

Abstract
The transmembrane receptor, Notch1 plays an important role during the differentiation of CD4 T cells into T helper (Th) subsets in the presence of appropriate cytokines, including differentiation into Th1 cells. MicroRNAs have also been shown to be important regulators of immune responses, including negatively regulating cytokine production by Th1 cells. The miR-29 family of microRNAs can act to inhibit tbx21 and ifng transcription, two important pro-inflammatory genes that are abundantly expressed in Th1 cells. Here we show that Notch1 may prime CD4 T cells to be responsive to Th1-polarizing cues through its early repressive effects on the miR-29 family of microRNAs. Using a combination of cell lines and primary cells, we demonstrate that Notch1 can repress miR-29a, miR-29b, and miR-29c transcription through a mechanism that is independent of NF-κB. We further show that this repression is mediated by canonical Notch signaling and requires active Mastermind like (MAML) 1, but this process is superseded by positive regulation of miR-29 in response to IFNγ at later stages of CD4 T cell activation and differentiation. Collectively, our data suggest an additional mechanism by which Notch1 signaling may fine-tune Th1 cell differentiation.
Keywords: Notch1, canonical Notch signaling, gamma-secretase inhibitor, GSI, MicroRNA-29, miR-29, NF-κB, IFNγ, CSL, DN-MAML
Graphical abstract
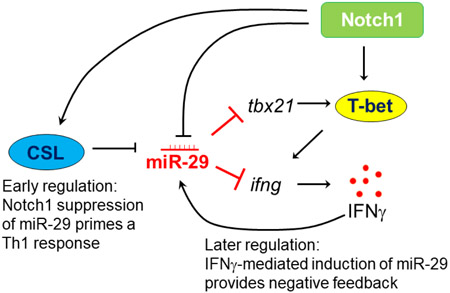
1. Introduction
Naïve CD4 T cells are activated in response to encountering antigens. Upon activation, these cells will proliferate and differentiate into T helper (Th) cells, facilitated by the cytokines present at the time of stimulation. Based on the expression of unique transcription factors and characteristic cytokines, Th cells can be broadly categorized into defined subsets, with Th1, Th2, and Th17 being the three most-studied to date1. For Th1 cells, the transcription factor, T-bet, which is encoded by the gene, tbx21, is a critical regulator of cell fate, and its expression ultimately leads to production of interferon gamma (IFNγ), a signature Th1 pro-inflammatory cytokine2,3. T-bet is a direct transcriptional target of the transmembrane receptor, Notch1, and is controlled by non-canonical Notch1 signaling during Th1 differentiation4,5.
The Notch family of transmembrane receptors, can be activated by its ligands, Delta-like-(DLL)-1,3,4 and Jagged (Jag)-1,26. Notch signaling plays an important role in cell fate determination. There are four Notch receptors (1-4) expressed in mammalian cells; however, only the expression of Notch1-3 has thus far been described in T cells. Following interaction with cognate ligands, Notch receptors undergo a series of proteolytic cleavage events. The final, S3 cleavage is mediated by a multi-protein γ-secretase complex, and generates the transcriptionally active, intracellular domain (NICD)7. NICD can translocate to the nucleus to interact with its DNA-binding partner, CBF1-Suppressor of Hairless-Lag1 (CSL)8. NICD binding to CSL results in further recruitment of transcriptional co-activators such as mastermind like-1 (MAML-1) and p3009,10. CSL-dependent Notch signaling is referred to as canonical Notch signaling. There are also reports indicating NICD can interact with proteins other than CSL, both in the nucleus and cytosol, to signal through non-canonical mechanisms11,12.
Studies have identified a crucial role for Notch signaling in Th cell differentiation, both through canonical and non-canonical signaling12-15. One of the first studies implicating Notch in Th1 differentiation, demonstrated that blocking the actions of γ-secretase, prevented Notch cleavage and attenuated the expression of T-bet and IFNγ in developing Th1 cells4. Expression of Th cell master transcription factors and proinflammatory cytokines must be tightly regulated. In the case of Th1 cells, dysregulated IFNγ expression can lead to chronic autoimmune disorders through its actions on other immune cells, resulting in a hyper-immune response16.
There are various cell-intrinsic mechanisms that have evolved to fine-tune cytokine expression, including those mediated by the actions of microRNAs (miRs). MiRs are short, single stranded non-coding RNAs that act as post-transcriptional inhibitors of gene expression. They silence their target mRNAs through complementary base pairing and by forming an RNA- induced silencing complex (RISC). Based on their complementarity, the target mRNA is either degraded or its translation into protein is greatly reduced. The miR-29 family of miRNAs consist of miR-29a, -b and -c, and directly targets the mRNA of Th1 signature molecules, tbx21 and ifng17-19. Interestingly, in response to IFNγ, miR-29 expression increases, thereby forming a negative feedback loop19. Furthermore, NF-κB, a non-canonical binding partner of Notch, can inhibit the expression of miR-2918,20,21, indicating miR-29 expression can be regulated through multiple mechanisms.
We previously showed Notch1 signaling contributes to Th1 differentiation, including regulation of T-bet and IFNγ production, as well as to the induction of NF-κB in activated T cells4,22. Here, we investigated whether Notch1 may also prime T cells for Th1 differentiation by affecting the expression of miR-29, and whether this proceeds through an NF-κB-dependent mechanism. In this study, we demonstrate that Notch1 can directly inhibit the expression of miR- 29 in Th1 cells, during the early stages of Th1 differentiation. Furthermore, we show that Notch1 represses miR-29 through an NF-κB-independent, CSL (CBF1-Suppressor of Hairless- Lag1)-mediated canonical signaling pathway. However, we also found that the early repressive effects of Notch1 on miR-29 are masked by IFNγ–induced expression of miR-29, during late stages of Th1 differentiation. Thus, we suggest a mechanism by which Notch1 and IFNγ may act in opposition to maintain a balance of miR-29 expression and fine-tune Th1 cell fate.
2. Materials & Methods
2.1. Mice
C57BL/6J mice were purchased from Jackson Laboratories (Bar Harbor, ME) and were bred, housed, and utilized in accordance with guidelines set forth by the Institutional Animal Care and Use Committee of the University of Massachusetts Amherst. Mice aged 8-12 weeks were used.
