

Abstract
Non-coding DNA variations play a critical role in increasing the risk for development of common complex diseases, and account for the majority of SNPs highly associated with cancer. However, it remains a challenge to identify etiologic variants and to predict their pathological effects on target gene expression for clinical purposes. Cis-overlapping motifs (COMs) are elements of enhancer regions that impact gene expression by enabling competitive binding and switching between transcription factors. Mutations within COMs are especially important when the involved transcription factors have opposing effects on gene regulation, like P53 tumor suppressor and cMYC proto-oncogene. In this study, genome-wide analysis of ChIP-seq data from human cancer and mouse embryonic cells identified a significant number of putative regulatory elements with signals for both P53 and cMYC. Each co-occupied element contains, on average, two COMs, and one common SNP every two COMs. Gene ontology of predicted target genes for COMs showed that the majority are involved in DNA damage, apoptosis, cell cycle regulation, and RNA processing. EMSA results showed that both cMYC and P53 bind to cis-overlapping motifs within a ChIP-seq co-occupied region in Chr12. In vitro functional analysis of selected co-occupied elements verified enhancer activity, and also showed that the occurrence of SNPs within three COMs significantly altered enhancer activity. We identified a list of COM-associated functional SNPs that are in close proximity to SNPs associated with common diseases in large population studies. These results suggest a potential molecular mechanism to identify etiologic regulatory mutations associated with common diseases.
Introduction
Transcription factors (TFs) encode for proteins that regulate target gene expression upon binding to regulatory sequences. This binding results in the changing of the chromatin structure and initiation of target gene transcription by RNA polymerase (1). TFs bind to conserved sites within regulatory elements, and do so with higher affinity to sequences more consistent with their consensus binding motifs (2,3). Many studies focus on the effect of DNA variants within the coding regions of functional genes, and how this may lead to Mendelian and common complex diseases (4). However, recent publications have shown that even a single nucleotide change within the noncoding, regulatory regions can result in pathologic phenotypes by significantly decreasing or in other cases increasing, target gene transcription (4–6).
Cis-overlapping motifs (COMs) are overlapping conserved binding sequences of multiple TFs that are located on the same DNA strand (7–9). This feature suggests a mechanism of gene regulation by competitive binding and inhibition, where binding of one factor occurs at the cost of another factor. TF preference binding at these sites is affected by several parameters, such as the binding affinity, the level of activated TFs, TF cooperativity, and number of TF binding sites present within COM vicinity (10–12). A good example of this mechanism is the overlapping cis-acting elements of SP1 and ERG1 within the promoter regions of tissue factor, platelet-derived growth factor B and C chains, and other genes (13,14). Competitive binding of transcription factors within regulatory regions is a conserved mechanism that has also been studied in embryonic development of the Drosophila model (12). Furthermore, a recent study showed that mutations at cis-overlapping of Ebox and AP-1-like sites impacted target gene expression significantly more than mutations at these binding sites when they are isolated within the enhancer element in human HCT116 cancer cells (15).
However, these studies focused on the role of cis-acting regulation of single individual genes rather than a mechanism of gene regulation at the genome-wide level. Such a mechanism for gene regulation may have significant impact on the dynamic of turn-on and turn-off of target genes if the competition is between cis-acting transcriptional activators and repressors. Activators are TFs that enhance gene expression upon binding at regulatory regions, while repressors suppress transcription by blocking activator binding via occupancy, making the chromatin more compact and less accessible, or by stalling the transcription machinery (12,16). The critical issue involving in the competitive inhibitory mechanism is the presence of DNA variants such as single nucleotide polymorphisms (SNPs) within these COMs. For instance, should these SNPs shift the motif away from the consensus sequence for one factor but not the other, homeostasis of this competitive binding may deviate from the acceptable range and subsequently alter gene expression and pattern. Carriers of SNPs at these particular sites in the genome may therefore be at risk of disease development and to put their progenies at higher risk to become affected.
In this study, we focused on the impact of COMs involving two important transcription factors that have opposing effects on cellular activities, P53 tumor suppressor and cMYC proto-oncogene, at the genome-wide level. Both factors P53 and cMYC function in cell development in addition to their roles in differentiated cells. While P53 is known mainly for its surveillance activity in somatic cells, where it regulates cell cycle exit and apoptosis in response to DNA damage, P53 activity has also been shown to induce differentiation and repress stem cell genes in embryonic cells (10,17,18). Likewise, cMYC is involved in cell cycle progression and apoptosis by interacting with various transcription factors such as Max to regulate expression of target genes (19). In contrast to P53, however, cMYC inhibits differentiation (20) and promotes embryonic stem cell pluripotency and self-renewal (21). Overrepresentation of several conserved motifs of different TFs within the vicinity of the P53 motif was previously reported, including the Ebox motif that basic helix-loop-helix (bHLH) transcription factors such as cMYC bind to it (2,22). Smeenk et al. (2) reported that 681 Ebox motifs were identified bioinformatically within 500 bp regions centered around 1546 peaks of P53 binding sites at the genome-wide level. Despite the significant number of Ebox motifs within the vicinity of P53 binding sites, no connection between P53 and cMYC for gene regulation has been proposed thus far.
While functional analysis of noncoding regions at the genome-wide level is tedious due to the large number of sequences within putative regulatory regions for which we lack knowledge of (23), we were able to use genomic databases and computer analysis in the identification of candidate sites for wet lab analysis. One of the difficulties of analyzing intergenic loci is the degeneracy of binding motifs that lay in regulatory elements (enhancers), where TFs may bind to motifs different from the consensus sequence. In addition, TFs do not always bind at every site that contains a consensus motif (1,2,24). To locate COMs that are likely to be functional binding sites within regulatory regions, we referred to published experimental data from previous genome-wide studies for P53 and cMYC, and then wrote a program that identified the peaks that overlapped. We selected studies conducted in two different organisms, human cancer and mouse embryonic cells, for which genomic data for P53 and cMYC are available. We searched for COMs using consensus P53 and cMYC motifs within the overlapping regions of binding peaks, and then identified common SNPs within these COMs. Presumptive target genes of the P53 and cMYC overlapping peaks were then identified using the online Genomic Regions Enrichment of Annotations Tool (GREAT) (25). After selection of COMs with high conservation and where presence of SNPs may potentially impact gene expression based on bioinformatics results and other criteria, we verified enhancer activity of six regions and determined if presence of common SNPs can alter enhancer activity as we predicted.
Results
Genome-wide analysis of P53 and cMYC binding sites yield significant number of overlaps
In both human cancer and mouse embryonic cells, a significant number of overlaps were found between P53 and cMYC binding sites. The bioinformatic process to locate these overlaps is illustrated in Figure 1. In human cancer cell analysis, 48 sets of overlapping ChIP peaks were identified out of the 2183 P53 ChIP peaks from Chang et al. (22) and 7054 cMYC ChIP peaks from Seitz et al. (26). About 2.19% (48/2183) of all reported P53 binding regions were involved in P53 and cMYC overlap, while 0.68% (48/7054) of all reported cMYC binding regions were involved in P53 and cMYC overlap. In mouse embryonic cells (mEC), 344 overlapping peaks were identified out of the 4785 P53 binding ChIP peaks from Kenzelmann Broz et al. (17) and 3422 cMYC binding ChIP peaks from Chen et al. (27). In comparison to cancer cell analysis, ∼7% (344/4785) P53 binding regions were involved in overlaps, while 10% of cMYC binding regions were involved (344/3422). It is interesting to note that there were far more overlaps in mECs in number of total binding sites involved (Supplementary Material, Table S1).
