

Abstract
Glycogen storage disease type II (GSDII) or Pompe disease is an autosomal recessive disorder caused by acid alpha-glucosidase (GAA) deficiency, leading to lysosomal glycogen accumulation. Affected individuals store glycogen mainly in cardiac and skeletal muscle tissues resulting in fatal hypertrophic cardiomyopathy and respiratory failure in the most severe infantile form. Enzyme replacement therapy has already proved some efficacy, but results remain variable especially in skeletal muscle. Substrate reduction therapy was successfully used to improve the phenotype in several lysosomal storage disorders. We have recently demonstrated that shRNA-mediated reduction of glycogen synthesis led to a significant reduction of glycogen accumulation in skeletal muscle of GSDII mice. In this paper, we analyzed the effect of a complete genetic elimination of glycogen synthesis in the same GSDII model. GAA and glycogen synthase 1 (GYS1) KO mice were inter-crossed to generate a new double-KO model. GAA/GYS1-KO mice exhibited a profound reduction of the amount of glycogen in the heart and skeletal muscles, a significant decrease in lysosomal swelling and autophagic build-up as well as a complete correction of cardiomegaly. In addition, the abnormalities in glucose metabolism and insulin tolerance observed in the GSDII model were corrected in double-KO mice. Muscle atrophy observed in 11-month-old GSDII mice was less pronounced in GAA/GYS1-KO mice, resulting in improved exercise capacity. These data demonstrate that long-term elimination of muscle glycogen synthesis leads to a significant improvement of structural, metabolic and functional defects in GSDII mice and offers a new perspective for the treatment of Pompe disease.
INTRODUCTION
Glycogen storage disease type II (GSDII), also termed Pompe disease (MIM 232300), is a rare autosomal recessive disorder caused by a defect in the gene encoding acid alpha-glucosidase (GAA; EC 3.2.1.20). The 110 kDa precursor of GAA is sequentially cleaved to produce a 95 kDa intermediate followed by cleavage within the lysosome to the 76 and 70 kDa mature forms (1). GAA catalyses the complete hydrolysis of its natural substrate glycogen by cleaving both α-1,4 and α-1,6 glycosidic linkages at an acidic pH, allowing glucose to be liberated into the cytoplasm. In the absence of GAA, glycogen accumulates within the lysosomes in various tissues, but mainly in cardiac and skeletal muscle. Clinically, Pompe disease manifests as a continuum of phenotypes which are largely dependent on the level of residual GAA activity (2). A near complete loss of GAA activity results in an infantile form, marked by a severe cardiomyopathy leading to death before 2 years of life due to heart failure. Partial enzymatic deficiency results in juvenile and adult-onset forms of GSDII characterized by a slowly progressive skeletal muscle weakness without cardiac involvement and death by respiratory failure. The overall incidence of Pompe disease is estimated around 1 in 40 000 live births, but the number varies in different ethnic populations (1,3). Mouse models of GSDII exhibit features of early and late forms of the disease, with massive storage of glycogen in the heart and progressive muscle wasting (4,5).
Enzyme replacement therapy (ERT) has been successfully applied to a number of lysosomal storage disorders and is now available for Pompe disease. Results of both preclinical (6,7) and clinical studies (8–12) showed that cardiac muscle responds well to recombinant human GAA derived from either rabbit milk or Chinese hamster ovary cells (Myozyme®). However, skeletal muscle receives very little of the recombinant enzyme because it is largely diverted to the liver; this contributes to the very limited glycogen clearance in skeletal muscle, especially in the late stages of the disease (10). A selective resistance of type II muscle fibers to ERT was demonstrated in mice (13,14). Another problem with the ERT may be that this approach relies on the cation-independent mannose-6-phosphate receptor (CI-MPR)-mediated delivery of the recombinant enzyme to the lysosome, a pathway which is relatively inefficient in muscle due to the low abundance of the CI-MPR in skeletal muscle (15,16) and abnormal GAA trafficking in type II fibers (7,17). Thus, alternative therapeutic strategies independent or complementary to ERT are warranted. Substrate reduction therapy (SRT) uses small acting molecules in order to inhibit the synthesis of compounds that cannot be degraded in lysosomes due to an enzymatic defect. SRT was successfully applied in combination with ERT or bone marrow transplantation to other lysosomal diseases (18). Advantages of SRT include (i) oral administration of the drug, which significantly improves the quality of life, (ii) lack of immunoreactivity and (iii) ability to cross the blood–brain barrier. Finally, this strategy is independent of the mannose-6-phosphate receptor pathway.
We have recently demonstrated, as a proof of principle, that SRT could be efficiently used to rescue the phenotype of Pompe disease by modulating glycogen synthesis (19). A lentiviral vector-mediated delivery of short hairpin RNA (shRNA) targeted to the muscle form of glycogen synthase (GYS1) allowed a specific knock-down of GYS1 expression, resulting in a reversal of the pathological glycogen accumulation in GAA-KO myofibers. A similar effect was observed after in vivo injection of AAV2/1-shRNAGYS in the gastrocnemius muscle of the GAA-KO mice. However, in this study, the transient expression of AAV vectors in vivo impaired the long-term evaluation of the effect of inhibiting glycogen synthesis in GAA-KO mice. To overcome this limitation, we have developed a novel GAA-KO model in which the GYS1 gene is completely inactivated genetically. This model was generated by breeding GAA-KO mice with the previously described GYS1-KO mice, also known as MGSKO mice (20–22). These GYS1-KO mice show a severe perinatal mortality with 90% of the GYS1-null pups dying soon after birth from impaired cardiac function (20). The surviving 10% GYS1-KO mice were devoid of glycogen in cardiac and skeletal muscle and the morphology of these tissues remained normal; the animals’ ability to exercise was not affected and they showed overall improved glucose tolerance (20–22).
In the present work, we analyzed the long-term effect of in vivo GYS1 inactivation on the pathological phenotype of GSDII mice. We demonstrate that blocking glycogen synthesis is associated with the lack of lysosomal glycogen accumulation and a complete rescue of cardiac and skeletal muscle function in GSDII mice. These results indicate that modulation of glycogen synthesis could complement or constitute an alternative to ERT in GSDII patients.
RESULTS
GAA-KO mice lacking GYS1 show perinatal mortality but normal growth development
In order to study the long-term effect of GYS1 inactivation on the Pompe disease phenotype, the GYS1 gene was disrupted in GAA-KO mice. GAA-KO mice were bred with heterozygous GYS1+/− mice to generate a novel mouse model with a dual inactivation of both GAA and GYS1 genes (GAA−/−/GYS1−/−) (see Materials and Methods). Inter-crosses allowed us to generate littermates of several genotypes: GAA-KO mice (GAA−/−/GYS1+/+), GYS1 heterozygous GAA-KO mice (GAA−/−/GYS1+/−) and double-KO mice (GAA−/−/GYS1−/−) (Table 1). Genotyping revealed that the number of newborn GAA/GYS1-KO pups was significantly less than what would be expected from a Mendelian inheritance (Table 1). Furthermore, the litter size of the GAA−/−/GYS1+/− inter-crosses was smaller compared with that of the GAA-KO breeders. Most of the GAA/GYS1-KO pups died right after birth, without taking a breath and in addition, significant mortality of GAA/GYS1-KO was observed during the first 3 days of life. Thus, 90% of GAA/GYS1-KO pups died before or shortly after birth; this percentage is similar to what was previously observed for GYS1-KO mice (20). The surviving 10% of GAA/GYS1-KO mice did not have any significant difference in the growth rate compared with normal (WT) or GAA-KO mice (Supplementary Material, Fig. S1).
Table 1.
Number of live GAA-KO mice according to their muscle glycogen synthase genotype
| Birth | Alive after 3 days of life | |
|---|---|---|
| GYSm−/− | 19 | 10 |
| GYSm+/− | 214 | 214 |
| GYSm+/+ | 119 | 119 |
| Total | 352 | 346 |
GAA-KO mice were genotyped for the GYS1 gene at birth or after 3 days of life.
