

Abstract
Expanded non-coding RNA repeats of CUG and CCUG are the underlying genetic causes for myotonic dystrophy type 1 (DM1) and type 2 (DM2), respectively. A gain-of-function of these pathogenic repeat expansions is mediated at least in part by their abnormal interactions with RNA-binding proteins such as MBNL1 and resultant loss of activity of these proteins. To study pathogenic mechanisms of CCUG-repeat expansions in an animal model, we created a fly model of DM2 that expresses pure, uninterrupted CCUG-repeat expansions ranging from 16 to 720 repeats in length. We show that this fly model for DM2 recapitulates key features of human DM2 including RNA repeat-induced toxicity, ribonuclear foci formation and changes in alternative splicing. Interestingly, expression of two isoforms of MBNL1, MBNL135 and MBNL140, leads to cleavage and concurrent upregulation of the levels of the RNA-repeat transcripts, with MBNL140 having more significant effects than MBNL135. This property is shared with a fly CUG-repeat expansion model. Our results suggest a novel mechanism for interaction between the pathogenic RNA repeat expansions of myotonic dystrophy and MBNL1.
INTRODUCTION
Myotonic dystrophy is the most common form of muscular dystrophy in adults with an incidence of 1 in ∼8000 worldwide (1). Major clinical features include myotonia, muscle wasting and multi-organ involvement (1,2). Two types of myotonic dystrophy with distinct genetic etiologies have been identified. Myotonic dystrophy type 1 (DM1) is caused by expansion of a trinucleotide CTG repeat expansion in the non-coding 3′ UTR region of DMPK (3–5). Myotonic dystrophy type 2, however, is caused by expansion of a tetranucleotide CCTG-repeat expansion in the first intron of ZNF9 (6). Despite clinical similarities, myotonic dystrophy types 1 and 2 have differences including the groups of muscles most affected, presence of congenital forms of DM1 and less-pronounced neurological involvement in DM2 generally (7,8).
Overwhelming evidence suggests that a gain-of-function activity owing to the accumulation of the expanded RNA underlies the multiple clinical features of the DM diseases (8–11). Both expanded repeat RNAs associated with the mutations form ribonuclear foci that interfere with RNA-binding proteins involved in the regulation of splicing such as Muscleblind-like (MBNL) proteins and CUG-BP1 (6,12–15). MBNL proteins belong to the family of zinc finger RNA-binding proteins and are evolutionarily conserved from flies to human (16–18). Expression of Muscleblind proteins is developmentally regulated and plays an essential role in the terminal differentiation of photoreceptors and muscles in flies (17,18). In mammals, MBNL proteins function as upstream splicing factors that orchestrate postnatal development and remodeling of heart and skeletal muscles through regulating alternative splicing of a key set of downstream pre-mRNAs (14,19–22). Recent studies also suggest that MBNL proteins repress embryonic stem cell-specific alternative splicing and thereby regulate stem cell pluripotency and differentiation (23). In addition, MBNL proteins are abundantly expressed in the cytoplasm of cells and are involved in the regulation of stability and localization of target mRNAs (24–27). In myotonic dystrophy, MBNL proteins are sequestered in the repeat-containing ribonuclear foci and reduction of MBNL protein levels is thought to shift splicing of target pre-mRNAs toward fetal isoforms and thereby mediate many pathogenic effects of expanded CUG or CCUG repeats (8,10,14,16). In support of this hypothesis, MBNL1 knockout mice develope features of myotonic dystrophy and MBNL1 upregulation rescues myotonia effects in a mouse DM1 model expressing a non-coding CTG expansion (28,29). Together, these findings highlight the importance of RNA-binding proteins such as MBNL1 in RNA metabolism under normal conditions, as well as pathological situations.
There are three closely related MBNL genes in human, MBNL1, MBNL2 and MBNL3 (12). The MBNL1 gene encompasses ten coding exons. While exons encoding the four CCCH zinc finger domains are constitutive (30), most of the other exons are alternatively spliced and nine MBNL1 splicing isoforms have been reported (31). Select MBNL1 isoforms are mainly expressed in fetal brain and muscles and are preferentially included in DM1-affected tissues (14,32). The alternatively spliced exon 4 encodes an alanine-rich linker between the second and the third CCCH zinc finger domain and is skipped in some of MBNL1 isoforms such as MBNL135 and MBNL136 (31). This linker domain is required for high binding affinity of MBNL1 to both expanded CUG-repeat RNA and pre-mRNA target sites (31,33). MBNL1 isoforms without the linker domain, such as MBNL135, can still be recruited to CUG ribonuclear foci, suggesting that the linker sequence is not required for MBNL1 to interact with CUG-repeat expansion, at least in ribonuclear foci, and the RNA-binding domain composed of the four CCCH zinc fingers may be sufficient to mediate the interaction (33).
To study toxicity of the CCUG-repeat expansion associated with DM2, we generated pure, uninterrupted CCTG expansions ranging from 16 to 720 repeats in length and created transgenic fly lines. Expression of non-coding CCUG-expanded RNA in flies led to formation of ribonuclear foci, changes in alternative splicing and length-dependent toxicity. Moreover, we demonstrate that MBNL isoforms mildly suppress CCUG-repeat toxicity in the fly model of DM2. Strikingly, both MBNL1 isoforms can mediate cleavage of CCUG-repeat RNA and concurrently increase the overall levels of repeat RNA. Together, our results revealed a novel interaction between the CUG/CCUG-repeat expansion and MBNL1.
RESULTS
A Drosophila model for DM2
In DM2, the size of the expanded allele ranges from 75 to 11 000 CCTG repeats, with a mean of ∼5000 repeats (6). Owing to the high GC content, these repeats are difficult to clone by traditional methods. Further, we were keen to maintain the repeat as a pure CCTG repeat, in case the pure repeat, when compared with an interrupted sequence, would yield different biology. We thus generated pure CCTG-repeat expansions de-novo through a sequential cloning approach (34,35). Like many pure trinucleotide repeats (36), CCTG tetranucleotide repeat expansions were not stable in Escherichia coli. Given this, the longest repeat expansions that we were able to generate by cloning methods using E. coli were ∼700 repeats based on sizing on agarose gels.
