

Abstract
Objectives
Several drug resistance and secondary mutations have been described in HIV-1 viruses from patients undergoing antiretroviral therapy. In this study, we assessed the impact of the protease substitution T74S on the phenotype and on the replicative fitness in HIV-1 subtypes B and C.
Methods
HIV-1 molecular clones carrying subtype B or C proteases had these coding regions subjected to site-directed mutagenesis to include T74S alone or in combination with four known protease inhibitor (PI) primary drug resistance mutations. All clones were used in a phenotypic assay to evaluate their susceptibility to most commercially available PIs. The impact of T74S on virus fitness was also assessed for all viruses through head-to-head competitions and oligonucleotide ligation assays to measure the proportion of each virus in culture.
Results
Viruses of both subtypes carrying T74S did not have their susceptibility altered to any tested PI. Viruses with the four resistance mutations showed strong resistance to most PIs with fold changes ranging from 5 to 300 times compared with their wild-type counterparts. Surprisingly, the addition of T74S to the multiresistant clones restored their susceptibilities to indinavir and ritonavir and partially to lopinavir, close to those of wild-type viruses. Most 74S-containing viruses were more fit than their 74T counterparts.
Conclusions
Our results suggest that T74S is not a major drug resistance mutation, but it resensitizes multiresistant viruses to certain PIs. T74S is a bona fide accessory mutation, restoring fitness of multidrug-resistant viruses in both subtypes B and C. T74S should be further studied in clinical settings and considered in drug resistance interpretation algorithms.
Keywords: antiretroviral, drug resistance, viral fitness
Introduction
Despite the successful worldwide antiretroviral (ARV) treatment policies implemented to contain the HIV/AIDS pandemic, the high plasticity of the HIV-1 genome results in rapid acquisition and fixation of drug resistance and associated mutations to all ARV classes. Mutations can be classified into primary (or major) and secondary (or accessory). Primary mutations confer decreased susceptibility to a given drug, while secondary changes are generally compensatory, to accommodate primary mutations and restore viral fitness, but can also further decrease drug susceptibility in combination with primary mutations.1
Over 25 amino acid substitutions have been identified as mutations selected under protease (PR) inhibitor (PI) therapy,2,3 and newly identified PI-associated mutations may play roles in functional compensation, drug resistance and even hypersusceptibility to PIs.4–6 Moreover, HIV genetic diversity (subtypes) is speculated to impact on the efficacy of ARV treatment, particularly in HIV-1 non-B subtypes. In vitro studies suggest that some non-B subtype samples have distinct patterns of susceptibility to certain ARV classes. For example, subtype G isolates have different susceptibility to some PIs compared with subtype B,7,8 while subtype C strains from Africa and Brazil are reported to be hypersusceptible to lopinavir.9 In addition, non-B subtypes carry several polymorphisms that are described as secondary resistance mutations for subtype B.10,11
In studies on HIV-1 subtype C-infected individuals in southern Brazil,12,13 we found that the PR T74S mutation was observed at higher frequencies in viruses from PI-treated versus untreated subjects. This was consistent with another, multicentric study, where T74S was shown to be selected to a higher frequency with PI treatment in subtype C- as compared with subtype B-infected patients.5 Furthermore, T74S is enriched in patients treated with nelfinavir,14 and appears to compensate for the primary PI resistance mutation M89I/V in subtype G clinical isolates.15 This perceived T74S compensation appears in the context of primary PI drug resistance mutations but alone this mutation does not confer PI resistance. These observations have prompted us to investigate the global compensation of T74S by measuring the replicative fitness of this mutation in the context of subtype B and C PR with or without primary PI resistance mutations. Pairwise competitions were performed to measure relative fitness through a novel oligonucleotide ligation assay (OLA).
Materials and methods
HIV-1 PR clones
We introduced the T74S mutation into the PR of four different molecular clones of HIV-1 by site-directed mutagenesis. The B.wt clone is the prototypical subtype B wild-type infectious clone pNL4-3.16 C.wt clone is pNL43-C6 PR, where the six consensus amino acid signatures of subtype C PR (I15V, M36I, R41K, H69K, L89M and I93L) were introduced into pNL4-3 by site-directed mutagenesis,9 thus turning it into a subtype C PR, indistinguishable from that of a clinical subtype C isolate from a drug-naive subject. The clones B.4 and C.4 are PR genes from pNL4-3 and pNL43-C6, respectively, where four PR primary resistance mutations (M46I, I54V, V82A and L90M) were inserted by site-directed mutagenesis.17 These mutations were selected in view of their high prevalence among PI-exposed, subtype B and C clinical isolates. In a recent search of the Stanford HIV Drug Resistance Database (http://hivdb.stanford.edu; accessed on 28 April 2009), subtype B and C viruses from PI-exposed individuals carrying T74S and combinations of one or more of the above-mentioned primary mutations corresponded to 93% (186/200) and 89% (55/62), respectively. PI-exposed viruses carrying T74S and four primary mutations corresponded to 6% of each subtype.