Pathological glycogen storage is reduced in GAA/GYS1-KO mice
Because the GYS1 isoform is expressed in skeletal and cardiac muscle, its absence is expected to eliminate glycogen synthesis in these tissues (20). In GAA-KO mice, the glycogen accumulation increased progressively with time in both cardiac (1.41 ± 0.2 mg glycogen/mg protein and 2.7 ± 0.3 mg glycogen/mg protein at 6- and 11-month-old, respectively) and skeletal muscles (gastrocnemius) (2.6 ± 0.33 mg glycogen/mg protein and 3.2 ± 0.33 mg glycogen/mg protein at 6- and 11-month-old, respectively), when compared with normal mice (0.04 ± 0.2 mg glycogen/mg protein and 0.12 ± 0.2 mg glycogen/mg protein in the heart; 0.21 ± 0.2 mg glycogen/mg protein and 0.1 ± 0.2 mg glycogen/mg protein in skeletal muscle) (Fig. 1A and B). In GAA/GYS1-KO mice, a significant decrease of glycogen accumulation was observed, reaching near normal values in the heart and skeletal muscle. As expected, a moderate but significant glycogen accumulation was found in the liver of GAA-KO when compared with WT mice and the glycogen levels remained unchanged in GAA/GYS1-KO (data not shown). These findings are explained by the existence of the liver-specific GYS2 isoform, which is responsible for hepatic glycogen synthesis.
Figure 1.

Prevention of glycogen accumulation in the heart and skeletal muscle of GAA-KO mice by glycogen synthesis inhibition. Glycogen accumulation in cardiac (A) and skeletal muscle (gastrocnemius) (B) was measured in 6-month-old mice. Results are expressed as the mean ± SEM (n ≥ 4). *P < 0.05 compared with WT mice and, #P < 0.05 compared with GAA/GYS1-KO mice. High resolution light microscopy (HRLM) sections of cardiac and skeletal muscle glycogen are presented in (C and D), respectively (PAS, periodic acid Schiff and Richardson's stain, ×600).
A dramatic decrease in glycogen accumulation in cardiac and skeletal muscle of GAA/GYS1-KO mice was confirmed by periodic acid Schiff (PAS) staining. An intense staining characteristic of lysosomal glycogen accumulation was clearly evident in the heart (Fig. 1C) and skeletal muscle (Fig. 1D) of GAA-KO mice, whereas no significant staining was observed in the heart and skeletal muscle of GAA/GYS1-KO mice.
Massive glycogen accumulation in GAA-KO mice is associated with the early development of cardiomegaly (Fig. 2A and B) and the later appearance of muscular atrophy (Fig. 2C). Cardiac hypertrophy was documented at birth as well as in the adult GAA-KO [at 6 months of age, the heart constitutes 0.79 ± 0.023% of the total body mass compared with 0.56% ± 0.02 in the WT mice (Fig. 2A and B)]. In contrast, both newborn and adult GAA/GYS1-KO mice have normal cardiac (0.52% ± 0.02 of the body mass in 6-month-old mice; Fig. 2A and B) and gastrocnemius size (0.46 ± 0.009%; Fig. 2C).
Figure 2.
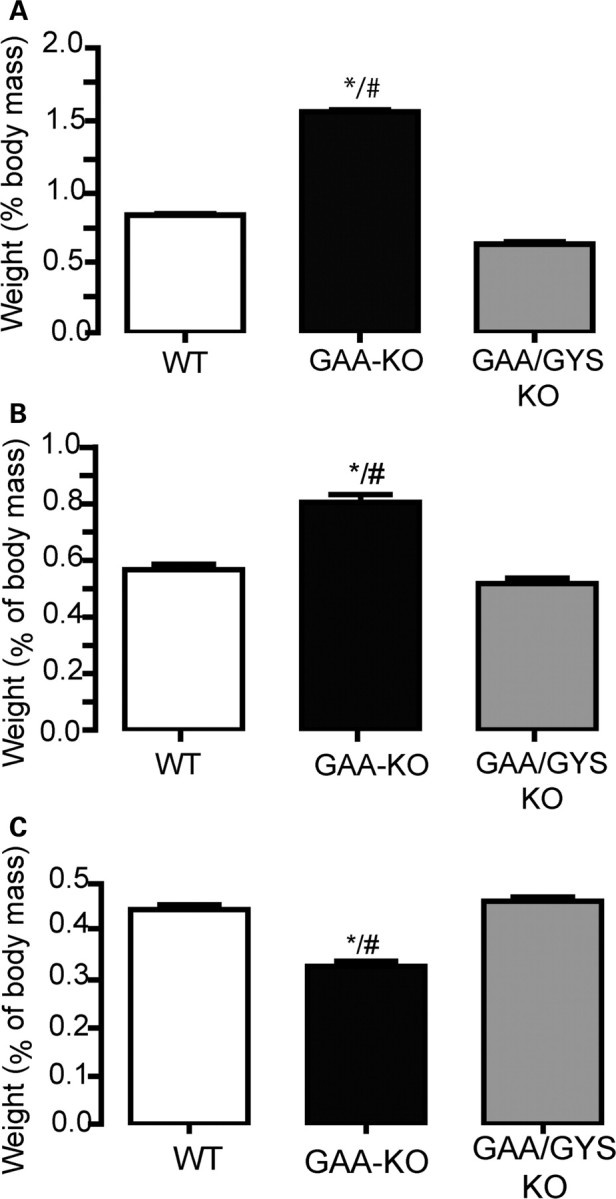
Prevention of cardiomegaly and skeletal muscle atrophy in GAA-KO mice by glycogen synthesis inhibition. The weight (% of body mass) of the heart [1-day-old in (A) and adult in (B)] and gastrocnemius muscle (6-month-old) (C) was measured. The weights are expressed as the mean ± SEM (n ≥ 4). *P < 0.05 compared with WT mice and #P < 0.05 compared with GAA/GYS1-KO mice.
Both the endosomal/lysosomal expansion and autophagic build-up are prevented in GAA/GYS1-KO mice
It is now hypothesized that in addition to the enlargement and rupture of the lysosomal/endosomal compartment, autophagic build-up represents a critical pathogenic mechanism responsible for muscle damage in GSDII. Therefore, the presence of autophagic build-up and the morphology of the lysosomal compartment were evaluated on single muscle fibers from a predominantly type II muscle (gastrocnemius) from 6-month-old mice. Confocal microscopy was used to analyze single muscle fibers after staining with the late endosomal/lysosomal marker lysosomal-associated membrane protein 1 (LAMP-1) (in red) along with the specific autophagosomal marker LC3 (in green). Clusters of LC3-positive autophagosomes were observed in the central regions of myofibers in GAA-KO mice (Fig. 3B), whereas LAMP-1 positive endosome/lysosomes were distributed throughout the myofibers (Fig. 3B and D). In contrast, autophagosomes were not detected in fibers from WT or GAA/GYS1-KO mice (Fig. 3A and C) and LAMP-1 staining revealed only tiny LAMP-1 positive vesicles. These results demonstrate that the two critical components, autophagic build-up and lysosomal enlargement, are prevented in GAA-KO mice by suppressing glycogen synthesis.
Figure 3.

Staining for LAMP-1 (late endosomes/lysosomes) and LC3 (autophagosomes) in type II myofibers from 6-month-old mice. The endosomal/lysosomal compartment (LAMP-1-positive structures in red) is enlarged in GAA-KO mice (B, D) when compared with WT (A) and GAA/GYS1-KO (C) mice. The autophagosomes (LC3-positive structures in green) are present in the core of the myofiber from GAA-KO (B), but not in WT (A) or in GAA/GYS1-KO mice (C).
Type II muscle fiber atrophy is reversed and myofiber subtype composition is normalized in GAA/GYS1-KO mice
It has previously been shown that the presence of autophagic build-up in type II fast-twitch myofibers (but not in type I slow-twitch) is associated with the resistance of these fibers to ERT in GSDII mice. The autophagic build-up may also contribute to the progressive muscle atrophy (Fig. 2C) observed in the gastrocnemius (type II myofibers, mainly type IIB), when compared with the soleus (majority of slow-oxidative type I myofibers, but also fast-oxidative type IIA fibers; data not shown) in 11-month-old GAA-KO versus WT mice. To address this issue, we analyzed the fiber size in predominantly fast and slow muscles in WT, GAA-KO and GAA/GYS1-KO mice. The fiber size was measured on dystrophin-stained sections of gastrocnemius, extensor digitorum longus (EDL; both fast) and soleus muscle (shown for gastrocnemius and EDL in Fig. 4) from 3-month-old mice. A physiological peripheral dystrophin staining was observed in type II muscle fibers (gastrocnemius and EDL) from normal mice (Fig. 4A and D). GAA-KO type II muscle exhibited strong, possibly non-specific staining, which may have colocalized with the autophagic area (Fig. 4B and E). This non-specific staining is localized in the center (Fig. 4G) of the fiber, but could spread (Fig. 4H) and fill completely the muscle fiber (Fig. 4I). In contrast, the dystrophin staining in GAA/GYS1-KO mice was similar to that seen in the WT (Fig. 4C and F).
