

Abstract
In response to virus infections, a cell autonomous, transcription-based antiviral program is engaged to create resistance, impair pathogen replication, and alert professional cells in innate and adaptive immunity. This dual phase antiviral program consists of type I interferon (IFN) production followed by the response to IFN signaling. Pathogen recognition leads to activation of IRF and NFκB factors that function independently and together to recruit cellular coactivators that remodel chromatin, modify histones and activate RNA polymerase II (Pol II) at target gene loci, including the well-characterized IFNβ enhanceosome. In the subsequent response to IFN, a receptor-mediated JAK-STAT signaling cascade directs the assembly of the IRF9-STAT1-STAT2 transcription factor complex called ISGF3, which recruits its own cohort of remodelers, coactivators, and Pol II machinery to activate transcription of a wide range of IFN-stimulated genes. Regulation of the IFN and antiviral gene regulatory networks is not only important for driving innate immune responses to infections, but also may inform treatment of a growing list of chronic diseases that are characterized by hyperactive and constitutive IFN and IFN-stimulated gene (ISG) expression. Here, gene-specific and genome-wide investigations of the chromatin landscape at IFN and ISGs is discussed in parallel with IRF- and STAT-dependent regulation of Pol II transcription.
Keywords: interferon, IRF, STAT, chromatin, transcription, antiviral
Graphical abstract
INTRODUCTION
Type I interferon (IFN) is the primary antiviral cytokine generated in response to a virus infection [1] and creates a cell intrinsic molecular barrier that prevents virus replication. In addition to this essential innate defense, IFN has diverse roles in innate and adaptive immunity and has therapeutic impacts on infectious diseases, intrinsic autoimmunity, immunosurveillance, and efficacy of established and contemporary cancer therapies [2-4]. Concerted IFN production and subsequent IFN signaling are critical in efficiently establishing a cell-autonomous antiviral program; establishment of this dual phase program is coordinately regulated by IRF and STAT transcription factors, namely IRF3/IRF7 for IFN production and STAT1-STAT2-IRF9 for IFN-stimulated gene (ISG) expression [5]. As these transcription factors are the ultimate arbiters of antiviral responses, the molecular mechanisms underlying their ability to recognize chromatin, modulate nucleosome structures, and interface with RNA polymerase as well as both specialized and general basal transcriptional apparatus are of fundamental biomedical interest.
For IFN production, non-self nucleic acid surveillance and detection by pathogen recognition receptors leads to activation of IRF and NFκB transcription factors [6]. These regulators act in concert and independently to activate the expression of type I IFN genes as well as numerous primary antiviral response gene targets [7-9].
Extracellular IFN secreted from infected cells binds to its transmembrane receptor, activating STAT1 and STAT2 by tyrosine phosphorylation and SH2-domain induced heterodimerization. The STATs associate with IRF9 to form ISGF3, thought to be the predominant transcription factor that regulates the activation of chromatinized ISG targets [10-13]. ISGF3 initiates ISG transcription by recruiting both general and specialized coactivators to remodel chromatin and assemble Pol II machinery.
Expression of IFN and IFN-stimulated gene effectors rapidly generates a broad and powerful cellular antiviral state. Central to the regulation of both IFN and ISG gene regulatory networks is the cooperation and interaction required to coordinate assembly of remodelers, co-activators, and Pol II machinery to activate IFN and ISG expression.
IRF3/IRF7 and NFκB: Master Regulators of Innate Antiviral Immunity
In response to a virus infection, pattern recognition receptors bind to specific virus-associated molecular pattern substrates [6, 14, 15]. For most RNA viruses, RIG-I-like receptors (RLRs) and Toll-like-receptors (TLRs) recognize features of non-self RNAs and induce a series of signaling events that converge in activation of the transcription factors NFκB and IRF3/IRF7, master regulators of immunity and inflammation. Specifically, the IKKα/β kinase complex phosphorylates the IκB inhibitor of NFκB, subjecting IκB to proteasome degradation [16]. NFκB is released to translocate into the nucleus and bind to target promoters featuring a κB target element [17]. Similarly, kinases TBK1 and IKKε phosphorylate IRF3/IRF7, promoting dimerization, nuclear translocation, and target gene engagement.
IRF3 and IRF7 are members of the IRF family of nine transcription factors including IRF1 through IRF9 and several of the IRFs are implicated in the type I IFN-mediated antiviral system. All IRF factors contain a highly conserved DNA binding domain [18] and a C-terminal association domain (IAD), that confers factor-specific protein-protein interactions [19]. IRF3 and IRF7 both contain multiple sites in their C-terminal domains that are phosphorylated by kinases, including TBK1 and IKKε, during infection. Phosphorylation relieves autoinhibition and favors dimerization, nuclear translocation, and DNA binding. IRF3 and IRF7 are thought to be the primary antiviral transcription regulators during virus-induced gene regulation, with IRF3 likely to constitute the major IRF dimer during the early infection stage driving type I IFN production because of low levels of IRF7 [20, 21]. Increased abundance following IFN-induced gene expression enables IRF7 to play a major role during later stages of infection alongside IRF3 to activate transcription of IFNβ and other target genes [20, 22].
Transcription of IFNβ
The human IFNβ gene represents one of the best-characterized gene promoters in mammals and serves as a general model for inducible transcription and chromatin regulation of gene expression (Figure 1). The human IFNβ gene promoter contains well-positioned +1 and −1 nucleosomes that define a nucleosome-deficient enhancer region [23, 24]. The position of the +1 nucleosome physically obscures the TATA box and prevents access to the transcription start site (TSS), and the nucleosome-deficient region is comprised of four adjacent cis regulatory units, termed positive regulatory domains (PRDs) I, II, III, and IV [24]. PRDI and PRDIII represent canonical IRF binding sites (5’-AANNGAAA-3’), and have been linked to IRF1, IRF3, and IRF7 [19, 25-27]. PRDII is a high affinity NFκB target [28], and PRDIV associates with an ATF2/cJUN dimer [29].
