. We pursued seizure histories in a directed fashion with the families in this study; seizure information was not always known during the forensic assessment of cause of death. One might further consider classifying the deaths of those children with history of any seizure and SUDP-HFM as SUDEP. Thus, the strong association of focal DB with epilepsy, the presence of HF malformation in various ages, and the presence of seizures in so many of the cases of SUDP-HFM is consistent with the hypothesis that seizure-related mechanisms contribute to the untimely deaths in some cases of SIDS and SUDC, just as in SUDEP. These associations, while compelling, do not allow us to predict which of the multitude infants and children with epilepsy (∼0.5%–1% of children) ( 45 ) and febrile seizure (2%–5% of children) ( 46 ) are at risk for sudden death. Large epidemiological studies are needed to address this issue. An additional consideration is that sudden death occurs in SUDP-HFM without a seizure in a manner analogous to monitored SUDEP occurring in 2 patients with epilepsy in which there were no epileptic seizures preceding death, but rather, varying patterns of respiratory and bradyarrhythmic cardiac dysfunction with profound EEG suppression ( 47 ).
An important question is why the abnormalities of the HF, arising during gestation and present at birth, manifest themselves as sudden death at different ages. We speculate that the timing is due to different developmental, environmental, and genetic risk factors combined with the underlying HF vulnerability. The different morphological patterns may make a child susceptible at a different age, given, for example that Pattern C occurs only in children ≥1 year of age. Hypoplasia of the arcuate nucleus associated in this cohort with SUDP-HFM has been reported previously in SIDS infants ( 48 , 49 ). This nucleus is considered the human homologue of the central respiratory fields for chemosensitivity at the ventral medullary surface ( 50 , 51 ). The association of arcuate and dentate gyral abnormalities may together augment the risk for sudden death in those cases in which both abnormalities occur simultaneously. Cerebral white matter and brainstem tegmental gliosis in the infant cases of the cohort may be related to hypoxia-ischemia sustained in the pre- and/or perinatal period because it mimics the topography of such injury ( 52 ); yet, we did not observe gliosis in the children who survived into childhood prior to death. The occurrence of widespread gliosis may put infants at risk for sudden death at the younger age bracket.
Prevailing age categories, i.e. before and after 1 year of age, also likely involve different maturational factors in the hippocampus/subiculum that orchestrate differential development, such as volume changes, neurogenesis and migration in the dentate gyrus, connectivity, and myelination ( 21 , 23 , 53 , 54 ). The undulations of the dentate gyrus and its molecular layer, particularly in the medial distribution, in Pattern D (hippocampal dysplasia) are reminiscent of the histopathologic dentate findings in the genetically engineered mutant mice which lack the non-receptor tyrosine kinase gene fyn , and demonstrate impaired long-term potentiation, spatial learning, seizures, and sudden death ( 55 , 56 ). The lack of fyn during hippocampal development in this mutant results in an increased number of GCs in the dentate gyrus, giving it a “scalloped” appearance, as seen in our Pattern D ( 55 ). The increase in cells was postulated to result from overproliferation, altered cell fates, or failure of cell death ( 55 ). In regard to the cluster of immature cells in the subgranular layer of the dentate gyrus observed by us in Patterns A, B, and D, in a prior publication, we demonstrated that they were present in infants with explained causes of deaths (controls), but that their frequency was unrelated to age across the infant period, and was increased in unexplained cases compared to controls ( 6 ). The frequency of immature (Tuj1-immunopositive [6]) neurons in the subgranular clusters from infancy into early childhood decreases, suggesting a morphological marker of changing pathology with age, and hence potentially changing susceptibilities to other risk factors. The association between sudden death and personal febrile seizures in young children likely reflects the vulnerability of the developing brain between 6 postnatal months and 6 years to fever due to brain maturational factors related to thermal sensitivity ( 57 ). Under 6 months of age, heightened thermal sensitivity may be present, but may manifest as central autonomic instability and increased risk for sudden unexplained death due to over-bundling, associated with SIDS, rather than a febrile seizure ( 57 ). The presence of antigenic stimulation in somatic organs in most cases may support a role for fever and inflammation in the pathogenesis, although it is not an uncommon autopsy finding in young children. In 6 SUDP-HFM cases, death certificates listed a specific, non-seizure related cause of death, including respiratory tract infection or sepsis, also evidence of antigenic stimulation. In our cases in which the listed cause of death was infection or cardiac channelopathy, hippocampal pathology was not recognized by the medical examiner, who concluded that the etiology was non-brain-related. Increased recognition of SUDP-HFM may further clarify the relationship between brain vulnerability and potential triggering or augmentation by infection, genetic susceptibilities to arrhythmias, or other factors.
