

Abstract
Multidrug resistance (MDR) is a major cause of failure in cancer chemotherapy. P-glycoprotein (P-gp), a promiscuous drug efflux pump, has been extensively studied for its association with MDR due to overexpression in cancer cells. Several P-gp inhibitors or modulators have been investigated in clinical trials in hope of circumventing MDR, with only limited success. Alternative strategies are actively pursued, such as the modification of existing drugs, development of new drugs, or combining novel drug delivery agents to evade P-gp-dependent efflux. Despite the importance and numerous studies, these efforts have mostly been undertaken without a priori knowledge of how drugs interact with P-gp at the molecular level. This perspective highlights and discusses progress toward and challenges impeding drug development for inhibiting or evading P-gp in the context of our improved understanding of the structural basis and mechanism of P-gp-mediated MDR.
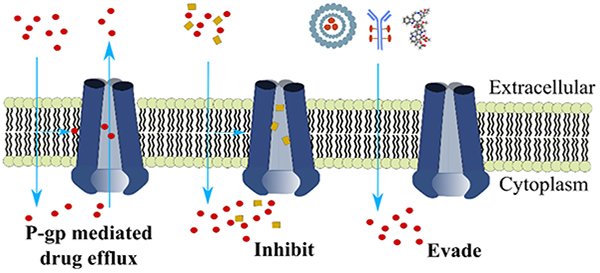
Graphical Abstract
Introduction
Many cancer cells acquire resistance to a broad spectrum of structurally and mechanistically distinct anticancer drugs by a phenomenon called multidrug resistance (MDR).1, 2 The development of MDR presents major challenges to cancer chemotherapy, especially in managing patients with metastatic cancers that are resistant to traditional therapies.3, 4 MDR occurs intrinsically in some cancers due to genetic and epigenetic alterations that affect drug sensitivity without previous exposure to chemotherapy agents. MDR can also be acquired during the course of chemotherapeutic treatment of cancers that were initially drug-sensitive but later recur in drug-resistant form. It is also recognized that cancers usually consist of a heterogeneous population of drug-sensitive and drug-resistant cells.5, 6 During the course of treatment, drug-sensitive cells are selectively removed, and resistant cells come to dominate the cancer cell population. The huge impact of chemotherapeutic drug resistance has led to extensive studies of the mechanistic aspects and strategies to understand, modulate or evade MDR.
A prevalent mechanism of cancer MDR is the expression of a class of energy-dependent efflux pumps called adenosine triphosphate (ATP)-binding cassette (ABC) transporters.7, 8 In humans, there are 48 members in this protein family, which are involved in diverse physiological functions such as transporting lipids, sterols, peptides, toxins, and ions.9 Among these members, at least three transporters, including P-glycoprotein (P-gp in short, also called multidrug resistance protein 1, MDR1, or ABCB1), MDR-associated protein 1 (MRP1 or ABCC1) and breast cancer resistance protein (BCRP or ABCG2), have been characterized in relation to multidrug resistance.10–12 These three transporters have broad drug specificity and transport an array of structurally diverse compounds, causing lowered drug accumulation inside cells and consequently diminished drug efficacy.
Discovered over 40 years ago, P-gp is the first identified and most characterized MDR transporter and has long been recognized as a viable target to overcome MDR in cancer.10 Expression of P-gp was detected in more than 50% of the NCI-60 tumor cell lines including all melanomas and central nervous system tumors and with high levels in renal and colon carcinomas.13, 14 The elevated P-gp expression in cancer cells has been linked to reduced chemotherapeutic responses and poor clinical outcome in various cancer types including both blood cancers and solid tumors. Some tumors with low levels of P-gp expression at baseline, such as leukemia and breast cancer, have shown upregulation of P-gp after disease progression following chemotherapy.15 A plethora of anticancer drugs that are central to many chemotherapeutic regimes are susceptible to P-gp-mediated efflux (Figure 1), such as the microtubule-targeting vinca alkaloids (e.g. vinblastine and vincristine) and taxanes (paclitaxel and docetaxel), the DNA-chelating anthracyclines (doxorubicin and daunorubicin), the topoisomerase inhibitors (topotecan and etoposide), and the tyrosine kinase inhibitors (dasatinib and gefitinib), among many others.2, 16–18 To overcome P-gp-mediated MDR, many small molecule drugs have been tested to modulate or inhibit the activity of P-gp.19, 20 However, these programs have been halted in recent years due mostly to failures in clinical trials. A major challenge with this strategy is the lack of potent and non-toxic inhibitors among other limiting factors, on which we will make further comments in the next sections of this perspective. Alternatively, many drug development programs now place an emphasis on the discovery of new compounds or strategies to bypass the activity of P-gp.
Figure 1.