Figure 1. Antiviral signaling and activation of IFNβ transcription.

Illustrations of the cell during steady state (left) and following RNA virus infection (right). Prior to virus infection (left), transcription factors IRF3 and NFκB are in their latent state, with NFκB bound to its inhibitor, IκB. Inside the nucleus (dotted line), the IFNβ promoter region is depicted with a −1 and +1 nucleosome flanking a nucleosome-free positive regulatory domain (PRD) depicting response elements PRDI-PRDIV. The +1 nucleosome obscures the transcriptional start site. Generic IRF3 and NFκB-driven targets are illustrated below. Following virus infection (right), viral-origin non-self nucleic acids are detected by RIG-I-like and Toll-like pattern recognition receptors resulting in IRF3 and NFκB activation and nuclear translocation. IRF3 homodimer and NFκB bind to their respective PRD sites along with ATF2/cJun to form the enhanceosome. Enhanceosome-mediated recruitment of chromatin modifying factors leads to eviction of the +1 nucleosome, revealing the IFN transcription start site and enabling Pol II assembly and IFNβ transcription. Individual IRF3 and NFκB target genes feature either recruited Pol II or paused Pol II, respectively.
Following virus infection, the IFNβ enhanceosome complex is formed from activated transcription factors bound to their respective PRD enhancer elements and HMG I(Y), which associates with PRDII and PRDIV and supports NFκB and cJun/ATF2 recruitment, respectively [30]. The enhanceosome dynamically recruits histone acetyltransferases (HATs), histone deacetylases (HDACs) and chromatin remodelers to remove the +1 nucleosome barrier, assemble a Pol II initiation complex, and mediate transcriptional elongation. Recruitment of HATs, GCN5, CBP and p300, catalyze the acetylation of the +1 nucleosome, promoting association with proteins containing acetyl-binding bromodomains including the SWI/SNF remodeling complex ATPase, BRG1 [31]. Engagement of the BRG1 bromodomain with the acetyl-rich histone environment [32] enables eviction of the +1 nucleosome from the TSS to expose the TATA box and assembly of the initiation complex, allowing Pol II binding and activation by CTD phosphorylation [24, 33]. Similarly, general transcription factor (GTF), TFIID, contains a bromodomain that can support its assembly at the IFNβ TSS [32]. Notably, acetylated histone residues H3K9 and H3K14 are attributed to strengthening the enhanceosome recruitment of TFIID, but the absolute requirement for HDACs in IFNβ transcription [34] suggests there is tightly controlled and coordinated recruitment of specialized chromatin modifying and remodeling machinery that lead to IFNβ gene transcription [24, 33].
The promoters of IFNα subtypes also feature PRD I- and PRD III-like elements recognized by IRF factors [20, 35], and nucleosome mapping revealed they also contain a +1 nucleosome occluding the TSS [33]. A prominent virus-induced loss of the +1 nucleosome was not observed at IFNα genes, but this likely reflects heterogenous transcriptional activity in the cell population. Cellular heterogeneity with respect to IFNβ production has been observed and estimated that even in cells exhibiting uniform virus infection, only 20% of cells are producing IFNβ [8]. Regulation of stochastic IFNβ expression is alleviated by NFκB activity, which is apparently rate-limiting for transcriptional activation. Genetic compensation for this lack of free NFκB is provided by interchromosomal interactions between the IFNβ promoter and transposable Alu-like repeat elements that are rich in NFκB binding sites [8]. Cross-chromosome interactions enable efficient NFκB delivery from the Alu site directly to the IFNβ enhancer. This transfer is mediated by cooperative association of transcription factor ThPOK and NFκB on Alu-like repeat elements [8, 9].
Histone Modification and Pol II Activation
Histone acetylation and IRF3 association with the HAT CBP/p300 activate IFNβ gene transcription. Following IRF3/IRF7 dimerization and activation, release of the IRF3 autoinhibitory state also exposes sites that promote CBP/p300 association [36]. The IRF3-CBP interaction site is only partially conserved among IRFs, and IRF7 binds to CBP/p300 using multiple sites [37].
In addition to the recruitment of HATs and HAT activity, HDACs have been shown to be required for IRF3-dependent transcription in an IFNβ luciferase-driven transcription assay [34]. RNA interference indicated that HDAC6 was needed to activate IRF3-dependent IFNβ promoter activity, while HDAC1 and HDAC8 repressed IFNβ activation. Knockdown of HDACs did not affect NFκB-driven transcription activity indicating HDAC regulation is IRF3-dependent and NFκB-independent. More recently, HDAC3, but not HDAC1, was shown to be recruited to IFNα subtypes [38]. HDAC3 had little effect on IFNβ, in agreement with a previous HDAC screen [34]. Recruitment of HDAC3 to the promoter of IFNα1, α2, or α14 correlated with deacetylation of H3K9 and H3K14 observed at IFNβ, and consequently, reverses the acetylation pattern established during IFN activation and bound by bromodomain proteins including BRG1. In contrast to the negative regulation typically associated with HDACs, HDAC6 was specifically found to be required to activate IFNβ transcription in response to Sendai virus or dsRNA stimulation via IRF3 [34]. However, the descriptions of HDAC6-mediated RIG-I deacetylation [39], and a virus-activated PKCα-HDAC6-β-catenin pathway that co-activates IRF3-mediated transcription indicate that non-nuclear roles for HDAC6 [40] may account for some of the defects induced by RNA interference.