To generate transgenic animals, we cloned the (CCTG)700 repeat expansion downstream of the UAS/mini-promoter in the pUAST transformation vector (Fig. 1A). No ATG start codon is present in front of the CCTG-repeat expansion, and the construct is predicted to express a CCUG-repeat RNA in flies in the presence of a GAL4 transactivator driver line. A short (CCTG)16 expansion was also cloned into pUAST for transgenesis, to use as a repeat length within the normal range, for control flies. We obtained a number of animals bearing transgene insertions. Southern blots revealed that the resultant transgenic fly lines generated from the long repeat comprised a set of flies with a range of repeat lengths, from 200 CCTG repeats to 720 CCTG repeats (Fig. 1B). Each fly line had an individual stable repeat length. The variability in repeat length between distinct fly lines is most likely due to the fact that the injected CCTG-repeat construct was not stable in E. coli such that the injected DNA mix was comprised of a pool of repeat expansions with a range of repeat lengths. We therefore obtained a set of fly lines, bearing a range of CCTG-repeat lengths.
Figure 1.
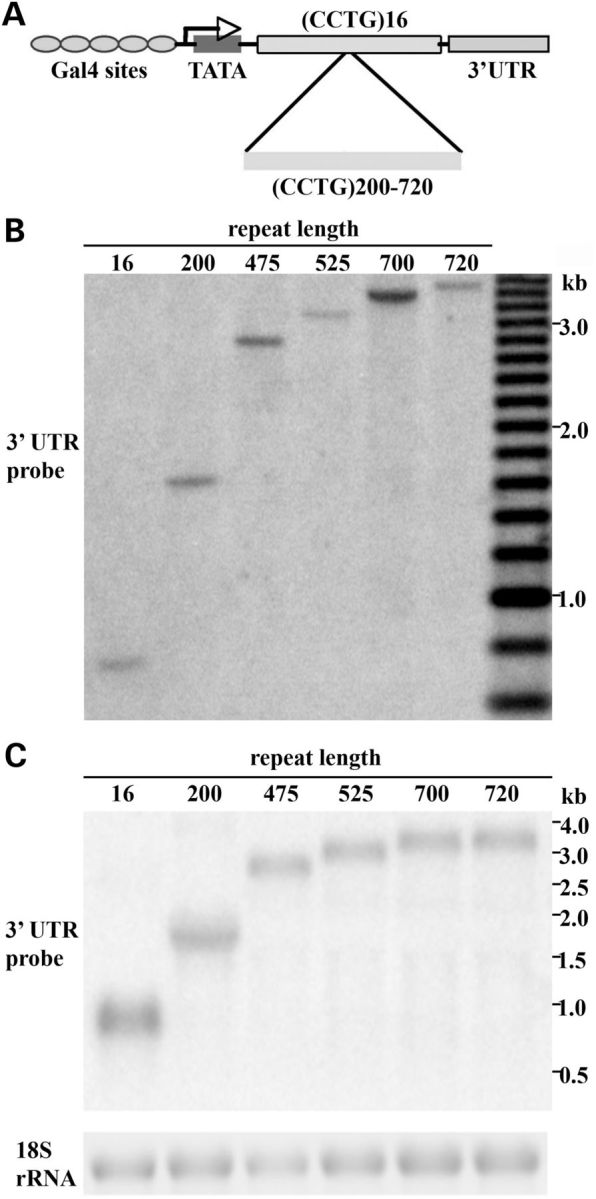
Characterization of transgenic flies expressing various-length CCUG RNA repeats. (A) Design of transgenic constructs. CCTG non-coding repeat expansions were expressed under the control of the GAL4/UAS system. The control contained 16 CCTG-repeat units whereas the expanded repeat ranged from 200 to 720 units. (B) DNA Southern blot to determine the CCTG-repeat length in six representative lines. From left to right, repeat lengths are 16, 200, 475, 520, 700 and 720, respectively. (C) Northern blots to determine RNA transcript levels in UAS-(CCTG) flies with various repeat lengths. Non-coding CCUG-repeat RNA was expressed with hs-gal4. The SV40 3′ UTR probe was used as for detection. These lines express similar levels of the CCUG-repeat transgene.
Analysis of the transgenic lines indicated that we did not observe significant CCTG-repeat instability or contraction detectable by Southern blots of flies (data not shown). Repeat lengths were stable over multiple generations, such that the repeat length of each line remained constant after the transgenic flies were passaged for more than ten generations on standard fly medium at room temperature. This is consistent with our earlier data that expanded repeats in Drosophila are generally stable intergenerationally, unless expressed in the germline (37). To compare repeat expression, we expressed the CCUG-repeat RNA under the hs-gal4 driver and performed northern analysis. From these studies, six independent fly lines with comparable expression were selected for subsequent study that carried repeat lengths of 16, 200, 475, 525, 700 and 720, as determined by Southern blot (Fig. 1C).
Non-coding CCUG-repeat expansions cause repeat length-dependent toxicity
With this set of fly lines with a range of repeat lengths, we asked whether expanded CCUG repeats are toxic to animals when expressed in different tissues. Transgenic expression of the expanded CCUG repeat with an eye-specific driver gmr-gal4 lead to abnormal pigmentation and a smooth eye surface indicative of disruption of the ommatidial structure (Fig. 2A). In addition, retinal depth was significantly reduced compared with control flies expressing a short control nontoxic (CCUG)16 repeat (Fig. 2B). The severity of the effect correlated with the length of the CCTG repeat in the animals (Fig. 2A and B): repeats of 16 and 200 were not toxic in this assay, but repeats of 475 and higher conferred toxicity.
Figure 2.

Repeat length-dependent CCUG toxicity in the fly eye. CCTG-repeat expansions of various lengths were expressed in the eye using the gmr-gal4 driver. 1d animals. (A) External eye and (B) internal retinal structure were examined. (A) Repeat RNA expression caused length-dependent toxicity as reflected by loss of pigmentation, necrotic patches and a degenerate eye surface indicative of disruption to the ommatidial organization (arrows). (B) Internally, the expanded repeat impacts depth and structure of the retina at repeat lengths of 475 and longer. Genotypes: gmr-gal4 in trans to UAS-(CCTG)16, UAS-(CCTG)200, UAS-(CCTG)475, UAS-(CCTG)525, UAS-(CCTG)700, UAS-(CCTG)720.
Expression of CCUG-expanded repeat RNA in muscle cells using the 24B-gal4 driver or in the nervous system using elav-gal4 also led to repeat length toxicity, conferring developmental lethality (Table 1). As mentioned earlier, flies bearing CCTG repeats of 16 and 200 were viable, but repeats of 475–720 conferred lethality. Together, these data suggest that expression of CCUG repeat causes length-dependent toxicity.
