

Abstract
We studied a consanguineous Palestinian Arab family segregating an autosomal recessive progressive myoclonus epilepsy (PME) with early ataxia. PME is a rare, often fatal syndrome, initially responsive to antiepileptic drugs which over time becomes refractory and can be associated with cognitive decline. Linkage analysis was performed and the disease locus narrowed to chromosome 19p13.3. Fourteen candidate genes were screened by conventional Sanger sequencing and in one, LMNB2, a novel homozygous missense mutation was identified that segregated with the PME in the family. Whole exome sequencing excluded other likely pathogenic coding variants in the linked interval. The p.His157Tyr mutation is located in an evolutionarily highly conserved region of the alpha-helical rod of the lamin B2 protein. In vitro assembly analysis of mutant lamin B2 protein revealed a distinct defect in the assembly of the highly ordered fibrous arrays typically formed by wild-type lamin B2. Our data suggests that disruption of the organisation of the nuclear lamina in neurons, perhaps through abnormal neuronal migration, causes the epilepsy and early ataxia syndrome and extends the aetiology of PMEs to include dysfunction in nuclear lamin proteins.
Introduction
Progressive myoclonus epilepsies (PMEs) are a rare group of predominantly recessive disorders that present with action myoclonus, tonic-clonic seizures and progressive neurological decline. Clinically, these disorders are divided into two groups based on preservation or loss of cognition. The former group includes Unverricht-Lundborg disease caused by mutations in CSTB (1), PME with or without glomerulosclerosis owing to mutations in SCARB2 (2) and PME plus early ataxia associated with mutation in GOSR2 (3) and KCNC1 (4). The latter group includes Lafora disease and neuronal ceroid lipofuscinoses (NCLs) that both have distinctive pathological diagnoses and for which at least ten genes have been causally linked [most reviewed in (5)]. PME genes encode proteins with diverse functions including lysosomal proteolysis (e.g. CSTB and CTSF) and vesicle trafficking (e.g. CLN3 and GOSR2). However, there are cases that are not explained by mutation of any known genes, suggesting further genetic heterogeneity. While substantial progress has been made in determining some of the genes associated with PMEs comparatively little is known about the mechanisms that underlie them.
Herein, we report a family segregating autosomal recessive PME with early ataxia. We mapped the disease to a novel locus on chromosome 19 and identified a homozygous missense mutation in the LMNB2 gene that encodes lamin B2, one of the four principal human nuclear lamin proteins (in germ cells special splice forms such as lamin B3 and lamin C2 are found). Our in vitro functional studies implicate this mutation in the pathogenesis of PME and early ataxia. In contrast to LMNA, for which over four hundred mutations have been found leading to a complex set of at least fourteen partially overlapping disease entities (6) including premature ageing (Hutchinson–Gilford progeria syndrome) and Emery–Dreifuss muscular dystrophy, no definitive pathogenic mutations have been reported in LMNB2. Interestingly, a mouse knockout model of LMNB2 has a neuronal migration defect resembling lissencephaly (7), and it was suggested that mutation of this gene might cause human neurologic disease (8,9).
Results
Clinical presentation
The proband (Fig. 1A, arrow) of the Palestinian Arab family was developmentally normal until age 7 years when myoclonic seizures began. By 9 years, her gait deteriorated, seizures were associated with falling and tonic-clonic seizures commenced. By 10 years, she required a wheelchair owing to frequent falls and seizures became resistant to anti-epileptic drugs. On examination, there was severe action myoclonus involving limbs and bulbar muscles. She had scoliosis and diffuse muscle wasting, and there was diffuse loss of subcutaneous fat. Cognition was normal. Severe action myoclonus continued through her teens, with tonic-clonic seizures occurring approximately monthly. Her cognition remained intact despite worsening epilepsy. She died at 20 years owing to status epilepticus. Post-mortem examination was not performed. MRI at 9 years was normal (Fig. 1B). An expansion in the known PME gene CSTB was excluded by direct sequencing.
Figure 1.

Family pedigree and magnetic resonance imaging. The six-generation Palestinian Arab family showing segregation of PME and ataxia in an autosomal recessive pattern. Double line consanguineous event; diagonal line deceased. (B) Normal brain MRI of proband at 9 years. (C) Brain MRI of younger affected sister at 18 months showing callosal agenesis, a large interhemispheric cyst and simplified gyration frontally.
The epilepsy of the proband's younger sister began at 6 years with morning myoclonus, followed by tonic-clonic seizures at 6.5 years with a very similar seizure pattern and course to the proband. However, she also had developmental delay, first talked at 20 months, walked at 2 years and only ever ran clumsily until her gait deteroriated with seizure onset in her seventh year. She attended a special school; though her intellect was low it remained stable until last evaluated at age 12 years. Examination showed she was small and wasted with little subcutaneous fat (18 kg at age 9 years); she had short thumbs, curved digits, a small tongue and scoliosis. MRI at 18 months showed complete agenesis of the corpus callosum, venticulomegaly, a left-sided interhemispheric cyst and simplified gyration frontally (Fig. 1C).