Figure 4.

Dystrophin staining of the gastrocnemius and EDL in 3-month-old mice of different genotypes. Gastrocnemius (A–C) and EDL (extensor digitorum longus) (D–I). WT (A, D), GAA-KO (B, E, G, H, I) and GAA/GYS1-KO mice (C, F).
We observed a significant atrophy of type II fast-twitch myofibers (gastrocnemius) when compared with type I slow-twitch and IIA myofibers (soleus) in 3-month-old GAA-KO mice (Fig. 5A and B). In the dystrophin-stained sections of the gastrocnemius muscle, the average areas of the myofibers in the gastrocnemius muscle were 1997 ± 104, 2684 ± 290 and 2540 ± 232 μm2 for GAA-KO, WT and GAA/GYS1-KO mice, respectively (Fig. 5A and C). A significant shift towards small myofibers was observed in fast gastrocnemius muscle from GAA-KO mice when compared with WT and GAA/GYS1-KO mice (Fig. 5C). In contrast, a moderate shift towards enlarged myofibers was observed in soleus muscle from GAA-KO when compared with WT and GAA/GYS1-KO mice (Fig. 5B and D).
Figure 5.

Quantitative analysis of myofiber composition in the gastrocnemius and soleus muscle from 3-month-old mice. Myofiber areas of the gastrocnemius (A, C) and soleus (B, D) were calculated after dystrophin staining. Results are expressed as the mean ± SEM (n ≥ 3). For each mouse, fiber area or percentage of fibers were measured on more than 500 fibers for the soleus and more than 2000 fibers for the gastrocnemius. The myofiber composition (E and F) was calculated after specific staining with antibodies against type I, IIA, IIB and IIX myosin. Values are expressed as the mean ± SEM (n ≥ 3). *P < 0.05 compared with WT mice and #P < 0.05 compared with GAA/GYS1-KO mice.
It is known that oxidative slow type I and IIA myofibers muscle display significant differences with respect to contraction, metabolism and susceptibility to exhaustion when compared with glycolytic fast type IIB muscle. To investigate the relative proportion of different fibers subtypes in predominantly type II gastrocnemius and type I soleus muscles from 3-month-old mice of different genotypes, sections were immunostained with antibodies against fast, slow or developmental myosin heavy chain. No significant shift in the various subtypes was observed in the gastrocnemius or the soleus from GAA-KO when compared with normal or GAA/GYS1-KO mice (Fig. 5E and F). Only a minor shift from type IIB to type IIA and IIX could be detected in the gastrocnemius of GAA-KO mice that was reversed to normal in GAA/GYS1-KO mice (Fig. 5E). These data suggest that the decrease in myofiber areas in the gastrocnemius of GAA-KO (Fig. 5B) results from atrophy rather than the rearrangement of the fiber subtypes.
Glucose and insulin tolerance were preserved in GAA/GYS1-KO mice
Because glycogen biosynthesis in liver and muscle is a very important step in glucose homeostasis, we then evaluated the regulation of glycemia in GAA-KO and GAA/GYS1-KO mice. After 12 h fasting, basal glucose (Glc) levels were reduced in GAA-KO mice (70 ± 7.2 mg/dl) when compared with WT and GAA/GYS1-KO mice (100 ± 5.8 and 106.2 ± 22 mg/dl, respectively) (Fig. 6A). The ability to clear glucose from the circulation in response to a glucose load was assessed by using a standard glucose tolerance test (GTT). Thirty minutes after glucose administration (i.p. injection of 2 mg Glc/g body weight), blood glucose reached 105 ± 9 mg/dl in GAA-KO mice, whereas higher values were observed in WT (169 ± 16 mg/dl) and GAA/GYS1-KO mice (165 ± 13 mg/dl) (Fig. 6B). Reuptake of blood glucose was slower in WT and in GAA/GYS1-KO mice when compared with GAA-KO mice suggesting a hyper-tolerance to glucose in these latter mice. In order to eliminate a possible modification of glucose absorption from the peritoneal cavity, GTT was also performed with orally administered glucose. As seen after intraperitoneal delivery, the GAA-KO mice disposed glucose more effectively (data not shown).
Figure 6.
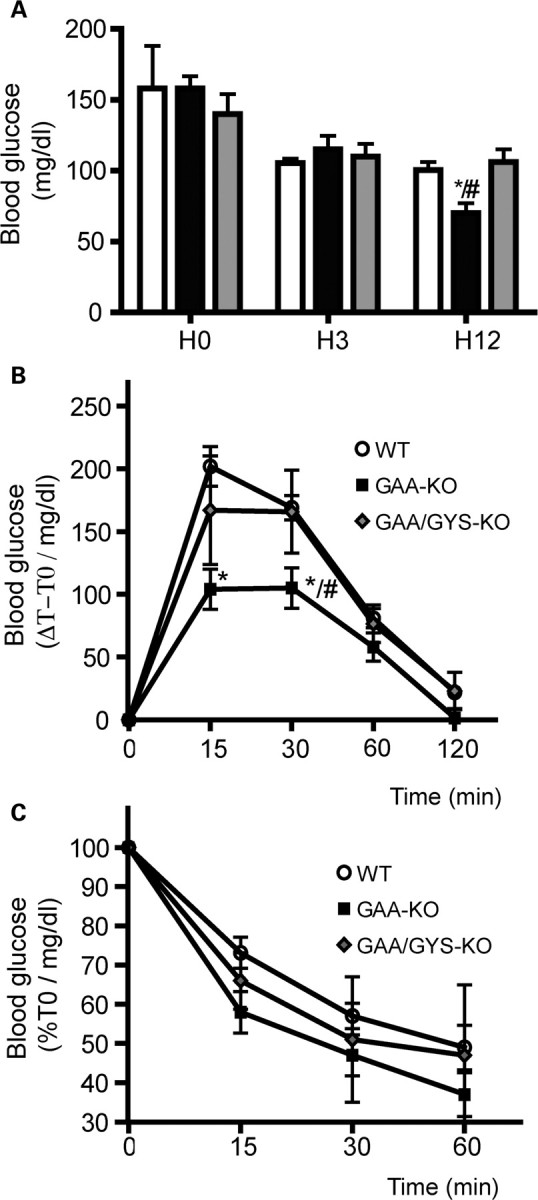
Blood glucose level, glucose and insulin tolerance of 3-month-old mice on a standard diet. Blood glucose level was measured after normal diet (H0), 3 h (H3) or 12 h (H12) of fasting (A). Glucose tolerance test (GTT) (B). Insulin tolerance test (ITT) (C). Values are expressed as the mean ± SEM (n ≥ 3). *P < 0.05 compared with WT mice and #P < 0.05 compared with GAA/GYS1-KO mice.
The capacity to respond to the hyperglycemic episode induced by a bolus of glucose depends on the integrity of complex metabolic mechanisms including (i) insulin secretion in response to glucose by the pancreatic β cells, (ii) hypoglycemic action of insulin in muscle and liver and (iii) reuptake of glucose at basal state. Our results suggest that either insulin secretion or clearance is modified in the GAA-KO mice and that both were restored after GYS1 inactivation in muscle. To discriminate between these possibilities, we performed insulin tolerance tests (ITT) by monitoring blood glucose after intra-peritoneal insulin injection. We observed a slight increase in glucose reuptake in GAA-KO mice when compared with WT and GAA/GYS1-KO mice after insulin administration (statistically insignificant) (Fig. 6C), possibly suggesting over-sensitiveness to insulin in the GAA-KO mice.
Insulin response pathway is deregulated in GAA and GAA/GYS1-KO mice
In skeletal muscle, glucose transport occurs at the plasma membrane through the facilitative glucose transporter GLUT1 which carries out the basal transport, and through the translocation of the insulin-regulatable glucose transporter GLUT4. We then analyzed insulin-mediated translocation of the GLUT4 glucose transporter at the myofiber cell surface membrane. After a 12 h fasting period, mice were challenged with insulin (intraperitoneal injection of 0.2 IU/kg). Five minutes later, tibialis anterior muscles were rapidly isolated and the accumulation of GLUT1 and GLUT4 to the plasma membrane were analyzed by western blot as shown in Fig. 7A. Insulin injection led to a significant translocation of the GLUT4 transporter to the cellular membrane in WT (3-fold increase) and GAA/GYS1-KO (2-fold increase) mice (Fig. 7B). In contrast, an already elevated GLUT4 level in the GAA-KO mice remained unchanged after insulin stimulation. The level of the insulin-insensitive GLUT1 glucose transporter was also elevated in GAA-KO mice when compared with WT and GAA/GYS1-KO. These results suggest that GAA-KO mice present an elevated basal glucose uptake in skeletal muscles that was corrected in GAA/GYS1-KO mice.
