

Abstract
Many studies have reported that CCN family protein 2 (also known as connective tissue growth factor) induces fibrotic response in skeletal muscle, thus emphasizing the pathological role of CCN2 in muscle tissues. However, the physiological role of CCN2 in myogenesis is still unknown. This study clarified the CCN2 functions during myogenesis. Recombinant CCN2 (rCCN2) promoted proliferation and MyoD production in C2C12 cells and primary myoblasts, but inhibited myogenin production. In accordance with these findings, the gene expression levels of myosin heavy chain, which is a marker of terminally differentiated myoblasts and desmin, which is the main intermediate filament protein of muscle cells, were decreased by rCCN2 treatment. In vivo analyses with Ccn2-deficient skeletal muscle revealed decreased proliferating cell nuclear antigen (PCNA)/MyoD double positive cells and muscle hypoplasia. Consistent with this finding, myogenic marker genes and myotube formation were repressed in Ccn2-deficient myoblasts. The protein production of CCN2 was increased in C2C12 myoblasts treated with tumor necrosis factor-α, which is a pro-inflammatory cytokine, suggesting its role in muscle regeneration after inflammation. These findings indicate that CCN2 promotes proliferation and early differentiation but inhibits the terminal differentiation of myoblasts, thus suggesting that CCN2 plays a physiological role in myogenesis.
Keywords: CCN2, CTGF, C2C12 cells, myoblasts, skeletal myogenesis, TNFα
Muscle development during embryogenesis is conducted by myogenic stem cells (1). These cells initially proliferate to produce myogenic precursor cells, called myoblasts, to regenerate the muscle (1, 2). These myoblasts then proliferate, increasing their number; and then, these cells undergo terminal differentiation and form multinucleate myotubes by cell fusion (1, 2). Eventually, new muscle fibers are produced, which finally mature into contracting muscle fibers (1, 2). The entire process is guided by various extracellular environments and is regulated by distinct intracellular signaling pathways, resulting in the activation of myogenic regulatory factors (MRFs) such as MyoD, Myf5, myogenin, and MRF4 (1, 2). MyoD and Myf5 are expressed in myoblasts; whereas myogenin and MRF4 induce terminal differentiation of myoblasts into the myotubes, which express important genes for muscle cell function, such as myosin heavy chain (MHC; 1, 2).
CCN2 (also known as connective tissue growth factor: CTGF) is a protein secreted from various cell types including chondrocytes, osteoblasts and vascular endothelial cells and its structure is characterized by four distinct modules, i.e. insulin-like growth factor binding protein-like, von Willebrand factor type C, thrombospondin type 1 repeat and a carboxyl terminal cystine knot (CT; 3–6). CCN2 belongs to the CCN family, which consists of six distinct proteins; i.e. Cysteine-rich 61 (Cyr61/CCN1), CTGF/CCN2, Nephroblastoma overexpressed (Nov/CCN3), from which the family name is derived; and Wnt-inducible secreted proteins 1, 2 and 3 (Wisp1-3/CCN4-6), having structures similar to those described above, except that Wisp2/CCN5 lacks the CT module (3–6). It has been reported that CCN family members are involved in a number of biological processes in development, including chondrogenesis, osteogenesis and angiogenesis (3–6). Therefore, it is suspected that CCN family members are potentially involved in musuclogenesis. It has been reported that excess CCN2 induces damage and fibrosis in skeletal muscle tissues (7–9) and that both CCN1 (10) and CCN3 (11) impair skeletal muscle differentiation. However, these findings only suggest the effects of overproduced CCN family members in skeletal muscle under the pathological condition. In fact, although overexpression of CCN2 causes fibrotic disorders in various tissues and organs, regulated expression of CCN2 is observed during the development of the same tissues and organs (12–14). Therefore, we hypothesized that CCN2 plays certain roles in skeletal myogenesis. Adult skeletal muscle possesses remarkable regeneration capabilities (1), and tissue regeneration is a recurrence of tissue development (2). In this study, to examine the validity of our hypothesis, we investigated the functions of CCN2 in skeletal muscle regeneration in the physiological state.
Materials and Methods
Materials
Dulbecco’s modified Eagle’s medium (DMEM) and fetal bovine serum (FBS) were purchased from Nissui Pharmaceutical Co. Ltd. (Tokyo, Japan) and Cancera International (Rexcalale, ON, Canada), respectively. Plastic dishes and multiwell plates were obtained from Greiner Bio-One (Frickenhausen, Germany). Anti-integrin α5 and anti-CCN2 antibodies were purchased from Chemicon International, Inc. (Billerica, MA, USA) and Abcam (Cambridge, UK), respectively. Anti-β-actin and anti-proliferating cell nuclear antigen (PCNA) were from Sigma (St. Louis, MO, USA), anti-hemagglutinin (HA) was from Covance (Princeton, NJ, USA), anti-desmin was from Nichirei (Tokyo, Japan) and both anti-MyoD and anti-myogenin antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA,USA). Recombinant tumor necrosis factor-α (TNFα) was purchased from Promega (Madison, WI, USA), and recombinant CCN2 (rCCN2) was purified as described previously (15).