The parents and siblings were phenotypically normal. There was no history of lipodystrophy, and full examination of the skin of the father and two siblings was unrevealing.
Linkage analysis reveals a single locus on chromosome 19
Using an autosomal recessive model, a single significant linkage peak was found extending 3 138 680 bp from the telomeric end of the p arm of chromosome 19 (19p13.3; 9.86 cM) with a maximum LOD score of 2.49 (Fig. 2A). No other regions with a LOD score of >1.0 were identified. As expected for a homozygous recessive model, haplotype analysis inferred that the affected siblings were homozygous for the region on 19p13.3 (Fig. 2B).
Figure 2.
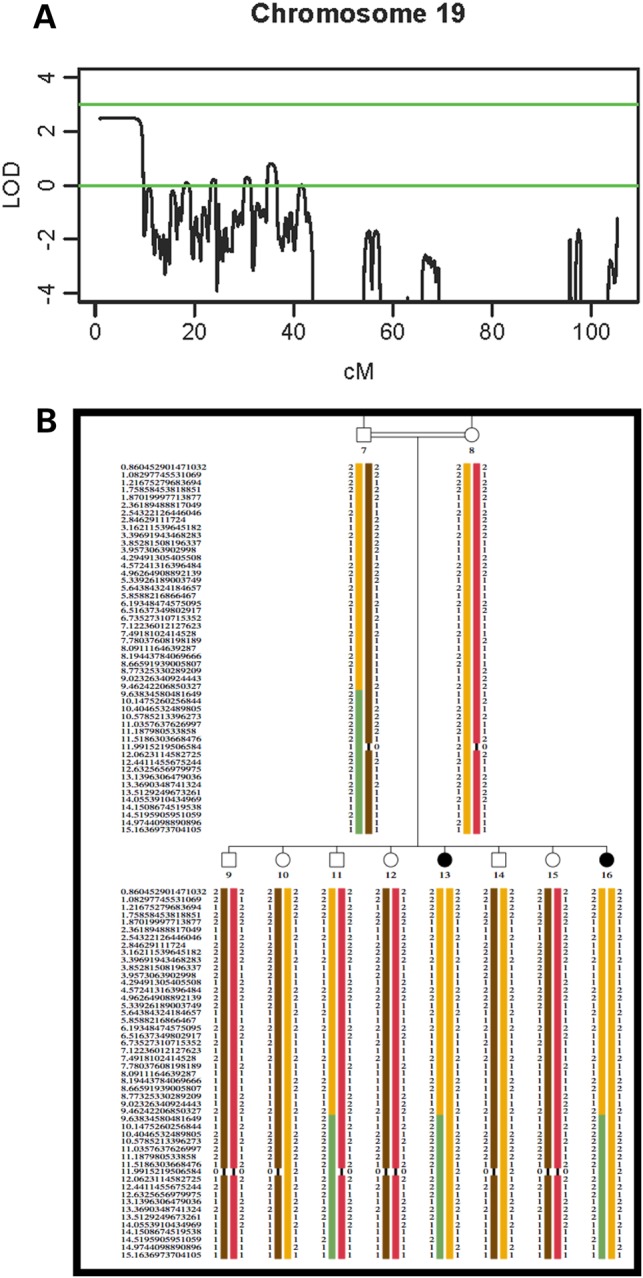
Linkage Mapping (A) LOD scores on chromosome 19. A maximum genome-wide significant LOD score of 2.49 was identified for a region of ∼9.86 cM on the p arm of chromosome 19. (B) Haplotype analysis of the Israeli Arab family. The disease haplotype is shown in orange.
Candidate gene screening at the chromosome 19 region
The linked region contains at least 80 genes and of these fourteen candidate genes (ATP8B3, ATP5D, CNN2, EFNA2, GAMT, GRIN3B, HCN2, LMNB2, NDUFS7, PALM, PLEKHJ1, POLRMT, POLR2E and TIMM13) were selected based on known association with a neurological disease, neural expression or function as an ion channel or transporter. The coding regions and splice sites of all genes were Sanger sequenced in the proband and her affected sister. A novel, homozygous missense mutation (c.469C>T, p.His157Tyr) was identified in the LMNB2 gene (RefSeq NM_032737). This is the most recent reference sequence for the gene—in a previous version, this residue was annotated His137 because the new sequence is 20 residues longer (Figs 1A and 3A). This mutation was absent from Arab (n = 104) and blood bank (n = 282) controls as well as publicly available databases. The p.His157Tyr variant was predicted to be damaging to the lamin B2 protein by pathogenicity prediction programs PolyPhen-2 (score = 0.823) and SIFT (score = 0). The His157 residue is highly conserved (Fig. 3B) and located in a coiled-coil domain important for stability of the protein.