Table 1.
Developmental lethality caused by expression in muscle or neurons of expanded CCUG repeats
| Repeat length | Muscle expression (B24-gal4 driver) | Neuronal expression (elav-gal4 driver) |
|---|---|---|
| 16 | Viable | Viable |
| 200 | Viable | Viable |
| 475 | Lethal | Lethal |
| 500 | Lethal | Lethal |
| 700 | Lethal | Lethal |
| 720 | Lethal | Lethal |
CCUG-repeat RNA forms nuclear foci and impacts alternative splicing
CCUG-repeat expansions form ribonuclear foci in DM2 muscle and neuronal cells and affect alternative splicing (6,9,15). We therefore examined whether flies expressing CCUG-repeat RNA recapitulated these features of disease. To circumvent the early developmental lethality caused by CCUG expanded repeat RNA in muscles, flies were maintained at 20°C to reduce transgene expression and toxicity. As shown in Figure 3A, discrete foci were observed throughout the nuclei in the larvae body-wall muscles of flies expressing expanded (CCTG)475. Interestingly, smaller but more densely populated ribonuclear foci were also observed in flies expressing (CCTG)200 with the 24B-gal4 muscle driver, although these flies did not show lethality or adult locomotor defects (Fig. 3A–C). In contrast, foci were not present in flies expressing the short control (CCTG)16 repeat. Consistent with previous findings (39,40), our data suggest nuclear foci formation and toxicity of CUG-repeat expansion can be decoupled, at least under certain situations.
Figure 3.

Expanded CCUG-repeat RNA accumulates in nuclear foci and impacts alternative splicing. (A) Confocal images of CCUG-repeat foci detected with (CAGG)5-FITC probe, nuclei stained with DAPI and merged images between ribonuclear foci and nuclei in body-wall muscles of third instar larvae. Genotypes: w1118, 24B-gal4 in trans to UAS-(CCTG)200, UAS-(CCTG)475, UAS-(CCTG)475/MBNL135. (B) Mean number of foci in each nucleus in flies obtained in three independent experiments. Genotypes: 24B-gal4 in trans to UAS-(CCTG)200, UAS-(CCTG)475, UAS-(CCTG)475/MBNL135 (**P < 0.05, ANOVA test). (C) Mean volume of foci obtained in three independent experiments. Genotypes: 24B-gal4 in trans to UAS-(CCTG)200, UAS-(CCTG)475, UAS-(CCTG)475/MBNL135 (**P < 0.05, ANOVA test). (D) Expression of expanded repeat CCUG in photoreceptor neurons promoted exclusion of the second exon of sTNI as determined by radioactive PCR. Shown in the top panel is a representative gel image for determining the densitometry ratio between 140-bp and 110-bp PCR products, corresponding to the splicing isoform with (140 bp) or without (110 bp) the second exon of sTNI minigene reporter, respectively (38). Shown in bottom panel is quantification of three independent experiments. With expression of expanded CCUG repeats, the ratio decreased, such that the second exon was preferentially excluded. Genotypes: rh1-gal4 UAS-hcTNT in trans to UAS-(CCTG)16, UAS-(CCTG)200, UAS-(CCTG)475, UAS-(CCTG)520, UAS-(CCTG)700. *P < 0.05, **P < 0.01 comparing to CCTG16 control (ANOVA followed by Dunnett's post-test).
To examine whether expression of RNA bearing CCUG expansions causes changes in alternative splicing, we co-expressed in photoreceptor neurons the CCUG-repeat expansion with a mingene sTNI (chicken skeletal Troponin I) splicing reporter (38). Alternative splicing of this minigene reporter responds to pathogenic CUG-repeat expansions in a fly model of DM1 and altered MBNL activities in mammalian cells (38,41). Expression of CCUG-repeat expansions of 475, 520 and 700 promoted exclusion of the second exon of sTNI as determined by radioactive reverse transcription RT–PCR, whereas repeats of 16 and 200 had no significant effect (Fig. 3D). These data suggest that expression of CCUG-repeat expansions in flies causes the formation of ribonuclear accumulation of the repeat-bearing RNA and can impact alternative splicing, as in DM2 disease.
Both MBNL135 and MBNL140 mediate cleavage of CCUG-repeat expansions with mild suppression of toxicity
Loss of MBNL function is thought to underlie many of the splicing defects in DM1 and DM2 (29). Therefore, we examined whether expression of human MBNL1 in the fly could modulate toxicity caused by the expression of the CCUG-repeat expansions. Moreover, as MBNL1 isoforms with and without the linker region between zinger fingers 2 and 3, MBNL140 and MBNL135, respectively, show differential affinity for CUG repeats as well as differential modification of CAG RNA-associated polyQ toxicity in fly models for polyglutamine disease (31,33,42), we were interested in assessing whether MBNL140 and MBNL135 behaved similarly or distinctly on the CCUG repeat. To this end, we co-expressed the CCUG-repeat expansions with MBNL140 or MBNL135 using the gmr-gal4 driver. Both MBNL140 and MBNL135 had mild effects to suppress CCUG toxicity, by improving the ommatidial organization externally and increasing the depth and structural integrity of the internal retina (Fig. 4A).
Figure 4.

MBNL135 and MBNL140 mildly suppress CCUG-repeat toxicity and mediate upregulation and cleavage of non-coding CUG/CCUG-repeat expansions. (A) Mild suppression of the external (arrows) and internal retinal toxicity of the CCUG repeat occurs upon expression of MBNL140 or MBNL135. Genotypes, from left to right, gmr-gal4 UAS-(CCTG)700 in trans to w1118, UAS-MBNL135, UAS-MBNL140. (B) Expression of MBNL135 and MBNL140 mediates upregulation and cleavage of expanded CCUG-repeat RNA. Genotype of flies (from the left lane to right lane): gmr-gal4, UAS-(CCTG)700 in trans to: w1118, UAS-MBNL135, UAS-MBNL140 and w1118 alone as the negative control. (C) Expression of MBNL135 and MBNL140 also mediate cleavage and upregulation of expanded CUG-repeat RNA. Genotype of flies (from the left lane to right lane): gmr-gal4, UAS-DsRed-(CTG)250 in trans to: w1118, UAS-MBNL135, UAS-MBNL140 and w1118 alone as the negative control. (D) In contrast to findings with MBNL1, expression of DNAJC17 does not lead to upregulation and cleavage of expanded CCUG-repeat RNA. Genotype of flies (from the left lane to right lane): gmr-gal4, UAS-(CCTG)700 in trans to: w1118, UAS-MBNL135, UAS-MBNL140 and dDNAJC17. (E) Integrity and level of coding CAG repeats are not dramatically affected by MBNL1. Genotype of flies (from the left lane to right lane): w1118 alone as the negative control and gmr-gal4 UAS-SCA3trQ78 in trans to: w1118, UAS-MBNL135 and UAS-MBNL140.