2.2. Plasmids and Cell lines
The pGL3-miR-29b1a promoter vectors were a kind gift from J. Mott20. The N1ICD parental pcDNA3 expression plasmids were a kind gift from A. Capobianco23 and subsequently cloned into pEGFP vector as described22. MAML-1 and DNMAML-1 vectors were provided in collaboration with L. Miele and generated as described24. Generation of DO11.10 T cell hybridoma cell lines with empty vector or N1ICD were described previously22.
2.3. T cell isolation, activation, and in vitro polarization
CD4 T cells were isolated from bulk splenocytes by magnetic bead separation using anti-CD4 magnetics particles (BD Biosciences, San Jose, CA) according to the manufacturer’s instruction. Cells were activated in vitro by plating 1 × 106 cells/ml/well of a 12-well plate pre-coated with anti-CD3ε and anti-CD28 (Biolegend, San Diego, CA) and crosslinked with anti-hamster IgG (Sigma-Aldrich Corp., St. Louis, MO). Cells were cultured in a 1:1 mixture of RPMI 1640:DMEM supplemented with 10% fetal bovine serum, L-Glutamine, Sodium Pyruvate, Penicillin/Streptomycin and β-mercaptoethanol. The following conditions were used for polarization. Th1: 10μg/ml of anti-IL-4 and 1ng/ml of recombinant mouse IL-12. Th2: 10μg/ml anti-IFNγ and 1ng/ml of recombinant mouse IL-4. For inhibitor assays, the CD4 T cells were pretreated with 25uM of GSI PF-03084014 (Medchem Express, Monmouth Junction, NJ) for 30 minutes. For NF-κB inhibition studies, the DO11.10 T cell hybridoma cell line was treated with 5μM of Bay11-7085 (Sigma-Aldrich Corp., St. Louis County, MO).
2.4. Protein detection by FACS analysis and immunoblotting
Activated CD4 T cells were harvested at indicated time points and restimulated with plate-bound anti-CD3ε for 5 hours with Golgi Stop (BD Biosciences). Intracellular staining was performed to detect Notch1, T-bet, and IFNγ using the FoxP3 staining buffer set, following the manufacturer’s instructions (eBioscience, Santa Clara, CA). Data were acquired on an LSR Fortessa (BD Biosciences) and analyzed using FlowJo Software (FlowJo LLC, Ashland, OR). For immunoblotting, cells were lysed in RIPA buffer (150mM NaCl, 1% IGEPAL-CA630, 0.1% SDS, 0.5% Sodium deoxycholate, 50mM Tris pH 8.0) with protease and phosphatase inhibitors (Bimake, Houston, TX). Lysates were resolved using SDS-PAGE and proteins were transferred to nitrocellulose membrane (GE Amersham, Pittsburgh, PA). Cleaved, intracellular domain of Notch1 was visualized with anti-cleaved Notch1 antibody (Val1744) (D3B8, Cell Signaling Technology, Danvers, MA) and anti-rabbit secondary antibody conjugated with horse radish peroxidase (GE Amersham)
2.5. Quantitative Real Time PCR
Stimulated CD4 T cell were harvested at indicated time points and total RNA was extracted using Quick-RNA Mini Prep kit (Zymo Research, Irvine, CA) following the manufacturer’s instruction. Total RNA was reverse-transcribed with oligo-dT or stem loop primers using MuLV Reverse Transcriptase. Stem loop primers for miRNA quantification were designed as described. Quantitative Real Time PCR was performed using 2x SYBR Green master mix (Bimake, Inc., Houston, TX). Transcripts were quantified using the 2−ΔΔCT method.
2.6. Luciferase Assay
NIH3T3 cells were co-transfected with pGL3-29b1a, N1ICD, MAML-1 or DNMAML-1 and control vectors using Xtremegene transfection reagent (Sigma-Aldrich Corp.). Luciferase assay was performed after 48 hours of transfection according to the manufacturer’s instruction. Firefly luciferase activity was measured using Luminometer 20/20n (Turner Biosystems-Promega, Madison, WI) and was normalized to Renilla luciferase activity to control for variances in transfection efficiency.
2.7. Chromatin Immunoprecipitation
Cells were treated with 1% formaldehyde to crosslink proteins to DNA, and treated with 125mM glycine to quench unreacted formaldehyde and lysed in SDS lysis buffer (1% SDS, 10mM EDTA and 50mM Tris pH8.1). Cells were sonicated for 90 seconds (5s ON/15s OFF) to shear the DNA, using Sonicator – XL2020 (Misonix Inc., Farmingdale, NY). Sonicated lysates were diluted 10-fold with ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2mM EDTA, 16.7mM Tris pH 8.1, 167mM NaCl) and precleared with Protein A/G PLUS agarose beads (Santa Cruz Biotechnology, Dallas, TX). Lysates were immunoprecipitated with 4ug anti-CSL (Santa Cruz Biotechnology), 1:100 anti-Notch1 (Cell Signaling Technology) or normal rabbit IgG control (Santa Cruz Biotechnology). Protein-DNA complex were recovered by adding agarose beads and washed as follows; low-salt buffer (0.1% SDS, 1% Triton X-100, 2mM EDTA, 20mM Tris pH8.1 and 150mM NaCl), high-salt buffer (0.1% SDS, 1% Triton X-100, 2mM EDTA, 20mM Tris pH 8.1), LiCl wash buffer (0.25M LiCl, 1% IGEPAL-CA630, 1% Sodium deoxycholate, 1mM EDTA and 10mM Tris pH8.1) and TE Buffer (1mM EDTA and 10mM Tris, pH 8.1). After eluting the DNA-protein complex, cross-linking was reversed overnight at 65 C. Following Proteinase K digestion, DNA was extracted using DNA extraction kit (Bio Basic, Inc., Markham, Ontario, Canada). The CSL/Notch1 binding region within the miR-29 promoter was amplified using specific primers as described18.
2.8. Statistical analysis
Data are the mean ± SD; all experiments were repeated at least three times. Unpaired, two-tailed Student’s t test and two-way ANOVA with Tukey’s multiple comparisons test were applied for statistical comparison using GraphPad Prism 5 software (GraphPad Software, Inc., San Diego, CA). P values of ≤ 0.05 were considered significant.