Site-directed mutagenesis and generation of recombinant viruses
PRs from all four clones described above were PCR amplified and cloned into the pCR4 TOPO vector with the TOPO TA cloning kit (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. Site-directed mutagenesis in each clone was conducted with the Quick Change Kit (Stratagene, La Jolla, CA, USA), using one pair of complementary primers to change the threonine residue at position 74 into a serine (primers 5′-GCTATAGGTTCAGTACTAGTAGGACCTACA-3′ and 5′-TGTAGGTCCTACTAGTACTGAACCTATAGC-3′; underlined bases represent changes introduced). All mutated clones were confirmed by DNA sequencing. Mutant PRs were then co-transfected by electroporation into CD4+ T lymphocytes (MT-4 cells) together with the plasmid pGEMT3ΔPR, which carries a defective HIV-1 HXB2 proviral DNA lacking the PR gene.18In vivo recombination led to the generation of chimeric viruses containing the modified PR genes.
Drug susceptibility phenotypic assays
All eight chimeric viruses (B.wt, C.wt, B.4, C.4, B.74, C.74, B.4.74 and C.4.74) had their 50% tissue culture infectious doses (TCID50) determined as described in the DAIDS Virology Manual for HIV Laboratories (http://aactg.s-3.com/labmanual.htm). Phenotypic susceptibility to the PIs amprenavir, indinavir, lopinavir, ritonavir, saquinavir and nelfinavir was determined by two independent 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)-based cell viability assay experiments in MT-4 cells as previously described.19 Each experiment was conducted in triplicate. Calculations to determine the inhibitory concentration for 50% of viral replication (IC50) were performed using the Analyze-It! v.1.62 program, inside the Microsoft Excel® statistical package and in SigmaPlot® software. Briefly, the percentage of cell viability for each virus and to each drug was plotted in a semi-log scale against the concentrations of each drug. A Hill's three-parameter non-linear regression was conducted and IC50 values were calculated for each virus and drug tested. Results are presented as fold changes (FCs), the ratio between the IC50 for each tested virus and the reference clone B.wt of a given drug.
Dual virus competitions
U87.X4 cell line cultures were infected individually and doubly (head-to-head competition) with the above-mentioned molecular clones at a multiplicity of infection (moi) of 0.0005. Cell supernatants were harvested at 14 days post-infection and an RT-nested PCR was conducted to amplify a 440 bp fragment spanning the PR coding region (outer primers, 5′-GGCTGTTGGAAATGTGG-3′ and 5′-TATGGATTTTCAGGCCC-3′; and inner primers, 5′-AGAGCAGACCAGAGCCAAC-3′ and 5′-ACTGGTACAGTCTCAATAGG-3′). OLAs were then performed with the generated PCR amplicons as described previously,20 and the quantification of ligated, radiolabelled products was performed in a Molecular Imager FX apparatus using Quantity One software (Bio-Rad). All subtype B and subtype C clones (see above) were competed against each other. In addition, the fittest variant of each subtype (B.wt and C.4.74) competed against all viruses of the other subtype (see the Results section).
In vitro fitness OLAs
The fitness of the eight recombinant viruses was assessed through head-to-head competition assays in cell culture, followed by detection of each competing form by an OLA, as described and validated by Lalonde et al.20 Briefly, specific upstream primers were designed to discriminate between the two competing variants from each competition (74T versus 74S, subtype B versus subtype C and wild-type versus multiresistant). To discriminate between subtypes B and C, PR position 15 (an isoleucine in subtype B and a valine in subtype C) was used; for discriminating between wild-type and multiresistant variants, PR position 54 (an isoleucine in wild-type viruses and a valine in resistant viruses) was assessed. All upstream and downstream primers are listed in Table 1. Upstream primers were labelled with [γ-32P]ATP (Perkin-Elmer) as previously described.20
Table 1.