Figure 1.

The flowchart illustrates the P53 and cMYC co-occupied regions found from selected ChIP-seq analysis of P53 and cMYC within different cell lines. A program written in python was then used to identify P53 and cMYC cis-overlapping motifs (COMs) located within the co-occupied regions, and the UCSC browser was used to identify SNPs located within these COMs. Further analysis with GREAT was used to predict potential target genes regulated by elements containing P53 and cMYC co-occupied regions, and predict their function via gene ontology. The programs CADD, GWAVA and rSNPBase were used to identify potentially significant SNPs.
In cancer cell analysis, 80 cis-overlapping motifs were identified within the 48 regions of these overlapping peaks, which means that there are on average 1.7 COMs within each of the identified overlapping ChIP peak. In mouse embryonic cell analysis, 642 COMs were identified within the 344 overlapping ChIP peaks (Supplementary Material, Table S2). Accordingly, 1.9 COMs are located on average within each overlapping ChIP peak in the mouse embryonic cells. Furthermore, 41 and 68 SNPs were found within the COMs of the human cancer and mouse embryonic cells, respectively.
Possible scenarios of P53 and cMYC overlap and its significance
A visual representation of the position weight matrices for cMYC and P53 motifs from JASPAR is depicted in Figure 2A and B, respectively. The multiple COMs within a set of overlapping ChIP peaks represent multiple possible binding sites for P53 and cMYC. Actual binding, as proven in past studies, depends on other variables in addition to conservation of the consensus sequence (28). Possible scenarios for three types of overlap involving P53 and cMYC are represented in Figure 2C.
Figure 2.
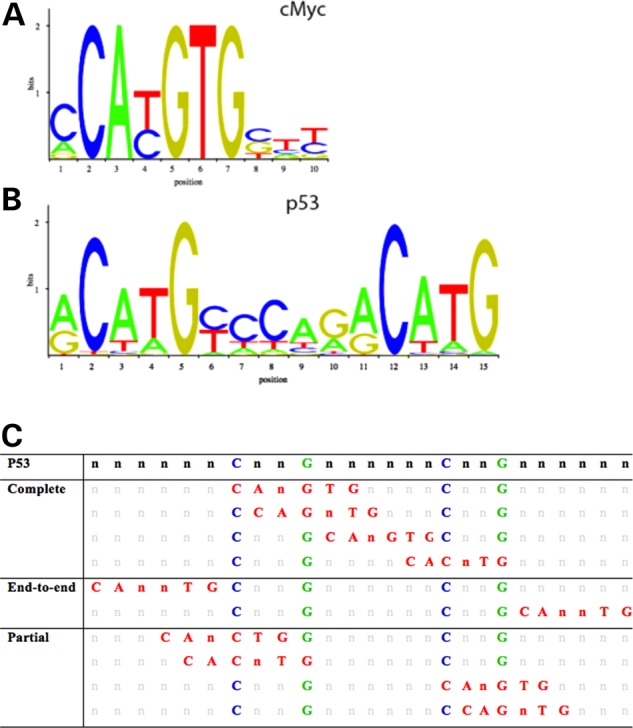
Position weight matrices (PWM) and all possible overlap scenarios of the P53 and cMYC. The two most highly conserved nucleotides for cMYC (A) and P53 (B) binding sites obtained from JASPAR CORE database derived from published collections of experimentally curated set of binding sites for eukaryotes. All possible scenarios of overlap involving the P53 (with conserved nucleotides in blue and green) and cMYC binding site (in red) overlap, while retaining the conserved nucleotides of the P53 motif (C). Complementary binding motifs on the anti-sense strand are not depicted.
Based on our analysis of COMs within ChIP peaks, we hypothesize that the amount of overlap involving the Ebox motif with the P53 motif will have different degrees of impact on the relative binding affinities of P53 and cMYC. COMs can be divided into complete, end-to-end, and partial overlap based on the number of nucleotides of each motif involved in the overlap, as shown in Figure 2C. We also suggest that end-to-end overlap of the two motifs will still have some effect on TF binding since the bulk of the bound co-factor proteins may affect binding of the other TF, but may not be nearly as disruptive.
Predicted target genes and gene ontology
Potential target genes of P53 and cMYC overlaps in human cancer cells and their functions are listed in Table 1, with further information in Supplementary Material, Table S3. Ultimately, analysis of ChIP peak overlaps in human cancer cells by GREAT (www.great.stanford.edu) predicted 22 genes with related GO terms to be regulated by the overlapping regions of P53 and cMYC binding sites. The listed gene function corresponding to each gene is defined by the Gene Cards (www.genecards.org) and Jackson database (www.jax.org) (29). Twenty-three out of 48 ChIP peak overlaps were predicted to be associated with functional gene and only 20 out of the 41 SNPs were identified within COMs (Table 1). Gene ontology (GO) analysis resulted in annotations for biological processes pertaining to DNA damage and repair. Disease ontologies and pathways such as Li–Fraumeni syndrome, tuberous sclerosis, and ataxia-telangiectasia mutated pathway are related to congenital defects that lead to uncontrolled tumor growth (Table 2).
Table 1.
Predicted target genes for P53 and cMYC co-occupied regions in human cancer cell analysis, with the number of regions potentially involved for each target gene listed
| Gene symbol | Overlaps involved | Gene function | Human SNPs within COMs |
|---|---|---|---|
| AEN | 1 | Apoptotic s enhancing nuclease | rs184449425, rs375618035 |
| BAX | 1 | Activation of cysteine-type endopeptidase Bcl-2 associated X protein complex | |
| CDKN1A | 1 | Cell cycle arrest, cyclin-dependent protein kinase activating | rs181272168 |
| DCP1B | 1 | Nonsense-mediated decay, coenzyme F390-A hydrolase activity | |
| DDIT3 | 1 | DNA-damage inducible transcript blood vessel maturation | rs191571963, rs367761860, rs375623461 |
| DDIT4 | 1 | Apoptotic process, brain development, 14-3-3 protein binding | |
| GDF15 | 1 | Growth factor activity | |
| MDM2 | 1 | E3 Ubiquitin ligase, DNA damage response, signal transduction by P53 | rs1135874, rs76264715 |
| NME1-NME2 | 1 | Cell differentiation, ATP binding, deoxyribonuclease activity | rs144896898 |
| PCNA | 1 | Base-excision repair, gap-filling, cellular response to DNA damage | |
| PLK3 | 1 | Cell cycle, ATP binding, and kinase activity | |
| POLH | 1 | Cellular response to DNA damage stimulus, DNA biosynthetic process | |
| PTP4A1 | 1 | Cell cycle, dephosphorylation, and protein tyrosine phosphatase | |
| RABGGTA | 1 | Rab geranylgeranyl transferase, prenyltransferase activity | rs374020827 |
| RAD51C | 1 | Holliday junction resolvase complex, ATP binding protein | rs28363301 |
| REV3L | 2 | DNA damage stimulus, DNA-dependent DNA polymerase | rs140357312 |
| RPS19 | 1 | Erythrocyte differentiation, maturation of SSU-rRNA | |
| SARS | 1 | Seryl-tRNA aminoacylation, ATP binding protein | |
| TNFRSF10B | 1 | Activation of cysteine-type endopeptidase activity, tumor necrosis factor receptor | |
| TRAF4 | 1 | Apoptotic process, multicellular organismal development, cell junction, TNF receptor | rs113012137 |
| TRIAP1 | 1 | Apoptotic process, P53 regulated inhibitor, phosphatidic acid transporter activity | rs145730860, rs149038744, rs373187248 |
| TYMS | 1 | dTMP biosynthetic process, thymidylate synthase | rs183205964, rs2853542, rs34743033, rs45445694 |
Gene ontology was used to predict gene function. SNPs associated with the P53 and cMYC COMs within each co-occupied region were identified using the UCSC Genome Browser.