3. Results
3.1. γ-Secretase-inhibitor-mediated inhibition of T-bet is post-transcriptional at early timepoints
Signaling through the Notch1 intracellular domain (N1ICD) contributes to the differentiation of multiple T helper cell subsets13,25. We previously demonstrated a cell-intrinsic role for Notch1 in Th1 differentiation, and that both tbx21 and ifng are transcriptional targets of Notch14,22,26. We have also shown that T-bet expression in Th1 cells can occur in the absence of the canonical Notch1 binding partner, CSL27. In contrast, inhibitory signals mediated by the miR-29 family of microRNAs have been shown to negatively regulate tbx21 and ifng, both17. To further investigate the detailed mechanism behind Notch1 regulation of Th1 differentiation, we explored the possibility of cross-talk between the Notch1 and miR-29 signaling pathways.
We began by using a novel gamma secretase inhibitor (GSI), PF-03084014, currently in Phase 1 clinical trials to treat patients with T-cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma28. This GSI prevents the intramembranous cleavage of Notch at the S3 site and the release of active NICD. We isolated CD4 T cells from bulk splenocytes, and cultured them under Th1 polarizing conditions for 4 days. We used an optimal concentration of GSI (25 μM) to inhibit the cleavage of Notch1, which is maximally expressed 48 hours after stimulation (Fig 1A). Inhibiting γ-secretase reduced expression of T-bet protein, as we have shown before4 (Fig 1B) but, surprisingly, we found the transcript levels of tbx21 were higher than vehicle-treated controls. Specifically, by 24 hours after activation, the GSI-treated samples showed an almost 2-fold increase in tbx21 transcripts compared to controls (Fig 1C) while protein expression remained lower than DMSO controls. These data suggested to us that Notch signaling may regulate T-bet within the first 24 hours of T cell stimulation by post-transcriptional mechanisms, without regulating T-bet protein levels to the same extent.
Figure 1. γ-Secretase-inhibitor-mediated inhibition of T-bet is post-transcriptional at early timepoints.
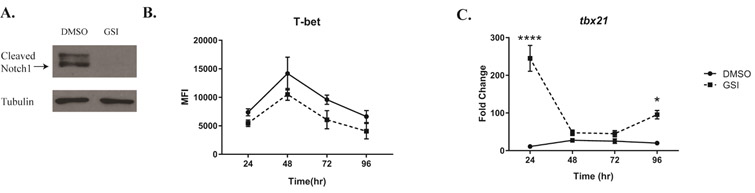
Splenic CD4 T cells isolated from C57BL/6 mice were pretreated with DMSO or GSI for 30 minutes and stimulated with plate bound anti-CD3ε and anti-CD28 under Th1 polarizing conditions for (A) 48 hours or (B-C) for indicated time. (A) Expression of cleaved Notch1 (Val1744) in DMSO- and GSI-treated samples were determined by immunoblotting.
Tubulin expression was used as a loading control. (B) T-bet protein, expressed at various time points after stimulation with anti-CD3ε and CD28 in DMSO- and GSI-treated CD4 T cells, was determined by using flow cytometry and quantifying Median Fluorescence Intensity (MFI). (C) Total RNA was extracted from CD4 T cells at the timepoints indicated, and relative expression of tbx21 level was determined in DMSO- and GSI-treated cells, and normalized to expression of β-actin. Fold change was determined relative to the unstimulated CD4 T cells. Results are representative of three independent experiments. Statistical analyses were performed using two-way ANOVA, with Tukey’s multiple comparisons test applied. *P<0.05; ****P<0.0001.
3.2. γ-Secretase-inhibitor-mediated inhibition of Notch leads to increased expression of miR-29 at earlier, but not at later, time intervals in Th1 cells.
The miR-29 family of miRNAs, miR-29a, -b and -c, can directly target the mRNA of Th1 signature molecules, tbx21 and ifng17-19. Based on the disparate levels we observed between T-bet protein and mRNA transcripts of the T-bet-encoding gene, tbx21, following GSI treatment, we asked whether Notch signaling might be negatively regulating miR-29. To determine whether Notch might inhibit miR-29 expression in murine Th1 cells we incubated CD4 T cells with DMSO, as a vehicle control, or with GSI for 30 minutes, then further cultured the cells under Th1 polarizing condition for 4 days. We used real time PCR to measure the transcript levels of individual miR-29 family members 24 hours into the Th1 differentiation process. We observed that, in cells treated with GSI, miR-29b expression was significantly higher than in cells treated with DMSO (Fig. 2A), while mature transcripts of miR-29a and miR-29c were less affected.
Figure 2. γ-Secretase-inhibitor-mediated inhibition of Notch leads to increased expression of miR-29 at earlier, but not at later, time intervals in Th1 cells.
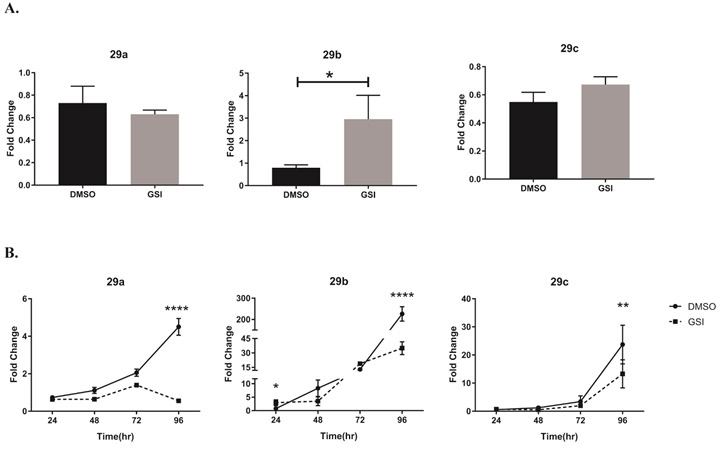
(A) Splenic CD4 T cells isolated from C57B/L6 mice were pretreated with DMSO or GSI for 30 minutes and stimulated with plate bound anti-CD3ε and anti-CD28 under Th1 polarizing condition. Total RNA was extracted at 24 hours, and relative expression of miR-29a, miR-29b, and miR-29c were determined using stem loop primers and real-time quantitative PCR, normalized to sno202. Fold changes were determined relative to unstimulated CD4 T cells. Results are the mean of triplicate wells + S.D. and are representative of three independent of experiments. Statistical analyses were performed using an unpaired Student t test, with statistical significance set at P<0.05. (B) Total RNA was extracted at indicated time intervals and miR-29a, miR-29b, and miR-29c was amplified as in (A). The relative expression of miR-29a, miR-29b, and miR-29c was normalized to expression of sno202. Fold changes were determined relative to unstimulated CD4 T cells. Results are the mean of triplicate wells + S.D. and are representative of three independent of experiments. Statistical analyses were performed using Student’s t test. *P<0.05; **P<0.01, ****P<0.0001.