Primers used in the OLAs for discriminating between variants tested in this study
| PR position | Upstream primer | OLA target | Downstream primer |
|---|---|---|---|
| 74 | 5′-CGGAUAUAAAGCTATAGGTA-3′ | 74T | 5′-CAGTAUTAGTAGGACCTACA-3′ |
| 5′-CGGAUAUAAAGCTATAGGTT-3′ | 74S | ||
| 15 | 5′-ACCCCTCGTCACAATAAAGA-3′ | subtype B (15I) | 5′-TAGGGGGGCAATTAAAGGAA-3′ |
| 5′-ACCCCTCGTCACAATAAAGG-3′ | subtype C (15V) | ||
| 54 | 5′-AGGGGGAATTGGAGGTTTTA-3′ | wild-type (54I) | 5′-TCAAAGTAAGACAGTATGAT-3′ |
| 5′-AGGGGGAATTGGAGGTTTTG-3′ | multiresistant (54V) |
Results
Impact of T74S in subtype B and C PR on susceptibility to PIs
PR clones B.wt, C.wt, B.4 and C.4 were all subjected to site-directed mutagenesis to introduce the mutation T74S into their PR coding regions. These four mutated clones were then introduced into a HXB2ΔPR proviral clone by homologous in vivo recombination (see the Materials and methods section). The four original clones (without 74S) were also subjected to the same procedure. Susceptibility to PIs was measured by adding an moi of 0.02 of each virus to microtitre plates containing 106 MT-4 cells and 10-fold dilutions of commercially available PIs. IC50s were calculated for each virus and for each drug at 6 days post-infection, and FCs in IC50 values were generated by dividing the IC50 of each drug for each virus by the IC50 of that drug for B.wt.
Table 2 summarizes all FC values obtained for the eight clones. Similar levels of baseline susceptibility were observed for the C.wt and B.wt viruses to all PIs, except lopinavir, for which C.wt showed hypersusceptibility. Wild-type viruses containing 74S (B.74 and C.74) again showed similar susceptibilities to all PIs but with C.74 displaying hypersusceptibility to lopinavir. Subtype B virus with four primary resistance mutations (B.4) was nearly non-susceptible to indinavir and ritonavir inhibition relative to the inhibition of B.wt, resulting in high FC values of 145 and 240, respectively. Similarly, C.4 had FC values of 192 and 303 for indinavir and ritonavir, respectively. These PI susceptibility results for viruses carrying the mutations 46I, 54V, 82A and 90M were consistent with previous studies. We did, however, observe lower levels of lopinavir resistance with B.4 (FC = 12.8, relative to B.wt) than the high lopinavir resistance with C.4 (FC = 189, relative to C.wt).
Table 2.
FC values for the HIV-1 subtype B and C clones tested in this study
| Clone | NFV (0.034)a (2.2)b | IDV (0.043) (2.4) | APV (0.126) (2.2) | SQV (0.049) (1.8) | LPV (0.053) (1.7) | RTV (0.088) (3.5) |
|---|---|---|---|---|---|---|
| B.wt (NL4-3)c | 1.00 ± 0.10 | 1.00 ± 0.17 | 1.00 ± 0.02 | 1.00 ± 0.04 | 1.00 ± 0.04 | 1.00 ± 0.09 |
| B.74 | 1.85 ± 0.16 | 1.00 ± 0.09 | 1.08 ± 0.02 | 2.67 ± 0.21 | 0.92 ± 0.03 | 1.11 ± 0.05 |
| B.4 | 7.13 ± 0.35 | 145.00 ± 4.15 | 1.00 ± 0.07 | 11.70 ± 0.67 | 12.80 ± 0.62 | 240.00 ± 6.95 |
| B.4.74 | 5.02 ± 0.19 | 1.72 ± 0.09 | 0.81 ± 0.02 | 2.02 ± 0.12 | 2.79 ± 0.20 | 7.04 ± 0.32 |
| C.wt | 0.66 ± 0.13 | 0.87 ± 0.06 | 0.97 ± 0.06 | 0.75 ± 0.14 | 0.34 ± 0.06 | 0.95 ± 0.08 |
| C.74 | 3.32 ± 0.12 | 1.30 ± 0.06 | 1.02 ± 0.02 | 0.77 ± 0.05 | 0.30 ± 0.03 | 0.875 ± 0.04 |
| C.4 | 34.00 ± 1.98 | 192.00 ± 2.58 | 1.10 ± 0.06 | 14.30 ± 0.87 | 189.00 ± 8.06 | 303.00 ± 9.73 |
| C.4.74 | 10.05 ± 1.51 | 4.41 ± 0.11 | 0.80 ± 0.02 | 1.63 ± 0.06 | 29.01 ± 0.46 | 25.70 ± 1.16 |
NFV, nelfinavir; IDV, indinavir; APV, amprenavir; SQV, saquinavir; LPV, lopinavir; RTV, ritonavir.
aIC50 values (μM) for the reference clone of each drug.
bNumbers depict biological cut-offs for each as defined by VIRCO.37 For ritonavir, the first generation is still used, as this drug is no longer used alone.
cB.wt was the reference clone used and therefore all its FC values are normalized to 1; all others are relative to that.