Figure 7.

Expression of GLUT4 and GLUT1 glucose transporters. Protein levels were analyzed by western blot on plasma membranes (30 μg) isolated from tibialis anterior muscles of mice challenged with 0.2 IU/kg body weight insulin for 5 min. Loading was normalized using anti-M-cadherin antibody (A). Quantification of three independent experiments (B). Data are expressed as the mean ± SEM (n ≥ 4). *P < 0.05 compared with WT mice and #P < 0.05 compared with GAA/GYS1-KO mice. NS: non-significant difference compared with WT in the presence of insulin.
Maximal muscle strength is restored in GAA/GYS1-KO mice
Since glycogen accumulation is associated with severe muscle dysfunction in GAA-KO mice, we analyzed the maximal tetanic force as well as the specific muscle tensions (mN/mm2) in mice of different genotypes. As shown in Fig. 8A, the maximal tetanic force was significantly reduced in soleus muscle of 11-month-old GAA-KO mice when compared with WT mice. In GAA/GYS1-KO mice, there was a significant improvement in muscle function. However, GAA/GYS1-KO mice showed a marked reduction in the resistance to fatigue compared with WT and GAA-KO mice (Fig. 8B) which could possibly be explained by the absence of glycogen in muscle.
Figure 8.
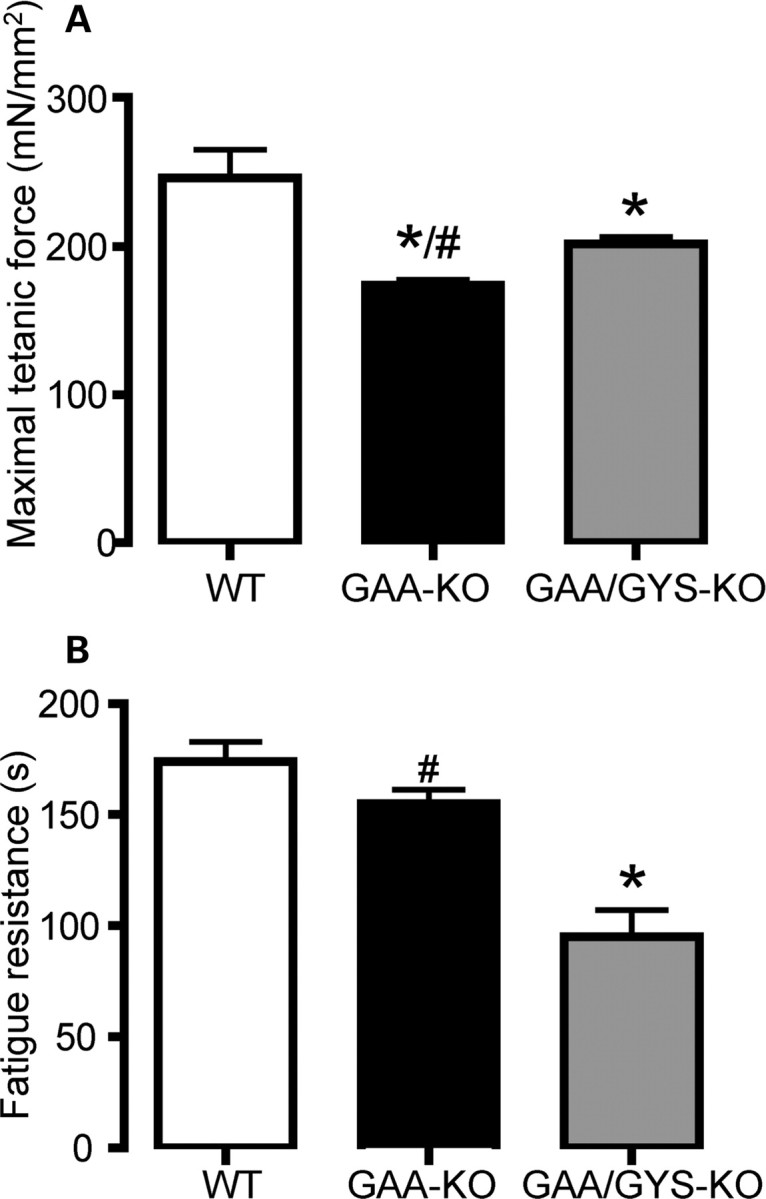
Muscle function in 11-month-old mice. Maximal tetanic force (A) and fatigue resistance (FR) were tested on the soleus. Results are expressed as the mean ± SEM (n ≥ 12). *P < 0.05 compared with WT mice and #P < 0.05 compared with GAA/GYS1-KO mice.
Because glycogen is consumed during sustained exercise, fed animals were subjected to treadmill running until exhaustion. We did not observed any obvious difference in exercise capacity between the different genotypes in 6-month-old mice (data not shown). However, 11-month-old GAA-KO mice presented a significant reduction in their locomotor activity when compared with normal mice (66.0 ± 3.04 and 33.1 ± 4.4 min in WT and GAA-KO mice, respectively), whereas in GAA/GYS1-KO mice the locomotor activity was largely preserved (57.7 ± 7.5 min).
DISCUSSION
The muscle isoform of glycogen synthase (GYS1) is a highly regulated enzyme that permits the elongation of the glycogen chain. Overexpression of glycogen synthase in skeletal muscle of the transgenic mice (GSL30) results in elevated muscle glycogen content (23). However, the limited activity of glycogen branching enzyme in these transgenic mice leads to the synthesis of abnormal less branched glycogen (24). Overexpression of glycogen synthase in skeletal muscle of the GAA-KO mice exacerbated the GAA-KO phenotype and resulted in severe early onset muscle wasting (25). These mice showed normal glycogen branching enzyme activity and accumulated abnormal polyglucosan bodies in skeletal muscle (25).
The disruption of the GYS1 gene in mice (GYS1-KO), on the other hand, resulted in undetectable glycogen levels in the heart and skeletal muscle (20). The most striking phenotype associated with the absence of GYS1 in mice is their poor survival rate, with ∼90% of newborn pups dying due to cardiac failure (20). It has been shown that in the fetal heart, glycogen can comprise up to 30% of the cell volume, whereas in adult only 2% of the cardiomyocyte volume is occupied by glycogen (26). The high level of glycogen during the embryonic development of cardiac tissue suggests its specific role, perhaps as an energy source for completion of certain developmental phases. Although the majority of the newborn pups died, the surviving 10% developed normally and did not show any major defects.
We reasoned that the suppression of glycogen synthesis in skeletal muscle of Pompe mice might rescue their phenotype. To meet this goal, we developed a novel murine model with both GAA and GYS1 disruption (GAA/GYS1-KO mice) after breeding GAA-KO and GYS1-KO mice. A complete reversal of glycogen storage was observed in GAA/GYS1-KO mice in cardiac and skeletal muscle when compared with GAA-KO from the same litter. In GAA-KO mice and early-onset GSDII patients, cardiomyocyte glycogen accumulation leads to a severe cardiomegaly. We found a complete reversal of the cardiomegaly in both newborn and adult GAA/GYS1-KO mice. Unlike cardiac muscle, abnormal long-term accumulation of glycogen in skeletal muscle leads to atrophy in type II fast muscle in GAA-KO mice. In GAA/GYS1-KO mice, we observed a normalization of the myofibers morphology with a reversal of the atrophy in the GAA/GYS1-KO.
Although high amounts of recombinant GAA enzyme are administered to GSDII patients during ERT, only a small fraction is targeted to muscle lysosomes. A 20- to 30-fold higher rhGAA dose is required for therapeutic efficiency when compared with ERT in Gaucher disease (up to 20–40 mg/kg every other week of rhGAA versus 1.0 mg/kg Cerezyme®). The resistance of muscle fibers to ERT is mainly due to (i) the low density of CI-MPRs (responsible for the uptake of the recombinant enzyme from the extracellular compartment to the lysosome) on skeletal muscle when compared with cardiac muscle and (ii) the disrupted downstream pathways of the endosomal/lysosomal system in GAA deficient myofibers. Furthermore, it was found that the cellular pathology in Pompe disease affects both endocytic (the route of the therapeutic enzyme) and autophagic (the presumed route of glycogen) pathways. The absence of glycogen accumulation in GAA/GYS1-KO mice seems to restrain both the pathological proliferation and enlargement of the endosomal/lysosomal compartment and the stimulation of the autophagic pathway.