Interestingly, after a full 96 hours of polarization, DMSO-treated Th1 cells expressed high levels of miR-29a, -b, and -c, while at this same timepoint, GSI-treated cells showed much lower miR- 29 expression (Fig. 2B). Primary CD4 T cells cultured under Th2-polarizing conditions did not display similar miR29 expression kinetics in DMSO- or in GSI-treated cells (Fig. S1), suggesting that Notch regulation of miR29 is likely to be highly cell-context dependent. Altogether, these observations suggested to us that miR-29 levels may be regulated differently during early and later stages of Th1 differentiation, and that Notch signaling may be involved in its regulation.
3.3. Notch1 represses miR-29 expression
Our previous studies have indicated that Notch1 can regulate IFNγ expression directly, as well as indirectly by modulating the expression of T-bet. Building on our initial, unexpected observations that after 24 hours of Th1 polarization, GSI treated cells showed low T-bet protein expression but high tbx21 transcripts (Fig. 1), coupled with the increased expression of miR-29b in GSI-treated cells at 24 hours (Fig. 2), we asked whether Notch1 could alter the expression of tbx21 by inhibiting miR-29 expression. The γ-secretase complex targets and cleaves numerous proteins, and it was not clear from our initial experiments whether Notch1, or another γ-secretase substrate, was responsible for the effects we observed. Therefore, to further refine our experimental question, we used the DO11.10 hybridoma T cell line, and retrovirally transduced it either with an empty vector or with a construct encoding the Notch1 intracellular domain (N1ICD; 1759-2555) to test our hypothesis that it is Notch1, and not another substrate of γ- secretase, that is repressing miR-29. As shown in Figure 3A, expressing N1ICD significantly reduced the mature transcripts of all miR-29 family members. Since we had observed miR-29b repression both in primary Th1 cells and in DO.11.10 cells expressing N1ICD, we sought to further confirm the repressive effects of N1ICD on miR-29b expression. We utilized a Firefly luciferase construct with a validated miR-29ab1 promoter sequence20 to address whether Notch1 may be regulating miR-29b expression by reducing its promoter activity. We co-transfected a Renilla luciferase construct to control for any differences in transfection efficiency between conditions. When we expressed the miR-29ab1 Firefly luciferase construct in 3T3 cells, together with N1ICD, we noted robustly decreased luciferase expression, suggesting that N1ICD can repress the promoter activity of miR-29 (Fig 3B, Fig. S2). Collectively, these data support the notion that Notch1 can also regulate tbx21, indirectly, by inhibiting miR-29 expression, especially within the first 24 hours of Th1 cell differentiation.
Figure 3. Notch1 represses miR-29 expression.
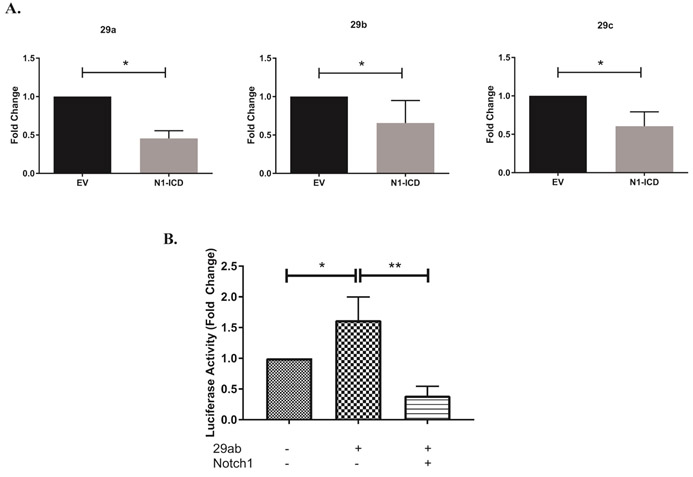
(A) DO11.10 hybridoma cells were stably transduced with empty vector (DO11.10-EV) or with Notch1-intracellular domain (DO11.10-N1-ICD). Total RNA was isolated and microRNAs were reverse transcribed using respective stem loop primers. The relative expression of miR-29 transcripts was normalized to expression of sno202. Fold changes in miR-29a, miR-29b and miR-29c are expressed relative to respective miR-29 levels in DO11.10-EV cells. (B) NIH3T3 cells were transfected with a construct containing the miR-29ab1 promoter, together with pRL-CMV, with or without N1-ICD. Relative firefly luciferase activity was measured (Firefly/Renilla) and normalized to the empty vector control. Results are the mean + S.D. of three independent experiments. Statistical analyses were performed using an unpaired Student’s t test. *P<0.05; **P<0.01.