We then assessed how MBNL140 and MBNL135 affected the expanded repeat RNA by examining transcript expression by northern blot. Surprisingly, we observed that a lower-molecular-weight CCUG-repeat smear was present in flies co-expressing either MBNL135 or MBNL140 (Fig. 4B). Moreover, co-expression of MBNL135 or MBNL140 leads to significant upregulation of the steady-state level of the overall RNA repeat transcript. To examine whether MBNL1 proteins have similar effects on a non-coding CUG-repeat expansion, we co-expressed MBNL135 or MBNL40 in the fly model of DM1, which expresses a non-coding CUG expansion in the 3′ UTR region of DsRed (38). As shown in Figure 4C, we also observed cleavage of full-length CUG-repeat expansion RNA into smaller fragments in flies expressing either MBNL135 or MBNL140. Like CCUG, the overall level of CUG-repeat expression was upregulated in these flies. We confirmed that this effect on the transcripts was selective to MBNL1 because expression of another RNA-binding protein, DNAJC17, did not cause upregulation or cleavage of expanded CCUG or CUG repeats (Fig. 4D and data not shown), suggesting that the effects of MBNL1 are specific. To probe specificity for repeat transcripts, we also examined whether CAG repeat expansions are subject to MBNL1-mediated cleavage and upregulation. However, the effect was specific to CUG and CCUG repeats, as neither MBNL140 nor MBNL135 leads to cleavage of the CAG repeats (Fig. 4E). Moreover, the level of the CAG-repeat transcript was only minimally affected by MBNL1 expression when compared with the CUG/CCUG-repeat expansions (Fig. 4E). Although differential effects of MBNL1 on CAG repeats in SCA3 and non-coding CUG and CCUG repeats could be due to a different sequence context, these data suggest that effects of MBNL1 may be specific to non-coding CUG/CCUG-repeat expansions. We also examined whether MBNL1 affected repeat foci formation in larval muscles and found that expression of MBNL135 had minimal effects on both the density and the volume of CCUG ribonuclear foci (Fig. 3A–C).
While the discrete full-length transcript was still present in flies co-expressing MBNL135, they were completely cleaved into smaller repeat fragments in flies expressing MBNL140. Moreover, the overall steady-state levels of the repeat transcripts were upregulated to a greater degree in flies expressing MBNL140 than MBNL135. As transgenic expression of MBNL135 and MBNL140 in the transgenic lines used in this study are comparable (42), differential effects of MBNL135 and MBNL140 on repeat regulation are likely due to the different isoforms of the MBNL1 protein. Together, these data suggest that MBNL140 has more significant effects in mediating cleavage and upregulation of the non-coding repeats than MBNL135 (Fig. 4B and C).
DISCUSSION
CCTG tetranucleotide expansions associated with DM2 are usually thousands of repeats in length with a mean length of 5000 repeat units and, like many other pathogenic repeat expansions, are typically pure and uninterrupted repeats (6). Interruption of the pure pathogenic repeats may lead to distinct phenotypic manifestations as demonstrated in the case of ATXN2, which is associated with parkinsonism or SCA2 depending upon whether the polyQ-coding CAG expansion within the gene is interrupted (43). Similarly, interruption of RNA repeat expansions may change RNA structure and protein-binding activities and thereby alter phenotypic outcome (44). Pure repeats expansions may also be subject to additional biological mechanisms of toxicity, including non-ATG-associated protein translation (45). Drosophila has proven an insightful and powerful model system for studying human diseases including myotonic dystrophy (40,46–51). A transgenic DM2 model has been developed in the mouse (52). To model DM2 in flies, we therefore created uninterrupted CCTG expansions of up to 700 repeats in length through molecular cloning approaches (34,35).
Similar to CUG expansions in fly models for DM1 (38,53,54), expression of non-coding CCUG-repeat expansion RNA causes repeat length-dependent toxicity in multiple tissues, including tissues highly relevant to DM2 such as neurons and muscles. Ribonuclear foci formation and misregulation of alternative splicing are two hallmark features of myotonic dystrophy and are both seen in flies expressing CUG repeats of DM1 (38). We also observed ribonuclear foci accumulation of the expanded CCUG-repeat RNA and alternative splicing defects in flies expressing CCUG-repeat expansions. Our results indicate that repeat length correlates with volume of foci and number of foci per nucleus: longer CCUG-repeat expansions seem to be more inclined to form fewer but larger foci. These data suggest that expression of non-coding CCUG RNA in flies recapitulates multiple key features of DM2 disease and will provide an important model for DM2-repeat-associated toxicity. Moreover, as the transgenic flies expressing non-coding CUG-repeat expansions and CCUG-repeat expansions showed these similar characteristics, the findings highlight the shared molecular mechanisms of toxicity between the two repeat expansions.
In line with previous findings that MBNL140 suppresses CUG-repeat toxicity in fly models for DM1 generated with interrupted repeat expansions (53,54), we show that both MBNL135 and MBNL140 suppress toxicity of CCUG expanded repeat RNA in the fly eye. Intriguingly, co-expression of either MBNL35 or MBNL40 leads to cleavage of these repeat RNAs as well as upregulation of overall steady-state level of the transcript in a specific manner (see Fig. 4D and E). Both fly muscleblind and human MBNL1 have been shown to stabilize and upregulate CUG-repeat expansion in a fly model of DM1 (55). Expanded CUG- and CCUG-repeat transcripts fold into stem-loop structures (56–60). Our results are in line with the possibility that MBNL1 proteins promote cleavage in the loop regions of the expanded CUG or CCUG stem-loop structures to generate two similarly sized repeat fragments. Such cleavage is most likely mediated by an endonuclease activity although further studies are needed to decipher the specific mechanism. Despite aforementioned effects on CCUG transcripts, MBNL135 expression does not seem to impact CCUG foci (see Fig. 3A–C). Thus, interactions between MBNL1 and CUG- and CCUG-repeat expansions may be more complex than previously appreciated. It remains to be determined whether upregulation and cleavage of the repeat expansions by MBNL1 are coupled or independent events. Further studies are also needed to examine whether MBNL1 has similar effects on CUG/CCUG-repeat expansions in human DM1 or DM2 cells.