We regard Patterns A-D as primary developmental lesions because they are not associated with gliosis and/or neuronal loss in the HF, except for hilar gliosis (see below). We speculate that abnormal folding of the hippocampus proper (Pattern B), subiculum (Pattern C), and GC and molecular layers of the dentate gyrus (Pattern D) originate as the hippocampus begins to form and fold at the end of the first trimester ( 21 ). The embryonic anlage of the HF, the cortical hem, which induces the formation of the hippocampus proper and subiculum within the adjacent cortical neuroepithelium ( 58 , 59 ) may be at fault in Patterns B, C, and D. GCs in the dentate gyrus are generated from stem cells in the subgranular layer throughout life, and integrate into the mature hippocampal circuitry, potentially participating in the formation of new memories ( 60 ). Because neurogenesis continues after birth into adulthood ( 31 , 61 ), Pattern A, consisting of DB only, could arise pre- and/or postnatally. GC dispersion, including DB, is hypothesized to be due to increased proliferation of progenitor cells from the subgranular zone ( 32 ), a defect in neuronal migration upwards from the subgranular zone ( 62–65 ), and/or inhibition of programmed GC death ( 66 ). We have examined the cohort histories for possible teratogens that could operate during early gestation, e.g. prenatal exposure to alcohol and cigarette smoke. Patterns A, B, and D combined demonstrated variable frequencies of borderline significance with exposure to prenatal alcohol, which is a known risk factor for SIDS ( 67 ), as is prenatal exposure to smoking ( 68 ). Pattern C with abnormal subicular folding was not associated with these exposures, suggesting that Pattern C may have a distinctive pathogenesis with a developmental vulnerability of the subiculum different from the hippocampus proper—a hypothesis for future testing in larger cohorts with more detailed exposure histories. The observation of subicular anomalies in children over 1 year only raise the question if they arise as the hippocampal shape continues to develop in size and shape after infancy ( 21 , 23 , 53 , 54 ).
Although the HF anomalies are the sine qua non of the SUDP-HFM entity that we describe, the presence of developmental pathology in the cerebral cortex and rhombic lip derivatives in the majority of SUDP-HFM suggests that a constellation of anomalies relates to common processes involved in the development of the different regional anlages. It is possible that these 3 embryonic anlages are developmentally and functionally linked, and pattern disruptions occur in the genetic regulation of a signaling molecule or transcription factor shared by all 3 embryonic anlages in SUDP-HM, at least in some cases. Many of the abnormalities inside and outside the hippocampus proper are consistent with neuronal migration defects, e.g. DB, FCD, heterotopia, peripheralization of olivary neurons, arcuate nucleus hyperplasia, and dispersed GCs in the molecular layer of the dentate gyrus and cerebellar cortex, suggesting that genetic disturbances in shared migration factors, e.g. reelin ( 62–65 , 69 ), may occur in different parts of the brain. The strong family history of febrile seizures and epilepsy in our cohort, (particularly the 2 fathers with refractory epilepsy and temporal lobe findings similar to their children who had died with SUDP-HFM), speak to a potential additional genetic risk.