Previous studies demonstrated that fast-twitch type II myofibers are resistant to ERT when compared with slow-twitch type I. This phenomenon may be explained by the low abundance of CI-MPR and proteins involved in lysosomal trafficking in type II when compared with type I fibers. It was also suggested that the autophagic clusters (referred to as ‘non-contractile material’), observed in type II muscle by electron microscopy, contribute to the decline in muscle contractile function and to the resistance to treatment in Pompe mice. We have found that the formation of LC3-positive autophagosomes was completely prevented in GYS1/GAA-KO mice. The role of the autophagic build-up in Pompe disease is still discussed (27). It was suggested that the failure of glycogen digestion within the lysosomes may result in a local starvation that stimulates an autophagic response. The newly formed autophagic vesicles are unable to fuse with and discharge their content into glycogen-filled lysosomes, leading to accumulation of autophagic debris and profound disorganization of the microtubules in skeletal muscle of Pompe mice. Therefore, efforts to provide energy to type II fibers should be sought. A reduction in the cytoplasmic glycogen synthesis could affect the level and the availability of intracellular glucose-6-phosphate. Thus, SRT could perhaps have a secondary advantage by increasing the energy status of the cell.
The role of skeletal muscle as a primary site for insulin-dependent glucose disposal is well established in humans (28). In mice, its role is less clear. A number of transgenic and knockout mouse models have been used to address this issue. Muscle-specific disruption of the insulin receptor causes skeletal muscle insulin resistance without clear diabetes (29,30). Elimination of muscle GLUT4 leads to a more severe phenotype, with impaired muscle glucose uptake and the development of diabetes in a subset of mice (31,32). GLUT1 overexpression in skeletal muscle of GAA-KO mice resulted in a severe disease with early-onset muscle pathology (25). However, glucose metabolism in basic GAA-KO was not clearly characterized. We found a hypoglycemia in the GAA-KO mice, a feature, which has not been reported in Pompe patients. It is, however, important to emphasize that glucose metabolism and glycogen distribution in humans and rodents are quite different—as a fraction of body weight, human muscle glycogen is 10-fold greater than mouse muscle glycogen (33).
GTT and ITT revealed low glucose amount and excessive glucose uptake after insulin injection in GAA-KO mice when compared with WT and GYS1/GAA-KO mice. Glucose homeostasis is highly dependent on the capacity of skeletal muscle to increase glucose uptake in response to insulin. In skeletal muscle, transport of glucose is mediated by GLUT4, whereas phosphorylation of glucose is catalyzed by hexokinase II (HKII). Insulin translocates an intracellular pool of GLUT4 to the plasma membrane and stimulates HKII transcription. We demonstrated here that the abundance of GLUT1 glucose transporter at the plasma membrane is higher in GAA-KO mice compared with WT. We also found an insulin-independent increase in the level of the GLUT4 transporter at the myofiber cell surface in these mice, suggesting that the flux of glucose was not limited by the amount of glucose transporters. The increased levels of GLUT1 and GLUT4 at the plasma membrane under basal conditions could partially explain the increased glucose reuptake in GAA-KO mice. This increased glucose uptake in the GAA-KO mice could, in turn, lead to the increase in glycogen synthesis and lysosomal storage in muscle. In contrast, the suppression of glycogen synthesis in GAA/GYS1-KO mice may contribute to the increase in the amount of available glucose. This excess of glucose may down-regulate the level of GLUT transporters at the membrane.
Glycogen storage is associated with lysosomal enlargement and disruption of the contractile unit in the myofiber leading to altered muscle contraction in GAA-KO mice. The suppression of muscle glycogen synthesis in these mice leads to an increased maximal force of the soleus in GAA/GYS1-KO mice and a global improvement of contraction capacity. Although the performance of the GAA/GYS1-KO mice on the treadmill was similar to that of the WT, the former group showed significantly less resistance to fatigue. Several factors such as global performance of different muscle groups (other than gastrocnemius) and cardiovascular performance may account for the apparent discrepancy.
In humans, the carbohydrate reserves are an important factor of endurance upon sustained exercise, and muscle glycogen has long been viewed as a reserve of critical energy during burst of activity, sustained muscle work and hypoxia (34–36). The depletion of muscle glycogen content results in excessive fatigue and muscle impairment (37–39). In patients with McArdle disease, a deficiency of muscle phosphorylase, the ineffective utilization of muscle glycogen associated with this disease, leads to impaired exercise tolerance (37). GYS2 deficiency, also known as glycogen storage disease type 0 presents with glucose homeostasis disturbance, intolerance to fasting and hypoglycemia (37). Muscle-specific glycogen storage disease type 0 has recently been described in four children from two different families (38,39). In these patients, a homozygous null mutation (R462X) or a homozygous 2 bp deletion in exon 2 were identified in the GYS1 gene leading to a truncated protein and a severe GYS1 enzymatic deficiency. Both families had occurrence of spontaneous abortions, stillbirth and early death. Two of these patients died from sudden cardiac arrest, whereas the other two presented with fatigability and cardiomyopathy. The disruption of GYS1 in mice leads to abnormal cardiac development and a severe perinatal mortality; 90% of GYS1-null pups die due to impaired cardiac function (20). These data highlight the role of muscular and especially cardiac glycogen as an energy reserve for fuel contraction and cardiac development. Therefore, only partial or muscle-targeted suppression of GYS1 can be considered as a therapeutic approach in Pompe disease.
We have previously demonstrated that a partial shRNA-mediated inhibition of GYS1 in muscle of GSDII mice was sufficient to significantly reduce the amount of stored glycogen (19). The development of a water-soluble pharmacological agent allowing partial and specific inactivation of the GYS1 enzyme would greatly facilitate the evaluation of this approach in GSDII mice. Chemical inhibitors present major advantages compared with AAV-mediated shRNAs such as oral administration and lack of immune reactivity (18). Furthermore, a molecule that can pass the blood–brain barrier could rescue the neurological involvement recently described in Pompe disease (40,41). Such molecules have already been used in combination with ERT for other lysosomal disorders like Gaucher disease (Zavesca®/Cerezyme®). This combination is recommended for patients with severe disease who have not achieved therapeutic goals with enzyme alone, or for those with a neuronopathic disease (42). Here we demonstrate that the genetic suppression of GYS1 prevents the pathological accumulation of lysosomal glycogen. Prevention of lysosomal glycogen accumulation was also observed by us in 2-day-old GAA-KO following shRNA-mediated inhibition of GYS1. Additional studies are required to evaluate the use of SRT alone or in combination with ERT to reverse the established pathology, especially in old GSDII mice.
In conclusion, we analyzed the long-term effect of suppressing glycogen synthesis in a murine model of Pompe disease. We demonstrated that this approach rescued the phenotype in Pompe mice as shown by the absence of lysosomal glycogen storage and the improvement in structural and functional muscle capacity. This innovative strategy could open novel perspectives that should be considered for the treatment of Pompe disease.
MATERIALS AND METHODS
Generation of GAA/GYS1-KO mice
Heterozygous GYS1+/−mice 129SV/C57Bl6 J (GAA+/+/GYS1+/−) and GAA-KO mice 129SV (GAA−/−/GYS1+/+) (F0) have previously been described (4,22). GYS1-KO mice were crossed with GAA-KO mice to generate F1 compound heterozygote animals (GAA+/−/GYS1+/−). Then, compound heterozygotes were crossed with GAA-KO mice to generate GAA−/−/GYS1+/−mice (F2). Finally, double knockouts (GAA−/−/GYS1−/−) (F3) were obtained by interbreeding the F2 generation. GAA-KO mice used as controls were the littermates of GAA/GYS1-KO mice in the F3 generation. WT mice (GAA+/+/GYS1+/+) were obtained after inter-crossing heterozygous GYS1+/−mice. GAA and GYS1-KO alleles were discriminated by PCR analysis using the following primers: GYS1-KO alleles: MGSer (C), AACATGCTGGACTCCTCAGACCCCATC; MGS12 (S12), GGCTCATAGTAGGAGG GGAAGA; BTKR12 (B) ATAAGTTGCTGGCCAGCTTACCTCCCGGT. GAA-KO alleles: exon 5 sense: CCTTTCTACCTGGCACTGGAGGAC; exon 7 antisense: GGACAATGGCGGTCGAGGAGTA.