Figure 3.

The LMNB2 mutation causes an amino acid change in a highly conserved domain of lamin B2. (A) Sequence chromatograms showing the wild-type sequence (upper line) and the c.469C>T (p.His157Tyr) mutation (lower line). The mutated nucleotide is marked in red. (B) Amino acid sequence alignment of the center eight heptads of coil 1B of lamin B2 for members of three different vertebrate classes: human (H. sapiens, accession NM_032737.3), chicken (G. gallus, accession NM_205285.1) and frog (X. laevis, accession NM_001087478.1). Identical amino acids are highlighted in turquois, positions where two amino acids are identical and the third is of homologous character are highlighted in yellow. Positions of the heptad repeat, mediating coiled-coil formation of a-helices, are indicated below the amino acid sequences. For the organization of subdomains in lamins, see (13,35) and references therein. The H157 residue is conserved from frog to man in an overall highly conserved segment of two and a half heptads. Note that through the introduction of a tyrosine in this position, two consecutive bulky aromatic residues are found in an a- and b-position, which is unfavorable for the stability of a coiled coil.
Whole exome sequencing to exclude other genes
To exclude other genes in the 19p13.3 region, we performed whole exome sequencing on the proband. In addition to the LMNB2 mutation, in the linked interval, we identified five coding variants in other genes that were all reported single-nucleotide polymorphisms (Table 1). This analysis combined with the absence of this variant in the Exome Aggregation Consortium (∼63 000 exomes), Exome Variant Server (∼6500 exomes) and 1000 Genomes data sets provides further evidence that the LMNB2 mutation was the cause of the PME and ataxia in this family.
Table 1.
Exome variants in genes at the chromosome 19 locus based on hg19
| Gene | Variant | Genomic site | Type | Validation |
|---|---|---|---|---|
| ABCA7 | G>C | 19:1056492 | Non-synonymous | rs3752246 |
| AP3D1 | T>C | 19:2109157 | Non-synonmous | rs25673 |
| BTBD2 | A>G | 19:1997363 | Synonymous | rs1610045 |
| LMNB2 | C>T | 19:2438462 | Non-synonmous | c.469C>T (p.His157Tyr) |
| MUM1 | G>A | 19:1360575 | Non-synonmous | rs3826942 |
| ZNF77 | C>A | 19:2934694 | Non-synonmous | rs76396690 |
| POLRE2 | C>T | 19:1094004 | Non-synonmous | rs12459404 |
| GRIN3B | G>A | 19:1003433 | Non-synonmous | rs367698479 |
Resequencing of LMNB2
We searched for other LMNB2 mutations in our PME cases (n = 87) but did not find any potential pathogenic mutations by Sanger or exome sequencing (4). In a previous study, it was shown that the Lmnb2 knockout mouse has a neuronal migration defect reminiscent of lissencephaly (7). Based on this, we Sanger-sequenced our cohort of patients with lissencephaly or similar brain malformations (n = 47). However, we did not find any LMNB2 mutations.
LMNB2 mutation disrupts fibrillar assembly
The assembly properties of higher vertebrate lamin B2 and domain-deleted versions thereof have been established with recombinant chicken protein under various assembly conditions (10,11). The conditions employed were orientated on their protocol and on previous assembly studies performed with rat lamin A, although both proteins do differ in their properties to some extent owing to their different charge distribution and isoelectrical point (12). Here, we tested different assembly protocols for wild-type and mutant p.His157Tyr lamin B2 (refer to Materials and Methods). In the most robust ‘basic’ standard ‘Mes-calcium’ protocol, clear-cut differences in the assembly of the mutant protein compared with the wild-type protein were evident already in overview images: whereas lamin B2 formed the regular and extended ‘needles’, i.e. laterally associated fibre arrays, the mutant protein organized into irregularly connected, network-type structures (compare Fig. 4A and B). At higher magnification, it was revealed that the principal 24.5-nm repeat pattern exhibited by lamin B2 was largely absent in the mutant protein fibre strands (Fig. 4C and D). Principally, the same results were obtained when assembly was carried out at pH 8.5 instead of pH 6.5 (Fig. 4E and F). Also here lamin B2 formed extended so-called paracrystalline arrays, i.e. regular lateral connections of multiple anti-parallel organized strands of head-to-tail associated coiled-coil dimers (Fig. 4E, filled arrow), whereas the mutant did lack this type of higher-order arrangement (Fig. 4F, open arrows). Only in some regions of the mutant protein fibre bundles, a ‘premature’ type of 24.5-nm repeat was observed (Fig. 4F, filled arrow). Very similar results were obtained at 120 min of assembly indicating the mutant protein also after extended assembly time did not achieve a stable cooperative lateral organisation, in contrast to the wild-type protein that appeared still well organized (data not shown).