The presence of focal DB in the resected temporal lobes of these fathers, coupled with our finding of DB in many children without known epilepsy, suggests that this lesion was present as a “precursor” rather than a consequence of seizures. AHS is the most common neuropathologic substrate of TLE ( 11 , 26 ), and classically is characterized by decreased neurons and gliosis in the GC layer of the dentate gyrus, the hilus of the dentate gyrus, and CA1 and CA3 of Ammon’s horn ( 11 , 12 ). Approximately 50% of cases with AHS demonstrate GCD, with DB in 10% of cases ( 13 ). The amount of neuronal injury appears to be progressive ( 70 , 71 ), and can be induced and propagated by seizures. If deceased children with HF maldevelopment had survived early childhood, they may have developed AHS in later life from recurrent clinical or subclinical seizures, leading to acquired gliosis and/or neuronal loss in the dentate gyrus, its hilus, CA1 and CA3. Children of all ages in the cohort of 32 cases had hilar gliosis in association with DB, possibly reflecting damage to the susceptible hilus by possible repetitive, sleep-related, subclinical seizures before the final lethal event.
A limitation of this study is that it is a retrospective review based mainly upon self-referred families, and is not population based. Because of the rarity of SUDC in particular (with an estimated incidence of 1.3/100 000 children between 1 and 3 years) ( 3 ), a national registry such as in Robert’s Program is essential for current research. Another limitation is that because autopsy tissue sampling in different medical examiner’s offices was not consistent, we were not able to analyze all brain regions in all cases. Nor was it possible to perform immunocytochemical studies in the majority of cases because the tissues were not all handled optimally for the technique to be applied. Nevertheless, we consider that valuable information was gained reviewing the available conventionally stained sections—the mainstay of all anatomic pathology and first step analysis in neuropathologic discovery.
In conclusion, this study defines neuropathology in the HF in sudden unexplained death in pediatrics, challenging age-related conventions in at least some cases. SUDP-HFM must be carefully identified in neuropathologic examination of sudden death in children by assessment of both hippocampi in more than 1 tissue section. We believe it is appropriate to designate SUDP-HFM as a distinct cause of death for the purposes of family counseling, vital statistics, and death certificates. In our opinion, the robustness of the clinicopathologic phenotype and the biologic plausibility of a fatal seizure-like event triggered by a set of exogenous factors in a critical developmental period justify this conclusion, and may lead to an increased understanding of antecedent risk factors for this entity.
Supplementary Material
ACKNOWLEDGEMENTS
We are deeply grateful to participating families for their courage and generosity in sharing the stories of their children for research. We also thank the clinicians, medical examiners, and pathologists who helped in the accrual of cases, including Drs. Gary Dale, Jeffrey Goldstein, Catherine Stoos, Marco M. Hefti, and Henry F. Krous. We thank Dr. Eugene E. Nattie for critical reading of the manuscript.
Financial support: This study was supported by Robert’s Program in Sudden Unexpected Death in Pediatrics, Barrett Edward Tallman Memorial Fund, Cooper Trewin Memorial SUDC Research Fund, Arcadia Family Fun Run, Nathan Lounsbury Foundation, CJ Foundation, First Candle, NINDS (K23NS069784 [AHP]), Eunice Kennedy Shriver National Institute of Child Health and Development (PO1-HD036379 [HCK]), and P30-HD18655 (Developmental Disabilities Research Center, Children’s Hospital Boston).
Declaration of Conflict of Interest: The authors have no duality or conflicts of interest to declare.
Supplementary Data can be found at http://www.jnen.oxfordjournals.org .