The most striking effect on PI susceptibility was, however, observed with the 74S amino acid linked to 46I, 54V, 82A and 90M in either the B or C PR (B.4.74 and C.4.74). Although 74S did not alter the pattern of susceptibility of these clones (compared with B.4 and C.4) to amprenavir and only slightly to nelfinavir, the 74S mutation antagonized most of the resistance to indinavir and ritonavir in both subtypes. In other words, B.4.74 and C.4.74 had susceptibilities close to B.wt and C.wt viruses to indinavir and ritonavir (Table 2). With lopinavir, levels of resistance fell to a lesser extent from an FC of 12.8 to 2.8 for subtype B and from 189 to 29 for subtype C.
Impact of T74S in subtype B and C PRs on replicative fitness
Although resistance was diminished for some PIs with T74S, viruses harbouring the four primary PI resistance mutations are likely to have lower replicative fitness (as measured in the absence of drug). In addition, the secondary T74S mutation is commonly selected during PI treatment in subtype C- as opposed to subtype B-infected patients. Based on these clinical observations, it was possible that T74S had a greater compensatory role in subtype C than in subtype B PR and in the context of the entire virus. To test this hypothesis, pairwise competitions were performed between the wild-type virus (.wt) and those harbouring the T74S mutation alone (.74), four primary mutations (.4) or the T74S mutation plus the four primary PI resistance mutations (.4.74) in the context of the subtype B or C PR. We also performed competitions with every virus (regardless of mutation and subtype) against B.wt and C.wt. Contrary to a previous report,21 but supported by other studies,22–24 all subtype C PR viruses (with or without the T74S or four primary mutations) were less fit than the B.wt virus. Consistent with this observation, all subtype B PR viruses (with or without the T74S or four primary mutations) were more fit than the C.wt virus (Figure 1). As described below, increased fitness of subtype B over subtype C virus also appears to be governed by PR. Subtype C PR harbouring T74S was more fit than without it. Interestingly, T74S not only improves the fitness of the C.4 PR (46I, 54V, 82A and 90M) but also the replicative fitness of the subtype C.wt (wild-type PR). Both C.4.74 and C.74 outcompeted C.4 and C.wt in direct competition (Figure 2b). In the case of subtype B PR, T74S reduced the fitness of the B wild-type and was fitness neutral in the context of B.4 (46I, 54V, 82A and 90M). Any compensation by T74S was likely minimal, which again reflects the low frequency of selection in PI-treated, subtype B-infected patients as opposed to higher frequency selection in PI-treated, subtype C-infected patients. As expected, subtype B isolates harbouring the four primary mutations were significantly less fit than B.wt in direct competition (Figure 2a). These data suggest that T74S in subtype C PR may be sufficient to restore fitness reduced by 46I, 54V, 82A and 90M while still maintaining PI resistance, albeit lower than without T74S. In the subtype B PR, reduced fitness conferred by 46I, 54V, 82A and 90M may require an alternative compensatory mutation to restore replicative fitness while maintaining drug resistance. As described below, this restoration in fitness in lieu of high-level PI resistance has been previously described with other PI drug mutational patterns.25,26
Figure 1.

Fitness difference (WD) among HIV clones calculated as reported previously.20 (a) Results from all competitions involving the wild-type clade B isolate (B.wt). (b–h) Results from all competitions involving the isolate in the graph title of each panel. Bars reflect fitness differences calculated from pairwise competitions between each isolate. WD < 1 denotes competitions in which the isolate in the x-axis was fitter. WD > 1 denotes competitions where the isolate in the graph title was fitter.
Figure 2.
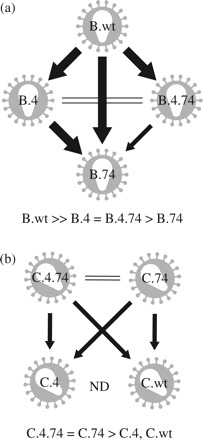
Diagrammatic illustration of fitness differences in pairwise competitions between studied clones. For any pairwise competition, calculation of fitness difference (WD) used the fitter virus in the numerator. Arrows point to isolates with lower fitness. Large arrows, WD > 20; small arrows, WD = 2–20; double lines, WD = 1–2; ND, not determined. (a) Fitness relationships between subtype B clones in the study. (b) Fitness relationships between subtype C clones in the study.