Table 2.
GO terms associated with predicted genes for P53 and cMYC co-occupied regions in human cancer cell line analysis
| GO term | Associated genes |
|---|---|
| Response to DNA damage | AEN, BAX, CDKN1A, DDIT3, MDM2, PCNA, POLH, RAD51C, REV3L, TRIAP1, TYMS |
| PCNA-P21 complex | CDKN1A, PCNA |
| Li–Fraumeni syndrome | BAX, CDKN1A, MDM2 |
| Tuberous sclerosis | BAX, DDIT4, PCNA |
| Ataxia-telangiectasia mutated (ATM) pathway | BAX, CDKN1A, DDIT4, GDF15, MDM2, PCNA, PLK3, TNFRSF10B, TRIAP1, TYMS |
| P53 signaling pathway | BAX, CDKN1A, MDM2, PCNA |
In comparison, analysis of ChIP peak overlaps in mouse embryonic cell using GREAT yielded 175 target genes with related GO terms, which are listed in Supplementary Material, Table S4. Fifty-three out of the 68 common SNPs found within COMs are also represented in Supplementary Material, Table S4. Gene ontology that represents overall functions of the predicted target genes in the mouse embryonic cells is presented in Table 3. Like the GO terms for human cancer cell analysis, GO terms for COMs in mouse embryonic cells also involve DNA damage and repair response and disease ontologies related to loss of this function, i.e. carcinoma. In addition to these terms, however, mouse embryonic cells also have GO terms related to ribosomal RNA and mRNA processing.
Table 3.
GO terms associated with predicted genes for P53 and cMYC co-occupied regions in mouse embryonic cell analysis
| GO term | Associated genes |
|---|---|
| Response to DNA damage stimulus | Aen, Bax, Bbc3, Btg2, Cdkn1a, Fan1, Foxo3, H2afx, Hus1, Mapk1, Mif, Mlh1, Msh2, Msh6, Phlda3, Rad50, Rps27l, Setx, TrP53, Zmat3 |
| Regulation of apoptosis | Aen, Bax, Bbc3, Capn10, Cdkn1a, Col18a1, Ctnnb1, Dffb, Foxo3, Gnb2l1, Map3k9, Mlh1, Msh2, Msh6, Myc, Phlda3, Rnf7, Rps27l, Sik1, Spdef, Tnfrsf10b, TrP53, Wnt7b, Zmat3 |
| Transcription initiation, DNA-dependent | Gtf2f2, Med17, Med30, Myc, Taf7, Ttf1 |
| rRNA processing | Mphosph10, Nol9, Pa2g4, Rpl14, Rps7, Rrp36, Utp20, Wdr36 |
| Carcinoma | Arid1a, Aurka, Bax, Bbc3, Btg2, Cdkn1a, Col18a1, Cstf1, Ctnnb1, Ece2, Eif4ebp1, Epha2, Esrra, Foxo3, Gdf15, Gja1, Gnb2l1, Gtf2i, H2afx, Hif1an, Hus1, Hmmr, Icam1, Lif, Mapk1, Mif, Mlh1, Msh2, Myc, Ncoa3, Nfe2l2, Nme1, Ppard, Ppia, Ptma, Ptp4a3, Rad50, Rps19, Sncg, Socs3, Sox1, Sp1, Tjp2, Tnfrsf10b, Traf4, TrP53, Wee1, Xpo1, Zap70 |
| c-MYC pathway | Bax, Cad, Cdkn1a, Foxo3, Ireb2, Myc, Nme1, Ptma, Sp1, Tjp2 |
| mRNA processing | Ccar1, Cpsf1, Cstf1, Gtf2f2, Mnat1, Nup214, Nup54, Pcbp1, Polr2l, Ptbp1 |
The reliability of GREAT analysis depends on the accuracy, appropriateness, and completion of the input data. In our case, our input was the set of overlapping regions that contain at least one COM. Therefore, 10 sets of overlapping ChIP peak coordinates from tumor and mouse embryonic cell analysis were randomly selected and uploaded to UCSC genome browser to validate their predicted target genes based on proximity (30).
Common genes in embryonic and cancer cells
Out of the 22 genes for human cancer cell line analysis and 175 genes for mouse embryonic cell analysis, there are 10 genes that are identical or members of the same family (Fig. 3A). This suggests that the DNA repair function in somatic cells is retained, while an additional function of cell development is seen in embryonic cells (Fig. 3B). Common genes regulated by P53 and cMYC COMs in different cell types suggests that these genes play an important role in maintaining cellular homeostasis, and that mutations affecting their expression may have profound repercussions (Fig. 3B).
Figure 3.

Number of genes that are common to both human cancer cell line analysis and mouse embryonic cell analysis (A). A list of target genes predicted for enhancer elements containing P53 and cMYC cis-overlapping motifs (B).
Enhancer activity of COM containing regions
Ultimately we are interested in locating COMs within regulatory elements that might be risk regions for common complex diseases. The putative target genes of COMs that were common to both human cancer and murine embryonic cells suggest a highly conserved mechanism of gene regulation in cellular function of both cell types, and that COMs may provide a possible sequence of concern regarding alteration of gene regulation. In addition, the number of common SNPs within COMs shows that there are many opportunities for shift in competitive binding to arise. Analysis of the COM-associated SNPs identified in human cancer cell lines with CADD, GWAVA, and rSNPBase strongly suggests functionality in many of these SNPs (Supplementary Material, Tables S5–S7).