In addition to the prominent IFNβ enhancer, IRF3 regulates many other target genes including chemokines (CXCL10, CCL5/RANTES) and ISGs (ISG15, IFIT1/ISG56, IFIT2/ISG54; Figure 3; [41]. In fact, IRF3 and NFκB bind to thousands of genomic targets following Sendai virus infection [7]. Significant colocalization of IRF3 and NFκB during virus infection demonstrates their extensive partnership beyond the IFNβ enhancer. In the same study, a high frequency of motifs corresponding to E-box regulatory elements, cAMP response elements, and ETS binding motifs were also uncovered at IRF3 target sites, suggesting IRF3 may collaborate with previously unrecognized auxiliary factors during the antiviral response.
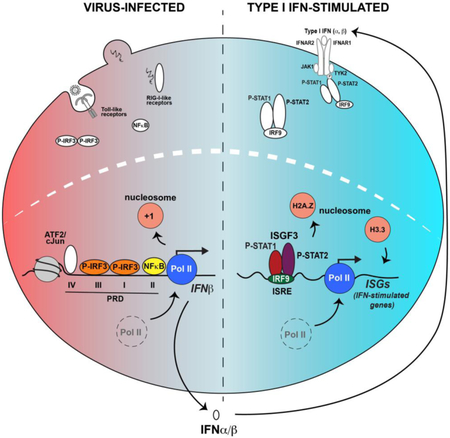
Figure 3. IRFs and STATs drive the IFN antiviral response.

Depiction of the two-phase antiviral response following virus infection (left), leading to IFN-mediated JAK-STAT signaling (right). The networks of both primary response genes activated by IRF3 and NFκB and IFN target genes activated by ISGF3, combine to produce a potent and coordinated response to infection. The ability of IRF proteins to recognize common core response elements leads to overlapping patterns of target gene expression, as exemplified by genes like ISG15 that are activated by IRF3 during virus infection and ISGF3 following IFN stimulation. In addition to the canonical IRF and ISGF3 factors, non-canonical STAT complexes are present both prior to and following IFN stimulation.
IRF3 also colocalized significantly with Pol II (total and elongating form) and the Mediator subunit, MED1 [7]. The enhanceosome mechanism of IFNβ transcriptional activation relies on regulated de novo recruitment of Pol II, but it has been widely observed that many inactive genes are pre-associated with RNA polymerase awaiting activation [42]. Release of the paused Pol II depends on phosphorylation of the elongation factor pTEFb to relieve association with the negative elongation factor, NELF. For Sendai virus-induced IRF3 target sites, evidence for both pause-release and de novo Pol II recruitment was uncovered, indicating distinct gene regulatory paradigms for IRF3 targets [7]. Highly inducible IRF3 targets were more likely to feature de novo Pol II recruitment, while NFκB targets were more frequently linked to paused Pol II [7]. Extensive IRF3 co-localization with Mediator subunit MED1 also suggests IRF3 may be more competent in recruiting Pol II. The MED1 subunit is a part of the middle module of the Mediator core and necessary for transcriptional regulation. Mediator is a multisubunit complex that physically bridges and relays site-specific transcription factor signals to Pol II machinery for gene activation and elongation [43] and this transcriptional division of labor is well-suited to rapid IFNβ gene expression, with IRF3 recruiting Pol II and NFκB providing rapid release and efficient elongation. In addition, the combined input from both IRF3 and NFκB provide checks and balances to distinguish the IFN antiviral response from a myriad of other pathways that activate either IRF or NFκB factors independently.
IFN-Stimulated JAK-STAT Signaling and ISGF3 Activation
Signal transduction downstream of type I IFN was the system used to discover Janus kinase (JAK) and Signal Transducer and Activator of Transcription (STAT) proteins [5], and is a powerful and well-studied example of cytokine-induced gene regulation [44, 45]. The IFN pathway culminates in ISGF3, that is unique among STAT factors. All of the other STAT factors act as homodimers or heterodimers leading to recognition of symmetrical GAS-like DNA elements, while ISGF3 is comprised of a heterotrimeric complex of STAT1, STAT2, and IRF9. The presence of IRF9 as the DNA-binding subunit of ISGF3 leads to recognition of the ISRE element [46], which is based on the core IRF-binding motif. STAT1 and STAT2 heterodimerize via IFN-induced SH2-phosphotyrosine interactions, and both STAT1 and STAT2 associate with IRF9 via interactions between the IRF C-terminal domain and the STAT coiled-coil domain [46-48]. This arrangement brings together the DNA binding specificity of IRF9 with the C-terminal transcriptional activation and signaling functions of the STATs. While STAT1 is an essential component of ISGF3 that incorporates signaling, imparts nuclear import, and stabilizes the complex, STAT2 provides the bulk of transcriptional activity, and can function in the absence of the STAT1 C-terminus [49], or when physically linked to IRF9 in a fusion protein [50]. This structure makes ISGF3 a mosaic of DNA binding, signaling, and transcription activation modules. In addition, modular associations between STATs and IRFs can contribute to non-canonical complexes such as those recently described by Decker and colleagues, consisting of IRF9 in complex with variations of unphosphorylated STAT1 or STAT2 [51]. While the ISGF3 paradigm can explain responses to acute activation by IFN, distinct sub-complexes may underlie basal and contextual IFN-like responses.
Chromatin Regulation of ISGs
In the nucleus, ISGF3 interactions with the IFN-stimulated response element (ISRE), a DNA sequence resembling 5’-AGTTTCNNTTTCNC/T-3’ that was initially characterized in the promoters of ISG15 and ISG54 [52, 53]. IRF9 recognizes the ISRE core sequence 5’-TTCNNTTT-3’, that resembles the IRF-E sequence (5’-AANNGAAA-3’) recognized by the conserved IRF DNA binding domain. STAT1 was found to preferentially cross-link to the 3’-TTT motif, and STAT2 exhibited more general association with central GC nucleotides [54]. More recent structural analysis strengthens this concept of ISGF3-DNA interaction and reveals more subtle binding relationships between STAT1, STAT2, and IRF9 that may uniquely regulate ISG subsets [47, 48, 54].