Discussion
This report describes the role of mutation T74S on the phenotype of HIV-1 subtype B and C PRs to PIs, and its impact on fitness restoration of multidrug-resistant viruses. The primary PI-associated mutations used here are the most commonly observed in HIV viruses from PI-failing subjects (with the exception of D30N, which is rarely selected in subtype C and therefore could not be compared). In addition, a high prevalence of those mutations is observed in conjunction with T74S in clinical isolates of PI-treated individuals (see the Materials and methods section for details).
We found out in our drug susceptibility assays that T74S is shown to decrease in vitro resistance to certain PIs, such as indinavir, ritonavir and partly lopinavir, when combined with other, primary PI-associated resistance mutations, despite being apparently selected by PI treatment. This decrease in PI resistance with T74S may be a trade-off to restore a possible drastic decrease in in vivo fitness. We are currently comparing the viral loads of subtype C-infected patients, who may or may not be receiving PIs but are harbouring PI resistance mutations in the presence or absence of T74S. We suspect that patients harbouring multiple primary PI resistance mutations will have lower viral loads than those patients with these mutations and T74S.
The mutation T74S alone failed to confer PI resistance in either subtype B or C backbone, i.e. characteristic of a primary resistance mutation. Even the hypersusceptibility of subtype C to lopinavir, which has been previously described by our group,9 was not altered by 74S. When T74S was introduced into viruses containing a backbone of primary PI resistance mutations, a surprising drop in resistance levels was observed, particularly to indinavir and ritonavir in both subtypes (Table 2). These findings were confirmed by multiple replicates and in different experiments, including instances using viral stocks produced and titred at different times. It is noteworthy that the drops in the FC values seen in the multiresistant clones are of clinical relevance. Recent clinical cut-offs (CCOs) have been described for most PIs based on clinical trials and cohort data.27 CCOs are defined as CCO1 or lower CCO, at which a given drug activity is lost by 20%, and CCO2 or upper CCO, where drug activity is lost by 80%. When the FC values obtained in our study are compared with the reported CCOs, we find that both subtype B and C multiresistant clones (B.4 and C.4) become fully clinically susceptible to saquinavir and indinavir (FC below CCO1) when 74S is present. For nelfinavir and lopinavir, B.4 becomes from fully to partially resistant (FC between CCO1 and CCO2). These observations highlight the relevance of the impact of T74S mutation in HIV PI resistance in the clinical setting.
It has been reported that nelfinavir exposure selects for T74S.14 It is possible that nelfinavir and indinavir/ritonavir exert an antagonistic selective pressure on the appearance of T74S, nelfinavir favouring it and indinavir/ritonavir preventing it. Examples of drug antagonism in HIV resistance have already been reported for both PR and reverse transcriptase inhibitors. The mutation M184V in HIV-1 reverse transcriptase is selected by lamivudine, but resensitizes the enzyme to thymidine analogues such as zidovudine and stavudine,28 and delays the appearance of specific mutations to these drugs.29 PR mutations I47A and V82T, selected, respectively, by indinavir and lopinavir, confer hypersusceptibility to saquinavir.30,31 A similar situation might be taking place with T74S and the use of nelfinavir followed by indinavir/ritonavir.
An important limitation of the current study relies on the fact that we have used constructs harbouring NL4-3 Gag-Pol (i.e. subtype B) precursors that are required to be cleaved by a subtype C PR. Recent reports have demonstrated the involvement of Gag sequence determinants on PI drug resistance.32,33 In this scenario, we might be misevaluating the extent of PI resistance in different HIV-1 subtypes, an issue that will require further evaluation.
An alternative explanation for T74S selection may relate to a restoration of fitness, originally lost or reduced with the appearance of primary PI resistance mutations. Multiresistant viruses commonly have decreased fitness due to the cost of maintaining drug resistance mutations, which are essential to virus survival in the presence of drugs.34 Indeed, our head-to-head competition experiments showed that T74S increases viral fitness of C.4, which carries multiple PI-related resistance mutations. All subtype C clones carrying 74S showed increased fitness when compared with their 74T counterparts (Figure 2b). Of most interest, subtype C PR carrying PI resistance mutations and 74S (C.4.74) was fitter than all other subtype C clones, including C.wt, showing that this mutation can increase fitness even above that of wild-type viruses. In fact, the low cost of 74S in subtype C isolates is evident from its higher natural frequency in this subtype (∼18% in drug-naive subtype C infections). A recent search of the University of Stanford HIV Drug Resistance Database (http://hibdb.stanford.edu; accessed on 28 April 2009) showed that, whereas the frequency of 74S in subtype B increases from 0% (n = 8445) among drug-naive isolates to 11% (n = 5250) in PI-treated persons, among subtype C-infected subjects the frequency of 74S increased from 8% (n = 2352) to 18% (n = 346).