Six of the identified co-occupied regions were tested for enhancer activity, and therefore potential impact on target gene expression, via luciferase assay. The enhancer activity of these six genomic regions was validated at least three times. Three of these COM containing regions, located in Chr1, Chr8, and Chr15, were selected based on high conservation of the DNA sequence across 44 mammalian species and enrichment of enhancer signatures, such as histone modifications and DNaseI footprints (Fig. 4). The selection of these regions was independent of the presence or absence of SNPs within the COMs. On the other hand, the other three COM containing regions, located on regions in Chr6, Chr7, and Chr12, were selected based on presence of a SNP that altered one or both conserved motifs within their respective COMs and based on their close proximity to SNPs that are significantly associated with different common complex diseases in large genome-wide association studies (Fig. 4). Specifically, the SNP rs140357312 in Chr6 disrupts the Ebox motif without affecting P53 binding site motif. Two SNPs occur in the COM located on Chr12, where the SNP rs373187248 disrupts one of the half-sites of the P53 binding motif, while the other SNP rs145730860 modifies the Ebox motif to the highest consensus sequence for cMYC binding, and potentially enhances its binding affinity. The rs143789306 SNP in Chr7 occurs within the spacer region between the two half-sites of P53, but does not disrupt the most conserved nucleotides of the binding motifs for either factor.
Figure 4.

UCSC genome browser screenshots of the P53 and cMYC co-occupied regions. The regions on the left were selected for luciferase assay based on presence of high conservation in 100 vertebrate species and signatures for putative regulatory element. The regions on the right were selected based on the presence of motif-altering SNPs, or location within a susceptibility locus for human disease.
All of the COMs possessed enhancer activity with different strength levels (Fig. 5). A potent enhancer was used as a positive control, while a control construct was used to determine the baseline activity in the luciferase experiment. Co-occupied regions in Chr1 and Ch8 showed the highest luciferase activity (Fig. 5). Remarkably, introduction of SNPs within COMs at Chr7 and Chr12 significantly increased the luciferase activity almost 2-fold in comparison to the regions with common sequence, while the SNP within COM of co-occupied region on Chr6 slightly affects luciferase expression (Fig. 5).
Figure 5.

Luciferase activity results for co-occupied genomic regions by P53 and cMYC. The bars represent the normalized luciferase activity driven by the co-occupied regions that contain COM elements compared with positive control construct with enhancer upstream of Luc reporter gene (black-white dots), and negative control construct with basic promoter (black-white dots). Strong luciferase activity was observed in Luc constructs driven by the co-occupied regions selected from Chr1, Chr8 and Chr15 (vertical stripes). Similarly, robust luciferase activity was observed Luc constructs driven by the co-occupied regions selected from Chr6, Chr12 and Chr7 (cross lines). After introduction of SNPs within the COMs, Luc driven by co-occupied regions from Chr12 and Chr7 showed higher activity (bricks), while the SNP within the COM in Chr6 slightly reduced luciferase activity (bricks).
Validation of P53 and cMYC binding at COMs
The binding of cMYC and P53 to COM within co-occupied region on Chr12 was confirmed with electrophoretic mobility gel shift assay (EMSA) (Fig. 6). Tagged probe with IR-700 was used to test the binding of cMYC and P53 recombinant proteins to COM sequence. Increasing concentrations of untagged probe that carry the SNP rs145730860 was used for competitive binding with the common sequence of Chr12 region. The SNP rs145730860 creates the highest consensus binding sequence for cMYC, and significantly competed with the consensus Ebox motif for cMYC binding at 100-fold concentration. On the other hand, the SNP rs373187248 disrupts a half-site of the P53 binding motif, which increased P53 binding when introduced at 10-fold concentration of the consensus sequence, but does not affect P53 binding at 100-fold concentration (Fig. 6). Using a monoclonal antibody against cMYC and P53 protein, we observed a slight super shift band (arrow and arrowhead), confirming the specificity the binding to the COM probe on Chr12.
Figure 6.

EMSA assay was used to test the binding of cMYC and P53 to COM of co-occupied region in Chr12. A unique shift band of recombinant cMYC protein and tagged probe was observed (arrow, lane 3) compared with free probe alone (lane 1) and probe with only reticulocyte extract mixture (lane 2). Competitive inhibition with nonlabeled probe (NLO) that contains the SNP rs145730860 abolished the unique shift band when competed with 100-fold increase of untagged probe (lane 4), while not effect was observed with 10-fold increase (lane 5). The binding of cMYC was confirmed by using monoclonal antibodies against cMYC protein (lane 6). Lanes 7–10 were performed similar to lanes 3–6 except using P53 recombinant protein instead of cMYC, and the nonlabeled probe (NLO) contained the SNP rs373187248 instead of rs145730860. The binding of P53 was confirmed by using monoclonal antibodies against P53 protein shown by the supershift band (arrowhead) (lane 10).
Discussion
While several studied have shown that transcription factors bind with higher affinity at conserved binding motifs, actual binding is largely dependent on cell type and context (11,28). Searching for TF binding sites based on experimental data yields more accurate results than searching for TF binding sites based on sequence motif alone. Because the P53 and cMYC sites used to derive COMs in this study were experimentally identified via ChIP-seq, the identified COMs are more reliably actual binding sites, with one verified via EMSA. Overrepresentation of P53 and cMYC COMs within 500 bp overlapping ChIP-seq peaks suggests conservation of the competitive binding of TFs. This also implies a mechanism that functions to buffer DNA variations (11). The large number of COMs within overlapping ChIP peaks may likely be due to the presence of multiple Ebox motifs around one P53 motif, since the Ebox motif is much shorter in length. These COMs within a set of overlapping ChIP peaks may be separate from one another, or can involve an overrepresentation of cMYC motifs around a single P53 motif. In our analysis, each cMYC and P53 motif overlap counts as a single COM, because each COM represents a separate site where variation may impact gene expression.
P53 and cMYC binding sites in human cancer cell lines were obtained from studies selected based on certain criteria. Both studies utilized the ChIP-seq technique to collect binding site data, a method which yields higher sensitivity and specificity compared with ChIP-chip analysis. In addition, the data sets were also publically available, and both studies had been published recently. However, the experiments were conducted in different cell types and under different growth conditions, which may limit our analysis to detect an accurate and representative number of co-occupied regions by cMYC and P53. Other papers on tumor cells that we considered were Wang et al. (31), Shaked et al. (32), Smeenk et al. (2), Ji et al. (33), and Zeller et al. (34).
In our search for published data on P53 and cMYC binding sites in embryonic cells, we considered studies that were carried out in human and murine cells. While human embryonic cell data is more ideal for our analysis, the only papers we were able to find for both P53 and cMYC data acceptable for this study were both carried out in murine embryonic cells. The studies we selected conducted by Kenzelmann Broz et al. (17) and Chen et al. (27) were carried out in murine embryonic cells harvested at very similar developmental time periods, i.e. E13.5 and E14, respectively, and used ChIP-seq analysis for identification of genomic binding sites. Other papers considered in murine embryonic cells include Krepelova et al. (21) Perna et al. (35) Lee et al. (36) Kidder et al. (20) and Li et al. (18). These papers were considered but were not used because they either did not provide binding site data, had few sites (<1000), or were conducted in cells from different species.