Unlike the well-studied IFNβ promoter, little is known about the interactions between ISGF3 and an individual chromatinized target promoter. Much data has accumulated by the investigation of promoter-reporters and subsets of well-behaved ISGs. The accumulated information is beginning to be revisited, verified, and expanded upon through the use of contemporary whole-genome techniques. Analysis of the IFN-stimulated nucleosome occupancy and chromatin accessibility in human and mouse cells indicates that this is a dynamic process that alters the native ISG chromatin landscape. [55, 56]. Increased chromatin accessibility after IFN treatment was demonstrated by complementary approaches, revealing that ISG promoters feature well-positioned nucleosomes at or near ISGF3 binding sites and throughout the ISG bodies that are mobilized in response to IFN stimulation. Basally-expressed ISGs such as IFIT1, IFIT3, and OAS2 were found to have lower TSS accessibility, suggesting a unique chromatin arrangement for basally-regulated ISGs [55]. Indeed, a prominent +1 nucleosome is positioned downstream the TSS for these tonic ISGs, but not for the neutral OAS3 [56]. These data demonstrate IFN induces chromatin alterations at ISG promoters that coincide with ISGF3 occupancy, and suggest that both general and gene-specific chromatin configurations contribute to basal and IFN-stimulated transcription.
In-depth examination of the ISG promoter nucleosome composition reveals the presence of core histones at steady state [56, 57]. Notably, while histone H2A is found within ISG gene bodies, it is absent from the ISRE-proximal promoters and replaced with histone variant H2A.Z [56]. H2A.Z nucleosomes appear to play a regulatory role in ISG transcription, as shRNA interference with H2A.Z results in greater ISGF3 occupancy, increased ISG mRNA production, and created a more potent antiviral state leading to more robust inhibition of virus replication [56]. IFN stimulation induces H2A.Z loss from the promoter, inversely correlating with ISGF3 (STAT2) occupancy. ISGF3 is required for IFN-induced H2A.Z depletion, and inhibition of either the HAT GCN5 or the bromodomain protein BRD2 disrupted both H2A.Z removal and ISG transcription.
Another histone important for ISG transcription is the histone H3 variant, H3.3. IFN treatment induces H3.3 deposition in the ISG body, and this deposition is essential for ISG transcription [57]. The H3.3 persists after IFN stimulation ceases and reflects a transcriptional sensitization or “memory” of prior stimulation [58]. Furthermore, H3.3 deposition has been described to inhibit linker histone H1 [59] and at ISGs, H1 occupancy decreases in response to IFN stimulation [60]. These results indicate ISG chromatin is subject to reorganization events that promote DNA accessibility and transcription. Despite the greater clarity of ISGF3 transcriptional regulation, it is important to note that well known coactivators including CBP, HDACs, and RVB1 were found to be dispensable for H2A.Z removal, but are nonetheless absolutely required for ISG transcription. Unraveling the nuances in coactivator requirements in ISG regulation will depend on integrating current and future findings.
HATs and HDACs
Both acetylation and deacetylation activities are required for ISG transcription activation [61-64]. STAT2 associates with CBP/p300 and GCN5, and these HATs have been implicated in ISG activation [62, 65]. Interference with either GCN5 or CBP inhibits ISG transcription, but H2A.Z eviction from ISG promoters depends only on GCN5, not CBP, demonstrating specificity rather than redundancy in HAT function [56], and reflecting the potential for multiple regulatory outcomes [56, 61, 62].
Complementarily, ISG transcription also requires HDAC activity; ISG transcription is blocked by both HDAC inhibitors and RNA interference with a subset of HDAC proteins [61, 63, 64, 66]. HDAC1 was found to associate with STAT2 while broad inhibition of HDAC activity had little effect on STAT phosphorylation, ISGF3 assembly, nuclear import, or DNA binding, yet potent loss of ISG expression was observed. While the exact roles for HDACs in ISG regulation are yet to be characterized, Pol II recruitment to ISGs was impaired by HDAC inhibitors [61, 63]. Perhaps related to the requirement for deacetylation, a component of the inhibitor of acetyltransferase (INHAT) complex, pp32, can influence the maximal levels of ISG expression [67, 68]. In the IFN response, pp32 associates with STAT2, but little is known about its mechanism of action [67]. The intricacies of ISG regulation by acetylation and deacetylation remain to be fully elucidated.
Chromatin Remodeling Factors
IFN-activated ISGF3 is also known to engage proteins involved in chromatin remodeling (Figure 2). Subunits of the SWI/SNF complex (or mammalian BAF/pBAF complex), including BAF47, BAF200 and the ATPase BRG1, are required for ISG transcription. Specifically, BRG1 interacts with STAT2 and is required for transcription of a subset of ISGs [69-72]. As observed in the IFNβ promoter, acetylated chromatin can enhance bromodomain recruitment, bringing BRG1 to ISGF3-proximal histones [31, 32, 73]. Likewise, chromatin remodelers can influence the histone acetylation status. For example, knockdown of BAF47 leads to decreased histone H4 acetylation at ISG promoters [71]. Several chromatin remodeling complexes, including SRCAP, TIP60, URI and INO80 have been described in association with the transcription co-factors RVB1 and RVB2. Both RVB1 and RVB2 were found to associate with STAT2 [74]. Interference with RVB1 prevents Pol II recruitment to ISG promoters, but the exact role of RVB proteins in mediating IFN-stimulated gene regulation remains at large. Thorough analysis demonstrated that chromatin remodeling complexes known to contain the RVB subunits, including SRCAP, TIP60, URI and INO80, were not required for ISG transcription, indicating an alternative role for RVB1 and RVB2 in IFN-stimulated transcription [74].
Figure 2. IFN-JAK-STAT signaling pathway.