REFERENCES
- 1. Beckwith JB. Defining the sudden infant death syndrome . Arch Pediatr Adolesc Med 2003. ; 157 : 286 – 90 [DOI] [PubMed] [Google Scholar]
- 2. Willinger M, James LS, Catz C. Defining the sudden infant death syndrome (SIDS): deliberations of an expert panel convened by the National Institute of Child Health and Human Development . Pediatr Pathol 1991. ; 11 : 677 – 84 [DOI] [PubMed] [Google Scholar]
- 3. Krous HF, Chadwick AM, Crandall L , et al. . Sudden unexpected death in childhood: A report of 50 cases . Pediatr Dev Pathol 2005. ; 8 : 307 – 19 [DOI] [PubMed] [Google Scholar]
- 4. Moon RY , Task Force on Sudden Infant Death Syndrome . SIDS and other sleep-related infant deaths: expansion of recommendations for a safe infant sleeping environment . Pediatrics 2011. ; 128 : 1030 – 9 [DOI] [PubMed] [Google Scholar]
- 5. Rodriguez M, McMillan K, Crandall LA , et al. . Hippocampal asymmetry and sudden unexpected death in infancy: a case report . Forensic Med Pathol Sci 2012. ; 8 : 441 – 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kinney HC, Cryan JB, Haynes RL , et al. . Dentate gyrus abnormalities in sudden unexplained death in infants: morphological marker of underlying brain vulnerability . Acta Neuropathol 2015. ; 129 : 65 – 80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kinney HC, Armstrong DL, Chadwick AE , et al. . Sudden death in young children associated with hippocampal abnormalities in the hippocampus: Five case studies . Pediatr Dev Pathol 2007. ; 10 : 208 – 23 [DOI] [PubMed] [Google Scholar]
- 8. Kinney HC, Chadwick AM, Crandall LA , et al. . Sudden death, febrile seizures, and hippocampal maldevelopment in young children: A new entity . Pediatr Dev Pathol 2009. ; 12 : 455 – 63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hefti MM, Kinney HC, Cryan JB , et al. . Sudden unexpected death in early childhood: general observations in a series of 151 cases: Part 1 of the investigations of the San Diego SUDC Research Project . Forensic Sci Med Pathol 2016. ; 12 : 4 – 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hefti MM, Cryan JB, Haas EA , et al. . Hippocampal malformation associated with sudden death in early childhood: a neuropathologic study: Part 2 of the investigations of the San Diego SUDC Research Project . Forensic Sci Med Pathol 2016. ; 12 : 14 – 25 [DOI] [PubMed] [Google Scholar]
- 11. Armstrong DD. Epilepsy-induced microarchitectural changes in the brain . Pediatr Dev Pathol 2005. ; 8 : 607 – 14 [DOI] [PubMed] [Google Scholar]
- 12. Armstrong DD. The neuropathology of temporal lobe epilepsy . J Neuropathol Exp Neurol 1993. ; 52 : 433 – 43 [DOI] [PubMed] [Google Scholar]
- 13. Houser CR. Granule cell dispersion in the dentate gyrus of humans with temporal lobe epilepsy . Brain Res 1990. ; 535 : 195 – 204 [DOI] [PubMed] [Google Scholar]
- 14. Blümcke I, Kistner I, Clusmann H , et al. . Towards a clinico-pathological classification of granule cell dispersion in human mesial temporal lobe epilepsies . Acta Neuropathol 2009. ; 117 : 535 – 44 [DOI] [PubMed] [Google Scholar]
- 15. Richerson GB, Buchanan GF. The serotonin-axis: shared mechanisms in seizures, depression, and SUDEP . Epilepsia 2011. ; 52 : 28 – 38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Moseley B, Bateman L, Millichap JJ , et al. . Autonomic epileptic seizures, autonomic effects of seizures, and SUDEP . Epilepsy Behav 2013. ; 26 : 375 – 85 [DOI] [PubMed] [Google Scholar]
- 17. Bermeo-Ovalle AC, Kennedy JD, Schuele SU. Cardiac and autonomic mechanisms contributing to SUDEP . J Clin Neurophysiol 2015. ; 32 : 21 – 9 [DOI] [PubMed] [Google Scholar]