In both models of cancer and embryonic cells, a number of cis-overlapping binding sites for P53 and cMYC were identified. In human cancer cell line analysis, 48 co-occupied regions by P53 and cMYC were identified, where most of the co-occupied regions contain at least one COM identified with the high consensus motif criteria for P53 and cMYC. Analysis of murine embryonic cells identified 344 co-occupied regions, which is a significantly larger number. Our bioinformatic analysis showed that a total of 80 COMs with 41 SNPs were located within the overlapping regions for human cancer cells, and 642 COMs with 68 SNPs in murine embryonic cells. Interestingly, the overlap of predicted target genes in the human cancer cells with predicted target genes in the mouse cells suggest that regulation involving P53 and cMYC COMs is conserved.
The GREAT online resource determines gene ontology of cis-regulatory elements by taking into account genes located within a certain distance of each element and selecting genes with related GO terms. The significance of these GO terms is determined by GREAT using both hypergeometric and binomial models (25). Coordinate of overlapping P53 and cMYC ChIP peaks were used as the main input data. In addition to the main input data set, GREAT also allows users to provide a set of background data against which to run the main input data. Because these overlapping ChIP peak coordinates were obtained from genome-wide studies and compared functions of two different transcription factor binding sites, we decided that annotation of the overlapping peaks against the whole genome is more appropriate. Only 22 genes were predicted for cancer cell lines and 175 for mouse embryonic cells, based on the relationship between their gene ontologies and how far each gene is located to the nearest COM. However, the majority of target genes predicted by GREAT are common to the target gene lists included in the source P53 and cMYC papers and other papers in both cell types (17,26,27,31,34,37).
We did not expect to find a large number of overlapping target genes between the two data sets because we were comparing COMs that eventually regulate fully differentiated cells to COMs that regulate embryonic cells. Furthermore, we were comparing data from the human genome to data from the mouse genome. As mentioned before, P53 functions as a surveillance protein in somatic cells, and induces differentiation and responds to damage in embryonic cells. cMYC functions as an amplifier for cell processes in somatic cells, including cell proliferation, and inhibits differentiation in embryonic cells. The GO terms that resulted from gene annotation analysis generally support these functions, since they primarily covered DNA damage and repair in cancer cells, and both DNA repair and other cellular processes such as transcription initiation and RNA processing in embryonic cells. However, the common target genes of COM containing regions predicted in both human cancer and murine embryonic cells suggest conservation of the competitive inhibitory model for gene regulation between different cell types.
Regions selected based on conservation and those selected based on potential disruptiveness of SNPs did not show any bias to enhancer activity strength compared with the controls. This suggests that selection of potential regulatory elements based on conservation is not the optimal strategy. Rather, the presence of ENCODE signatures, transcription factors binding signals and presence of COMs should be considered for identifying novel enhancer elements. Knowledge of the mechanism behind transcription factor interactions may aid in prediction of the type of impact DNA variations can have on target gene activation or repression, as well as the extent of this impact. This is in contrast to conservation scores and enhancer signatures, which may indicate potential functional impact, but does not detail how the mechanism impacts gene expression.
Our findings concerning P53 and cMYC co-occupied regions and cis-overlapping motifs suggest that the two transcription factors interact and regulate gene expression through competitive binding (Fig. 7). Investigation of six different COM-containing regions via luciferase assay showed that all elements possess enhancer activity. Furthermore, introduction of SNPs at different locations within the COMs had different effects on enhancer activity. Introduction of the SNP rs140357312 at the co-occupied region on Chr6, which disrupts the Ebox motif but not the P53 motif, did not change luciferase expression significantly. This SNP is near TRAF3IP2-AS1 on the corresponding sense strand, and TRAF3 on the anti-sense strand. TRAF3 encodes for a protein that regulates the responses of NF-κB transcription factor family, which is expected to be regulated by P53 and cMYC. Furthermore, the SNP rs373187248 introduced on Chr12 only disrupted the P53 binding site. This SNP is near TRIAP1, which encodes for TP53 regulated inhibitor of apoptosis1. Because P53 generally inhibits cell cycle and promotes apoptosis, it can be expected that binding of P53 would normally downregulate TRIAP1 expression. Introduction of rs373187248 subsequently caused a significant increase in luciferase expression, as expected in the case of impaired P53 binding. The SNP rs143789306 is located in the spacer of the two P53 half-sites at Chr7, near TNRC18 that encodes for Trinucleotide Repeat Containing 18. The notable increase in luciferase expression after introduction of the SNP may be due to a de novo binding site of an activator or a disruption of a repressor binding site. Finally, EMSA results using the co-occupied region at Chr12 as the test region confirmed that P53 and cMYC bound specifically at the identified COM region, and that SNP introduction directly caused a change in the binding affinity of cMYC and P53 which may explain the observed changes in luciferase expression. Decrease in band intensity was observed when the cMYC protein with the consensus tagged probe was competed with the SNP-containing untagged probes.
Figure 7.
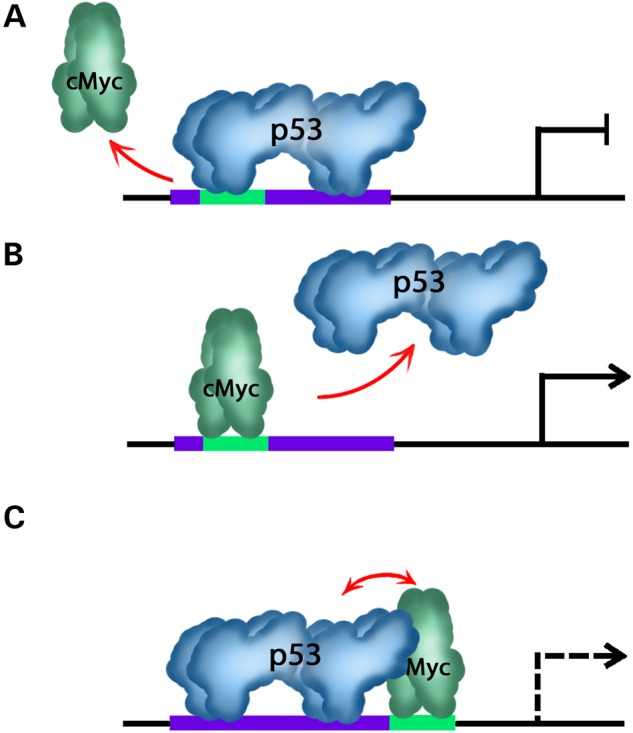
An illustrative diagram explains different possibilities of the competitive inhibitory mechanism at COMs. In case of a complete or partial overlap among P53 and cMYC binding sites, P53 protein binds and prevents cMYC from binding if P53 protein is more abundant and has higher binding affinity. Consequently the expression of target gene is turned-off (A). In the second scenario, cMYC is more abundant and has higher binding affinity and subsequently drives gene expression by inhibition of P53 binding (B). In the third scenario where P53 and cMYC binding sites are head-to-head, both factors will try to compete for binding to its motif and depending on several parameters and cofactors, quenching could take place between both factors (C).