Glycogen measurement
Muscles, heart and liver were removed and immediately frozen. Tissues (10 mg) were homogenized at −80°C in 1 ml of acetate buffer (0.2 m, pH 4.5). Homogenized fractions (100 μl) of each sample (cells or tissues) were incubated at 54°C for 2 h in the presence or absence of Aspergillus niger amylo-α-1,4-α-1,6 glucosidase (5 U/ml; Roche, Mannheim, Germany) which converts glycogen to glucose. Glycogen from bovine liver (Sigma-Aldrich, St Louis, MO, USA) hydrolyzed in the same conditions is used as a standard curve. Samples were centrifuged and the glucose level in the supernatant was determined using Glucose Assay Reagent (Sigma-Aldrich) according to the manufacturer's instructions.
PAS staining and high resolution light microscopy
Samples were prepared for high resolution light microscopy according to the procedures of Lynch et al. (43). Briefly, tissues were fixed in 3% glutaraldehyde in 0.2% cacodylate buffer and embedded in epon resin. One micron sections were cut on a Leica Ultracut E ultramicrotome (Leica Instruments GmbH, Germany) and placed onto adhesion coated slides. Slides were first placed in water, then in 1% periodic acid, followed by several rinses in water; the slides were then exposed to Schiff's reagent (Surgipath, Richmond, IL, USA) and rinsed again in water. The sections were then counterstained in 1:10 diluted Richardson's stain and rinsed in water. Slides were dried overnight in a 60°C oven and coverslipped with mounting medium. This protocol results in intense pink-to-purple staining of well-preserved glycogen against a light blue background.
Isolation of fixed single muscle fibers and immunofluorescence confocal microscopy
Gastrocnemius muscles were removed from WT, GAA-KO and GAA/GYS1-KO mice immediately after sacrifice and pinned to Sylgaard-coated dishes for fixation with 2% paraformaldehyde in 0.1 m phosphate buffer for 1 h, followed by fixation in methanol at −20°C for 6 min. Single fibers were obtained by manual teasing. Fibers were placed in a 24-well plate in blocking reagent (Vector Laboratories, Burlingame, CA, USA) for 1 h. Myofibers were then permeabilized and incubated with the primary antibody overnight at 4°C. After washing, myofibers were incubated with the secondary antibody for 2 h, washed, and mounted in Vectashield (Vector Laboratories) on a glass slide. Cells were stained with rat anti-mouse primary antibodies against LAMP-1 (BD Pharmingen, San Diego, CA, USA) or LC3 (microtubules associated protein 1 light chain 3) followed by AlexaFluor 680-labeled anti-rat secondary antibody (Invitrogen, Carlsbad, CA, USA). Slides were mounted in Vectashield mounting medium (Vector Laboratories) and analyzed by confocal microscopy (Zeiss LSM 510 META; Zeiss, Thornwood, NY, USA).
Immunohistochemistry and western blot analysis
Dystrophin and myosin staining
After sacrifice, muscles (gastrocnemius and soleus) were immediately frozen and sectioned; cryosections (7 μm) were stored at −80°C. Before use, slides were thawed and endogenous peroxidase was blocked with hydrogen peroxide (3% H2O2 in 1× PBS, 0.1% Tween). After washing with 0.1% PBS/Tween, serial sections were incubated with anti-slow, anti-fast type -IIA, anti-fast type-IIB or anti-fast type-IIX myosin mouse primary antibodies overnight at 4°C, as previously described (26). Slides were washed again and incubated at 10°C for 1 h with the secondary goat anti-mouse biotinylated antibody. Visualization was achieved using the Vector ELITE ABC Vectastain detection kit according to the manufacturer's instructions (Vector Laboratories) and then dehydrated and mounted on a glass slide.
For dystrophin staining, thawed cross-sections were incubated with 0.1 m glycine in HCl (pH 2.8) for 15 min at 20°C. Slides were washed and blocked with 10% goat serum in PBS buffer and incubated overnight at 4°C with the primary anti-dystrophin antibody, as previously described (27). The sections were washed and incubated 30 min with an anti-mouse IgG Alexa secondary antibody (Invitrogen).
Western blot analysis of GLUT1 and GLUT4 glucose transporters
Plasma membranes were isolated according to Guillet-Deniau et al. (44). Muscles were minced in extraction buffer (10 mm Tris–HCl, pH 7.5, 10 mm sodium chloride, 3 mm magnesium chloride, 180 mm potassium chloride, 0.5% Nonidet P40 (m/v), 50 mm sodium fluoride, 0.1 mm sodium orthovanadate, 1 mm phenylmethylsulfonyl fluoride, 2 μg/ml pepstatin A, 2 μg/ml leupeptin, 5 μg/ml aprotinin, 2.5 μg/ml benzamidine) and crushed for 30 s (Ultra-Turax). Unbroken fibers, debris and nuclei were discarded by centrifugation at 15 000g for 20 min at 4°C. Supernatants were incubated for 30 min in the presence of 0.8 m KCl at 4°C, then ultracentrifuged at 120 000g for 90 min at 4°C. Membrane pellets were suspended in membrane buffer (10 mm Tris–HCl, pH 7.5, 250 mm sucrose, 1 mm EDTA, 5 mm disodium phosphate, 5 mm NaH2PO4, 0.1 mm Na3VO4, 1 mm PMSF, 2 μg/ml pepstatin A, 2 μg/ml leupeptin and 5 μg/ml aprotinin) and separated by SDS–PAGE gel electrophoresis. Polyclonal antibodies were used to detect GLUT4 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and GLUT1 (Chemicon/Millipore, Billerica, MA, USA).
Measurement of the contraction force
The isometric contractile properties of soleus muscles were studied in vitro. Measurements were performed according to the previously described methods (45). The muscles were dissected free from adjacent connective tissue and soaked in an oxygenated Krebs solution (95% O2 and 5% CO2) containing 118 mm NaCl, 25 mm NaHCO3, 5 mm KCl, 1 mm KH2PO4, 2.5 mm CaCl2, 1 mm MgSO4 and 5 mm glucose, and maintained at a temperature of 20°C. Muscles were connected at one end to an electromagnetic puller and at the other end to a force transducer. After equilibration (30 min), electrical stimulation was delivered through electrodes running parallel to the muscle. Isometric contractions were recorded at the length at which maximal isometric tetanic force is observed (L0). Absolute maximal isometric tetanic force (P0, mN) was measured (usual frequency of 125 Hz, train of stimulation of 1500 ms). Specific maximal tetanic force (sP0, mN/mm2) was calculated by dividing the force by the estimated cross-sectional area (CSA) of the muscle. Assuming muscles have a cylindrical shape and a density of 1.06 mg/mm3, muscle CSA corresponds to the wet weight of the muscle divided by its fiber length (Lf). The fiber length to L0 ratio of 0.70 was used to calculate Lf. The fatigue protocol consists of multiple isometric tetanus at 50 Hz every 2 s. The time to reach 80% of the initial force was then calculated (FR). Muscles were weighed, and either flash frozen in liquid nitrogen or isopentane pre-cooled in liquid nitrogen. Samples were stored at −80°C for histological and biochemical analyses.
Exercise protocol
For exercise studies, mice were acclimatized and trained for 2 days on a treadmill during 15 min. The incline was set to 20° and the speed from 5 to 15 cm/s. For the experimental run, fed mice were placed on a treadmill set at 20° with an initial belt speed of 5 cm/min. After 5 min of warming run, the test began. The speed was increased over the test to 10 cm/s after 5 min, 15 cm/s after 10 min, 25 cm/s after 30 min, 35 cm/s after 45 min and 45 cm/s after 60 min. A mild electrical stimulus was applied to mice that stepped off of the treadmill belt. Mice were run to exhaustion.
GTT and ITT
For the GTT, mice were fasted overnight for 12 h. We tested two glucose administration ways: (i) oral gavage of 1 mg/g body weight (OGTT) and intraperitoneal injection of 2 mg/g body weight (IPGTT). Blood samples were collected from the tail at the indicated times (0, 5, 15, 30, 60 and 120 min after glucose administration), and blood glucose was measured using a glucometer (Glucotrend®, Roche Diagnostics, Basel, Switzerland). For the ITT, mice were fasted for 3 h followed by an intraperitoneal insulin injection of 0.2 IU/kg (Novolet Insuline®, Novo Nordisk, Princeton). Blood samples were collected from the tail vein at 0, 5, 15, 30 and 60 min after insulin injection and blood glucose was measured as described above.