3.4. IFNγ masks the effects of Notch1 on miR-29
It has been reported that pro-inflammatory, IFNγ signaling can induce miR-29 expression forming a negative feedback loop that act to temper IFNγ expression. However, in certain autoimmune diseases, such as multiple sclerosis, Th cell activity is dysregulated. Circulating proinflammatory cytokines are often high, suggesting disruption in pathways that would otherwise negatively regulate their expression. In support of this, memory T cells isolated from multiple sclerosis patients display an active Th1 response, and a greater than two-fold increase in miR-29b, even in the presence of high IFNγ levels19. We previously showed that Notch1 induces ifng expression in Th1 cells, producing high IFNγ levels by 96 hours in culture. Furthermore, treating Th1 cells with GSI during the polarizing process limits IFNγ production4. Data from the present study showed elevated miR-29b levels in GSI-treated Th1 cells at the 24 hours’ timepoint, but reduced miR-29b levels in GSI-treated Th1 cells at the 96 hours’ timepoint (Fig. 2). Viewing these data collectively, we hypothesized that IFNγ signaling might be “out-competing” any Notch1-mediated repressive effects, to upregulate miR-29 expression, especially at later timepoints when Notch1 levels begin to decrease. To address this potential for hierarchical regulation of miR-29 by IFNγ over Notch1, we asked what happens to miR-29 levels if we blocked the ability of IFNγ to act on the cells and, further, if we also inhibited Notch1 signaling in these cells. To do this, we cultured CD4 T cells in with DMSO or GSI for 4 days under neutral, non-polarizing conditions. We added an IFNγ-neutralizing antibody to some cells during the first 48 hours of culture. We observed that blocking IFNγ signaling reduced miR-29 expression (Fig 4A), as has been previously reported19, but GSI-treatment did not significantly alter miR-29 expression in these -non-polarized cells. We next asked whether an IFNγ-Notch regulatory axis of miR-29 might be at work in Th1 cells, in the form of hierarchical signaling between IFNγ and Notch1 during the later stages of Th1 differentiation, a time at which IFNγ levels are high and Notch1 levels wane. We predicted that, if this were the case, removing IFNγ during the later stages of Th1 cell differentiation would abrogate miR-29 expression in these, while inhibiting Notch1 activity in the absence of IFNγ signaling, would de-repress miR-29 levels. To test this hypothesis, we cultured CD4 T cells under Th1-polarizing conditions for 48 hours. After this initial culture period, we lifted the cells, replated them in fresh media to remove any secreted IFNγ, and cultured them for an additional 48 hours in the presence of DMSO or GSI. Under these conditions, in which Th1 cells are rapidly deprived of exogenous IFNγ, we observed that there was little to no expression of miR-29 in our DMSO-treated samples (Fig 4B). In contrast, we noted consistently higher levels of miR-29 in Th1 cells treated with GSI, lending further support to the notion that Notch signaling represses miR-29 expression in the absence of IFNγ. Collectively, these data provide evidence that miR-29 expression may be tightly regulated by Notch1-mediated repression and IFNγ-mediated induction, to fine-tune Th1 polarization, and that a hierarchical regulatory axis between Notch and IFNγ may exist.
Figure 4. IFNγ masks the effects of Notch1 on miR-29.
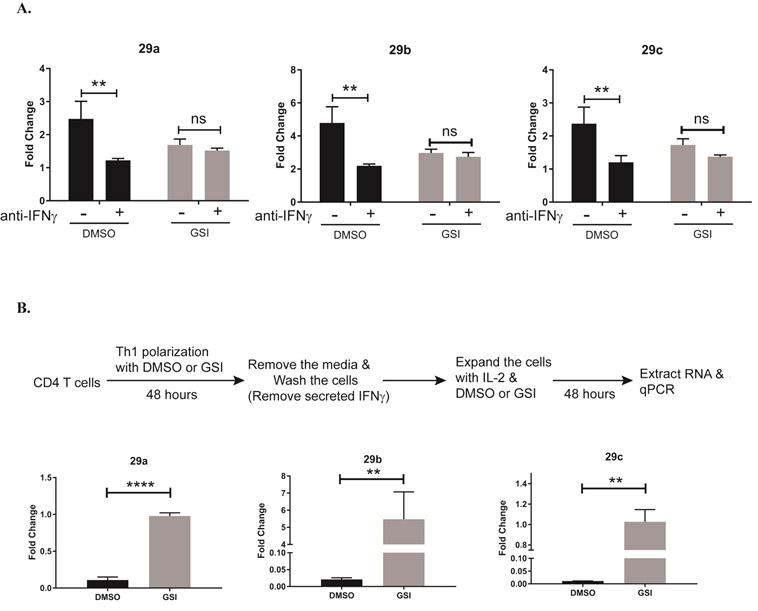
(A) Splenic CD4 T cells isolated from C57BL/6 mice were pretreated with DMSO or GSI for 30 minutes and stimulated with plate-bound anti-CD3ε and anti-CD28 for 72 hours under nonpolarizing conditions without or with IFNγ-neutralizing antibody. Total RNA was isolated and microRNAs were reverse transcribed using respective stem loop primers. The relative expression of miR-29 transcripts was normalized to expression of sno202. Fold changes are expressed relative to unstimulated CD4 T cells. Statistical analyses were performed using two-way ANOVA with Tukey’s multiple comparisons test applied. (B) Splenic CD4 T cells isolated from C57BL/6 mice were pretreated with DMSO or GSI for 30 minutes and stimulated with plate-bound anti-CD3ε and anti-CD28 under Th1 polarizing conditions for 48 hours. Cells were harvested and cultured with fresh media, with rmIL-2 added, in the presence of DMSO or GSI for an additional 48 hours. Total RNA was isolated and microRNAs were reverse transcribed using respective stem loop primers. The relative expression of miR-29 transcripts was normalized to expression of sno202. Fold changes are expressed relative to DMSO-treated CD4 T cells. Results are the mean of triplicate wells + S.D. and are representative of three independent of experiments. Statistical analyses were performed using an unpaired Student’s t test. **P<0.01; ****P<0.0001
3.5. Notch1-mediated inhibition of miR-29 is NF-κB-independent
To further elucidate the mechanism behind Notch1-mediated repression of miR-29, we focused on NF-κB, which is a non-canonical binding partner of Notch1 and is also known to suppress miR-29 expression18,20,21. The NF-κB family of transcriptional regulators are normally held in an inactive complex in the cytosol of resting T cells. Following stimulation, a series of signaling events that are initiated at the T cell receptor, culminate in the phosphorylation, and subsequent degradation, of NF-κB inhibitory proteins, including that of Inhibitor of NF-κB alpha (IκBa).