Conclusions
Currently, a number of single nucleotide variants have been identified as potentially linked to certain traits or diseases via genome-wide association studies (GWAS). However, genome-wide association studies associate these variants to certain traits or diseases based only on their frequency of occurrence in individuals carrying that condition compared to those without it, and does not indicate a particular variant or gene as etiologic to the condition. This means that GWAS SNPs are linked to a condition, but are not necessarily functional or etiologic of that condition. Furthermore, the SNPs we tested at Chr6, 7, and 12 using luciferase assay are located near genes containing GWAS SNPs (Table 4) (38–43). If these tested SNPs are within the same haplotype with the GWAS SNPs listed in Table 4, they may very well be the functional SNPs of a particular condition associated with that haplotype. The results of our study suggest a potential molecular mechanism that can be used to identify etiologic DNA variants in regulatory elements that increase the risk for common complex diseases. This provides a novel predictive approach to expedite the identification of functional non-coding DNA variations to improve the risk assessment and prognosis of disease development, and for enhancing targeted therapies in patients with cancer.
Table 4.
SNPs within cis-overlapping motifs of cMYC and P53 that are in close proximity to GWAS SNPs associated with common complex diseases
| SNPs within COMs | Target gene | Associated common diseases |
|---|---|---|
| rs140357312 | TRAF3IP2-AS1/TRAFIP2 | Xerostomia, Systemic Lupus Erythematosus, Psoriasis (90 kb, GWAS SNP rs33980500) (38,39) |
| rs143789306 | TNRC18 | Left/right asymmetry of hand skill disorder, Ulcerative Colitis (85 kb, GWAS SNP rs10216189) (42) |
| rs373187248 rs145730860 |
TRIAP1 | Insulin resistance (2 kb, GWAS SNP rs17431357) (40) |
| rs375618035 | AEN | 0.5 kb, eQTL SNP rs11858257 (41, 43) |
Materials and Methods
Data acquisition
For human cancer cell lines analysis, the experimental ChIP peak coordinates for P53 and cMYC binding regions were obtained from published data (22,26). Similarly for mouse embryonic cells, coordinates of P53- and cMYC-bound sites were obtained from previous studies (17,27). ChIP peak coordinates of P53 and cMYC were expanded to 500 bp in each direction from the center of each peak with the assumption that the coordinates reported by these article represented the middle of each ChIP peak. All of these studies used ChIP-seq approach to identify TF binding sites.
Mapping COMs within ChIP-seq elements
To map COMs within experimental ChIP elements, our motif criteria were based on the consensus binding sequences of P53 and cMYC. These position weight matrices were determined from collections of published data based on past SELEX studies and experimentally identified binding of regulatory regions (http://jaspar.genereg.net/) (44). According to previously published papers, the accepted motif for the P53 binding site is RRRCWWGYYY-spacer-RRRCWWGYYY (2,37), where R represents purines, W represents A or T, and Y represents pyrimidines. The spacer ranges from 0 to 13 unspecified nucleotides, although it is most commonly 0. The consensus motif for the cMYC binding site is the Ebox motif CANNTG, where N represents unspecified nucleotides. Other basic helix-loop-helix transcription factors (bHLH) such as Twist1, Hand2, and MyoD bind to this site as well (3,20).
The sequences represented by the overlapping ChIP peak coordinates were obtained to search for overlap of P53 and cMYC consensus motifs within these regions. A computer program written in Python code was used to locate P53 and cMYC overlapping binding motifs. While these motifs are conserved across species, past studies have demonstrated the degeneracy of these sequences (2,3,24). Therefore, the accepted sequence criteria for successfully identified binding motif of P53 is less stringent for the periphery nucleotides, with the P53 motif as CWWG-spacer-CWWG, and for the cMYC motif as CANNTG. N represents unspecified nucleotides, and the ‘spacer’ within the P53 motif consisted of 6–24 unspecified nucleotides. Because these conserved motifs are palindromic, the program accepts the complement sequence as motifs (i.e. GWWC-spacer-GWWC is also recognized as the P53 motif). The program generated the resulting COMs data in the BED format that is required for uploading the data to the online UCSC Genome Browser for further analysis (45).
Template conversions and non-coding DNA variants
In the analysis of P53 and cMYC binding in cancer cell lines, cMYC ChIP peak coordinates was presented in GRCh37/hg19 build (released in 2009) (26), while P53 ChIP peak coordinates was presented in human genome build NCBI36/hg18 (released in 2006) (22). Coordinate data of P53 therefore had to be converted using the Batch Coordinate Conversion tool (LiftOver tool) provided by the UCSC Genome Browser to GRCh37/hg19 prior to any analysis. In mouse embryonic cell lines, P53 ChIP peak coordinates was presented in mm9 build (17), while cMYC ChIP peak coordinates was presented in mm8 build (27). Again, the UCSC LiftOver tool was used to convert the mm8 build data to mm9 build prior to any analysis. We used the same programs written in Python code to analyze human tumor cell lines and mouse embryonic cells to map overlapping P53 and cMYC ChIP peaks and COMs within these regions. After determining the coordinates for P53 and cMYC COMs, the UCSC Table Browser tool was used to locate SNPs within these COMs for both cell types (30).
Gene annotation and analysis of regulatory DNA variants
To annotate genes that are near COMs regions and potentially regulated by them, the coordinates of overlapping ChIP peaks were uploaded to Genomic Regions Enrichment Annotations Tool (GREAT). This is in contrast to the coordinates used to locate SNPs, which must be COM coordinates. The GREAT used in this analysis corrects for variations in region input size, predicts genes affected by regulatory element input using two different tests that are hypergeometric and binomial probability tests and incorporates ontologies from many different databases during analysis (25). We used different tools to analyze the role of regulatory SNPs within COMs and association to susceptibility loci. The analysis was done for human DNA variants only because analysis of SNPs associated with animals is not available. We used the Combined Annotation-Dependent Depletion (CADD) program that scores the pathogenicity of short insertions-deletions based on integrations of allelic diversity, annotations of functionality and pathogenicity, and other experimentally measured effects (46). Genome Wide Annotation of Variants (GWAVA), a tool maintained by Sanger which uses annotations of non-coding elements (provided in large by ENCODE/GENCODE) and other properties to predict the functional impact of non-coding genetic variants such as SNPs (47), was also used in our analysis. rSNPBase is an online tool that was also used to similarly predict potential regulatory function of SNPs based on complete positional weight matrix (PWM) scores and statistical predictions of the impact of SNPs (43).
Site-directed mutagenesis and luciferase assay in cell culture
For cell transfection, a 96 well plate with a glass bottom was used to grow HEK293 cells in DMEM, 10% FBS, and no antibiotics medium at 37°C as previously described (5). HEK293 cells were selected for transactivation experiments because they are biologically relevant and they grow and transfect well. The cells were transfected 2 h after plating using lipofectamine 2000 (Life Technology, CA) with pGL3-basic-Luc and pGL3-enh-Luc as negative and positive controls, respectively, and with the co-occupied regulatory regions fused upstream of luciferase gene. The pGL3-SV40-Renilla plasmid served as an internal control for transfection efficiency. Site-directed mutagenesis was used to introduce the identified SNPs within COMs as previous described using PCR and DpnI digestion enzyme (5).