Figure 4.

LMNB2 protein assembly. Wild-type lamin B2 (A, C, E) and the mutant H157T-lamin B2 (B, D, F) were assembled in (A–D) 25 mm Mes-NaOH (pH 6.5), 150 mm NaCl, 25 mm CaCl2; and (E and F) 25 mm Tris–HCl (pH 8.5), 150 mm NaCl, 25 mm CaCl2, at room temperature for 45 min (A and B) and 60 min (C–F), respectively. The open arrows in (F) point to extended regions within the filamentous strands formed by the mutant protein that do not exhibit any ordered lateral organization; the filled arrows in (E) point to regions exhibiting the typical 24.5-nm repeat pattern (11) that on occasion is also seen within restricted regions in the mutant polymer (F). Refer to Results section for further explanation. Scale bars: (A and B) 250 nm, (C–F) 100 nm.
Hence, the longitudinal assembly was not affected by the mutation His157Tyr, in agreement with the fact that this amino acid is not close to either end of the α-helical central ‘rod’ domain, and it is these end segments of the rod that mediate the ‘head-to-tail’ overlap in the process of longitudinal annealing of dimeric coiled-coil chains (13). After 2 h of assembly, the mutant protein did not achieve a stable cooperative lateral organization, in contrast to the wild-type protein, which appeared still well organized, particularly evident in thinner paracrystal sheets. Moreover, when assembly was carried out at higher pH (e.g. Tris–HCl buffer pH 8.5), which Heitlinger and colleagues (10,11) pointed out as a meaningful way to follow structure formation by lamin B2, the wild-type protein generated regular associations whereas the mutant again failed to laterally associate regularly (data not shown).
Discussion
Nuclear lamins are divided into two classes according to their amino acid sequence and biochemical behaviour: A-type (lamins A, C and C2 encoded by the LMNA gene) and B-type (lamin B1 encoded by LMNB1; lamins B2 and B3 encoded by the LMNB2 gene). Both types of proteins form a scaffolding meshwork at the inner nuclear membrane known as the nuclear lamina; however, they topologically segregate nearly completely (14). LMNB2 is expressed ubiquitously with highest expression in brain cells (www.proteinatlas.org). Lamin gene mutations have been reported in an array of human diseases including muscular dystrophy (15) and leukodystrophy (16). The vast majority of these mutations have been reported in LMNA. Duplication of LMNB1 results in myelin defects in leukodystrophy (16). This is the first report linking LMNB2 mutation to epilepsy. In mice, homozygous LMNB2 knockout induces a dramatic neuronal layering defect in the cerebral cortex and cerebellum resembling lissencephaly (7). Lamin B2 protein has also been shown to localize to mitochondria in axons and prevent axonal degeneration by maintaining mitochondrial function and axonal integrity (17). Together these data suggest that lamin B2 is a nucleoskeletal protein required for mammalian nuclear migration. There is also evidence that B-type lamins are important for neuronal survival (8,9), which could relate to the neurodegenerative aspects of the PME phenotype reported here.
The family with PME and early ataxia segregated a novel homozygous LMNB2 missense mutation (c.469C>T, p.His157Tyr). This missense mutation was not observed in Arab and Caucasian controls and was not present in publicly available databases including >60 000 exomes. Furthermore, software programs SIFT and PolyPhen 2.0 predicted the change to be damaging to the LMNB2 protein, the His157 residue is highly conserved and the LMNB2 gene is intolerant to change (18). In addition, our in vitro functional assays clearly depict an assembly defect owing to the His157Tyr mutation under multiple assembly modes and experimental conditions, similar to those we have recently reported for LMNA (19). The differences in protein assembly are evident after 60 min of dialysis, with the mutant exhibiting irregular longitudinal fibre organization compared with wild-type fibres, which formed extended, regular paracrystaline structures. The His157 residue is located in the coil 1B domain in a b-position of the heptad pattern. Notably, a phenylalanine is located immediately upstream in the a-position. The presence of these two bulky amino acids side by side in a coiled-coil sequence is highly unusual and is likely to disrupt the coiled-coil structure. Consistent with this, a number of pathogenic mutations have been discovered in the equivalent coil 1B domain of the closely related LMNA protein. For example, the p.Leu140Pro and p.Glu145Lys mutations, both involving residues conserved between these proteins in the coil 1B domain, are reported to cause Emery–Dreifuss muscular dystrophy (20) and progeria (21), respectively.
We performed either Sanger or exome sequencing (4) of a further 87 PME patients without finding a second mutation in LMNB2. While a positive finding would have been confirmatory genetic evidence for pathogeneicity, the negative finding likely reflects the known genetic heterogeneity of PMEs. As lamin A and lamin B are proposed to have a role in glial-guided neuronal migration and the mouse model shows abnormal cortical development (7,8), we also investigated a cohort of 47 patients with cortical malformations but found no sequence variants of interest. The possibility remains that large copy number changes were missed by these approaches. LMNB2 has seven known interacting protein partners (17), and these are also potential candidates for PME.