- 18. Kennedy JD, Seyal M. Respiratory pathophysiology with seizures and implications for sudden unexpected death in epilepsy . J Clin Neurophysiol 2015. ; 32 : 10 – 3 [DOI] [PubMed] [Google Scholar]
- 19. Morse AM, Kothare SV. Pediatric sudden unexpected death in epilepsy . Pediatr Neurol 2016. ; 57 : 7 – 16 . [DOI] [PubMed] [Google Scholar]
- 20. Surges R, Thijs RD, Tan HL , et al. . Sudden unexpected death in epilepsy: risk factors and potential pathomechanisms . Nat Rev Neurol 2009. ; 5 : 492 – 504 [DOI] [PubMed] [Google Scholar]
- 21. Duvernoy H, Cattin F, Risold P-Y. The Human Hippocampus: Functional Anatomy, Vascularization and Serial Sections with MRI . 4th ed. , Heidleberg: : Springer; 2013. : 1 – 237 [Google Scholar]
- 22. Goldstein RD, Kinney HC, Willinger M. Sudden unexpected death in fetal life through early childhood . Pediatrics 2016. ; 137 : pii: e20154661. doi: 10.1542/peds.2015-4661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Insausti R, Cebada-Sanchez S, Marcos P. Postnatal Development of the Human Hippocampal Formation . Berlin: , Springer; , 2010. , p. 1 – 892 [PubMed] [Google Scholar]
- 24. Kennedy JD, Hardin KA, Parikh P , et al. . Pulmonary edema following generalized tonic clonic seizures is directly associated with seizure duration . Seizure 2015. ; 27 : 19 – 24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Blinkow SM, Glezer I. (eds). The Human Brain in Figures and Tables: A Quantitative Handbook . New York: , Plenum Press; , 1968. , p. 335 [Google Scholar]
- 26. Babb TL, Brown WJ. Pathologic findings in epilepsy . In: Engel J, Jr. , ed. Surgical Treatment of the Epilepsies . New York: : Raven Press; 1987. : 511 – 40 [Google Scholar]
- 27. Kasper BS, Stefan H, Buchfelder M , et al. . Temporal lobe microgenesis in epilepsy versus control brains . J Neuropathol Exp Neurol 1999. ; 58 : 22 – 8 [DOI] [PubMed] [Google Scholar]
- 28. Harper RM. State-related physiological changes and risk for the sudden infant death syndrome . Aust Pediatr J 1986. ; 22 : 55 – 8 [PubMed] [Google Scholar]
- 29. Liu M, Pleasure SJ, Collins AE , et al. . Loss of BET2/NeuroD leads to malformation of the dentate gyrus and epilepsy . PNAS 2000. ; 97 : 865 – 70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stafstrom CE. The role of the subiculum in epilepsy and epileptogenesis . Epilepsy Curr 2005. ; 5 : 121 – 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Parent JM, Fron MM. Neurogenesis and Epilepsy . In: Noebels JL, Avoli M, Rogawski MA , et al. ., eds. Jasper’s Basic Mechanisms of the Epilepsies (Internet) , 4th edition.Bethesda (MD) : National Center for Biotechnology Information (US)2012 [PubMed] [Google Scholar]
- 32. Jessberger S, Parent JM. Epilepsy and adult neurogenesis . Cold Spring Harb Perspect Biol 2015. ; 7 : pii: a020677. doi: 10.1101/cshperspect.a020677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pun RY, Rolle IJ, Lasarge CL , et al. . Excessive activation of mTOR in postnatally generated granule cells is sufficient to cause epilepsy . Neuron 2012. ; 75 : 1022 – 34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Edlow BL, McNab JA, Witzel T , et al. . The structural connectome of the human central homeostatic network . Brain Connect 2016. ; 6 : 187 – 200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hajek MA, Buchanan GF. Influence of vigilance state on physiologic consequences of seizures and seizure-induced death in mice . J Neurophysiol 2016. ; 115 : 2286 – 93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Noebels J. , Commentary Hippocampal abnormalities and sudden childhood death . Forensic Sci Med Pathol 2016. ; 12 : 198 – 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Benbadis SR, Wolgamuth BR, Goren H , et al. . Value of tongue biting in the diagnosis of seizures . Arch Intern Med 1995. ; 155 : 2346 – 9 [PubMed] [Google Scholar]