Metamorph analysis and determination of fiber area
Fiber area was analyzed by incubating muscle sections with an anti-dystrophin Dys2 Novocastra mouse antibody (Leica Microsystems, Wetzlar, Germany), as described above. Representative fields were photographed and acquired with a DXM 1200 camera on a Nikon Eclipse 600 light microscope. More than 400 fields were analyzed per muscle. For each muscle, the distribution of fiber CSA was determined by using Metamorph software version 2.56.
Statistical analysis
All data are presented as mean ± standard deviation. One- or two-way ANOVA were performed on each group. P-value of <0.05 was considered to be statistically significant.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
Conflict of Interest statement. None declared.
FUNDING
This work was supported by INSERM and the Association Vaincre les Maladies Lysosomales (VML). E.R. was supported by post-doctoral fellowships from VML and the Association Française contre les Myopathies (AFM). G.D.G. was supported by doctoral fellowship from AFM.
Supplementary Material
REFERENCES
- 1.Hirschhorn R., Reuser A.J. Glycogen storage disease type II: acid alpha-glucosidase (acid maltase) deficiency. In: Scriver C.R., Beaudet A.L., Sly W.S., Valle D., editors. The Metabolic and Molecular Bases of Inherited Disease. Vol. 3. New York: McGraw Hill; 2001. pp. 3389–3420. [Google Scholar]
- 2.Kishnani P.S., Steiner R.D., Bali D., Berger K., Byrne B.J., Case L.E., Crowley J.F., Downs S., Howell R.R., Kravitz R.M., et al. Pompe disease diagnosis and management guideline. Genet. Med. 2006;8:267–288. doi: 10.1097/01.gim.0000218152.87434.f3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ausems M.G., ten Berg K., Kroos M.A., van Diggelen O.P., Wevers R.A., Poorthuis B.J., Niezen-Koning K.E., van der Ploeg A.T., Beemer F.A., Reuser A.J., et al. Glycogen storage disease type II: birth prevalence agrees with predicted genotype frequency. Community Genet. 1999;2:91–96. doi: 10.1159/000016192. [DOI] [PubMed] [Google Scholar]
- 4.Raben N., Nagaraju K., Lee E., Kessler P., Byrne B., Lee L., LaMarca M., King C., Ward J., Sauer B., et al. Targeted disruption of the acid alpha-glucosidase gene in mice causes an illness with critical features of both infantile and adult human glycogen storage disease type II. J. Biol. Chem. 1998;7:53–62. doi: 10.1074/jbc.273.30.19086. [DOI] [PubMed] [Google Scholar]
- 5.Bijvoet A.G., van de Kamp E.H., Kroos M.A., Ding J.H., Yang B.Z., Visser P., Bakker C.E., Verbeet M.P., Oostra B.A., Reuser A.J., van der Ploeg A.T. Generalized glycogen storage and cardiomegaly in a knockout mouse model of Pompe disease. Hum. Mol. Genet. 1998;7:53–62. doi: 10.1093/hmg/7.1.53. [DOI] [PubMed] [Google Scholar]
- 6.Raben N., Jatkar T., Lee A., Lu N., Dwivedi S., Nagaraju K., Plotz P.H. Glycogen stored in skeletal but not in cardiac muscle in acid alpha-glucosidase mutant (Pompe) mice is highly resistant to transgene-encoded human enzyme. Mol. Ther. 2002;6:601–608. [PubMed] [Google Scholar]
- 7.Raben N., Danon M., Gilbert A.L., Dwivedi S., Collins B., Thurberg B.L., Mattaliano R.J., Nagaraju K., Plotz P.H. Enzyme replacement therapy in the mouse model of Pompe disease. Mol. Genet. Metab. 2003;80:159–169. doi: 10.1016/j.ymgme.2003.08.022. [DOI] [PubMed] [Google Scholar]
- 8.Van den Hout J.M., Reuser A.J., de Klerk J.B., Arts W.F., Smeitink J.A., Van der Ploeg A.T. Enzyme therapy for Pompe disease with recombinant human alpha-glucosidase from rabbit milk. J. Inherit. Metab. Dis. 2001;24:266–274. doi: 10.1023/a:1010383421286. [DOI] [PubMed] [Google Scholar]
- 9.Amalfitano A., Bengur A.R., Morse R.P., Majure J.M., Case L.E., Veerling D.L., Mackey J., Kishnani P., Smith W., McVie-Wylie A., et al. Recombinant human acid alpha-glucosidase enzyme therapy for infantile glycogen storage disease type II: results of a phase I/II clinical trial. Genet. Med. 2001;3:132–138. [PubMed] [Google Scholar]
- 10.Koeberl D.D., Kishnani P.S., Chen Y.T. Glycogen storage disease types I and II: treatment updates. J. Inherit. Metab. Dis. 2007;30:159–164. doi: 10.1007/s10545-007-0519-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reuser A.J., Van Den Hout H., Bijvoet A.G., Kroos M.A., Verbeet M.P., Van Der Ploeg A.T. Enzyme therapy for Pompe disease: from science to industrial enterprise. Eur. J. Pediatr. 2002;161(Suppl. 1):S106–S111. doi: 10.1007/s00431-002-1015-8. [DOI] [PubMed] [Google Scholar]
- 12.Thurberg B.L., Lynch C.M., Vaccaro C., Afonso K., Chun-Hui Tsai A., Bossen E., Kishnani P.S., O’Callaghan M. Characterization of pre- and post-treatment pathology after enzyme replacement for Pompe disease. Lab. Invest. 2006;86:1208–1220. doi: 10.1038/labinvest.3700484. [DOI] [PubMed] [Google Scholar]
- 13.Raben N., Fukuda T., Gilbert A.L., de Jong D., Thurberg B.L., Mattaliano R.J., Meikle P., Hopwood J.J., Nagashima K., Nagaraju K., et al. Replacing acid alpha-glucosidase in Pompe disease: recombinant and transgenic enzymes are equipotent, but neither completely clears glycogen from type II muscle fibers. Mol. Ther. 2005;11:48–56. doi: 10.1016/j.ymthe.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 14.Hawes M.L., Kennedy W., O'Callaghan M.W., Thurberg B.L. Differential muscular glycogen clearance after enzyme replacement therapy in a mouse model of Pompe disease. Mol. Genet. Metab. 2007;91:343–351. doi: 10.1016/j.ymgme.2007.04.018. [DOI] [PubMed] [Google Scholar]
- 15.Funk B., Kessler U., Eisenmenger W., Hansmann A., Kolb H.J., Kiess W. Expression of the insulin-like growth factor-II/mannose-6-phosphate receptor in multiple human tissues during fetal life and early infancy. J. Clin. Endocrinol. Metab. 1992;75:424–431. doi: 10.1210/jcem.75.2.1379254. [DOI] [PubMed] [Google Scholar]
- 16.Sklar M.M., Kiess W., Thomas C.L., Nissley S.P. Developmental expression of the tissue insulin-like growth factor II/mannose 6-phosphate receptor in the rat. Measurement by quantitative immunoblotting. J. Biol. Chem. 1989;264:16733–16738. [PubMed] [Google Scholar]
- 17.Fukuda T., Ahearn M., Roberts A., Mattaliano R.J., Zaal K., Ralston E., Plotz P.H., Raben N. Autophagy and mistargeting of therapeutic enzyme in skeletal muscle in Pompe disease. Mol. Ther. 2006;14:831–839. doi: 10.1016/j.ymthe.2006.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cox T., Lachmann R., Hollak C., Aerts J., van Weely S., Hrebicek M., Platt F., Butters T., Dwek R., Moyses C., et al. Novel oral treatment of Gaucher's disease with N-butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesis. Lancet. 2000;355:1481–1485. doi: 10.1016/S0140-6736(00)02161-9. [DOI] [PubMed] [Google Scholar]
- 19.Douillard-Guilloux G., Raben N., Takikita S., Batista L., Caillaud C., Richard E. Modulation of glycogen synthesis by RNA interference: towards a new therapeutic approach for glycogenosis type II. Hum. Mol. Genet. 2008;17:3876–3886. doi: 10.1093/hmg/ddn290. [DOI] [PubMed] [Google Scholar]