Cell culture
Mouse myoblast cell line C2C12 was purchased from the European collection of cell cultures (ECACCs, Salisbury, UK). The cells were inoculated at a density of 1.5 × 104 cells/cm2 in DMEM supplemented with 10% FBS and were cultured at 37°C under 5% CO2 in air. Primary mouse myoblasts were isolated from the gastrocnemius muscle of Ccn2-deficient mice at embryonic day (E) 18.5 (16). The isolated tissues were digested with 0.2% collagenase (Wako Co., Osaka, Japan) for 40 min at 37°C, and then filtered through a 40 µm cell strainer (BD Biosciences; Bedford, MA, USA; 17, 18). After genotyping, these cells were passaged, inoculated at a density of 1.5 × 104 cells/cm2, and cultured in DMEM containing 10% FBS at 37°C under 5% CO2 until they reached 70–80% confluence. For induction of differentiation, the culture medium for both C2C12 cells and primary myoblasts was changed to serum-free DMEM containing 1 mg/ml bovine serum albumin (BSA; Sigma), 10 µg/ml bovine insulin (I; Wako Co.), 5 µg/ml human transferrin (T; Sigma) and 1 × 10−8 M sodium selenite (S; Sigma); and the cells were then cultured at 37°C under 5% CO2 for 4–7 days (19). The Animal Committee of Okayama University Graduate School of Medicine, Dentistry, and Pharmaceutical Sciences had approved all of the procedures.
Quantitative RT–PCR analysis
Total RNA was isolated from C2C12 cells and primary myoblasts by using ISOGEN reagent (Nippon Gene, Tokyo, Japan). First-strand cDNA was synthesized by use of a Takara RNA PCR kit (AMV) Version 3.0 (Takara Shuzo, Tokyo, Japan), and amplification reactions were performed with a SYBR® Green Real-time PCR Master Mix (Toyobo, Tokyo, Japan) and a StepOnePlus™ Real-time PCR System (Applied Biosystems, Basel, Switzerland; 20, 21). The primer sequences are indicated in Table I.
Table I.
Sense (S) and antisense (AS) primers used for PCR.
| Genes | Accession Number | Primer sequence |
|---|---|---|
| MyoD | NM_010866 | (S) 5′-CGCTCCAACTGCTCTGAT-3′ |
| (AS) 5′-AGATGCGCTCCACTATGC-3′ | ||
| Desmin | NM_010043 | (S) 5′-CTTGACTCAGGCAGCCAATAA-3′ |
| (AS) 5′-AGCTCCCTCATCTGCCTCAT-3′ | ||
| MHC | NM_080728 | (S) 5′-CGCAATGCAGAGTCAGTGAA-3′ |
| (AS) 5′-TTGCGGAACTTGGACAGGTT-3′ | ||
| Myostatin | NM_ 010834 | (S) 5′-AATCCACCACGGTGCTAATG-3′ |
| (AS) 5′-CTCCCGCTGTGCTAGAGTTG-3′ | ||
| Myogenin | NM_ 031189 | (S) 5′-CCTTGCTCAGCTCCCTCAA-3′ |
| (AS) 5′-CTGGGTTGGGACCGAACT-3′ | ||
| Gapdh | NM_008084 | (S) 5′-GCCAAAAGGGTCATCATCTC -3′ |
| (AS) 5′-GTCTTCTGGGTGGCAGTGAT-3′ |
Transfection with CCN2 expression plasmid and western blot analysis
C2C12 cells were transfected with a CCN2 expression plasmid bearing an HA-tag (22) by using Fugene6 reagent (Roche Applied Science, Mannheim, Germany). After 4 days, the cell lysate was collected; and then western blot analysis was performed as described previously (23).
Cell proliferation assay
For measurement of cell proliferation, C2C12 cells were inoculated into each well of a 96-well multiwell plate at a density of 1 × 104 cells/well and cultured in DMEM containing 10% FBS. Then, the next day, the medium was replaced with DMEM without FBS; and the cells were treated with rCCN2 for 16 h. The effect of CCN2 on the proliferation of C2C12 cells was determined by use of a Cell proliferation ELISA, BrdU (5-bromo-2′-deoxyuridine) colorimetric immunoassay (Roche Applied Science; 24).
Immunohistochemistry
Ccn2-deficient mice at E 18.5 were euthanized to obtain hind limbs, which were fixed in 10% formalin overnight at 4°C before being embedded in paraffin. Five-micrometre sections were mounted on glass slides and deparaffinized with xylene, rehydrated and treated with hyaluronidase (25 mg/ml; Sigma) for 30 min at room temperature for antigen retrieval. Immunofluorescence staining of these sections was performed with both anti-PCNA and anti-MyoD antibodies, or anti-desmin (Nichirei) alone as described previously (25).
Indirect immunofluorescence analysis
Myoblasts from WT and Ccn2-deficient mice were cultured until they had reached 70–80% confluence. Then, the culture medium was exchanged for serum-free DMEM containing 1 mg/ml BSA and insulin–transferrin–selenite (ITS). After 7 days, the cultures were fixed with 3.5% paraformaldehyde for 30 min at room temperature, and made permeable with 0.1% NP-40 in PBS. Indirect immunofluorescence analysis was performed as described previously (26).
Statistical analysis
Unless otherwise specified, all experiments were repeated at least twice, and similar results were obtained. Statistics were calculated using Student’s t test or one-way ANOVA.