Distinct HIV-1 subtypes may have different phenotypic responses to the acquisition of given drug resistance mutations. Although subtype C is known to be hypersusceptible to lopinavir, the introduction of four primary drug resistance mutations (C.4) actually results in increased levels of resistance in subtype C versus subtype B PR (compare C.4 with B.4; Table 2). Although T74S appears to have a similar effect in both subtypes B and C, our in vitro data suggesting higher replicative capacity with the 74S in subtype C supports the observation of higher prevalence in subtype C compared with subtype B.5 Further investigation is still necessary to define the impact of specific resistance mutations in different HIV-1 subtypes.
Finally, we also observed in this study a decreased fitness of subtype C versus subtype B PR in the context of the NL4-3 genome. Contrary to a previous report21 but supported by other studies,22–24 all subtype C PR viruses (with or without T74S or four primary mutations) were less fit than B.wt virus. We have previously described22–24 that most group M primary HIV-1 isolates, including those of subtype B, have higher replicative fitness than subtype C HIV-1 isolates. This increased fitness was attributed to entry efficiency and mapped to the env gene,23,24 but in this study, the reduced fitness of subtype C may also be associated with reduced activity of HIV-1 PR. Indeed, the involvement of the 5′ half of the HIV genome in virus fitness has been recently suggested.35 Again here, the involvement of the Gag precursor in viral fitness, which has been recently demonstrated, is not being evaluated in our constructs.36 We are currently testing the PR fitness of various subtypes and will determine whether an observed decrease in PR activity (in the context of replicative fitness) may be associated with cleavage of autologous (same subtype) versus heterologous Gag-Pol precursors.
In summary, we have shown in this report that T74S is a bona fide fitness-restoring mutation in both subtypes B and C. Most importantly, it restores susceptibility to indinavir and ritonavir down to levels below CCO1. Although the PIs tested here are of the first generation, this work constitutes a proof of principle, which may also be proved to be true for other, more recently introduced PIs. Determining the phenotypic impact of resistance mutations in different subtypes can help in designing more rational uses of ARVs, particularly in Africa, where PIs of first and second generations have achieved a low cost and are to be widely used, and non-B subtypes of HIV-1 predominate. Although the clinical relevance of these findings is not yet elucidated, the impact of this mutation in PI resistance and its inclusion in HIV drug resistance algorithms should be considered.
Funding
This work was supported by CAPES (E. A. S. and L. M. G. PhD fellowships), CNPq grant no. 403589/2004-5 and A. F. S. PhD fellowship, FAPERJ grant no. E-26/170.545/2004, Brazilian Ministry of Health and the Case Western Reserve University Center for AIDS Research. A. F. S. has received an international fellowship from the CFAR at Case Western Reserve University, USA.
Transparency declarations
None to declare.
Acknowledgements
We acknowledge the NIH AIDS Research and Reference Reagent Program for providing the antiretroviral compounds used in this work. We are also in debt to Dr Charles Boucher (University of Utrecht Medical Center, Utrecht, The Netherlands) for providing us with the pHXBΔPR backbone vector.
References
- 1.Ridky T, Leis J. Development of drug resistance to HIV-1 protease inhibitors. J Biol Chem. 1995;270:29621–3. doi: 10.1074/jbc.270.50.29621. [DOI] [PubMed] [Google Scholar]
- 2.Garcia-Lerma JG, Heneine W. Resistance of human immunodeficiency virus type 1 to reverse transcriptase and protease inhibitors: genotypic and phenotypic testing. J Clin Virol. 2001;21:197–212. doi: 10.1016/s1386-6532(00)00163-3. [DOI] [PubMed] [Google Scholar]
- 3.Johnson VA, Brun-Vezinet F, Clotet B, et al. Update of the drug resistance mutations in HIV-1. Top HIV Med. 2008;16:138–45. [PubMed] [Google Scholar]