EMSA
To test the binding of cMYC and P53 proteins to cis-overlapping motif within a co-occupied region in Chr12, sense oligo was tagged with IR-700 at 5′ ends (IDT, Coralville, Iowa) while the complimentary probe was synthesized without labeling. The EMSA assay was performed using Li-COR binding protocol and a recombinant protein of cMYC and P53 were produced using the reticulocyte TNT kit from Promega as previously described (5). Sequences of oligos used in EMSA this study are 5′-IR700- TTAGGGTGAACACCAAATGAACCAAAGGATG (sense); 5′-CATCCTTTGGTTCATTTGGTGTTCACCCTAA (anti-sense); untagged oligos 5′-SNP rs145730860 TTAGGGTGAACACCAAGTGAACCAAAGGATG (sense); 5′-CATCCTTTGGTTCACTTGGTGTTCACCCTAA (anti-sense); untagged oligos 5′- SNP rs373187248 TTAGGGTGAATACCAAATGAACCAAAGGATG (sense); 5′-CATCCTTTGGTTCATTTGGTATTCACCCTAA (anti-sense); Mouse antibodies against cMYC and P53 were used to validate the specificity of the binding of the each protein to labeled probe.
Funding
This study was supported by start-up funding to W.D.F. from University of Texas Health Science Center at Houston School of Dentistry, and from the Research Office at the University of Texas Health Science Center at Houston School of Dentistry to K.K.
Supplementary Material
Acknowledgements
We thank Dr Wenjin Zheng from the Bioinformatics Core Facility at University of Texas Health Science Center at Houston for reviewing our manuscript. We are also grateful to the help that Dr Xiaojing Dai provided with the luciferase assay.
Conflict of Interest statement. None declared.
References
- 1. Yang A., Zhu Z., Kapranov P., McKeon F., Church G.M., Gingeras T.R., Struhl K. (2006) Relationships between p63 binding, DNA sequence, transcription activity, and biological function in human cells. Mol. Cell, 24, 593–602. [DOI] [PubMed] [Google Scholar]
- 2. Smeenk L., van Heeringen S.J., Koeppel M., van Driel M.A., Bartels S.J., Akkers R.C., Denissov S., Stunnenberg H.G., Lohrum M. (2008) Characterization of genome-wide p53-binding sites upon stress response. Nucleic Acids Res., 36, 3639–3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Eilers M., Eisenman R.N. (2008) Myc's broad reach. Genes Dev., 22, 2755–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Epstein D.J. (2009) Cis-regulatory mutations in human disease. Brief. Funct. Genomic. Proteomic., 8, 310–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fakhouri W.D., Rahimov F., Attanasio C., Kouwenhoven E.N., Ferreira De Lima R.L., Felix T.M., Nitschke L., Huver D., Barrons J., Kousa Y.A. et al. (2014) An etiologic regulatory mutation in IRF6 with loss- and gain-of-function effects. Hum. Mol. Genet., 23, 2711–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cowie P., Ross R., MacKenzie A. (2013) Understanding the dynamics of gene regulatory systems; characterisation and clinical relevance of cis-regulatory polymorphisms. Biology (Basel), 2, 64–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Raynal J.F., Dugast C., Le Van Thai A., Weber M.J. (1998) Winged helix hepatocyte nuclear factor 3 and POU-domain protein brn-2/N-oct-3 bind overlapping sites on the neuronal promoter of human aromatic L-amino acid decarboxylase gene. Brain Res. Mol. Brain Res., 56, 227–237. [DOI] [PubMed] [Google Scholar]
- 8. Guerin S.L., Leclerc S., Verreault H., Labrie F., Luu-The V. (1995) Overlapping cis-acting elements located in the first intron of the gene for type I 3 beta-hydroxysteroid dehydrogenase modulate its transcriptional activity. Mol. Endocrinol., 9, 1583–1597. [DOI] [PubMed] [Google Scholar]
- 9. Gupta M.P., Gupta M., Zak R. (1994) An E-box/M-CAT hybrid motif and cognate binding protein(s) regulate the basal muscle-specific and cAMP-inducible expression of the rat cardiac alpha-myosin heavy chain gene. J. Biol. Chem., 269, 29677–29687. [PubMed] [Google Scholar]
- 10. Akdemir K.C., Jain A.K., Allton K., Aronow B., Xu X., Cooney A.J., Li W., Barton M.C. (2013) Genome-wide profiling reveals stimulus-specific functions of p53 during differentiation and DNA damage of human embryonic stem cells. Nucleic Acids Res., 42, 205–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Erceg J., Saunders T.E., Girardot C., Devos D.P., Hufnagel L., Furlong E.E. (2014) Subtle changes in motif positioning cause tissue-specific effects on robustness of an enhancer's activity. PLoS Genet., 10, e1004060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nibu Y., Senger K., Levine M. (2003) CtBP-independent repression in the Drosophila embryo. Mol. Cell. Biol., 23, 3990–3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang P., Tchou-Wong K.M., Costa M. (2007) Egr-1 mediates hypoxia-inducible transcription of the NDRG1 gene through an overlapping Egr-1/Sp1 binding site in the promoter. Cancer Res., 67, 9125–9133. [DOI] [PubMed] [Google Scholar]
- 14. Alfonso-Jaume M.A., Mahimkar R., Lovett D.H. (2004) Co-operative interactions between NFAT (nuclear factor of activated T cells) c1 and the zinc finger transcription factors Sp1/Sp3 and Egr-1 regulate MT1-MMP (membrane type 1 matrix metalloproteinase) transcription by glomerular mesangial cells. Biochem. J., 380, 735–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tang J., Luo Z., Zhou G., Song C., Yu F., Xiang J., Li G. (2011) Cis-regulatory functions of overlapping HIF-1alpha/E-box/AP-1-like sequences of CD164. BMC Mol. Biol., 12, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ryu J.R., Arnosti D.N. (2003) Functional similarity of Knirps CtBP-dependent and CtBP-independent transcriptional repressor activities. Nucleic Acids Res., 31, 4654–4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kenzelmann Broz D., Spano Mello S., Bieging K.T., Jiang D., Dusek R.L., Brady C.A., Sidow A., Attardi L.D. (2013) Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev., 27, 1016–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li M., He Y., Dubois W., Wu X., Shi J., Huang J. (2012) Distinct regulatory mechanisms and functions for p53-activated and p53-repressed DNA damage response genes in embryonic stem cells. Mol. Cell, 46, 30–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nie Z., Hu G., Wei G., Cui K., Yamane A., Resch W., Wang R., Green D.R., Tessarollo L., Casellas R. et al. (2012) c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell, 151, 68–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kidder B.L., Yang J., Palmer S. (2008) Stat3 and c-Myc genome-wide promoter occupancy in embryonic stem cells. PLoS One, 3, e3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Krepelova A., Neri F., Maldotti M., Rapelli S., Oliviero S. (2014) Myc and max genome-wide binding sites analysis links the Myc regulatory network with the polycomb and the core pluripotency networks