Results
CCN2 promotes the proliferation of C2C12 cells
Firstly, we performed a BrdU incorporation assay on C2C12 cells stimulated by rCCN2. As shown in Fig. 1A, rCCN2 significantly stimulated the cell proliferation of C2C12 cells in a dose-dependent manner. Next, we performed western blot analysis of cell lysates of C2C12 cells treated with rCCN2 at a concentration of 50 and 100 ng/ml by using antibody against PCNA, which is a marker of cell proliferation. As shown in Fig. 1B, the signal for PCNA was increased by the treatment with rCCN2 in a dose-dependent manner. To support the data of Fig. 1B, we transfected C2C12 cells with an HA epitope-tagged full-length Ccn2 (HA-Ccn2) expression plasmid (22). After 4 days we collected the cell lysates and performed western blot analysis with anti-HA and PCNA antibodies. As a result, an HA-specific signal was clearly detected in C2C12 cells transfected with HA-Ccn2 expression plasmid, and the intensity of the band of PCNA was increased in the cells transfected with the HA-Ccn2 expression plasmid (Fig. 1C). These findings indicate that CCN2 promotes the proliferation of C2C12 cells.
Fig. 1.

CCN2 promotes the cell proliferation of C2C12 cells. (A) C2C12 cells were stimulated for 16 h with rCCN2 at the indicated concentrations in differentiation medium. Then, cell proliferation assay was performed. Each bar shows the mean value and standard deviation of the results from nine wells. **P < 0.01 indicates significant difference from control by one-way ANOVA. (B) C2C12 cells were treated with rCCN2 at the indicated concentrations for 24 h. Western blot analysis was then performed by using anti-PCNA and anti-β-actin antibodies. (C) C2C12 cells were transiently transfected with CCN2 expression plasmid containing an HA tag, and the cells were cultured for 4 days. Western blot analysis was performed by using antibodies against the indicated antigens.
CCN2 promotes the myoblast differentiation at an early stage but inhibits differentiation at late stage
We next investigated the effect of CCN2 on the differentiation of myoblasts. C2C12 cells were inoculated into 35-mm dishes, and cultured until they reached 70–80% confluence. Then, the medium was changed to serum-free DMEM containing ITS (differentiation medium) and rCCN2. After 24 h, total RNA was collected; and quantitative RT–PCR analysis was then performed by using specific primers for MyoD, which is a marker of early-stage myoblast differentiation. As shown in Fig. 2A left panel, the gene expression level of MyoD was increased in the cells treated with rCCN2 at the concentration of 100 and 250 ng/ml. To confirm this result at the protein level, cell lysates were collected after 2 days of stimulation with rCCN2; and western blot analysis was then performed by using anti-MyoD antibody. As shown in Fig. 2A right panel, MyoD production was increased by rCCN2 in a dose-dependent manner. Next, to investigate the effect of CCN2 on the myoblast differentiation at the late-stage, we performed quantitative RT–PCR analysis and western blot analysis after 4 and 5 days of stimulation with rCCN2, respectively. As shown in Fig. 2B, the gene expression and protein production levels of myogenin, which is a marker of terminally differentiated myoblasts (1, 2), were oppositely decreased by rCCN2 in a dose-dependent manner. Because it was reported that MyoD up-regulates the gene expression level of Myogenin (27), we examined how CCN2 regulated the MyoD and myogenin production during myoblast differentiation. Cell lysate of C2C12 cells at days 2, 5 and 7 after treatment with rCCN2 was collected; and western blot analysis was performed by using anti-MyoD and myogenin antibodies. In the early stage of differentiation, CCN2 induced MyoD production, which was, however, rather repressed by CCN2 in the late stage. Consistent with these results, myogenin production was repressed in the late stage by CCN2 under the support of less MyoD (Fig. 2C). To further investigate CCN2 functions during myoblast differentiation, we performed quantitative RT–PCR analysis by using specific primers to evaluate late-stage myoblast differentiation. As shown in Fig. 3A and B, the gene expression levels of Desmin and MHC, which are markers of differentiated myoblasts at the late stage (1, 2), was significantly decreased by the addition of rCCN2. Interestingly, the gene expression level of Myostatin, which is an important regulator of muscle growth (28, 29), was oppositely increased in C2C12 cells treated with rCCN2 in a dose-dependent manner (Fig. 3C). Taken together, these findings indicate that CCN2 up-regulates the MyoD production at the early-stage of differentiation, but down-regulates the MyoD and myogenin production at the late-stage of differentiation. Consequently, CCN2 down-regulated the gene expression of late stage markers, thus suggesting that CCN2 promotes the early-stage differentiation but inhibits the late-stage differentiation of myoblasts.
Fig. 2.

CCN2 regulates the gene expression and protein production of MyoD and myogenin in C2C12 cells. (A) C2C12 cells were stimulated with rCCN2 at the indicated concentrations in differentiation medium. After 1 and 2 days, the gene expression and protein production of MyoD were examined, respectively. *P < 0.05 indicates significant difference by one-way ANOVA. (B) C2C12 cells were treated with rCCN2 in differentiation medium. After 4 and 5 days, the gene expression and protein production of myogenin were evaluated, respectively. *P < 0.05 indicates significant difference by one-way ANOVA. (C) C2C12 cells were stimulated with rCCN2 in differentiation medium. At the indicated time after stimulation, cell lysate was collected and western blot analysis was performed.
Fig. 3.

CCN2 regulates the gene expression of Desmin (A), MHC (B), and Myostatin (C) in C2C12 cells. C2C12 cells were stimulated with rCCN2 at the concentrations indicated for 4 days, and real-time PCR analysis was performed. The amounts of these gene products were normalized to that of Gapdh with respect to the control sample (ratio = 1.0). Bars represent means and standard deviations of results obtained from eight to nine different cultures for each sample. *P < 0.05 and **P < 0.01 indicate significant differences from control by one-way ANOVA.