- 20.Pederson B.A., Chen H., Schroeder J.M., Shou W., DePaoli-Roach A.A., Roach P.J. Abnormal cardiac development in the absence of heart glycogen. Mol. Cell. Biol. 2004;24:7179–7187. doi: 10.1128/MCB.24.16.7179-7187.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pederson B.A., Schroeder J.M., Parker G.E., Smith M.W., DePaoli-Roach A.A., Roach P.J. Glucose metabolism in mice lacking muscle glycogen synthase. Diabetes. 2005;54:3466–3473. doi: 10.2337/diabetes.54.12.3466. [DOI] [PubMed] [Google Scholar]
- 22.Pederson B.A., Cope C.R., Schroeder J.M., Smith M.W., Irimia J.M., Thurberg B.L., DePaoli-Roach A.A., Roach P.J. Exercise capacity of mice genetically lacking muscle glycogen synthase: in mice, muscle glycogen is not essential for exercise. J. Biol. Chem. 2005;280:17260–17265. doi: 10.1074/jbc.M410448200. [DOI] [PubMed] [Google Scholar]
- 23.Manchester J., Skurat A.V., Roach P., Hauschka S.D., Lawrence J.C., Jr Increased glycogen accumulation in transgenic mice overexpressing glycogen synthase in skeletal muscle. Proc. Natl Acad. Sci. USA. 1996;93:10707–10711. doi: 10.1073/pnas.93.20.10707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pederson B.A., Csitkovits A.G., Simon R., Schroeder J.M., Wang W., Skurat A.V., Roach P.J. Overexpression of glycogen synthase in mouse muscle results in less branched glycogen. Biochem. Biophys. Res. Commun. 2003;305:826–830. doi: 10.1016/s0006-291x(03)00862-3. [DOI] [PubMed] [Google Scholar]
- 25.Raben N., Danon M., Lu N., Lee E., Shliselfeld L., Skurat A.V., Roach P.J., Lawrence J.C., Jr, Musumeci O., Shanske S., DiMauro S., Plotz P. Surprises of genetic engineering: a possible model of polyglucosan body disease. Neurology. 2001;56:1739–1745. doi: 10.1212/wnl.56.12.1739. [DOI] [PubMed] [Google Scholar]
- 26.Jue T., Rothman D.L., Shulman G.I., Tavitian B.A., DeFronzo R.A., Shulman R.G. Direct observation of glycogen synthesis in human muscle with 13C NMR. Proc. Natl Acad. Sci. USA. 1989;86:4489–4491. doi: 10.1073/pnas.86.12.4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raben N., Roberts A., Plotz P.H. Role of autophagy in the pathogenesis of Pompe disease. Acta Myol. 2007;26:45–48. [PMC free article] [PubMed] [Google Scholar]
- 28.DeFronzo R.A., Jacot E., Jequier E., Maeder E., Wahren J., Felber J.P. The effect of insulin on the disposal of intravenous glucose. Results from indirect calorimetry and hepatic and femoral venous catheterization. Diabetes. 1981;30:1000–1007. doi: 10.2337/diab.30.12.1000. [DOI] [PubMed] [Google Scholar]
- 29.Bruning J.C., Michael M.D., Winnay J.N., Hayashi T., Horsch D., Accili D., Goodyear L.J., Kahn C.R. A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol. Cell. 1998;2:559–569. doi: 10.1016/s1097-2765(00)80155-0. [DOI] [PubMed] [Google Scholar]
- 30.Kim J.K., Michael M.D., Previs S.F., Peroni O.D., Mauvais-Jarvis F., Neschen S., Kahn B.B., Kahn C.R., Shulman G.I. Redistribution of substrates to adipose tissue promotes obesity in mice with selective insulin resistance in muscle. J. Clin. Invest. 2000;105:1791–1797. doi: 10.1172/JCI8305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zisman A., Peroni O.D., Abel E.D., Michael M.D., Mauvais-Jarvis F., Lowell B.B., Wojtaszewski J.F., Hirshman M.F., Virkamaki A., Goodyear L.J., et al. Targeted disruption of the glucose transporter 4 selectively in muscle causes insulin resistance and glucose intolerance. Nat. Med. 2000;6:924–928. doi: 10.1038/78693. [DOI] [PubMed] [Google Scholar]
- 32.Kim J.K., Zisman A., Fillmore J.J., Peroni O.D., Kotani K., Perret P., Zong H., Dong J., Kahn C.R., Kahn B.B., et al. Glucose toxicity and the development of diabetes in mice with muscle-specific inactivation of GLUT4. J. Clin. Invest. 2001;108:153–160. doi: 10.1172/JCI10294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kasuga M., Ogawa W., Ohara T. Tissue glycogen content and glucose intolerance. J. Clin. Invest. 2003;111:1282–1284. doi: 10.1172/JCI18526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bergstrom J., Hermansen L., Hultman E., Saltin B. Diet, muscle glycogen and physical performance. Acta Physiol. Scand. 1967;71:140–150. doi: 10.1111/j.1748-1716.1967.tb03720.x. [DOI] [PubMed] [Google Scholar]
- 35.Ivy J.L. Role of carbohydrate in physical activity. Clin. Sports Med. 1999;18:469–484. doi: 10.1016/s0278-5919(05)70162-9. [DOI] [PubMed] [Google Scholar]
- 36.Karlsson J., Saltin B. Diet, muscle glycogen, and endurance performance. J. Appl. Physiol. 1971;31:203–206. doi: 10.1152/jappl.1971.31.2.203. [DOI] [PubMed] [Google Scholar]
- 37.Chen Y.T. Glycogen storage diseases. In: Scriver C.R., Beaudet A.L., Sly W.S., Valle D., editors. The Metabolic and Molecular Bases of Inherited Disease. New-York: McGraw Hill; 2001. pp. 1521–1551. [Google Scholar]
- 38.Kollberg G., Tulinius M., Gilljam T., Ostman-Smith I., Forsander G., Jotorp P., Oldfors A., Holme E. Cardiomyopathy and exercise intolerance in muscle glycogen storage disease 0. N. Engl. J. Med. 2007;357:1507–1514. doi: 10.1056/NEJMoa066691. [DOI] [PubMed] [Google Scholar]
- 39.Cameron J.M., Levandovskiy V., Mackay N., Utgikar R., Ackerley C., Chiasson D., Halliday W., Raiman J., Robinson B.H. Identification of a novel mutation in GYS1 (muscle-specific glycogen synthase) resulting in sudden cardiac death, that is diagnosable from skin fibroblasts. Mol. Genet. Metab. 2009;98:378–382. doi: 10.1016/j.ymgme.2009.07.012. [DOI] [PubMed] [Google Scholar]
- 40.Sidman R.L., Taksir T., Fidler J., Zhao M., Dodge J.C., Passini M.A., Raben N., Thurberg B.L., Cheng S.H., Shihabuddin L.S. Temporal neuropathologic and behavioral phenotype of 6neo/6neo Pompe disease mice. J. Neuropathol. Exp. Neurol. 2008;67:803–818. doi: 10.1097/NEN.0b013e3181815994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.DeRuisseau L.R., Fuller D.D., Qiu K., DeRuisseau K.C., Donnelly W.H., Jr, Mah C., Reier P.J., Byrne B.J. Neural deficits contribute to respiratory insufficiency in Pompe disease. Proc. Natl Acad. Sci. USA. 2009;106:9419–9424. doi: 10.1073/pnas.0902534106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Elstein D., Dweck A., Attias D., Hadas-Halpern I., Zevin S., Altarescu G., Aerts J.F., van Weely S., Zimran A. Oral maintenance clinical trial with Miglustat for type I Gaucher disease: switch from or combination with intravenous enzyme replacement. Blood. 2007;110:2296–2301. doi: 10.1182/blood-2007-02-075960. [DOI] [PubMed] [Google Scholar]
- 43.Lynch C.M., Johnson J., Vaccaro C., Thurberg B.L. High resolution light Microscopy (HRLM) and digital analysis of Pompe disease pathology. J. Histochem. Cytochem. 2005;53:63–73. doi: 10.1177/002215540505300108. [DOI] [PubMed] [Google Scholar]
- 44.Guillet-Deniau I., Leturque A., Girard J. Expression and cellular localization of glucose transporters (GLUT1, GLUT3, GLUT4) during differentiation of myogenic cells isolated from rat foetuses. J. Cell Sci. 1994;107:487–496. [PubMed] [Google Scholar]
- 45.Vignaud A., Fougerousse F., Mouisel E., Guerchet N., Hourde C., Bacout F., Butler-Brown G.S., Chatonnet A., Ferry A. Genetic inactivation of acetylcholinesterase causes functional and structural impairment of mouse soleus muscles. Cell Tissue Res. 2005;333:289–296. doi: 10.1007/s00441-008-0640-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.