MBNL isoforms without the linker region between the second and the third zinc finger such as MBNL35 have previously shown to display attenuated affinity for CUG-repeat expansions and its target pre-mRNA-binding sites (31,33). However, these isoforms can still be recruited to ribonuclear foci suggesting that the linker region is not required for the interaction of MBNL35 with CUG-repeat expansions (33). It is likely that a physical interaction of MBNL1 and non-coding CUG/CCUG repeats is a prerequisite for MBNL1-mediated cleavage and upregulation of the steady-state levels of the repeat-bearing transcript. Although experimental evidence is needed to exclude the possibility that MBNL1 harbors nuclease activity itself, in-silicon motif prediction of MBNL135 and MBNL140 fails to identify a nuclease domain within these proteins (61). A nuclease activity may instead be recruited to the expanded repeat RNA by MBNL1. MBNL has been shown to affect subcellular localization of select mRNAs and plays important role in local translation of these transcripts (26). An alternative possibility is that MBNL1, through interacting with CUG and CCUG expansions, changes the subcellular localization of the RNA bearing the repeat expansions to make them accessible to nuclease cleavage. Together, these results reveal that MBNL1 may lead to cleavage and upregulation of steady levels of non-coding CUG/CCUG repeats in select cells and situations; this may represent a novel interaction between the pathogenic RNA repeat expansions and MBNL1.
MATERIALS AND METHODS
Generation of non-interrupted CCTG expansions
Sense and antisense oligos that contain 16 CCTG repeats, namely, (CCTG)16, flanked by BsaI, EcoRI on one end and BbsI, KpnI on the other end, were chemically synthesized (IDT, Inc., Coralville, Iowa, USA). The oligonucleotides were annealed to form double-stranded DNA that was subsequently cloned into a low-copy plasmid, pACYC177, and transformed into E. coli HB101. CCTG repeats were then extended through sequential dimerization (34,35). Briefly, plasmids carrying (CCTG)16 were digested with EcoRI and BsaI to generate a linearized cloning vector that contains (CCTG)16. A second aliquot of the plasmid was digested with EcoRI and BbsI, and the ∼80-bp fragment containing (CCTG)16 repeats was gel-purified. As BsaI and BbsI cut within the first and the last CCTG repeat produce complementary ends, subsequent cloning of the ∼80-bp fragment into the linearized vector resulted in a recombinant a plasmid containing 31 non-interrupted CCTG repeats flanked by the same restriction enzyme sites as the parental plasmid. The plasmid containing 31 CCTG repeats was then used as the new parental plasmid for the next round of dimerization cloning to make a repeat expansion of (CCTG)61 and so on until the repeat became empirically too unstable to maintain in E. coli.
Fly lines
General stocks were maintained at 25°C on standard medium unless otherwise indicated. The CCTG-repeat expansion constructs were cloned under the control of UAS elements in the transformation vector pUAST. The construct was injected into w1118 embryos for transgenesis. Repeat lengths in the resultant transgenic flies were determined by Southern blotting.
Southern and Northern blots
Standard techniques were used. For Southern blots, genomic DNA was extracted from ∼50 flies using the Gentra Puregene Cell Kit (Qiagen), and 5 µg of genomic DNA was fully digested with 200 units of EcoRI and XbaI for Southern blots. For northern blots to characterize transgene expression between the various lines, flies bearing CCTG-repeat expansion insertions were crossed to the hs-gal4 driver line. Progeny flies were heat-shocked at 37°C for 30 min and allowed to recover at 25°C for 20 h. Total RNA was extracted using Trizol Reagent (Invitrogen) from either whole flies (for comparing transgene levels among various lines) or heads. Two micrograms of total RNA was loaded on 1% denaturing formaldehyde/MOPS agarose gels. The SV40 3′ UTR probe was PCR-amplified using primers: forward 5′ TGT GGT GTG ACA TAA TTG GAC A 3′ and reverse 5′ AGA TGG CAT TTC TTC TGA GCA 3′, purified using QIAquick Gel Extraction Kit (Qiagen) and labeled using the High Prime DNA Labeling Kit (Roche Applied Science). For northern blots to examine the effect of MBNL1 proteins on repeat RNA expansion, r(CAG)7 probes, r(CUG)7 probes, r(CAGG)5 probes and 18S rRNA control probes were labeled with P32 α-CTP using the MAXIscript Kit (Ambion) from the annealed double-stranded DNA template containing T7 promoter. Band densitometry was quantified using Image J (NIH). Oligo sequences used for annealing were: T7 promoter forward oligo: 5′ GAT AAT ACG ACT CAC TAT AGG GAG A 3′; 18S rRNA: 5′ AGG GAG CCT GAG AAA CGG CTA CCA CAT CTA AGG AAT CTC CCT ATA GTG AGT CGT ATT ATC 3′; r(CAG)7: 5′ GGG CAG CAG CAG CAG CAG CAG CAG TCT CCC TAT AGT GAG TCG TAT TAT 3′; r(CUG)7 probe: 5′ GGG CTG CTG CTG CTG CTG CTG CTG TCT CCC TAT AGT GAG TCG TAT TAT C 3′; r(CCTG)5 probe: 5′ GGG CCT GCC TGC CTG CCT GCC TGT CTC CCT ATA GTG AGT CGT ATT ATC 3′. Ladders used to assess sizing were from NEB catalog no. N0362S (9, 7, 5, 3, 2, 1 and 0.5 kb) and Invitrogen catalog no. AM7151 (0.5, 1, 1.5, 2, 2, 5, 3, 4, 5, 6 and 9 kb).
Ribonuclear foci
In-situ hybridization using 5′ FITC-labeled 2-O′-methyl-(CAGG)5 RNA oligo was performed as described (38). After staining, a minimum of three larvae were imaged per genotype using a Leica inverted DM6000B confocal microscope. 3D movie projections for each z-series were developed using the Leica Application Suite (LAS) microscope software to confirm that foci were only observed within the nucleus. To count foci and measure the volume, z-stacked images were analyzed using the 3D object counter plugin available for FIJI freeware (62,63). The scale was set in FIJI based on information from the LAS microscope software. Only particles that were fully imaged were included in the analysis.