CCN2 production is increased in C2C12 cells treated with TNFα
Then, to clarify whether or not the CCN2 production was up-regulated when an inflammatory response that damages tissue occurred in skeletal muscle, we investigated the CCN2 production in C2C12 cells stimulated by the proinflammatory cytokine TNFα in a time course. As shown in Fig. 4A and C, CCN2 production was increased at 48 h after the treatment with TNFα at a concentration of 20 ng/ml. Next, we evaluated whether the production level of CCN2 was increased by treatment with TNFα in a dose-dependent manner. As shown in Fig. 4B, the promotion of CCN2 production was confirmed at the protein level in C2C12 cells treated with TNFα, which increase occurred at a concentration of 5 ng/ml. These results indicate that CCN2 production is increased after 48 h of stimulation with TNFα, also suggesting that CCN2 is induced after inflammation to be engaged in muscle regeneration.
Fig. 4.

CCN2 production is increased in C2C12 cells treated with TNFα. (A) C2C12 cells were stimulated by 20 ng/ml TNFα in differentiation medium. At the indicated time the cell lysates were collected and western blot analysis was performed by using anti-CCN2 and β-actin antibodies. (B) C2C12 cells were treated with TNFα at the indicated concentrations for 2 days. Then, western blot analysis was similarly performed. (C) Quantification of the results obtained from the experiments for panel A at 48 h in triplicates. *P < 0.05 indicates significant difference from control by Student’s t-test.
Cell proliferation and myotube formation are impaired in Ccn2-deficient myoblasts
Next, to investigate CCN2 functions in primary myoblasts, we isolated from wild-type (WT) and Ccn2-deficient myoblasts. Firstly, we examined the role of CCN2 in the production of MyoD. As shown in Fig. 5A, although the production of MyoD was increased in WT myoblasts treated with rCCN2 in a dose-dependent manner, MyoD production in Ccn2-deficient myoblasts was decreased compared with that in WT myoblasts, and the reduced MyoD production in Ccn2-deficient cells was not increased by rCCN2 treatment (Fig. 5A). To investigate why MyoD production level was not increased by treatment with rCCN2 in Ccn2-deficient cells, we examined the production of integrin α5, which has an important role in myogenesis (30, 31) and interacts with CCN2, in WT and Ccn2-deficient myoblasts. As shown in Fig. 5B, the production level of integrin α5 in Ccn2-deficient cells was lower than that in WT cells. These findings suggest that the impaired response of Ccn2-deficient cells by rCCN2 may be due to decreased integrin α5. Moreover, we investigated the effect of CCN2 on the myotube formation using WT and Ccn2-deficient myoblasts. Immunofluorescence analysis by using anti-desmin antibody was performed to detect the formation of myotubes. As shown in Fig. 5C, immunoreactivity for desmin was detected in both WT and Ccn2-deficient myotubes, but the number of myotube formation derived from Ccn2-deficient cells was decreased compared with that derived from WT cells. These findings suggest that myoblast differentiation is impaired in Ccn2-deficient cells. Next, to clarify the direct effect of CCN2 on the myoblast differentiation, we examined the gene expression levels of myoblast differentiation markers such as MyoD and MHC by determining mRNA expression levels in WT and Ccn2-deficient myoblasts. As shown in Fig. 5D, the expression levels of these genes were decreased in Ccn2-deficient myoblasts compared with those in the WT cells. Interestingly, gene expression of Myostatin in Ccn2-deficient cells was increased at a marginally significant level (P = 0.051). Finally, to confirm the effect of CCN2 functions on the proliferation and myotube formation of myoblasts in vivo, we performed immunofluorescence double staining with anti-PCNA and anti-MyoD antibodies and single staining with an anti-desmin antibody of WT littermates and Ccn2-deficient skeletal muscle tissues at E18.5. In skeletal muscle, myoblasts, which are localized at the periphery of myofibers, proliferate, differentiate and then fuse with each other to form multinucleate myotubes during regeneration. Indeed, PCNA and MyoD double immunoreactivity was found in the nuclei of cells at the periphery of myofibers in both WT and Ccn2-deficient tissues, whereas the number of the immunoreaction was significantly reduced in the Ccn2-deficient muscle (Fig. 6A, graph), suggesting decreased regeneration potential in Ccn2-deficient muscle. Immunoreactivity for desmin, a cytoskeletal intermediate filament protein, was detected in muscle fibers of both WT and Ccn2-deficient skeletal muscle tissues, whereas the transversal area of muscle fibers was decreased in the Ccn2-deficient mice (Fig. 6B, graph). These findings indicate that the skeletal muscle from Ccn2-deficient mice showed reduced PCNA/MyoD staining and impaired muscle integrity, thus suggesting that both proliferation of myoblasts and muscle formation had been impaired in Ccn2-deficient skeletal muscle tissues in vivo.
Fig. 5.