- 4.Brann TW, Dewar RL, Jiang MK, et al. Functional correlation between a novel amino acid insertion at codon 19 in the protease of human immunodeficiency virus type 1 and polymorphism in the p1/p6 Gag cleavage site in drug resistance and replication fitness. J Virol. 2006;80:6136–45. doi: 10.1128/JVI.02212-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kantor R, Katzenstein DA, Efron B, et al. Impact of HIV-1 subtype and antiretroviral therapy on protease and reverse transcriptase genotype: results of a global collaboration. PLoS Med. 2005;2:e112. doi: 10.1371/journal.pmed.0020112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paolucci S, Baldanti F, Dossena L, et al. Amino acid insertions at position 35 of HIV-1 protease interfere with virus replication without modifying antiviral drug susceptibility. Antivir Res. 2006;69:181–5. doi: 10.1016/j.antiviral.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 7.Descamps D, Apetrei C, Collin G, et al. Naturally occurring decreased susceptibility of HIV-1 subtype G to protease inhibitors. AIDS. 1998;12:1109–11. [PubMed] [Google Scholar]
- 8.Santos AF, Abecasis AB, Vandamme AM, et al. Discordant genotypic interpretation and phenotypic role of protease mutations in HIV-1 subtypes B and G. J Antimicrob Chemother. 2009;63:593–9. doi: 10.1093/jac/dkn526. [DOI] [PubMed] [Google Scholar]
- 9.Gonzalez LM, Brindeiro RM, Tarin M, et al. In vitro hypersusceptibility of human immunodeficiency virus type 1 subtype C protease to lopinavir. Antimicrob Agents Chemother. 2003;47:2817–22. doi: 10.1128/AAC.47.9.2817-2822.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fonjungo PN, Mpoudi EN, Torimiro JN, et al. Human immunodeficiency virus type 1 group M protease in Cameroon: genetic diversity and protease inhibitor mutational features. J Clin Microbiol. 2002;40:837–45. doi: 10.1128/JCM.40.3.837-845.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vicente AC, Agwale SM, Otsuki K, et al. Genetic variability of HIV-1 protease from Nigeria and correlation with protease inhibitors drug resistance. Virus Genes. 2001;22:181–6. doi: 10.1023/a:1008123508416. [DOI] [PubMed] [Google Scholar]
- 12.Soares EA, Martinez AM, Souza TM, et al. HIV-1 subtype C dissemination in southern Brazil. AIDS. 2005;19(Suppl 4):S81–6. doi: 10.1097/01.aids.0000191497.00928.e4. [DOI] [PubMed] [Google Scholar]
- 13.Soares EA, Santos RP, Pellegrini JA, et al. Epidemiologic and molecular characterization of human immunodeficiency virus type 1 in southern Brazil. J Acquir Immune Defic Syndr. 2003;34:520–6. doi: 10.1097/00126334-200312150-00012. [DOI] [PubMed] [Google Scholar]
- 14.Deforche K, Silander T, Camacho R, et al. Analysis of HIV-1 pol sequences using Bayesian networks: implications for drug resistance. Bioinformatics. 2006;22:2975–9. doi: 10.1093/bioinformatics/btl508. [DOI] [PubMed] [Google Scholar]
- 15.Gonzalez LM, Santos AF, Abecasis AB, et al. Impact of HIV-1 protease mutations A71V/T and T74S on M89I/V-mediated protease inhibitor resistance in subtype G isolates. J Antimicrob Chemother. 2008;61:1201–4. doi: 10.1093/jac/dkn099. [DOI] [PubMed] [Google Scholar]
- 16.Adachi A, Gendelman HE, Koenig S, et al. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol. 1986;59:284–91. doi: 10.1128/jvi.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gonzalez LM, Aguiar RS, Afonso A, et al. Biological characterization of human immunodeficiency virus type 1 subtype C protease carrying indinavir drug-resistance mutations. J Gen Virol. 2006;87:1303–9. doi: 10.1099/vir.0.81517-0. [DOI] [PubMed] [Google Scholar]
- 18.Maschera B, Furfine E, Blair ED. Analysis of resistance to human immunodeficiency virus type 1 protease inhibitors by using matched bacterial expression and proviral infection vectors. J Virol. 1995;69:5431–6. doi: 10.1128/jvi.69.9.5431-5436.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hertogs K, de Bethune MP, Miller V, et al. A rapid method for simultaneous detection of phenotypic resistance to inhibitors of protease and reverse transcriptase in recombinant human immunodeficiency virus type 1 isolates from patients treated with antiretroviral drugs. Antimicrob Agents Chemother. 1998;42:269–76. doi: 10.1128/aac.42.2.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lalonde MS, Troyer RM, Syed AR, et al. Sensitive oligonucleotide ligation assay for low-level detection of nevirapine resistance mutations in human immunodeficiency virus type 1 quasispecies. J Clin Microbiol. 