Radioactive PCR
Radioactive PCR for alternative splicing analysis was performed as described (38).
FUNDING
This research received funding from the Muscular Dystrophy Association (to N.M.B.).
Acknowledgments
Conflict of Interest statement. None declared.
REFERENCES
- 1.Harper P.S. Myotonic Dystrophy. 3rd edn. London: WB Saunders; 2001. [Google Scholar]
- 2.Udd B., Meola G., Krahe R., Wansink D.G., Bassez G., Kress W., Schoser B., Moxley R. Myotonic dystrophy type 2 (DM2) and related disorders report of the 180th ENMC workshop including guidelines on diagnostics and management 3-5 December 2010, Naarden, The Netherlands. Neuromuscul. Disord. 2011;21:443–450. doi: 10.1016/j.nmd.2011.03.013. [DOI] [PubMed] [Google Scholar]
- 3.Fu Y.H., Pizzuti A., Fenwick R.G., King J., Rajnarayan S., Dunne P.W., Dubel J., Nasser G.A., Ashizawa T., Dejong P., et al. An unstable triplet repeat in a gene related to myotonic muscular-dystrophy. Science. 1992;255:1256–1258. doi: 10.1126/science.1546326. [DOI] [PubMed] [Google Scholar]
- 4.Mahadevan M., Tsilfidis C., Sabourin L., Shutler G., Amemiya C., Jansen G., Neville C., Narang M., Barcelo J., O'Hoy K., et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science. 1992;255:1253–1255. doi: 10.1126/science.1546325. [DOI] [PubMed] [Google Scholar]
- 5.Brook J.D., McCurrach M.E., Harley H.G., Buckler A.J., Church D., Aburatani H., Hunter K., Stanton V.P., Thirion J.-P., Hudson T., et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell. 1992;68:799–808. doi: 10.1016/0092-8674(92)90154-5. [DOI] [PubMed] [Google Scholar]
- 6.Liquori C.L., Ricker K., Moseley M.L., Jacobsen J.F., Kress W., Naylor S.L., Day J.W., Ranum L.P. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. 2001;293:864–867. doi: 10.1126/science.1062125. [DOI] [PubMed] [Google Scholar]
- 7.Minnerop M., Weber B., Schoene-Bake J.C., Roeske S., Mirbach S., Anspach C., Schneider-Gold C., Betz R.C., Helmstaedter C., Tittgemeyer M., et al. The brain in myotonic dystrophy 1 and 2: evidence for a predominant white matter disease. Brain. 2011;134:3530–3546. doi: 10.1093/brain/awr299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ranum L.P., Cooper T.A. RNA-mediated neuromuscular disorders. Ann. Rev. Neurosci. 2006;29:259–277. doi: 10.1146/annurev.neuro.29.051605.113014. [DOI] [PubMed] [Google Scholar]
- 9.Day J.W., Ranum L.P. RNA pathogenesis of the myotonic dystrophies. Neuromuscul. Disord. 2005;15:5–16. doi: 10.1016/j.nmd.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 10.Wheeler T.M., Thornton C.A. Myotonic dystrophy: RNA-mediated muscle disease. Curr. Opin. Neurol. 2007;20:572–576. doi: 10.1097/WCO.0b013e3282ef6064. [DOI] [PubMed] [Google Scholar]
- 11.Poulos M.G., Batra R., Charizanis K., Swanson M.S. Developments in RNA splicing and disease. Cold Spring Harb. Perspect. Biol. 2011;3:a000778. doi: 10.1101/cshperspect.a000778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fardaei M., Rogers M.T., Thorpe H.M., Larkin K., Hamshere M.G., Harper P.S., Brook J.D. Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Hum. Mol. Genet. 2002;11:805–814. doi: 10.1093/hmg/11.7.805. [DOI] [PubMed] [Google Scholar]
- 13.Jiang H., Mankodi A., Swanson M.S., Moxley R.T., Thornton C.A. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum. Mol. Genet. 2004;13:3079–3088. doi: 10.1093/hmg/ddh327. [DOI] [PubMed] [Google Scholar]
- 14.Lin X., Miller J.W., Mankodi A., Kanadia R.N., Yuan Y., Moxley R.T., Swanson M.S., Thornton C.A. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum. Mol. Genet. 2006;15:2087–2097. doi: 10.1093/hmg/ddl132. [DOI] [PubMed] [Google Scholar]
- 15.Mankodi A., Teng-Umnuay P., Krym M., Henderson D., Swanson M., Thornton C.A. Ribonuclear inclusions in skeletal muscle in myotonic dystrophy types 1 and 2. Ann. Neurol. 2003;54:760–768. doi: 10.1002/ana.10763. [DOI] [PubMed] [Google Scholar]
- 16.Miller J.W., Urbinati C.R., Teng-Umnuay P., Stenberg M.G., Byrne B.J., Thornton C.A., Swanson M.S. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. Embo J. 2000;19:4439–4448. doi: 10.1093/emboj/19.17.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Begemann G., Paricio N., Artero R., Kiss I., Perez-Alonso M., Mlodzik M. Muscleblind, a gene required for photoreceptor differentiation in Drosophila, encodes novel nuclear Cys3His-type zinc-finger-containing proteins. Development. 1997;124:4321–4331. doi: 10.1242/dev.124.21.4321. [DOI] [PubMed] [Google Scholar]
- 18.Artero R., Prokop A., Paricio N., Begemann G., Pueyo I., Mlodzik M., Perez-Alonso M., Baylies M.K. The muscleblind gene participates in the organization of Z-bands and epidermal attachments of Drosophila muscles and is regulated by Dmef2. Dev. Biol. 1998;195:131–143. doi: 10.1006/dbio.1997.8833. [DOI] [PubMed] [Google Scholar]
- 19.Kalsotra A., Xiao X., Ward A.J., Castle J.C., Johnson J.M., Burge C.B., Cooper T.A. A postnatal switch of CELF and MBNL proteins reprograms alternative splicing in the developing heart. Proc. Natl Acad. Sci. USA. 2008;105:20333–20338. doi: 10.1073/pnas.0809045105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanadia R.N., Johnstone K.A., Mankodi A., Lungu C., Thornton C.A., Esson D., Timmers A.M., Hauswirth W.W., Swanson M.S. A muscleblind knockout model for myotonic dystrophy. Science. 2003;302:1978–1980. doi: 10.1126/science.1088583. [DOI] [PubMed] [Google Scholar]