Illustrations of the canonical ISGF3 signaling system at steady state (left) and following type I IFN-stimulation (right). Prior to IFN stimulation, transmembrane IFNAR1/2 receptor chains are associated with TYK2 and JAK1 kinases, and latent factors STAT1 and STAT2 are present in the cytoplasm. IRF9 associates with STAT2 in the cytoplasm and also shuttles into the nucleus. Histones H2A.Z and H3.3 occupy transcriptionally silent ISG promoters. IFN binding to the receptor complex (right) induces oligomerization and phosphorylation of the receptor chains, generating docking sites for the latent STAT proteins’ SH2 domains, resulting in phosphorylation of STAT2 Y690 and STAT1 Y701. Phosphorylated STAT1 and STAT2 undergo SH2-mediated dimerization and along with IRF9 form the ISGF3 complex. ISGF3 translocates into the nucleus, where it binds to the ISRE at ISG promoters and recruits coactivators relevant to histone modification, chromatin remodeling, and Pol II activation, including GCN5 and BRD2 that remodel the H2A.Z-containing nucleosome, MED14 to recruit Pol II for activating ISG transcription, and histone H3.3 is deposited at ISG gene bodies. The roles for essential co-activators RVB1, RVB2, CBP, BRG1, and HDACs are poorly understood.
Recruitment of Mediator and Pol II to ISGs
Specific interactions between ISGF3 and the Pol II transcriptional machinery include STAT2 associations with both Mediator and general transcription factor subunits. The Mediator subunits, MED14 and MED17 (previously called DRIP150 and DRIP77), interact with ISGF3 via the STAT2 C-terminal transcriptional activation domain [75], and MED14 was found to enhance ISRE-driven transcription activity following IFN stimulation. As MED14 connects the Mediator head, middle and tail modules that support transcription regulation and Pol II assembly, STAT2 associates with a key Mediator component to harness and activate the Pol II complex.
Among the first steps for Pol II assembly at gene promoters is binding of the TATA-binding protein (TBP) to the TATA box in the promoter region as a part of the TFIID complex. Interestingly, TBP was found to be dispensable for ISG transcription, despite the presence of a TATA box at many ISG promoters [62]. This transcriptional nuance for ISGs is thought to reflect an evolutionary response to selective pressure from viral pathogens such as poliovirus, which is known to destroy TBP. Instead, ISGF3 can use a TBP-free transcription complex consisting of TAFII130 in conjunction with the HAT GCN5 [62].
Evaluation of Pol II recruitment to ISGs generally indicates de novo Pol II recruitment following IFN stimulation, but pause-release mechanisms are also used to regulate ISG transcription [7, 42, 55]. Pol II recruited to ISGs is activated by phosphorylation of its C-terminal domain by pTEFb through association with the bromodomain protein BRD4 [42, 76]. This results in the release of the Pol II-associated repressors NELF and DSIF, which negatively regulate ISG transcription, and enables ISG mRNA elongation [76].
SUMMARY
The cell-autonomous innate antiviral response is an interconnected, transcription-mediated program (Figure 3). The interplay between the native chromatin and IRF and STAT transcription factor complexes initiates genetic reorganization, assembly of the RNA polymerase holoenzyme complex, and activation of transcriptional elongation that drives the expression of IFN and ISG effectors that create and reinforce the cellular antiviral environment. In addition to the dynamic IRF and NFκB components of the IFNβ enhanceosome and the trimeric IFN-responsive ISGF3 complex, non-canonical IRF and STAT complexes as well as auxiliary transcription factors contribute to basal and secondary ISG expression; and these factors undoubtedly provide gene-specific, cell type-specific, and context-specific plasticity to the overall antiviral network. These IFN responses also modulate the innate and adaptive immune processes that underlie immune-related diseases and drive immunotherapy and antitumor therapeutic strategies. Thus, greater understanding of the mechanisms underlying or contributing to the control of this gene expression network will not only generate fundamental insights into the processes that govern inducible gene regulation in higher eukaryotes, but will also provide clinically relevant diagnostic and therapeutic tools.
Highlights.
Type I interferon (IFN) production and response to IFN establishes a protective antiviral cellular environment by driving gene expression programs
IRF3, NFkB, and ISGF3 are essential conductors of the dual phase antiviral transcription program
Dynamic chromatin regulation governs both the production of and response to IFN
Collaboration between transcription factors, coactivators, Pol II, and chromatin regulates the coordinated induction of IFN and IFN-stimulated genes
Biography

Nancy Au-Yeung obtained her B.A. in Biochemistry from University of Colorado at Boulder and recently completed her Ph.D. from the Northwestern University Interdisciplinary Biological Sciences graduate program. Her research focuses on the transcriptional and chromatin regulation of type I interferon-stimulated genes.

Curt Horvath is a Professor in the Department of Molecular Biosciences at Northwestern University. His lab works on mechanistic cell biology in the field of IFN-JAK-STAT signal transduction and mechanisms of gene regulation. These interests include virus–host interactions, protein–RNA interactions, and the molecular basis of IFN production and cellular antiviral transcription.