Our findings strongly indicate LMNB2 is the causative gene for PME and ataxia in this family, evidenced by: (i) a single linkage peak above LOD score 1.0 (at 19p13.3) under a consanguineous autosomal recessive model; (ii) a single novel, homozygous pathogenic mutation in one gene in the linkage region and (iii) a protein assembly assay showing a defect in fibrillar formation owing to the mutation. LMNB2 adds to the genetic heterogeneity of PMEs and introduces a new biochemical pathway for further investigation into this syndrome.
Materials and Methods
Subjects
This study was approved by the Institutional Review Board of the Tel Aviv Sourasky Medical Centre and the Human Research Ethics Committee of Austin Health, Melbourne, Australia. Following informed consent, a six-generational consanguineous Israeli Arab family segregating PME and early ataxia was studied (Fig. 1). Affected individuals underwent detailed phenotyping where all previous medical records, electroencephalography and neuroimaging data were analysed. Ten millilitres of whole blood was obtained and genomic DNA extracted as described previously (22).
SNP genotyping and linkage analysis
Ten family members (Fig. 1) were genotyped for 660 000 SNPs using the Illumina 660 Quad Beadchip array (San Diego, CA, USA) at the Australian Genome Research Facility (AGRF, Melbourne, Australia). Genotyping data were pre-processed by Linkdatagen (23) for parametric multipoint linkage analysis using MERLIN (24). This linkage analysis was performed specifying a fully penetrant recessive disease model and rare disease allele frequency.
Whole exome sequencing
To sequence the exome, 3 μg of genomic DNA from the proband was sonicated to ∼200-bp fragments and adaptor-ligated to make a library for paired-end sequencing. Following amplification, the libraries were hybridized to biotinylated complementary RNA oligonucleotide baits from the SureSelect Human All Exon 50-Mb Kit (Agilent Technologies, Santa Clara, CA, USA) and purified using streptavidin-bound magnetic beads as described previously (25). A 12-cycle amplification was conducted prior to sequencing on the SOLiD v4 system (Applied Biosystems, Grand Island, NY, USA). The captured sample was run on one quadrant of a SOLiD plate. The exome design covers 50 Mb of human genome that includes all exons annotated in the GENCODE project (reference annotation for the ENCODE project; Wellcome Trust Sanger Institute) and consensus CDS database (CCDS—March 2009) as well as 10 base pairs of flanking sequence for each targeted region and small non-coding RNAs from miRBase (v.13) and Rfam.
For analysis of sequencing data, the Genome Analysis Toolkit (GATK) (26) was used that includes (i) alignment, (ii) post-processing alignment and quality control, (iii) calling variants and genotyping, (iv) variant annotation and finally, (v) in silico variant effect prediction. The alignment to the human reference genome (hg19, GRCh37) was performed using Novoalign version 2.08.01 (http://novocraft.com). Alignment post-processing that includes removing duplicate reads, local realignment and base quality recalibration was performed using GATK and Picard (http://picard.sourceforge.net) tools. Variants were called using the Unified Genotyper implemented within GATK and the mpileup method in the SAMtools package (27,28). Variants were manually filtered based on variant allele frequency and coverage. Annotation of the variants was performed using snpEff (29). Functional impact of the coding variants was predicted using the PolyPhen-2 (30) and SIFT (31) online tools.
PCR and sanger sequencing
Candidate genes and exome variants were amplified using gene-specific primers (LMNB2 gene, Table 2; oligonucleotides for other genes available on request) designed to reference human gene transcripts (RefSeq NM_032737.3). Amplification reactions were cycled using a standard protocol on a Veriti Thermal Cycler (Applied Biosystems, Carlsbad, CA, USA). Bidirectional sequencing of all exons and flanking regions was completed with a BigDye™ v3.1 Terminator Cycle Sequencing Kit (Applied Biosystems), according to the manufacturer's instructions. Sequencing products were resolved using a 3730xl DNA Analyzer (Applied Biosystems). All sequencing chromatograms were compared with published cDNA sequence (Ensembl; www.ensembl.org); nucleotide changes were detected using Codon Code Aligner (CodonCode Corporation, Dedham, MA, USA). Coding variants were checked against Exome Aggregation Consortium (Cambridge, MA, USA), Exome Variant Server and 1000 Genomes.
Table 2.