- 21.Pascual M., Vicente M., Monferrer L., Artero R. The Muscleblind family of proteins: an emerging class of regulators of developmentally programmed alternative splicing. Differentiation. 2006;74:65–80. doi: 10.1111/j.1432-0436.2006.00060.x. [DOI] [PubMed] [Google Scholar]
- 22.Squillace R.M., Chenault D.M., Wang E.H. Inhibition of muscle differentiation by the novel muscleblind-related protein CHCR. Dev. Biol. 2002;250:218–230. doi: 10.1006/dbio.2002.0798. [DOI] [PubMed] [Google Scholar]
- 23.Han H., Irimia M., Ross P.J., Sung H.K., Alipanahi B., David L., Golipour A., Gabut M., Michael I.P., Nachman E.N., et al. MBNL proteins repress ES-cell-specific alternative splicing and reprogramming. Nature. 2013;498:241–245. doi: 10.1038/nature12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Du H., Cline M.S., Osborne R.J., Tuttle D.L., Clark T.A., Donohue J.P., Hall M.P., Shiue L., Swanson M.S., Thornton C.A., et al. Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nat. Struct. Mol. Biol. 2010;17:187–193. doi: 10.1038/nsmb.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adereth Y., Dammai V., Kose N., Li R., Hsu T. RNA-dependent integrin alpha3 protein localization regulated by the Muscleblind-like protein MLP1. Nat. Cell Biol. 2005;7:1240–1247. doi: 10.1038/ncb1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang E.T., Cody N.A., Jog S., Biancolella M., Wang T.T., Treacy D.J., Luo S., Schroth G.P., Housman D.E., Reddy S., et al. Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell. 2012;150:710–724. doi: 10.1016/j.cell.2012.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Masuda A., Andersen H.S., Doktor T.K., Okamoto T., Ito M., Andresen B.S., Ohno K. CUGBP1 and MBNL1 preferentially bind to 3′ UTRs and facilitate mRNA decay. Sci. Rep. 2012;2:209. doi: 10.1038/srep00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poulos M.G., Batra R., Li M., Yuan Y., Zhang C., Darnell R.B., Swanson M.S. Progressive impairment of muscle regeneration in muscleblind-like 3 isoform knockout mice. Hum. Mol. Genet. 2013;22:3547–3558. doi: 10.1093/hmg/ddt209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kanadia R.N., Shin J., Yuan Y., Beattie S.G., Wheeler T.M., Thornton C.A., Swanson M.S. Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy. Proc. Natl Acad. Sci. USA. 2006;103:11748–11753. doi: 10.1073/pnas.0604970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Teplova M., Patel D.J. Structural insights into RNA recognition by the alternative-splicing regulator muscleblind-like MBNL1. Nat. Struct. Mol. Biol. 2008;15:1343–1351. doi: 10.1038/nsmb.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kino Y., Mori D., Oma Y., Takeshita Y., Sasagawa N., Ishiura S. Muscleblind protein, MBNL1/EXP, binds specifically to CHHG repeats. Hum. Mol. Genet. 2004;13:495–507. doi: 10.1093/hmg/ddh056. [DOI] [PubMed] [Google Scholar]
- 32.Dhaenens C.M., Schraen-Maschke S., Tran H., Vingtdeux V., Ghanem D., Leroy O., Delplanque J., Vanbrussel E., Delacourte A., Vermersch P., et al. Overexpression of MBNL1 fetal isoforms and modified splicing of Tau in the DM1 brain: two individual consequences of CUG trinucleotide repeats. Exp. Neurol. 2008;210:467–478. doi: 10.1016/j.expneurol.2007.11.020. [DOI] [PubMed] [Google Scholar]
- 33.Tran H., Gourrier N., Lemercier-Neuillet C., Dhaenens C.M., Vautrin A., Fernandez-Gomez F.J., Arandel L., Carpentier C., Obriot H., Eddarkaoui S., et al. Analysis of exonic regions involved in nuclear localization, splicing activity, and dimerization of Muscleblind-like-1 isoforms. J. Biol. Chem. 2011;286:16435–16446. doi: 10.1074/jbc.M110.194928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Osborne R.J., Thornton C.A. Cell-free cloning of highly expanded CTG repeats by amplification of dimerized expanded repeats. Nucl. Acids Res. 2008;36:e24. doi: 10.1093/nar/gkn025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim S.H., Cai L., Pytlos M.J., Edwards S.F., Sinden R.R. Generation of long tracts of disease-associated DNA repeats. Biotechniques. 2005;38:247–253. doi: 10.2144/05382RR01. [DOI] [PubMed] [Google Scholar]
- 36.Wells R.D., Dere R., Hebert M.L., Napierala M., Son L.S. Advances in mechanisms of genetic instability related to hereditary neurological diseases. Nucl. Acids Res. 2005;33:3785–3798. doi: 10.1093/nar/gki697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jung J., Bonini N. CREB-binding protein modulates repeat instability in a Drosophila model for polyQ disease. Science. 2007;315:1857–1859. doi: 10.1126/science.1139517. [DOI] [PubMed] [Google Scholar]
- 38.Yu Z., Teng X., Bonini N.M. Triplet repeat-derived siRNAs enhance RNA-mediated toxicity in a Drosophila model for myotonic dystrophy. PLoS Genet. 2011;7:e1001340. doi: 10.1371/journal.pgen.1001340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mahadevan M.S., Yadava R.S., Yu Q., Balijepalli S., Frenzel-McCardell C.D., Bourne T.D., Phillips L.H. Reversible model of RNA toxicity and cardiac conduction defects in myotonic dystrophy. Nat. Genet. 2006;38:1066–1070. doi: 10.1038/ng1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu Z., Bonini N.M. Modeling human trinucleotide repeat diseases in Drosophila. Int. Rev. Neurobiol. 2011;99:191–212. doi: 10.1016/B978-0-12-387003-2.00008-2. [DOI] [PubMed] [Google Scholar]