Differentiation of myoblasts from Ccn2-deficient mice is impaired. (A) Primary myoblasts isolated from gastrocnemius muscle of WT and Ccn2-deficient (KO) mice at E18.5 were treated with rCCN2 at the concentration indicated for 7 days. Western blot analysis was performed by using anti-MyoD and anti-β-actin antibodies. (B) WT and KO myoblasts were induced to differentiate for 7 days in differentiation medium. Then, western blot analysis was performed by using anti-integrin α5 and anti-β-actin antibodies. (C) WT and KO myoblasts were induced to differentiate for 7 days. The cells were thereafter fixed, and immunofluorescence analysis by using anti-desmin antibody was performed. Bar represents 100 µm. (D) WT and KO myoblasts were induced to differentiate for 6 days, and real-time RT–PCR analysis was performed. The amounts of MyoD, MHC, and Myostatin mRNAs were normalized to that of Gapdh mRNA and the bars represent means and standard deviations of four sets of independent cultures. Exact P-values are determined by Student’s t test.
Fig. 6.
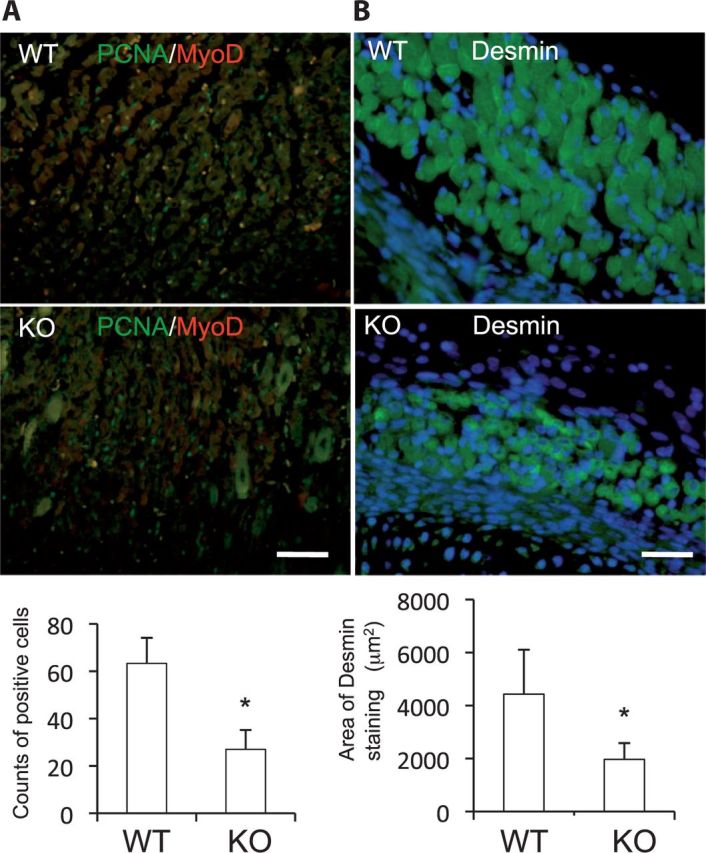
Both proliferation and differentiation of Ccn2-deficient (KO) myoblasts is impaired in vivo. (A) Double immunoreactivity for PCNA and MyoD was detected in the sections of the skeletal muscle from E18.5 WT and KO littermates by immunofluorescence analysis, and the bottom graph shows the means and standard deviations of the number of double positive cells from three different views. (B) Immunoreactivity for desmin was detected in the sections from WT and KO skeletal muscle tissues, and the bottom graph shows the means and standard deviations of areas of muscle fibers stained with anti-desmin antibody. *P < 0.05 indicates significant difference by Student’s t test. Bar represents 50 µm.
Discussion
Because many studies concerning the role of CCN2 in skeletal muscle almost always focused on pathological states such as fibrosis, little is known concerning the physiological significance of CCN2 in skeletal muscle. In this study, we showed that CCN2 promoted the proliferation of C2C12 cells and increased the MyoD production in both C2C12 cells and primary myoblasts at an early-stage of differentiation (Figs 2, 3 and 5A). Furthermore, we clarified that myogenin production and the gene expression levels of Myogenin, Desmin and MHC, which are the markers of terminally differentiated myoblasts, were decreased by the addition of rCCN2 to cultures of C2C12 cells at a later-stage of differentiation (Figs 2–4). These findings indicate that CCN2 promoted cell proliferation and early differentiation of myoblasts, whereas inhibited their terminal differentiation, suggesting that CCN2 plays an important role in early stages of myoblast differentiation. To confirm the physiological role of CCN2 in myoblast differentiation, using WT and Ccn2-deficient skeletal muscle, we performed immunofluorescence analysis with anti-PCNA/MyoD and anti-desmin antibodies. As a result, skeletal muscle fibers of Ccn2-deficient mice showed hypoplasia, and the number of PCNA and MyoD double positive cells was decreased in the skeletal muscle of these mice (Fig. 6). Moreover, the gene expression levels of MRFs, such as MyoD, and those of MRF-regulated MHC, were found to have decreased in the Ccn2-deficient myoblasts (Fig. 5D). Previously, it was reported that inhibition of CCN2 expression by small interfering RNA (siRNA) treatment decreases the myogenic differentiation of human rhabdomyosarcoma cells (32). Our results indicating impaired proliferation and differentiation of Ccn2-deficient myoblasts are consistent with those obtained with human rhabdomyosarcoma cells treated with siRNA against CCN2. Both results may have been due to the inhibition of the early stage of differentiation of myoblasts via decreased MyoD production caused by depletion or knockdown of CCN2. Taken together, these findings suggest that CCN2 plays an important role in both the proliferation and early differentiation of myoblasts in the physiological state.