Footnotes
Conflict of Interest
Neither of the authors has any conflicts of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Isaacs A, Lindenmann J, Valentine RC, Virus interference. II. Some properties of interferon, Proc R Soc Lond B Biol Sci 147(927) (1957) 268–73. [DOI] [PubMed] [Google Scholar]
- [2].Parker BS, Rautela J, Hertzog PJ, Antitumour actions of interferons: implications for cancer therapy, Nat Rev Cancer 16(3) (2016) 131–44. [DOI] [PubMed] [Google Scholar]
- [3].Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G, Type I interferons in anticancer immunity, Nat Rev Immunol 15(7) (2015) 405–14. [DOI] [PubMed] [Google Scholar]
- [4].Gonzalez-Navajas JM, Lee J, David M, Raz E, Immunomodulatory functions of type I interferons, Nat Rev Immunol 12(2) (2012) 125–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Stark GR, Darnell JE Jr., The JAK-STAT pathway at twenty, Immunity 36(4) (2012) 503–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bruns AM, Horvath CM, Activation of RIG-I-like receptor signal transduction, Crit Rev Biochem Mol Biol 47(2) (2012) 194–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Freaney JE, Kim R, Mandhana R, Horvath CM, Extensive cooperation of immune master regulators IRF3 and NFkappaB in RNA Pol II recruitment and pause release in human innate antiviral transcription, Cell Rep 4(5) (2013) 959–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Apostolou E, Thanos D, Virus Infection Induces NF-kappaB-dependent interchromosomal associations mediating monoallelic IFN-beta gene expression, Cell 134(1) (2008) 85–96. [DOI] [PubMed] [Google Scholar]
- [9].Nikopoulou C, Panagopoulos G, Sianidis G, Psarra E, Ford E, Thanos D, The Transcription Factor ThPOK Orchestrates Stochastic Interchromosomal Interactions Required for IFNB1 Virus-Inducible Gene Expression, Mol Cell 71(2) (2018) 352–361 e5. [DOI] [PubMed] [Google Scholar]
- [10].Borden EC, Sen GC, Uze G, Silverman RH, Ransohoff RM, Foster GR, Stark GR, Interferons at age 50: past, current and future impact on biomedicine, Nat Rev Drug Discov 6(12) (2007) 975–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Fu XY, Kessler DS, Veals SA, Levy DE, Darnell JE Jr., ISGF3, the transcriptional activator induced by interferon alpha, consists of multiple interacting polypeptide chains, Proc Natl Acad Sci U S A 87(21) (1990) 8555–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Levy DE, Kessler DS, Pine R, Darnell JE Jr., Cytoplasmic activation of ISGF3, the positive regulator of interferon-alpha-stimulated transcription, reconstituted in vitro, Genes Dev 3(9) (1989) 1362–71. [DOI] [PubMed] [Google Scholar]
- [13].Platanitis E, Demiroz D, Capelle C, Schneller A, Hartl M, Gossenreiter T, Mueller M, Novatchkova M, Decker T, Homeostatic and Interferon-induced gene expression represent different states of promoter-associated transcription factor ISGF3, bioRxiv 377275 (2018). [Google Scholar]
- [14].Kawai T, Akira S, Toll-like receptors and their crosstalk with other innate receptors in infection and immunity, Immunity 34(5) (2011) 637–50. [DOI] [PubMed] [Google Scholar]
- [15].Chow KT, Gale M Jr., Loo YM, RIG-I and Other RNA Sensors in Antiviral Immunity, Annu Rev Immunol 36 (2018) 667–694. [DOI] [PubMed] [Google Scholar]
- [16].Wan F, Lenardo MJ, Specification of DNA binding activity of NF-kappaB proteins, Cold Spring Harb Perspect Biol 1(4) (2009) a000067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chen FE, Huang DB, Chen YQ, Ghosh G, Crystal structure of p50/p65 heterodimer of transcription factor NF-kappaB bound to DNA, Nature 391(6665) (1998) 410–3. [DOI] [PubMed] [Google Scholar]
- [18].De Ioannes P, Escalante CR, Aggarwal AK, Structures of apo IRF-3 and IRF-7 DNA binding domains: effect of loop L1 on DNA binding, Nucleic Acids Res 39(16) (2011) 7300–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Yanai H, Negishi H, Taniguchi T, The IRF family of transcription factors: Inception, impact and implications in oncogenesis, Oncoimmunology 1(8) (2012) 1376–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Honda K, Takaoka A, Taniguchi T, Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors, Immunity 25(3) (2006) 349–60. [DOI] [PubMed] [Google Scholar]
- [21].Marie I, Durbin JE, Levy DE, Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7, EMBO J 17(22) (1998) 6660–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, Taniguchi T, Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction, Immunity 13(4) (2000) 539–48. [DOI] [PubMed] [Google Scholar]
- [23].Thanos D, Maniatis T, Virus induction of human IFN beta gene expression requires the assembly of an enhanceosome, Cell 83(7) (1995) 1091–100. [DOI] [PubMed] [Google Scholar]
- [24].Agalioti T, Lomvardas S, Parekh B, Yie J, Maniatis T, Thanos D, Ordered recruitment of chromatin modifying and general transcription factors to the IFN-beta promoter, Cell 103(4) (2000) 667–78. [DOI] [PubMed] [Google Scholar]
- [25].Miyamoto M, Fujita T, Kimura Y, Maruyama M, Harada H, Sudo Y, Miyata T, Taniguchi T, Regulated expression of a gene encoding a nuclear factor, IRF-1, that specifically binds to IFN-beta gene regulatory elements, Cell 54(6) (1988) 903–13. [DOI] [PubMed] [Google Scholar]
- [26].Escalante CR, Nistal-Villan E, Shen L, Garcia-Sastre A, Aggarwal AK, Structure of IRF-3 bound to the PRDIII-I regulatory element of the human interferon-beta enhancer, Mol Cell 26(5) (2007) 703–16. [DOI] [PubMed] [Google Scholar]