2007;45:2604–15. doi: 10.1128/JCM.00431-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Velazquez-Campoy A, Todd MJ, Vega S, et al. Catalytic efficiency and vitality of HIV-1 proteases from African viral subtypes. Proc Natl Acad Sci USA. 2001;98:6062–7. doi: 10.1073/pnas.111152698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arien KK, Abraha A, Quinones-Mateu ME, et al. The replicative fitness of primary human immunodeficiency virus type 1 (HIV-1) group M, HIV-1 group O, and HIV-2 isolates. J Virol. 2005;79:8979–90. doi: 10.1128/JVI.79.14.8979-8990.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ball SC, Abraha A, Collins KR, et al. Comparing the ex vivo fitness of CCR5-tropic human immunodeficiency virus type 1 isolates of subtypes B and C. J Virol. 2003;77:1021–38. doi: 10.1128/JVI.77.2.1021-1038.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marozsan AJ, Moore DM, Lobritz MA, et al. Differences in the fitness of two diverse wild-type human immunodeficiency virus type 1 isolates are related to the efficiency of cell binding and entry. J Virol. 2005;79:7121–34. doi: 10.1128/JVI.79.11.7121-7134.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nijhuis M, van Maarseveen NM, Boucher CA. Antiviral resistance and impact on viral replication capacity: evolution of viruses under antiviral pressure occurs in three phases. Handb Exp Pharmacol. 2009:299–320. doi: 10.1007/978-3-540-79086-0_11. [DOI] [PubMed] [Google Scholar]
- 26.van Maarseveen NM, de Jong D, Boucher CA, et al. An increase in viral replicative capacity drives the evolution of protease inhibitor-resistant human immunodeficiency virus type 1 in the absence of drugs. J Acquir Immune Defic Syndr. 2006;42:162–8. doi: 10.1097/01.qai.0000219787.65915.56. [DOI] [PubMed] [Google Scholar]
- 27.Winters B, Montaner J, Harrigan PR, et al. Determination of clinically relevant cutoffs for HIV-1 phenotypic resistance estimates through a combined analysis of clinical trial and cohort data. J Acquir Immune Defic Syndr. 2008;48:26–34. doi: 10.1097/QAI.0b013e31816d9bf4. [DOI] [PubMed] [Google Scholar]
- 28.Larder BA, Kemp SD, Harrigan PR. Potential mechanism for sustained antiretroviral efficacy of AZT-3TC combination therapy. Science. 1995;269:696–9. doi: 10.1126/science.7542804. [DOI] [PubMed] [Google Scholar]
- 29.Kuritzkes DR, Quinn JB, Benoit SL, et al. Drug resistance and virologic response in NUCA 3001, a randomized trial of lamivudine (3TC) versus zidovudine (ZDV) versus ZDV plus 3TC in previously untreated patients. AIDS. 1996;10:975–81. doi: 10.1097/00002030-199610090-00007. [DOI] [PubMed] [Google Scholar]
- 30.de Mendoza C, Valer L, Bacheler L, et al. Prevalence of the HIV-1 protease mutation I47A in clinical practice and association with lopinavir resistance. AIDS. 2006;20:1071–4. doi: 10.1097/01.aids.0000222084.44411.cc. [DOI] [PubMed] [Google Scholar]
- 31.Martinez-Picado J, Savara AV, Shi L, et al. Fitness of human immunodeficiency virus type 1 protease inhibitor-selected single mutants. Virology. 2000;275:318–22. doi: 10.1006/viro.2000.0527. [DOI] [PubMed] [Google Scholar]
- 32.Dam E, Quercia R, Glass B, et al. Gag mutations strongly contribute to HIV-1 resistance to protease inhibitors in highly drug-experienced patients besides compensating for fitness loss. PLoS Pathog. 2009;5:e1000345. doi: 10.1371/journal.ppat.1000345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parry CM, Kohli A, Boinett CJ, et al. Gag determinants of fitness and drug susceptibility in protease inhibitor resistant HIV-1. J Virol. 2009 doi: 10.1128/JVI.02356-08. doi:10.1128/JVI.02356-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Andreoni M. Viral phenotype and fitness. New Microbiol. 2004;27:71–6. [PubMed] [Google Scholar]
- 35.Rodriguez MA, Ding M, Ratner D, et al. High replication fitness and transmission efficiency of HIV-1 subtype C from India: implications for subtype C predominance. Virology. 2009;385:416–24. doi: 10.1016/j.virol.2008.12.025. [DOI] [PubMed] [Google Scholar]
- 36.Ho SK, Coman RM, Bunger JC, et al. Drug-associated changes in amino acid residues in Gag p2, p7(NC), and p6(Gag)/p6(Pol) in human immunodeficiency virus type 1 (HIV-1) display a dominant effect on replicative fitness and drug response. Virology. 2008;378:272–81. doi: 10.1016/j.virol.2008.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Verlinden Y, Vermeiren H, Lecocq P, et al. Assessment of the antivirogram® performance over time including a revised definition of biological test cut-off values. Antivir Ther. 2005;10(Suppl 2):S51. [Google Scholar]