It was reported that myostatin belonged to the TGF-β superfamily, and was a negative regulator of myoblast proliferation and differentiation (33, 34). However, it has been reported that myostatin stimulates the proliferation of C2C12 cells and promotes the terminal differentiation of embryonic muscle progenitors, thus suggesting that myostatin plays a context-dependent role in skeletal muscle to maintain its homeostasis (28, 29, 33, 34). In this study, we demonstrated that the gene expression level of Myostatin was increased in C2C12 cells by rCCN2 in a dose-dependent manner, whereas it was increased in Ccn2-deficient myoblasts compared with that in WT cells with a marginally significant difference (P = 0.051). Since skeletal muscle mass of Ccn2-deficient mice was decreased compared with that of WT mice, these findings suggest that CCN2 may play an important role in maintaining muscle homeostasis at least in part through regulating myostatin expression.
Unexpectedly, the suppressed MyoD production in Ccn2-deficient myoblasts was not recovered to normal by the addition of exogenous rCCN2 (Fig. 5A). To address this point, we performed western blot analysis by using anti-integrinα5 antibody on WT and Ccn2-deficient myoblasts. As shown in Fig. 5B, the production of integrin α5 was decreased in Ccn2-deficient cells. It was reported that integrin α5 acts to regulate myoblast proliferation and differentiation (30, 31), and we also showed that CCN2 regulated chondrocytes via binding with this molecule (35, 36). These findings indicate that, because the level of integrin α5, which functions as a CCN2 receptor, is decreased, the effect of rCCN2 remains transient and not sufficient to redeem MyoD production in Ccn2-deficient cells.
It was previously reported that over-expression of CCN2 inhibited myoblast differentiation and induced a fibrotic response in skeletal muscle (7–9). Based on these findings, the authors suggest that CCN2 plays major roles in the pathogenesis of Duchenne muscular dystrophy. However, in this study, we showed that Ccn2-deficient skeletal muscle tissues were hypoplastic (Fig. 6). In addition, we revealed that CCN2 promoted the production of MyoD in both C2C12 cells and primary myoblasts, although CCN2 inhibited the late stage of myoblast differentiation (Fig. 2). These results indicate that proper myoblast differentiation under physiological condition requires CCN2. Therefore, we suspect that CCN2 may be cleaved at the late stage of myoblast differentiation to allow the completion of myoblast differentiation. Zimowska et al. (37) reported that matrix metalloproteinase (MMP)-9 activity was present during all stages of myoblast differentiation but MMP-2 activity reached its highest level during myoblast fusion. Furthermore, it was previously reported that MMP-2 cleaved CCN2 into approximately half-sized fragments (38). Taken together, these findings suggest that CCN2 promotes the myoblast proliferation and the production of MyoD at the early stage of myoblast differentiation and that harmonious skeletal muscle regeneration may progress via conditional CCN2 cleavage by MMP-2 at the late differentiation stage.
In summary, this study showed that CCN2 plays an important role in myoblast differentiation in the physiological state. However, further investigation is needed to entirely clarify CCN2 functions during myoblast differentiation, particularly at late stages.
Acknowledgements
This work was supported in part by grants from the programs Grants-in-Aid for Scientific Research (C) to T.N. (Nos. 23592732, 26462810) and to S.K. (No. 25462886), and (B) to M.T. (No. 24390415) from Japan Society for the Promotion of Sciences, Japan. D.J. is a recipient of a Postdoctoral Fellowship from Japan Society for the Promotion of Science. They thank Drs Takako Hattori and Mitsuhiro Hoshijima for helpful suggestions and Miss Yoshiko Miyake for secretarial assistance.
Glossary
Abbreviations
- BrdU
5-bromo-2′-deoxyuridine
- BSA
bovine serum albumin
- DMEM
Dulbecco’s modified Eagle’s medium
- FBS
fetal bovine serum
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- HA
hemagglutinin
- ITS
insulin–transferrin–selenite
- KO
Ccn2-deficience
- MHC
myosin heavy chain
- PCNA
proliferating cell nuclear antigen
- rCCN2
recombinant CCN2 protein
- TNFα
tumor necrosis factor-α
- WT
wild-type
Conflict of Interest
None declared.