in mouse embryonic stem cells. PLoS One, 9, e88933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chang G.S., Chen X.A., Park B., Rhee H.S., Li P., Han K.H., Mishra T., Chan-Salis K.Y., Li Y., Hardison R.C. et al. (2014) A comprehensive and high-resolution genome-wide response of p53 to stress. Cell Rep., 8, 514–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Won K.J., Ren B., Wang W. (2010) Genome-wide prediction of transcription factor binding sites using an integrated model. Genome Biol., 11, R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Veerla S., Ringner M., Hoglund M. (2010) Genome-wide transcription factor binding site/promoter databases for the analysis of gene sets and co-occurrence of transcription factor binding motifs. BMC Genomics, 11, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McLean C.Y., Bristor D., Hiller M., Clarke S.L., Schaar B.T., Lowe C.B., Wenger A.M., Bejerano G. (2010) GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol., 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Seitz V., Butzhammer P., Hirsch B., Hecht J., Gutgemann I., Ehlers A., Lenze D., Oker E., Sommerfeld A., von der Wall E. et al. (2011) Deep sequencing of MYC DNA-binding sites in Burkitt lymphoma. PLoS One, 6, e26837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen X., Xu H., Yuan P., Fang F., Huss M., Vega V.B., Wong E., Orlov Y.L., Zhang W., Jiang J. et al. (2008) Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell, 133, 1106–1117. [DOI] [PubMed] [Google Scholar]
- 28. Wei C.L., Wu Q., Vega V.B., Chiu K.P., Ng P., Zhang T., Shahab A., Yong H.C., Fu Y., Weng Z. et al. (2006) A global map of p53 transcription-factor binding sites in the human genome. Cell, 124, 207–219. [DOI] [PubMed] [Google Scholar]
- 29. Blake J.A., Bult C.J., Eppig J.T., Kadin J.A., Richardson J.E. and Mouse Genome Database Group. (2014) The Mouse Genome Database: integration of and access to knowledge about the laboratory mouse. Nucleic Acids Res., 42, D810–D817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. (2002) The human genome browser at UCSC. Genome Res., 12, 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang B., Niu D., Lam T.H., Xiao Z., Ren E.C. (2014) Mapping the p53 transcriptome universe using p53 natural polymorphs. Cell Death Differ., 21, 521–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shaked H., Shiff I., Kott-Gutkowski M., Siegfried Z., Haupt Y., Simon I. (2008) Chromatin immunoprecipitation-on-chip reveals stress-dependent p53 occupancy in primary normal cells but not in established cell lines. Cancer Res., 68, 9671–9677. [DOI] [PubMed] [Google Scholar]
- 33. Ji H., Wu G., Zhan X., Nolan A., Koh C., De Marzo A., Doan H.M., Fan J., Cheadle C., Fallahi M. et al. (2011) Cell-type independent MYC target genes reveal a primordial signature involved in biomass accumulation. PLoS One, 6, e26057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zeller K.I., Zhao X., Lee C.W., Chiu K.P., Yao F., Yustein J.T., Ooi H.S., Orlov Y.L., Shahab A., Yong H.C. et al. (2006) Global mapping of c-Myc binding sites and target gene networks in human B cells. Proc. Natl Acad. Sci. USA, 103, 17834–17839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Perna D., Faga G., Verrecchia A., Gorski M.M., Barozzi I., Narang V., Khng J., Lim K.C., Sung W.K., Sanges R. et al. (2011) Genome-wide mapping of Myc binding and gene regulation in serum-stimulated fibroblasts. Oncogene, 31, 1695–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee K.H., Li M., Michalowski A.M., Zhang X., Liao H., Chen L., Xu Y., Wu X., Huang J. (2009) A genomewide study identifies the Wnt signaling pathway as a major target of p53 in murine embryonic stem cells. Proc. Natl Acad. Sci. USA, 107, 69–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Idogawa M., Ohashi T., Sasaki Y., Maruyama R., Kashima L., Suzuki H., Tokino T. (2014) Identification and analysis of large intergenic non-coding RNAs regulated by p53 family members through a genome-wide analysis of p53-binding sites. Hum. Mol. Genet., 23, 2847–2857. [DOI] [PubMed] [Google Scholar]
- 38. Hayashi M., Hirota T., Saeki H., Nakagawa H., Ishiuji Y., Matsuzaki H., Tsunemi Y., Kato T., Shibata S., Sugaya M. et al. (2014) Genetic polymorphism in the TRAF3IP2 gene is associated with psoriasis vulgaris in a Japanese population. J. Dermatol. Sci., 73, 264–265. [DOI] [PubMed] [Google Scholar]
- 39. Perricone C., Ciccacci C., Ceccarelli F., Di Fusco D., Spinelli F.R., Cipriano E., Novelli G., Valesini G., Conti F., Borgiani P. (2013) TRAF3IP2 gene and systemic lupus erythematosus: association with disease susceptibility and pericarditis development. Immunogenetics, 65, 703–709. [DOI] [PubMed] [Google Scholar]
- 40. Irvin M.R., Wineinger N.E., Rice T.K., Pajewski N.M., Kabagambe E.K., Gu C.C., Pankow J., North K.E., Wilk J.B., Freedman B.I. et al. (2011) Genome-wide detection of allele specific copy number variation associated with insulin resistance in African Americans from the HyperGEN study. PLoS One, 6, e24052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Westra H.J., Peters M.J., Esko T., Yaghootkar H., Schurmann C., Kettunen J., Christiansen M.W., Fairfax B.P., Schramm K., Powell J.E. et al. (2013) Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet., 45, 1238–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Brandler W.M., Morris A.P., Evans D.M., Scerri T.S., Kemp J.P., Timpson N.J., St Pourcain B., Smith G.D., Ring S.M., Stein J. et al. (2013) Common variants in left/right asymmetry genes and pathways are associated with relative hand skill. PLoS Genet., 9, e1003751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Guo L., Du Y., Chang S., Zhang K., Wang J. (2014) rSNPBase: a database for curated regulatory SNPs. Nucleic Acids Res., 42, D1033–D1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mathelier A., Zhao X., Zhang A.W., Parcy F., Worsley-Hunt R., Arenillas D.J., Buchman S., Chen C.Y., Chou A., Ienasescu H. et al. (2013) JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res., 42, D142–D147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Karolchik D., Hinrichs A.S., Furey T.S., Roskin K.M., Sugnet C.W., Haussler D., Kent W.J. (2004) The UCSC Table Browser data retrieval tool. Nucleic Acids Res., 32, D493–D496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kircher M., Witten D.M., Jain P., O'Roak B.J., Cooper G.M., Shendure J. (2014) A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet., 46, 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ritchie G.R., Dunham I., Zeggini E., Flicek P. (2014) Functional annotation of noncoding sequence variants. Nat. Methods, 11, 294–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.