- 38. Panigrahy A, Filiano JJ, Sleeper LA , et al. . Decreased serotonergic receptor binding in rhombic lip-derived regions of the medulla oblongata in the sudden infant death syndrome . J Neuropathol Exp Neurol 2000. ; 59 : 377 – 84 [DOI] [PubMed] [Google Scholar]
- 39. Duncan JR, Paterson DS, Hoffman JM , et al. . Brainstem serotonergic deficiency in the sudden infant death syndrome . jama 2010. ; 303 : 430 – 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kinney HC, Randall LL, Sleeper LA , et al. . Serotonergic brainstem abnormalities in Northern Plains Indians with sudden infant death syndrome . J Neuropathol Exp Neurol 2003. ; 62 : 1178 – 91 [DOI] [PubMed] [Google Scholar]
- 41. Paterson DS, Thompson EG, Belliveau RA , et al. . Multiple abnormalities in the brainstem serotonergic system in sudden infant death syndrome . JAMA 2006a. ; 296 : 2124 – 32 [DOI] [PubMed] [Google Scholar]
- 42. Azmitia EC, Gannon PJ. The primate serotonergic system: a review of human and animal studies and a report on Macaca fascicularis. [Review] . Adv Neurol 1986. ; 43 : 407 – 68 [PubMed] [Google Scholar]
- 43. Zhan Q, Buchanan GF, Motelow JE , et al. . Impaired serotonergic brainstem function during and after seizures . J Neurosci 2016. ; 36 : 2711 – 22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fisher RS, Acevedo C, Arzimanoglou A , et al. . ILAE official report: a practical clinical definition of epilepsy . Epilepsia 2014. ; 55 : 475 – 82 [DOI] [PubMed] [Google Scholar]
- 45. Hauser WA, Beghi E. First seizure definitions and worldwide incidence and mortality . Epilepsia 2008. ; 49 : 8 – 12 [DOI] [PubMed] [Google Scholar]
- 46. Nelson KB, Ellenberg JH. Predictors of epilepsy in children who have experienced febrile seizures . N Engl J Med 1976. ; 295 : 1029 – 33 [DOI] [PubMed] [Google Scholar]
- 47. Lhatoo SD, Nei M, Raghavan M , et al. . Nonseizure SUDEP: Sudden unexpected death in epilepsy without preceding epileptic seizures . Epilepsia 2016. ; 57 : 1161 – 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Filiano JJ, Kinney HC. Arcuate nucleus hypoplasia in the sudden infant death syndrome . J. Neuropathol Exp Neurol 1992. ; 51 : 394 – 403 [DOI] [PubMed] [Google Scholar]
- 49. Matturri L, Biondo B, Mercurio P , et al. . Severe hypoplasia of medullary arcuate nucleus: quantitative analysis in sudden infant death syndrome . Acta Neuropathol 2000. ; 99 : 371 – 5 [DOI] [PubMed] [Google Scholar]
- 50. Filiano JJ, Choi JC, Kinney HC. Candidate cell populations for respiratory chemosensitive fields in the human infant medulla . J Comp Neurol 1990. ; 293 : 448 – 65 [DOI] [PubMed] [Google Scholar]
- 51. Paterson DS, Thompson EG, Kinney HC. Serotonergic and glutamatergic neurons at the ventral medullary surface of the human infant: Observations relevant to central chemosensitivity in early human life . Auto Neurosci 2006b. ; 124 : 112 – 24 [DOI] [PubMed] [Google Scholar]
- 52. Kinney HC, Armstrong DD. Perinatal neuropathology . In: Graham DI, Lantos PL , eds. Greenfield’s Neuropathology . London: : Arnold; 2002. : 519 – 606 . [Google Scholar]
- 53. Benes FM, Turtle M, Khan Y , et al. . Myelination of a key relay zone in the hippocampal formation occurs in the human brain during childhood, adolescence, and adulthood . Arch Gen Psychiatry 1994. ; 51 : 477 – 84 [DOI] [PubMed] [Google Scholar]
- 54. Milesi G, Garbelli R, Zucca I , et al. . Assessment of human hippocampal developmental neuroanatomy by means of ex-vivo 7 T magnetic resonance imaging . Int J Dev Neurosci 2014. ; 34 : 33 – 41 [DOI] [PubMed] [Google Scholar]