- [27].Lin R, Genin P, Mamane Y, Hiscott J, Selective DNA binding and association with the CREB binding protein coactivator contribute to differential activation of alpha/beta interferon genes by interferon regulatory factors 3 and 7, Mol Cell Biol 20(17) (2000) 6342–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Escalante CR, Shen L, Thanos D, Aggarwal AK, Structure of NF-kappaB p50/p65 heterodimer bound to the PRDII DNA element from the interferon-beta promoter, Structure 10(3) (2002) 383–91. [DOI] [PubMed] [Google Scholar]
- [29].Du W, Maniatis T, An ATF/CREB binding site is required for virus induction of the human interferon beta gene [corrected], Proc Natl Acad Sci U S A 89(6) (1992) 2150–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Du W, Thanos D, Maniatis T, Mechanisms of transcriptional synergism between distinct virus-inducible enhancer elements, Cell 74(5) (1993) 887–98. [DOI] [PubMed] [Google Scholar]
- [31].Zeng L, Zhou MM, Bromodomain: an acetyl-lysine binding domain, FEBS Lett 513(1) (2002) 124–8. [DOI] [PubMed] [Google Scholar]
- [32].Agalioti T, Chen G, Thanos D, Deciphering the transcriptional histone acetylation code for a human gene, Cell 111(3) (2002) 381–92. [DOI] [PubMed] [Google Scholar]
- [33].Freaney JE, Zhang Q, Yigit E, Kim R, Widom J, Wang JP, Horvath CM, High-density nucleosome occupancy map of human chromosome 9p21-22 reveals chromatin organization of the type I interferon gene cluster, J Interferon Cytokine Res 34(9) (2014) 676–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Nusinzon I, Horvath CM, Positive and negative regulation of the innate antiviral response and beta interferon gene expression by deacetylation, Mol Cell Biol 26(8) (2006) 3106–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ryals J, Dierks P, Ragg H, Weissmann C, A 46-nucleotide promoter segment from an IFN-alpha gene renders an unrelated promoter inducible by virus, Cell 41(2) (1985) 497–507. [DOI] [PubMed] [Google Scholar]
- [36].Qin BY, Liu C, Srinath H, Lam SS, Correia JJ, Derynck R, Lin K, Crystal structure of IRF-3 in complex with CBP, Structure 13(9) (2005) 1269–77. [DOI] [PubMed] [Google Scholar]
- [37].Yang H, Lin CH, Ma G, Baffi MO, Wathelet MG, Interferon regulatory factor-7 synergizes with other transcription factors through multiple interactions with p300/CBP coactivators, J Biol Chem 278(18) (2003) 15495–504. [DOI] [PubMed] [Google Scholar]
- [38].Genin P, Lin R, Hiscott J, Civas A, Recruitment of histone deacetylase 3 to the interferon-A gene promoters attenuates interferon expression, PLoS One 7(6) (2012) e38336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Choi SJ, Lee HC, Kim JH, Park SY, Kim TH, Lee WK, Jang DJ, Yoon JE, Choi YI, Kim S, Ma J, Kim CJ, Yao TP, Jung JU, Lee JY, Lee JS, HDAC6 regulates cellular viral RNA sensing by deacetylation of RIG-I, EMBO J 35(4) (2016) 429–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zhu J, Coyne CB, Sarkar SN, PKC alpha regulates Sendai virus-mediated interferon induction through HDAC6 and beta-catenin, EMBO J 30(23) (2011) 4838–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Grandvaux N, Servant MJ, tenOever B, Sen GC, Balachandran S, Barber GN, Lin R, Hiscott J, Transcriptional profiling of interferon regulatory factor 3 target genes: direct involvement in the regulation of interferon-stimulated genes, J Virol 76(11) (2002) 5532–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Adelman K, Lis JT, Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans, Nat Rev Genet 13(10) (2012) 720–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Malik S, Roeder RG, Dynamic regulation of pol II transcription by the mammalian Mediator complex, Trends Biochem Sci 30(5) (2005) 256–63. [DOI] [PubMed] [Google Scholar]
- [44].Ivashkiv LB, Donlin LT, Regulation of type I interferon responses, Nat Rev Immunol 14(1) (2014) 36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Au-Yeung N, Mandhana R, Horvath CM, Transcriptional regulation by STAT1 and STAT2 in the interferon JAK-STAT pathway, JAKSTAT 2(3) (2013) e23931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Paul A, Tang TH, Ng SK, Interferon Regulatory Factor 9 Structure and Regulation, Front Immunol 9 (2018) 1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Horvath CM, Stark GR, Kerr IM, Darnell JE Jr., Interactions between STAT and non-STAT proteins in the interferon-stimulated gene factor 3 transcription complex, Mol Cell Biol 16(12) (1996) 6957–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Rengachari S, Groiss S, Devos JM, Caron E, Grandvaux N, Panne D, Structural basis of STAT2 recognition by IRF9 reveals molecular insights into ISGF3 function, Proc Natl Acad Sci U S A 115(4) (2018) E601–E609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Muller M, Laxton C, Briscoe J, Schindler C, Improta T, Darnell JE Jr., Stark GR, Kerr IM, Complementation of a mutant cell line: central role of the 91 kDa polypeptide of ISGF3 in the interferon-alpha and -gamma signal transduction pathways, EMBO J 12(11) (1993) 4221–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kraus TA, Lau JF, Parisien JP, Horvath CM, A hybrid IRF9-STAT2 protein recapitulates interferon-stimulated gene expression and antiviral response, J Biol Chem 278(15) (2003) 13033–8. [DOI] [PubMed] [Google Scholar]
- [51].Majoros A, Platanitis E, Kernbauer-Holzl E, Rosebrock F, Muller M, Decker T, Canonical and Non-Canonical Aspects of JAK-STAT Signaling: Lessons from Interferons for Cytokine Responses, Front Immunol 8 (2017) 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Levy DE, Kessler DS, Pine R, Reich N, Darnell JE Jr., Interferon-induced nuclear factors that bind a shared promoter element correlate with positive and negative transcriptional control, Genes Dev 2(4) (1988) 383–93. [DOI] [PubMed] [Google Scholar]