We utilized a pharmacological inhibitor of NF-κB, Bay11-7085, which acts to irreversibly prevent phosphorylation and degradation of IκBα, to determine whether Notch1 repression of miR-29 required active NF-κB signaling. We treated the DO11.10-EV and DO11.10-N1ICD cell lines with Bay11-7085 (5μM) for 24 hours, harvested RNA, and determined the expression of miR-29. Consistent with previous reports suggesting NF-κB can suppress miR-29 levels, we observed increased miR-29a and miR-29b expression in DO11.10-EV cells, which do not express Notch1, when we inhibited NF-κB activity with Bay11 (Fig. 5A). Surprisingly, in DO11.10 cells that constitutively express N1ICD, inhibiting NF-κB with Bay11 did not alter the expression of miR-29a or miR-29b expression (Fig. 5B). Thus, in the absence of Notch1, NF-κB repressed mirR-29 expression. However, these results also confirm that when N1ICD is expressed, it can act to repress miR-29 expression, and that this process does not require NF-κB activity.
Figure 5. Notch1-mediated inhibition of miR-29 is NF-κB-independent.
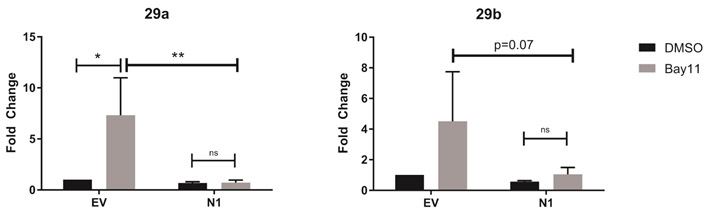
The DO11.10 T cell hybridoma cell line was stably transduced with empty vector (DO11.10-EV) or with Notch1-intracellular domain (DO11.10-N1-ICD). Transduced cells were treated with DMSO or the NF-κB inhibitor, Bay11-7085, for 24 hours. Total RNA was isolated and microRNAs were reverse transcribed using respective stem loop primers. The relative expression of miR-29 transcripts was normalized to expression of sno202. Fold-change is expressed relative to DMSO treatment. Results are the mean of three independent experiments + S.D. Statistical analyses were performed using two-way ANOVA with Tukey’s multiple comparisons test applied. *P<0,05; **P<0.01.
3.6. Canonical Notch1 signaling represses miR-29 expression
Within the promoter of several Notch-regulated genes, CSL binding sites have been found to be “nested” within larger NF-κB response elements29. This finding may explain how, during non-canonical Notch signaling, CSL proteins may be “competed off’ of these “nested” sites, as NF-κB levels increase. In the report by Cao and colleagues, mutating one, but not the other, of two NF-κB-containing binding sites within the miR-29ab1 promoter resulted in miR-29 de-repression18. A closer examination of these two NF-κB binding sites revealed that, only within the mutated site that resulted in de-repressed miR-29 levels, was there a nested CSL binding site. During canonical Notch signaling, CSL will bind first to a promoter region. It then recruits Notch and additional transcriptional activators, such as mastermind-like-1 (MAML1) and p300, to positively mediate transcription. To investigate the potential contribution of non-canonical regulation of miR-29, we over-expressed MAML1 or dominant-negative MAML1 (DN-MAML1), together with N1ICD and a luciferase construct driven by the miR-29ab1 promoter, in NIH3T3 cells. We observed that overexpressing N1ICD alone or with MAML1 decreased the promoter activity of miR-29ab1 (Fig. 6A, Fig. S3A). By contrast, over-expressing DN-MAML1 together with N1ICD, had the reverse effect, and abrogated Notch1-mediated repression of miR-29ab1 (Fig 6B, Fig. S3B). To further confirm a possible role of canonical Notch signaling in regulating miR-29, we performed chromatin immunoprecipitation assays using antibodies specific for CSL or Notch1. This revealed that CSL and Notch1, both, occupy the can be found at this nested CSL binding site on the miR-29ab1 promoter (Fig 6B). Altogether, these data suggest that Notch1 may repress miR-29 by reducing its promoter activity through an active, CSL-dependent, canonical Notch signaling mechanism that requires MAML1.
Figure 6: Notch inhibits miR-29 expression by canonical signaling.
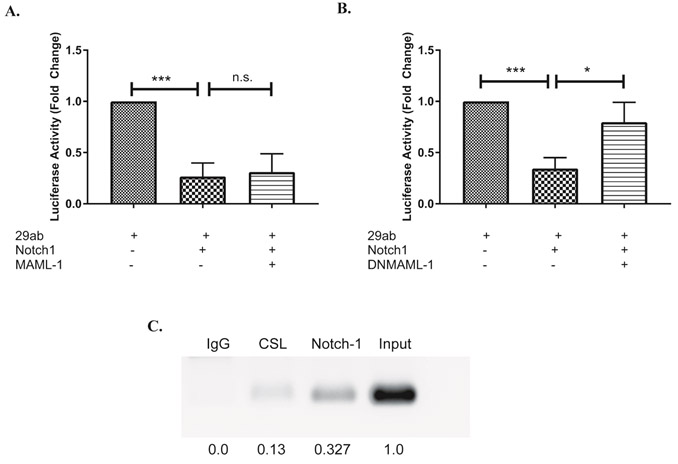
(A, B) NIH3T3 cells were transfected with a construct containing the miR-29ab1 promoter, together with pRL-CMV, with or without constructs expressing N1ICD, Mastermind like-1 (MAML-1; A) or a dominant-negative MAML-1 (DNMAML-1; B). Relative firefly luciferase activity was measured (Firefly/Renilla) and normalized to the expression of 29ab1. Results are the mean + S.D. of three independent experiments. (C) Splenic CD4 T cells isolated from C57BL/6 mice were stimulated with plate bound anti-CD3ε and anti-CD28 for 72 hours under Th1 polarizing conditions. Anti-CSL or anti-Notch1 was used to precipitate the respective proteins bound to DNA. DNA in complex was amplified with primers specific for CSL binding region in miR-29 promoter. Densitometric values were normalized to the input control using ImageJ software. Statistical analyses were performed using an unpaired Student’s t test. *P<0.05; ***P< 0.001.
4. Discussion
miRNAs have been identified as essential regulators of immune responses, including during T cell proliferation and differentiation. However, how these miRNAs, themselves, are regulated is not well-understood. In this report, we investigated a role for Notch1 signaling in regulating miR-29, a microRNA known to modulate the expression of two pro-inflammatory transcripts, tbx21 and ifng17-19. We have previously demonstrated that Notch1 influences T helper cell differentiation and regulates IFNγ and T-bet, the protein, product of tbx21, in Th1 cells4,26. This led us to further investigate whether Notch1 might also contribute to Th1 differentiation through its effects on miR-29 expression. Using both pharmacological and genetic approaches, we show that Notch1 can repress miR-29 in a CSL-dependent manner during the early stages of Th1 cell differentiation and, thus, prime CD4 T cells for Th1 differentiation.