References
- 1.Karalaki M, Fili S, Philippou A, Koutsilieris M. Muscle regeneration: cellular and molecular events. In vivo. 2009;23:779–796. [PubMed] [Google Scholar]
- 2.Francetic T, Li Q. Skeletal myogenesis and Myf5 activation. Transcription. 2011;2:109–114. doi: 10.4161/trns.2.3.15829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takigawa M, Nakanishi T, Kubota S, Nishida T. Role of CTGF/HCS24/ecogenin in skeletal growth control. J. Cell. Physiol. 2003;194:256–266. doi: 10.1002/jcp.10206. [DOI] [PubMed] [Google Scholar]
- 4.Perbal B, Takigawa M. CCN proteins: A new family of cell growth and differentiation regulators. London: Imperial College Press; 2005. pp. 1–311. [Google Scholar]
- 5.Kubota S, Takigawa M. Role of CCN2/CTGF/Hcs24 in bone growth. Int. Rev. Cytol. 2007;257:1–41. doi: 10.1016/S0074-7696(07)57001-4. [DOI] [PubMed] [Google Scholar]
- 6.Takigawa M. CCN2: a master regulator of the genesis of bone and cartilage. J. Cell Commun. Signal. 2013;7:191–201. doi: 10.1007/s12079-013-0204-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vial C, Zúñiga LM, Cabello-Verrugio C, Cañón P, Fadic R, Brandan E. Skeletal muscle cells express the profibrotic cytokine Connective Tissue growth factor (CTGF/CCN2), which induces their dedifferentiation. J. Cell. Physiol. 2008;215:410–421. doi: 10.1002/jcp.21324. [DOI] [PubMed] [Google Scholar]
- 8.Cabello-Verrugio C, Morales MG, Cabrera D, Vio CP, Brandan E. Angiotensin II receptor type 1 blockade decreases CTGF/CCN2-mediated damage and fibrosis in normal and dystrophic skeletal muscles. J. Cell Mol. Med. 2012;16:752–764. doi: 10.1111/j.1582-4934.2011.01354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morales MG, Cabello-Verrugio C, Santander C, Cabrera D, Goldschmeding R, Brandan E. CTGF/CCN-2 over-expression can directly induce features of skeletal muscle dystrophy. J. Pathol. 2011;225:490–501. doi: 10.1002/path.2952. [DOI] [PubMed] [Google Scholar]
- 10.Du J, Klein JD, Hassounah F, Zhang J, Zhang C, Wang XH. Aging increases CCN1 expression leading to muscle senescence. Am. J. Physiol. Cell. Physiol. 2014;306:C28–C36. doi: 10.1152/ajpcell.00066.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calhabeu F, Lafont J, Le Dreau G, Laurent M, Kazazian C, Schaeffer L, Martinerie C, Dubois C. Nov/CCN3 impairs muscle cell commitment and differentiation. Exp. Cell. Res. 2006;312:1876–1889. doi: 10.1016/j.yexcr.2006.02.027. [DOI] [PubMed] [Google Scholar]
- 12.Rachfal AW, Brigstock DR. Connective tissue growth factor (CTGF/CCN2) in hepatic fibrosis. Hepatol. Res. 2003;26:1–9. doi: 10.1016/s1386-6346(03)00115-3. [DOI] [PubMed] [Google Scholar]
- 13.Huang G, Brigstock DR. Regulation of hepatic stellate cells by connective tissue growth factor. Front. Biosci. 2012;17:2495–2507. doi: 10.2741/4067. [DOI] [PubMed] [Google Scholar]
- 14.Shi-Wen X, Leask A, Abraham D. Regulation and function of connective tissue growth factor/CCN2 in tissue repair, scarring and fibrosis. Cytokine Growth Factor Rev. 2008;19:133–144. doi: 10.1016/j.cytogfr.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 15.Nishida T, Nakanishi T, Shimo T, Asano M, Hattori T, Tamatani T, Tezuka K, Takigawa M. Demonstration of receptors specific for connective tissue growth factor on a human chondrocytic cell line (HCS-2/8) Biochem. Biophys. Res. Commun. 1998;247:905–909. doi: 10.1006/bbrc.1998.8895. [DOI] [PubMed] [Google Scholar]
- 16.Ivkovic S, Yoon BS, Popoff SN, Safadi FF, Libuda DE, Stephenson RC, Daluiski A, Lyons KM. Connective tissue growth factor coordinates chondrogenesis and angiogenesis during skeletal development. Development. 2003;130:2779–2791. doi: 10.1242/dev.00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Uezumi A, Fukada S, Yamamoto N, Takeda S, Tsuchida K. Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat. Cell Biol. 2010;12:143–152. doi: 10.1038/ncb2014. [DOI] [PubMed] [Google Scholar]
- 18.Leblanc E, Trensz F, Haroun S, Drouin G, Bergeron E, Penton CM, Montanaro F, Roux S, Faucheux N, Grenier G. BMP-9-induced muscle heterotopic ossification requires changes to the skeletal muscle microenvironment. J. Bone Miner. Res. 2011;26:1166–1177. doi: 10.1002/jbmr.311. [DOI] [PubMed] [Google Scholar]
- 19.Yoshida S, Fujisawa-Sehara A, Taki T, Arai K, Nabeshima Y. Lysophosphatidic acid and bFGF control different modes in proliferating myoblasts. J. Cell Biol. 1996;132:181–193. doi: 10.1083/jcb.132.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nishida T, Emura K, Kubota S, Lyons KM, Takigawa M. CCN family 2/connective tissue growth factor (CCN2/CTGF) promotes osteoclastogenesis via induction of and interaction with dendritic cell-specific transmembrane protein (DC-STAMP) J. Bone Miner. Res. 2011;26:351–363. doi: 10.1002/jbmr.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nishida T, Kubota S, Aoyama E, Takigawa M. Impaired glycolytic metabolism causes chondrocyte hypertrophy-like changes via promotion of phospho-Smad1/5/8 translocation into nucleus. Osteoarthritis Cartilage. 2013;21:700–709. doi: 10.1016/j.joca.2013.01.013. [DOI] [PubMed] [Google Scholar]