- [53].Reich N, Evans B, Levy D, Fahey D, Knight E Jr., Darnell JE Jr., Interferon-induced transcription of a gene encoding a 15-kDa protein depends on an upstream enhancer element, Proc Natl Acad Sci U S A 84(18) (1987) 6394–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Qureshi SA, Salditt-Georgieff M, Darnell JE Jr., Tyrosine-phosphorylated Stat1 and Stat2 plus a 48-kDa protein all contact DNA in forming interferon-stimulated-gene factor 3, Proc Natl Acad Sci U S A 92(9) (1995) 3829–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Mostafavi S, Yoshida H, Moodley D, LeBoite H, Rothamel K, Raj T, Ye CJ, Chevrier N, Zhang SY, Feng T, Lee M, Casanova JL, Clark JD, Hegen M, Telliez JB, Hacohen N, De Jager PL, Regev A, Mathis D, Benoist C, C. Immunological Genome Project, Parsing the Interferon Transcriptional Network and Its Disease Associations, Cell 164(3) (2016) 564–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Au-Yeung N, Horvath CM, Histone H2A.Z Suppression of Interferon-Stimulated Transcription and Antiviral Immunity is Modulated by GCN5 and BRD2, iScience 6 (2018) 68–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Tamura T, Smith M, Kanno T, Dasenbrock H, Nishiyama A, Ozato K, Inducible deposition of the histone variant H3.3 in interferon-stimulated genes, J Biol Chem 284(18) (2009) 12217–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Kamada R, Yang W, Zhang Y, Patel MC, Yang Y, Ouda R, Dey A, Wakabayashi Y, Sakaguchi K, Fujita T, Tamura T, Zhu J, Ozato K, Interferon stimulation creates chromatin marks and establishes transcriptional memory, Proc Natl Acad Sci U S A (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Braunschweig U, Hogan GJ, Pagie L, van Steensel B, Histone H1 binding is inhibited by histone variant H3.3, EMBO J 28(23) (2009) 3635–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Kadota S, Nagata K, Silencing of IFN-stimulated gene transcription is regulated by histone H1 and its chaperone TAF-I, Nucleic Acids Res 42(12) (2014) 7642–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Nusinzon I, Horvath CM, Interferon-stimulated transcription and innate antiviral immunity require deacetylase activity and histone deacetylase 1, Proc Natl Acad Sci U S A 100(25) (2003) 14742–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Paulson M, Press C, Smith E, Tanese N, Levy DE, IFN-Stimulated transcription through a TBP-free acetyltransferase complex escapes viral shutoff, Nat Cell Biol 4(2) (2002) 140–7. [DOI] [PubMed] [Google Scholar]
- [63].Chang HM, Paulson M, Holko M, Rice CM, Williams BR, Marie I, Levy DE, Induction of interferon-stimulated gene expression and antiviral responses require protein deacetylase activity, Proc Natl Acad Sci U S A 101(26) (2004) 9578–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Sakamoto S, Potla R, Larner AC, Histone deacetylase activity is required to recruit RNA polymerase II to the promoters of selected interferon-stimulated early response genes, J Biol Chem 279(39) (2004) 40362–7. [DOI] [PubMed] [Google Scholar]
- [65].Bhattacharya S, Eckner R, Grossman S, Oldread E, Arany Z, D'Andrea A, Livingston DM, Cooperation of Stat2 and p300/CBP in signalling induced by interferon-alpha, Nature 383(6598) (1996) 344–7. [DOI] [PubMed] [Google Scholar]
- [66].Kelly RD, Cowley SM, The physiological roles of histone deacetylase (HDAC) 1 and 2: complex co-stars with multiple leading parts, Biochem Soc Trans 41(3) (2013) 741–9. [DOI] [PubMed] [Google Scholar]
- [67].Kadota S, Nagata K, pp32, an INHAT component, is a transcription machinery recruiter for maximal induction of IFN-stimulated genes, J Cell Sci 124(Pt 6) (2011) 892–9. [DOI] [PubMed] [Google Scholar]
- [68].Kutney SN, Hong R, Macfarlan T, Chakravarti D A signaling role of histone-binding proteins and INHAT subunits pp32 and Set/TAF-Ibeta in integrating chromatin hypoacetylation and transcriptional repression, J Biol Chem 279(29) (2004) 30850–5. [DOI] [PubMed] [Google Scholar]
- [69].Huang M, Qian F, Hu Y, Ang C, Li Z, Wen Z, Chromatin-remodelling factor BRG1 selectively activates a subset of interferon-alpha-inducible genes, Nat Cell Biol 4(10) (2002) 774–81. [DOI] [PubMed] [Google Scholar]
- [70].Chi T, A BAF-centred view of the immune system, Nat Rev Immunol 4(12) (2004) 965–77. [DOI] [PubMed] [Google Scholar]
- [71].Cui K, Tailor P, Liu H, Chen X, Ozato K, Zhao K, The chromatin-remodeling BAF complex mediates cellular antiviral activities by promoter priming, Mol Cell Biol 24(10) (2004) 4476–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Yan Z, Cui K, Murray DM, Ling C, Xue Y, Gerstein A, Parsons R, Zhao K, Wang W, PBAF chromatin-remodeling complex requires a novel specificity subunit, BAF200, to regulate expression of selective interferon-responsive genes, Genes Dev 19(14) (2005) 1662–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Lomvardas S, Thanos D, Modifying gene expression programs by altering core promoter chromatin architecture, Cell 110(2) (2002) 261–71. [DOI] [PubMed] [Google Scholar]
- [74].Gnatovskiy L, Mita P, Levy DE, The human RVB complex is required for efficient transcription of type I interferon-stimulated genes, Mol Cell Biol 33(19) (2013) 3817–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Lau JF, Nusinzon I, Burakov D, Freedman LP, Horvath CM, Role of metazoan mediator proteins in interferon-responsive transcription, Mol Cell Biol 23(2) (2003) 620–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Patel MC, Debrosse M, Smith M, Dey A, Huynh W, Sarai N, Heightman TD, Tamura T, Ozato K, BRD4 coordinates recruitment of pause release factor P-TEFb and the pausing complex NELF/DSIF to regulate transcription elongation of interferon-stimulated genes, Mol Cell Biol 33(12) (2013) 2497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]