- 41.Ho T.H., Charlet B.N., Poulos M.G., Singh G., Swanson M.S., Cooper T.A. Muscleblind proteins regulate alternative splicing. Embo. J. 2004;23:3103–3112. doi: 10.1038/sj.emboj.7600300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li L.B., Yu Z., Teng X., Bonini N.M. RNA toxicity is a component of ataxin-3 degeneration in Drosophila. Nature. 2008;453:1107–1111. doi: 10.1038/nature06909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Charles P., Camuzat A., Benammar N., Sellal F., Destee A., Bonnet A.M., Lesage S., Le Ber I., Stevanin G., Durr A., et al. Are interrupted SCA2 CAG repeat expansions responsible for parkinsonism? Neurology. 2007;69:1970–1975. doi: 10.1212/01.wnl.0000269323.21969.db. [DOI] [PubMed] [Google Scholar]
- 44.Braida C., Stefanatos R.K., Adam B., Mahajan N., Smeets H.J., Niel F., Goizet C., Arveiler B., Koenig M., Lagier-Tourenne C., et al. Variant CCG and GGC repeats within the CTG expansion dramatically modify mutational dynamics and likely contribute toward unusual symptoms in some myotonic dystrophy type 1 patients. Hum. Mol. Genet. 2010;19:1399–1412. doi: 10.1093/hmg/ddq015. [DOI] [PubMed] [Google Scholar]
- 45.Zu T., Gibbens B., Doty N.S., Gomes-Pereira M., Huguet A., Stone M.D., Margolis J., Peterson M., Markowski T.W., Ingram M.A., et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl Acad. Sci. USA. 2011;108:260–265. doi: 10.1073/pnas.1013343108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Muqit M.M., Feany M.B. Modelling neurodegenerative diseases in Drosophila: a fruitful approach? Nat. Rev. Neurosci. 2002;3:237–243. doi: 10.1038/nrn751. [DOI] [PubMed] [Google Scholar]
- 47.Bilen J., Bonini N.M. Drosophila as a model for human neurodegenerative disease. Annu. Rev. Genet. 2005;39:153–171. doi: 10.1146/annurev.genet.39.110304.095804. [DOI] [PubMed] [Google Scholar]
- 48.Fortini M.E., Bonini N.M. Modeling human neurodegenerative diseases in Drosophila: on a wing and a prayer. Trends Genet. 2000;16:161–167. doi: 10.1016/s0168-9525(99)01939-3. [DOI] [PubMed] [Google Scholar]
- 49.Sang T.K., Jackson G.R. Drosophila models of neurodegenerative disease. NeuroRx. 2005;2:438–446. doi: 10.1602/neurorx.2.3.438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bakhoum M.F., Jackson G.R. Demise of the flies: why Drosophila models still matter. Prog. Mol. Biol. Transl. Sci. 2011;100:483–498. doi: 10.1016/B978-0-12-384878-9.00011-X. [DOI] [PubMed] [Google Scholar]
- 51.Zoghbi H.Y., Botas J. Mouse and fly models of neurodegeneration. Trends Genet. 2002;18:463–471. doi: 10.1016/s0168-9525(02)02729-4. [DOI] [PubMed] [Google Scholar]
- 52.Salisbury E., Schoser B., Schneider-Gold C., Wang G.L., Huichalaf C., Jin B., Sirito M., Sarkar P., Krahe R., Timchenko N.A., et al. Expression of RNA CCUG repeats dysregulates translation and degradation of proteins in myotonic dystrophy 2 patients. Am. J. Pathol. 2009;175:748–762. doi: 10.2353/ajpath.2009.090047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Garcia-Lopez A., Monferrer L., Garcia-Alcover I., Vicente-Crespo M., Alvarez-Abril M.C., Artero R.D. Genetic and chemical modifiers of a CUG toxicity model in Drosophila. PLoS One. 2008;3:e1595. doi: 10.1371/journal.pone.0001595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.de Haro M., Al-Ramahi I., De Gouyon B., Ukani L., Rosa A., Faustino N.A., Ashizawa T., Cooper T.A., Botas J. MBNL1 and CUGBP1 modify expanded CUG-induced toxicity in a Drosophila model of myotonic dystrophy type 1. Hum. Mol. Genet. 2006;15:2138–2145. doi: 10.1093/hmg/ddl137. [DOI] [PubMed] [Google Scholar]
- 55.Houseley J.M., Wang Z., Brock G.J., Soloway J., Artero R., Perez-Alonso M., O'Dell K.M., Monckton D.G. Myotonic dystrophy associated expanded CUG repeat muscleblind positive ribonuclear foci are not toxic to Drosophila. Hum. Mol. Genet. 2005;14:873–883. doi: 10.1093/hmg/ddi080. [DOI] [PubMed] [Google Scholar]
- 56.Tian B., White R.J., Xia T., Welle S., Turner D.H., Mathews M.B., Thornton C.A. Expanded CUG repeat RNAs form hairpins that activate the double-stranded RNA-dependent protein kinase PKR. RNA. 2000;6:79–87. doi: 10.1017/s1355838200991544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Michalowski S., Miller J.W., Urbinati C.R., Paliouras M., Swanson M.S., Griffith J. Visualization of double-stranded RNAs from the myotonic dystrophy protein kinase gene and interactions with CUG-binding protein. Nucl. Acids Res. 1999;27:3534–3542. doi: 10.1093/nar/27.17.3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mooers B.H., Logue J.S., Berglund J.A. The structural basis of myotonic dystrophy from the crystal structure of CUG repeats. Proc. Natl Acad. Sci. USA. 2005;102:16626–16631. doi: 10.1073/pnas.0505873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dere R., Napierala M., Ranum L.P., Wells R.D. Hairpin structure-forming propensity of the (CCTG.CAGG) tetranucleotide repeats contributes to the genetic instability associated with myotonic dystrophy type 2. J. Biol. Chem. 2004;279:41715–41726. doi: 10.1074/jbc.M406415200. [DOI] [PubMed] [Google Scholar]
- 60.Sobczak K., de Mezer M., Michlewski G., Krol J., Krzyzosiak W.J. RNA structure of trinucleotide repeats associated with human neurological diseases. Nucl. Acids Res. 2003;31:5469–5482. doi: 10.1093/nar/gkg766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Obenauer J.C., Cantley L.C., Yaffe M.B. Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucl. Acids Res. 2003;31:3635–3641. doi: 10.1093/nar/gkg584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bolte S., Cordelieres F.P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]