- 22.Maeda A, Nishida T, Aoyama E, Kubota S, Lyons KM, Kuboki T, Takigawa M. CCN family 2/connective tissue growth factor modulates BMP signalling as a signal conductor, which action regulates the proliferation and differentiation of chondrocytes. J. Biochem. 2009;145:207–216. doi: 10.1093/jb/mvn159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nishida T, Kubota S, Fukunaga T, Kondo S, Yosimichi G, Nakanishi T, Takano-Yamamoto T, Takigawa M. CTGF/Hcs24, hypertrophic chondrocyte-specific gene product, interacts with perlecan in regulating the proliferation and differentiation of chondrocytes. J. Cell. Physiol. 2003;196:265–275. doi: 10.1002/jcp.10277. [DOI] [PubMed] [Google Scholar]
- 24.Nishida T, Kubota S, Aoyama E, Janune D, Maeda A, Takigawa M. Effect of CCN2 on FGF2-induced proliferation and MMP9 and MMP13 productions by chondrocytes. Endocrinology. 2011;152:4232–4241. doi: 10.1210/en.2011-0234. [DOI] [PubMed] [Google Scholar]
- 25.Nishida T, Kondo S, Maeda A, Kubota S, Lyons KM, Takigawa M. CCN family 2/connective tissue growth factor (CCN2/CTGF) regulates the expression of Vegf through Hif-1α expression in a chondrocytic cell line, HCS-2/8, under hypoxic condition. Bone. 2009;44:24–31. doi: 10.1016/j.bone.2008.08.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kubota S, Hattori T, Shimo T, Nakanishi T, Takigawa M. Novel intracellular effects of human connective tissue growth factor expressed in Cos-7 cells. FEBS Lett. 2000;474:58–62. doi: 10.1016/s0014-5793(00)01573-8. [DOI] [PubMed] [Google Scholar]
- 27.Kitzmann M, Carnac G, Vandromme M, Primig M, Lamb NJC, Fernandez A. The muscle regulatory factors MyoD and Myf-5 undergo distinct cell cycle-specific expression in muscle cells. J. Cell Biol. 1998;142:1447–1459. doi: 10.1083/jcb.142.6.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Williams NG, Interlichia JP, Jackson MF, Hwang D, Cohen P, Rodgers BD. Endocrine actions of myostatin: systemic regulation of the IGF and IGF binding protein axis. Endocrinology. 2011;152:172–180. doi: 10.1210/en.2010-0488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodgers BD, Wiedeback BD, Hoversten KE, Jackson MF, Walker RG, Thompson TB. Myostatin stimulates, not inhibits, C2C12 myoblast proliferation. Endocrinology. 2014;155:670–675. doi: 10.1210/en.2013-2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Enomoto M, Boettiger D, Menko AS. Alpha 5 integrin is a critical component of adhesion plaques in myogenesis. Dev. Biol. 1993;155:180–197. doi: 10.1006/dbio.1993.1017. [DOI] [PubMed] [Google Scholar]
- 31.Sastry SK, Lakonishok M, Thomas DA, Muschler J, Horwitz AF. Integrin α subunit ratios, cytoplasmic domains, and growth factor synergy regulate muscle proliferation and differentiation. J. Cell Biol. 1996;133:169–184. doi: 10.1083/jcb.133.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Croci S, Landuzzi L, Astolfi A, Nicoletti G, Rosolen A, Sartori F, Follo MY, Oliver N, De Giovanni C, Nanni P, Lollini P-L. Inhibition of connective tissue growth factor (CTGF/CCN2) expression decreases the survival and myogenic differentiation of human rhabdomyosarcoma cells. Cancer Res. 2004;64:1730–1736. doi: 10.1158/0008-5472.can-3502-02. [DOI] [PubMed] [Google Scholar]
- 33.Manceau M, Gros J, Savage K, Thomé V, McPherron A, Paterson B, Marcelle C. Myostatin promotes the terminal differentiation of embryonic muscle progenitors. Genes Dev. 2008;22:668–681. doi: 10.1101/gad.454408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Elliott B, Renshaw D, Getting S, Mackenzie R. The central role of myostain in skeletal muscleand whole body homeostasis. Acta Physiol. 2012;205:324–340. doi: 10.1111/j.1748-1716.2012.02423.x. [DOI] [PubMed] [Google Scholar]
- 35.Nishida T, Kawaki H, Baxter RM, Deyoung RA, Takigawa M, Lyons KM. CCN2 (connective tissue growth factor) is essential for extracellular matrix production and integrin signaling in chondrocytes. J. Cell Commun. Signal. 2007;1:45–58. doi: 10.1007/s12079-007-0005-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoshijima M, Hattori T, Inoue M, Araki D, Hanagata H, Miyauchi A, Takigawa M. CT domain of CCN2/CTGF directly interacts with fibronectin and enhances cell adhesion of chondrocytes through integrin α5β1. FEBS Lett. 2006;580:1376–1382. doi: 10.1016/j.febslet.2006.01.061. [DOI] [PubMed] [Google Scholar]
- 37.Zimowska M, Brzoska E, Swierczynska M, Streminska W, Moraczewski J. Distinct patterns of MMP-9 and MMP-2 activity in slow and fast twitch skeletal muscle regeneration in vivo. Int. J. Dev. Biol. 2008;52:307–314. doi: 10.1387/ijdb.072331mz. [DOI] [PubMed] [Google Scholar]
- 38.Dean RA, Butler GS, Hamma-Kourbali Y, Delbé J, Brigstock DR, Courty J, Overall CM. Identification of candidate angiogenic inhibitors processed by matrix metalloproteinase 2 (MMP-2) in cell-based proteomic screens: disruption of vascular endothelial growth factor (VEGF)/heparin affin regulatory peptide (pleiotrophin) and VEGF/connective tissue growth factor angiogenic inhibitory complexes by MMP-2 proteolysis. Mol. Cell Biol. 2007;27:8454–8465. doi: 10.1128/MCB.00821-07. [DOI] [PMC free article] [PubMed] [Google Scholar]