- 55. Grant SGN, O’Dell TJ, Karl KA , et al. . Impaired long-term potentiation, spatial learning, and hippocampal development in fyn mutant mice . Science 1992. ; 258 : 1903 – 10 [DOI] [PubMed] [Google Scholar]
- 56. Kojima N, Ishibashi H, Obata K , et al. . Higher seizure susceptibility and enhanced tyrosine phosphorylation of N-Methyl-D-Aspartate receptor subunit 2B in fyn transgenic mice . Learning Mem 1998. ; 5 : 429 – 45 [PMC free article] [PubMed] [Google Scholar]
- 57. Hoppenbrouwers T. Sudden infant death syndrome, sleep, and seizures . J Child Neurol 2015. ; 30 : 904 – 11 [DOI] [PubMed] [Google Scholar]
- 58. Mangale VS, Hirokawa KE, Satyaki PR , et al. . Lhx2 selector activity specifies cortical identity and suppresses hippocampal organizer fate . Science 2008. ; 319 : 304 – 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Abellán A, Desfilis E, Medina L. Combinatorial expression of Lef1, Lhx2, Lhx5, Lhx9, Lmo3, Lmo4, and Prox1 helps to identify comparable subdivisions in the developing hippocampal formation of mouse and chicken . Front Neuroanat 2014. ; 8 : 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Encinas JM, Vaahtokari A, Enikolopov G. Fluoxetine targets early progenitor cells in the adult brain . Proc Natl Acad Sci USA 2006. ; 103 : 8233 – 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Knoth R, Singec I, Ditter M , et al. . Murine features of neurogenesis in the human hippocampus across the lifespan from 0 to 100 years . PLoS One 2010. ; 5 : e8809 doi:10.1371/journal.pone.0008809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Frotscher M, Haas CA, Förster E. Reelin controls granule cell migration in the dentate gyrus by acting on the radial glial scaffold . Cereb Cortex 2003. ; 13 : 634 – 40 [DOI] [PubMed] [Google Scholar]
- 63. Gong C, Wang TW, Huang HS , et al. . Reelin regulates neuronal progenitor migration in intact and epileptic hippocampus . J Neurosci 2007. ; 27 : 1803 – 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Müller MC, Osswald M, Tinnes S , et al. . Exogenous reelin prevents granule cell dispersion in experimental epilepsy . Exp Neurol 2009. ; 216 : 390 – 7 [DOI] [PubMed] [Google Scholar]
- 65. Heinrich C, Nitta N, Flubacher A , et al. . Reelin deficiency and displacement of mature neurons, but not neurogenesis, underlie the formation of granule cell dispersion in the epileptic hippocampus . J Neurosci 2006. ; 26 : 4701 – 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Diaz SL, Narboux-Nême N, Trowbridge S , et al. . Paradoxical increase in survival of newborn neurons in the dentate gyrus of mice with constitutive depletion of serotonin . Eur J Neurosci 2013. ; 38 : 2650 – 8 [DOI] [PubMed] [Google Scholar]
- 67. Iyasu S, Randall LL, Welty TK , et al. . Risk factors for sudden infant death syndrome among Northern Plains Indians . JAMA 2002. ; 288 : 2717 – 23 [DOI] [PubMed] [Google Scholar]
- 68. Mitchell EA, Milerad J. Smoking and the sudden infant death syndrome . Rev Environ Health 2006. ; 21 : 81 – 103 [DOI] [PubMed] [Google Scholar]
- 69. Fatermi SH. (ed). Reelin Glycoprotein: Structure, Biology and Roles in Health and Disease . New York: : Springer; 2008 [DOI] [PubMed] [Google Scholar]
- 70. Dam AM. Epilepsy and neuronal loss in the hippocampus . Epilepsia 1980. ; 21 : 617 – 29 [DOI] [PubMed] [Google Scholar]
- 71. Mathern GW, Babb TL, Leite JP , et al. . The pathogenic and progressive features of chronic human hippocampal epilepsy . Epilepsy Res 1996. ; 26 : 151 – 61 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.