We utilized the novel γ-secretase inhibitor PF-03084014, to inhibit Notch signaling during Th1 polarization, and found T-bet protein levels in GSI-treated Th1 cells were comparable to those of DMSO-treated cells 24 hours after polarization. However, transcripts of tbx21, were significantly higher in cells treated with GSI for 24 hours, than in DMSO-treated cells harvested at this same timepoint. This suggested to us that post-transcriptional regulation of tbx21 may be at work during the early stages of Th1 differentiation. In parallel, we also observed increased expression of miR-29b in GSI-treated cells, suggesting miR-29 may be involved in early regulation of tbx21. The 3’untranslated region (UTR) of labile transcripts, such as cytokines, contain binding sites for microRNAs, allowing their direct association and negative regulation of mRNAs, either by destabilizing mRNA or by preventing translation initiation. However, in proliferating cells these 3’UTRs are often edited to remove the microRNA binding sites, relieving micro-RNA-mediated inhibition and stabilizing transcripts30. In GSI-treated T cells, we noted high levels of tbx21 transcripts 24 hours after stimulation, although protein levels remained similar to DMSO-treated cells. Thus, one benefit of Notch1 repression of miR-29 during early stages of Th1 cell differentiation, would be to rapidly prevent miR-29 binding and subsequent tbx21 and ifng inhibition, before 3’UTR shortening occurs. However, additional studies would be necessary to test this model.
Since GSI treatment can also affect other cellular substrates of γ-secretase, including other Notch family members and Notch ligands, we expressed the Notch1 intracellular domain in different cell lines to conclusively show that Notch1 can repress miR-29. From our study, we conclude that miR-29 exerts its effects on tbx21 expression by inhibiting its translation, as has been shown for CDK231. In contrast, a previous study indicated miR-29 regulates the expression of tbx21 by destabilizing and degrading the transcript17. This may be a result of over-expression of miR-29 and may not reflect actual, physiological conditions. We did not observe significant differences in T-bet protein expression 24 hours after GSI-treatment, even though tbx21 expression was significantly higher in GSI-treated cells. Thus, another possible explanation for these contradictory data may be that miR-29 inhibits translation of tbx21 before inducing its degradation, as was observed for miR-430 targets identified by ribosome profiling during zebrafish development32.
During Th1 polarization assays we observed opposing effects at early and later stages of differentiation. Since IFNγ can induce the expression of miR-29 and initiate a negative feedback loop, it is possible that Notch-mediated effects are “eclipsed” by IFNγ signaling. By blocking IFNγ signaling and removing IFNγ from the extracellular environment we showed that IFNγ indeed masks the repressive effects of Notch1 on miR-29. This suggests that Notch1 may inhibit the expression of miR-29 at the very early stages of polarization, when IFNγ levels are low, and acts to prime CD4 T cells to adopt a Th1 response. IFNγ in turn, increases the expression of miR-29 at later stages of differentiation and further modulates proinflammatory responses. Therefore, Notch1 and IFNγ may act as a “yin and yang” of miR-29 regulation to fine-tune Th1 responses. Furthermore, we did not observe a similar de-repression of miR-29 in GSI-treated Th2 cells, suggesting that Notch1 regulation of miR-29 is likely to be highly cell-context-dependent, as is the case with many documented Notch signaling effects.
In CD4 T cells, miR-29 expression has been shown to be inhibited by NFκB, a non-canonical binding partner of Notch118. However, our study revealed that Notch1 represses miR-29 through an NF-κB-independent pathway. Notch1 lacks a DNA binding domain and requires a chromatin binding partner to regulate the transcription of its target genes. In addition to c-Myc, YY1, and hedgehog binding sites20,21, the miR-29 promoter also has a putative CSL binding site nested within the NF-κB binding sequence. We used a dominant-negative mutant of the transcriptional co-activator, MAML1, to determine that Notch1 can repress miR-29 through the CSL-dependent, canonical Notch signaling pathway. Furthermore, results of the chromatin immunoprecipitation assays we performed provide additional evidence that CSL and Notch1, both, are associated with the miR-29b1a promoter. NF-κB, as well as Notch-CSL complexes, primarily act as transcriptional activators. However, previous studies have reported that NF-κB and Notch-CSL complexes, both, can act to repress transcription through their interactions with HDAC proteins and YY1, respectively33,34. Additional studies are necessary to determine whether YY1 also associates with this high molecular weight Notch-CSL-MAML1 to repress miR-29.
In conclusion, here we have determined that Notch1 can represses miR-29 expression in CD4 T cells during the early stages of Th1 cell differentiation, and this proceeds through a CSL-dependent, canonical Notch signaling mechanism. However, further studies are needed to determine how the Notch-CSL-MAML1 complex may act on miR-29 in cases of dysregulated Th1 responses, such as those seen in autoimmune conditions. Aberrant Notch1 expression has been linked to autoimmune responses35, as has down regulation of miR-2919. Gaining a greater understanding of how Notch1 may regulate miRNAs during Th cell differentiation may reveal novel therapeutic targets for immune regulation.
Supplementary Material
Highlights.
Notch1 represses miR-29 expression, during early stages of T helper type 1 cell differentiation
Notch1-miR-29 repression is NF-κB-independent
Canonical Notch signaling mediates early Notch1-miR-29 repression and requires MAML1
IFNγ signaling upregulates miR-29 to override Notch1-miR-29 repression
Acknowledgements
The authors wish to acknowledge Dr. Amy Burnside, Director, for critical input and for use of the Flow Cytometry Core Facility at the University of Massachusetts Amherst. Support for this work was provided by the National Institutes of Health (NIH 5P01CA166009).