Oligonucleotides used to sequence the LMNB2 gene
| Exon | Forward (5′–3′) | Reverse (5′–3′) |
|---|---|---|
| 1 | GGCGGTCGGACTACCAGT | GAAACCCCGCGGAAAC |
| 2 | CTCTCTCTCTGCGTGCCAGT | AGAGAGGATGGGAGCTGTGA |
| 3 | CACTTGCCCTCTTGACTTTCA | GTGCCAGGATCAGGGTGAC |
| 4 | CCCAGCAGGGTATATGCAAC | AGGGTGGACTTTGCGTTTC |
| 5 | GTGCACAAGGAACCAGGAAT | GTCCAGCTGTGGGGAGAC |
| 6 | AGCTGGAGCAGACCTACCAG | GATGCCAAGAGGCCAGAG |
| 7 | ATGGGCTGCCCTGACCTT | CCTCACTCTGTGCTCCCAAG |
| 8 | TGTCTGGGGCTCCCACTA | GGTCACCTTGTCCGAGTTGT |
| 9 | AGGTGTGTGCCGTGTCTTC | CAAGTCCTGTGCCTCCAGTC |
| 10 | GAAGGGGGCTTTGTGGTAG | CCTCCCCATTCTCATTCTCA |
| 11 | AGTCCTGGGCAGGGTCTG | GTGGGCACAGGGGTCCTAC |
| 12(1)a | CCTTCCTTCCTTCCTTCCTG | ACGCCTGGATTCTGAATTTG |
| 12(2)a | CCCACTCCTCATCTCACCAT | TTCTTCCTTCCCCAGGTCTT |
aExon divided into two amplimers.
In vitro protein assays
The full-length cDNA clone coding for human lamin B2 [see (32)] was sub-cloned into the bacterial expression vector pET24a, and protein was expressed in E. coli as described for chicken lamin B2 and Caenorhabditis elgans B-type lamin, respectively (10,33). The human lamin B2 clone is 20 amino acids longer than the one originally described in databases; eventually, the full-length version was independently verified and published in Pubmed (NCBI Reference Sequence: NP_116126.3). Isolation from inclusion bodies and standard ion-exchange chromatography in buffers containing 8 m urea yielded highly pure proteins, which were stored in 8 m urea at −80°C (34).
For assembly studies, we prepared two independent preparations of both wild-type lamin B2 and the mutant H157Y-lamin B2 protein. From each bacterial suspension used to generate the four protein isolates, aliquots were taken for plasmid purification and subsequent complete sequence analysis. We employed two different protocols to study assembly: (i) ‘Direct dialysis’—the proteins (0.1–0.2 mg/ml) were dialyzed into (A) ‘buffer 1’ (50 mm Mes-NaOH, pH 6.5); (B) ‘buffer 1’ with 150 mm NaCl; (C) ‘buffer 1’ with 150 mm NaCl and 25 mm CaCl2; (D) 25 mm Mes-NaOH, pH 6.5, 150 mm NaCl and 25 mm CaCl2. (ii) ‘Stepwise dialysis’—the proteins (0.1–0.2 mg/ml) were equilibrated first by dialysis first into ‘buffer 2’ (25 mm Tris–HCl, pH 8.5, 150 mm NaCl, 1 mm DTT) and then by further dialysis into (A) 25 mm Tris–HCl, pH 8.5, 150 mm NaCl, 25 mm CaCl2; (B) 25 mm Mes-NaOH, pH 6.5, with 150 mm NaCl, 25 mm CaCl2; (C) 25 mm Mes-NaOH, pH 6.5. At appropriate time points, samples were taken out of the dialysis bag and applied to freshly glow discharged, carbon-coated copper grids. After 1 min of adsorption, the grids were washed in a drop of distilled water, stained with uranyl acetate and washed again with distilled water. At least two independent and complete assembly experiments were performed with each of the protein isolates mentioned earlier. Electron microscopy was performed at a ZEISS 900 transmission electron microscope (Carl Zeiss, Oberkochen, Germany) and images recorded with a CCD camera.
Authors’ Contributions
S.F.B. initiated the project. J.A.D., M.S.H. and S.F.B. directed the project. J.A.D., T.A., H.H.M.D. and M.S.H. completed molecular genetics analyses. M.S.H., A.E.S., M.M., A.E.L. and R.J.H.S. performed exome sequencing. M.B. performed linkage analysis. N.H. and K.M. conducted exome sequencing analysis. Z.A., A.M., K.L.O., R.J.L., I.E.S., A.D.K. and S.F.B. conducted clinical phenotyping. H.H. and M.M. performed in vitro protein assays. J.A.D., M.S.H. and S.F.B. wrote the paper. All authors discussed the results and commented on the manuscript.
Funding
This work was supported by National Health and Medical Research Council (NHMRC) Program Grant (628952) to S.F.B. and I.E.S., an Australia Fellowship (466671) to S.F.B., a Practitioner Fellowship (1006110) to I.E.S and a Career Development Fellowship (1063799) to M.S.H. M.B. was supported by an Australian Research Council (ARC) Future Fellowship (FT100100764) and NHMRC Program Grant (APP1054618). This work was also supported by Victorian State Government Operational Infrastructure Support and Australian Government NHMRC IRIISS funding. H.H. received funding from the German Research Foundation (DFG HE 1853/11-1, FOR 1228).