Footnotes
Conflict of Interest Statement
The authors declare they have no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Luckheeram RV, Zhou R, Verma AD & Xia B CD4(+)T cells: differentiation and functions. Clin Dev Immunol 2012, 925135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Afkarian M et al. T-bet is a STAT1-induced regulator of IL-12R expression in naive CD4+ T cells. Nat Immunol 3, 549–557 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Szabo SJ et al. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 100, 655–669 (2000). [DOI] [PubMed] [Google Scholar]
- 4.Minter LM et al. Inhibitors of gamma-secretase block in vivo and in vitro T helper type 1 polarization by preventing Notch upregulation of Tbx21. Nat Immunol 6, 680–688 (2005). [PubMed] [Google Scholar]
- 5.Bailis W et al. Notch simultaneously orchestrates multiple helper T cell programs independently of cytokine signals. Immunity 39, 148–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Artavanis-Tsakonas S, Rand MD & Lake RJ Notch signaling: cell fate control and signal integration in development. Science 284, 770–776 (1999). [DOI] [PubMed] [Google Scholar]
- 7.Bray SJ Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol 7, 678–689 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Mumm JS & Kopan R Notch signaling: from the outside in. Dev Biol 228, 151–165 (2000). [DOI] [PubMed] [Google Scholar]
- 9.Petcherski AG & Kimble J Mastermind is a putative activator for Notch. Curr Biol 10, R471–473 (2000). [DOI] [PubMed] [Google Scholar]
- 10.Maillard I, Adler SH & Pear WS Notch and the immune system. Immunity 19, 781–791 (2003). [DOI] [PubMed] [Google Scholar]
- 11.Andersen P, Uosaki H, Shenje LT & Kwon C Non-canonical Notch signaling: emerging role and mechanism. Trends Cell Biol 22, 257–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Minter LM & Osborne BA Canonical and non-canonical Notch signaling in CD4(+) T cells. Curr Top Microbiol Immunol 360, 99–114. [DOI] [PubMed] [Google Scholar]
- 13.Amsen D, Helbig C & Backer RA Notch in T Cell Differentiation: All Things Considered. Trends Immunol 36, 802–814. [DOI] [PubMed] [Google Scholar]
- 14.Sun J, Krawczyk CJ & Pearce EJ Suppression of Th2 cell development by Notch ligands Deltal and Delta4. J Immunol 180, 1655–1661 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maekawa Y et al. Delta1-Notch3 interactions bias the functional differentiation of activated CD4+ T cells. Immunity 19, 549–559 (2003). [DOI] [PubMed] [Google Scholar]
- 16.Pollard KM, Cauvi DM, Toomey CB, Morris KV & Kono DH Interferon-gamma and systemic autoimmunity. Discov Med 16, 123–131. [PMC free article] [PubMed] [Google Scholar]
- 17.Steiner DF et al. MicroRNA-29 regulates T-box transcription factors and interferon-gamma production in helper T cells. Immunity 35, 169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma F et al. The microRNA miR-29 controls innate and adaptive immune responses to intracellular bacterial infection by targeting interferon-gamma. Nat Immunol 12, 861–869. [DOI] [PubMed] [Google Scholar]
- 19.Smith KM et al. miR-29ab1 deficiency identifies a negative feedback loop controlling Th1 bias that is dysregulated in multiple sclerosis. J Immunol 189, 1567–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mott JL et al. Transcriptional suppression of mir-29b-1/mir-29a promoter by c-Myc, hedgehog, and NF-kappaB. J Cell Biochem 110, 1155–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang H et al. NF-kappaB-YY1-miR-29 regulatory circuitry in skeletal myogenesis and rhabdomyosarcoma. Cancer Cell 14, 369–381 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shin HM et al. Notch1 augments NF-kappaB activity by facilitating its nuclear retention. EMBO J 25, 129–138 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jeffries S & Capobianco AJ Neoplastic transformation by Notch requires nuclear localization. Mol Cell Biol 20, 3928–3941 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hao L et al. Notch-1 activates estrogen receptor-alpha-dependent transcription via IKKalpha in breast cancer cells. Oncogene 29, 201–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Osborne BA & Minter LM Notch signalling during peripheral T-cell activation and differentiation. Nat Rev Immunol 7, 64–75 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Roderick JE et al. Therapeutic targeting of NOTCH signaling ameliorates immune-mediated bone marrow failure of aplastic anemia. J Exp Med 210, 1311–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dongre A et al. Non-Canonical Notch Signaling Drives Activation and Differentiation of Peripheral CD4(+) T Cells. Front Immunol 5, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Papayannidis C et al. A Phase 1 study of the novel gamma-secretase inhibitor PF-03084014 in patients with T-cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma. Blood Cancer J 5, e350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shirakata Y, Shuman JD & Coligan JE Purification of a novel MHC class I element binding activity from thymus nuclear extracts reveals that thymic RBP-Jkappa/CBF1 binds to NF-kappaB-like elements. J Immunol 156, 4672–4679 (1996). [PubMed] [Google Scholar]
- 30.Pai AA, Baharian G, Ariane Pagé Sabourin AP, Brinkworth JF, Nédélec Y, Foley JW, Jean-Christophe Grenier J-C, Siddle KJ, Dumaine A, Yotova V, Johnson ZP, Lanford RE, Burge CB,. & Barreiro LB Widespread Shortening of 3’ Untranslated Regions and Increased Exon Inclusion Are Evolutionarily Conserved Features of Innate Immune Responses to Infection. PLoS Genet 12, e1006338 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xiao L et al. miR-29b represses intestinal mucosal growth by inhibiting translation of cyclin-dependent kinase 2. Mol Biol Cell 24, 3038–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bazzini AA, Lee MT & Giraldez AJ Ribosome profiling shows that miR-430 reduces translation before causing mRNA decay in zebrafish. Science 336, 233–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Elsharkawy AM et al. The NF-kappaB p50:p50:HDAC-1 repressor complex orchestrates transcriptional inhibition of multiple pro-inflammatory genes. J Hepatol 53, 519–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yeh TS, Lin YM, Hsieh RH & Tseng MJ Association of transcription factor YY1 with the high molecular weight Notch complex suppresses the transactivation activity of Notch. J Biol Chem 278, 41963–41969 (2003). [DOI] [PubMed] [Google Scholar]
- 35.Matesic LE, Copeland NG & Jenkins NA Itchy mice: the identification of a new pathway for the development of autoimmunity. Curr Top Microbiol Immunol 321, 185–200 (2008). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.