Acknowledgements
We thank the family for their participation in this study. Elena Aleksoska (Epilepsy Research Centre) is acknowledged for technical support and sample management. We also thank Stephen Young (UCLA) for his comments on the manuscript.
Conflict of Interest statement. None declared.
References
- 1. Lafreniere R.G., Rochefort D.L., Chretien N., Rommens J.M., Cochius J.I., Kalviainen R., Nousiainen U., Patry G., Farrell K., Soderfeldt B., et al. (1997) Unstable insertion in the 5′ flanking region of the cystatin B gene is the most common mutation in progressive myoclonus epilepsy type 1, EPM1. Nat. Genet., 15, 298–302. [DOI] [PubMed] [Google Scholar]
- 2. Berkovic S.F., Dibbens L.M., Oshlack A., Silver J.D., Katerelos M., Vears D.F., Lullmann-Rauch R., Blanz J., Zhang K.W., Stankovich J., et al. (2008) Array-based gene discovery with three unrelated subjects shows SCARB2/LIMP-2 deficiency causes myoclonus epilepsy and glomerulosclerosis. Am. J. Hum. Genet., 82, 673–684.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Corbett M.A., Schwake M., Bahlo M., Dibbens L.M., Lin M., Gandolfo L.C., Vears D.F., O'Sullivan J.D., Robertson T., Bayly M.A., et al. (2011) A mutation in the golgi Qb-SNARE Gene GOSR2 causes progressive myoclonus epilepsy with early ataxia. Am. J. Hum. Genet., 88, 657–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Muona M., Berkovic S.F., Dibbens L.M., Oliver K.L., Maljevic S., Bayly M.A., Joensuu T., Canafoglia L., Franceschetti S., Michelucci R., et al. (2015) A recurrent de novo mutation in KCNC1 causes progressive myoclonus epilepsy. Nat. Genet., 47, 39–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kousi M., Lehesjoki A.E., Mole S.E. (2012) Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Hum. Mutat., 33, 42–63 [DOI] [PubMed] [Google Scholar]
- 6. Young S.G., Jung H.J., Lee J.M., Fong L.G. (2014) Nuclear lamins and neurobiology. Mol. Cell. Biol., 34, 2776–2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Coffinier C., Chang S.Y., Nobumori C., Tu Y., Farber E.A., Toth J.I., Fong L.G., Young S.G. (2010) Abnormal development of the cerebral cortex and cerebellum in the setting of lamin B2 deficiency. Proc. Natl Acad. Sci. USA, 107, 5076–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Coffinier C., Jung H.J., Nobumori C., Chang S., Tu Y., Barnes R.H., 2nd, Yoshinaga Y., de Jong P.J., Vergnes L., Reue K., et al. (2011) Deficiencies in lamin B1 and lamin B2 cause neurodevelopmental defects and distinct nuclear shape abnormalities in neurons. Mol. Biol. Cell, 22, 4683–4693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Young S.G., Jung H.J., Coffinier C., Fong L.G. (2012) Understanding the roles of nuclear A- and B-type lamins in brain development. J. Biol. Chem., 287, 16103–16110 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Heitlinger E., Peter M., Haner M., Lustig A., Aebi U., Nigg E.A. (1991) Expression of chicken lamin B2 in Escherichia coli: characterization of its structure, assembly, and molecular interactions. J. Cell. Biol., 113, 485–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Heitlinger E., Peter M., Lustig A., Villiger W., Nigg E.A., Aebi U. (1992) The role of the head and tail domain in lamin structure and assembly: analysis of bacterially expressed chicken lamin A and truncated B2 lamins. J. Struct. Biol., 108, 74–89. [DOI] [PubMed] [Google Scholar]
- 12. Aebi U., Cohn J., Buhle L., Gerace L. (1986) The nuclear lamina is a meshwork of intermediate-type filaments. Nature, 323, 560–564. [DOI] [PubMed] [Google Scholar]
- 13. Kapinos L.E., Schumacher J., Mucke N., Machaidze G., Burkhard P., Aebi U., Strelkov S.V., Herrmann H. (2010) Characterization of the head-to-tail overlap complexes formed by human lamin A, B1 and B2 ‘half-minilamin’ dimers. J. Mol. Biol., 396, 719–731. [DOI] [PubMed] [Google Scholar]
- 14. Shimi T., Pfleghaar K., Kojima S., Pack C.G., Solovei I., Goldman A.E., Adam S.A., Shumaker D.K., Kinjo M., Cremer T., et al. (2008) The A- and B-type nuclear lamin networks: microdomains involved in chromatin organization and transcription. Genes. Dev., 22, 3409–3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lassuthova P., Barankova L., Kraus J., Marikova T., Seeman P. (2009) Emery-Dreifuss muscular dystrophy: a novel mutation in the LMNA gene. Pediatr. Neurol., 41, 127–130. [DOI] [PubMed] [Google Scholar]
- 16. Padiath Q.S., Saigoh K., Schiffmann R., Asahara H., Yamada T., Koeppen A., Hogan K., Ptacek L.J., Fu Y.H. (2006) Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat. Genet., 38, 1114–1123. [DOI] [PubMed] [Google Scholar]
- 17. Yoon B.C., Jung H., Dwivedy A., O'Hare C.M., Zivraj K.H., Holt C.E. (2012) Local translation of extranuclear lamin B promotes axon maintenance. Cell, 148, 752–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Petrovski S., Wang Q., Heinzen E.L., Allen A.S., Goldstein D.B. (2013) Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet., 9, e1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Alastalo T.P., West G., Li S.P., Keinanen A., Helenius M., Tyni T., Lapatto R., Turanlahti M., Heikkila P., Kaariainen H., et al. (2015) LMNA mutation c.917T>G (p.L306R) leads to deleterious hyper-assembly of lamin A/C and associates with severe right ventricular cardiomyopathy and premature aging. Hum. Mutat., 27, 22793. [DOI] [PubMed] [Google Scholar]
- 20. Boriani G., Gallina M., Merlini L., Bonne G., Toniolo D., Amati S., Biffi M., Martignani C., Frabetti L., Bonvicini M., et al. (2003) Clinical relevance of atrial fibrillation/flutter, stroke, pacemaker implant, and heart failure in Emery-Dreifuss muscular dystrophy: a long-term longitudinal study. Stroke, 34, 901–908. [DOI] [PubMed] [Google Scholar]
- 21. Eriksson M., Brown W.T., Gordon L.B., Glynn M.W., Singer J., Scott L., Erdos M.R., Robbins C.M., Moses T.Y., Berglund P., et al. (2003) Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature, 423, 293–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grimberg J., Nawoschik S., Belluscio L., McKee R., Turck A., Eisenberg A. (1989) A simple and efficient non-organic procedure for the isolation of genomic DNA from blood. Nucl. Acids. Res., 17, 8390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bahlo M., Bromhead C.J. (2009) Generating linkage mapping files from Affymetrix SNP chip data. Bioinformatics., 25, 1961–1962. [DOI] [PubMed] [Google Scholar]
- 24. Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. (2002) Merlin--rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet., 30, 97–101. [DOI] [PubMed] [Google Scholar]
- 25. Zheng J., Miller K.K., Yang T., Hildebrand M.S., Shearer A.E., DeLuca A.P., Scheetz T.E., Drummond J., Scherer S.E., Legan P.K., et al. (2011) Carcinoembryonic antigen-related cell adhesion molecule 16 interacts with alpha-tectorin and is mutated in autosomal dominant hearing loss (DFNA4). Proc. Natl Acad. Sci. USA, 108, 4218–4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M., et al. (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet., 43, 491–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li H. (2011) Improving SNP discovery by base alignment quality. Bioinformatics, 27, 1157–1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cingolani P., Platts A., Wang le L., Coon M., Nguyen T., Wang L., Land S.J., Lu X., Ruden D.M. (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin), 6, 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. (2010) A method and server for predicting damaging missense mutations. Nat. Methods., 7, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kumar P., Henikoff S., Ng P.C. (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc., 4, 1073–1081. [DOI] [PubMed] [Google Scholar]
- 32. Schumacher J., Reichenzeller M., Kempf T., Schnolzer M., Herrmann H. (2006) Identification of a novel, highly variable amino-terminal amino acid sequence element in the nuclear intermediate filament protein lamin B(2) from higher vertebrates. FEBS. Lett., 580, 6211–6216. [DOI] [PubMed] [Google Scholar]
- 33. Foeger N., Wiesel N., Lotsch D., Mucke N., Kreplak L., Aebi U., Gruenbaum Y., Herrmann H. (2006) Solubility properties and specific assembly pathways of the B-type lamin from Caenorhabditis elegans. J. Struct. Biol., 155, 340–350. [DOI] [PubMed] [Google Scholar]
- 34. Herrmann H., Kreplak L., Aebi U. (2004) Isolation, characterization, and in vitro assembly of intermediate filaments. Methods Cell. Biol., 78, 3–24. [DOI] [PubMed] [Google Scholar]
- 35. Meier M., Padilla G.P., Herrmann H., Wedig T., Hergt M., Patel T.R., Stetefeld J., Aebi U., Burkhard P. (2009) Vimentin coil 1A-A molecular switch involved in the initiation of filament elongation. J. Mol. Biol., 390, 245–261. [DOI] [PubMed] [Google Scholar]