Examples of frontline anticancer drugs that are susceptible to P-gp-mediated efflux.
In addition to the role of P-gp in cancer which is a focus of this article, P-gp plays a pivotal role in normal physiological detoxification and host protection processes by transporting numerous exogenous and endogenous substrates.2 P-gp is distributed in the epithelial cell surfaces where it is expressed at high levels, such as in the mucosa of the gastrointestinal tract, in biliary epithelium of the liver, in proximal tubules of the kidney, in the adrenal cortex and in blood-tissue barriers.21–23 The latter include the placenta, endometrium, testicular tissue and the endothelial component of the blood-brain barrier. Subject to imperfections in P-gp detection methods and quantification errors, the amount of P-gp expression in adrenal and kidney has been shown as high as or higher than levels detected in some MDR tumor cell lines.22, 24 The presence of P-gp protects hematopoietic progenitor cells of bone marrow against the cytotoxic effects of chemotherapeutic agents.25 Additionally, P-gp expressed in the intestinal epithelia has a major impact on the bioavailability of orally administered drugs due to its capacity to alter tissue absorption and elimination of the drugs into bile and urine.26 As such, the evaluation of drug candidates for their P-gp susceptibility has become an important step in the development of novel therapeutics in the pharmaceutical industry.2, 27 The US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) now mandate documentation of drug interactions with P-gp and several other transporters for approval of any new drug.28 Thus, studies of P-gp and drug interactions are exceedingly important not only for cancer treatment but also in a broad range of drug discovery programs.
Conceivably, these medicinal chemistry efforts involving P-gp can greatly benefit from a detailed understanding of the structure and mechanism of the underlying transporter. Significant progress has been made in recent years in characterizing the structure of P-gp at near-atomic resolution, and these structures have provided a first glimpse of the polyspecific drug binding sites of P-gp. In this perspective, we will present the state of knowledge of P-gp structures, and in that context we will review and discuss the progress and impeding challenges in the drug development to inhibit or evade P-gp in cancer. As understanding of P-gp and its drug interactions continues to improve, we can anticipate that a rational approach may be undertaken in future drug designs.
Structural insights into P-gp-mediated MDR
Overall architecture and conformations of P-gp
P-gp has been a highly sought-after target for structural determinations, yet with formidable challenges due to its conformational flexibility and inherent water insolubility as an integral membrane protein. The first high-resolution structure of mouse P-gp was reported in 2009, determined by X-ray crystallography at a resolution of 3.8 Å.29 This success was achieved after decades of community efforts for biochemical characterizations and for establishment of large-scale expression and purification of quality protein materials. Mouse P-gp shares 87% amino acid identity with human P-gp, yet is more stable and can be expressed in yeast with good yield.30 Several X-ray structures of mouse P-gp have been determined in recent years at marginally improved resolutions (up to 3.3 Å) in addition to a P-gp structure from Caenorhabditis elegans, showing overall similar structural features (Figure 2).31–34 These recent structures resolved the issue of transmembrane (TM) registry in previous lower resolution structures.35
Figure 2.

X-ray structures of mouse P-gp solved in various inward-facing conformations. Shown below the structure cartoons are the PDB codes together with the distances measured between the N607 and T1262 Cα positions (marked by red dots) in the separated NBDs. The N- and C-terminal TMD-NBD halves are depicted in blue and green, respectively. A short flexible linker, unresolved in these X-ray structures, is depicted by black dashes in the first structure that connect the end of NBD1 and the start of TMD2. Shown on far right is a structure (5KOY) with ATP (rendered in spheres) bound in NBD1. Estimated lipid bilayer positions are indicated in grey lines.
The structure of P-gp displays the canonical ABC transporter fold consisting of two pseudo-symmetric halves, each containing a long transmembrane domain (TMD) and a cytosolic ABC or nucleotide binding domain (NBD). The two NBD domains, which are largely conserved in ABC proteins, dimerize to bind and hydrolyze ATP at the interface. To date all X-ray structures of P-gp have been solved in inward-facing conformations with inverted V-shaped architecture and the NBDs separated to varying degrees (Figure 2).29–35 Two bundles of TM helices, formed with a characteristic domain swapping (TM4/TM5 in domain I and TM10/TM11 in domain II cross over and associate with the opposing monomer, respectively) within the B-subfamily of ABC transporters, enclose a large cavity of > 6000 Å3 in the inward-facing structures.29 Drugs are thought to enter this cavity for binding through portals open to the cytoplasm and the inner leaflet of the membrane. Not represented in the crystal structures is a flexible linker of ~60–70 amino acids which connects the two halves of P-gp in a single polypeptide chain. This linker region contains several sites for phosphorylation but with no defined role in the expression and drug transport of P-gp.36
P-gp undergoes dynamic conformational changes through which the drug binding sites alternately access the two sides of the membrane, binding the cargo on one side and releasing it on the other. Distinct outward facing conformations, with the TM-enclosed drug-binding cavity open to the extracellular side, have been determined for several homologous bacterial ABC transporters in the presence of bound nucleotide.37, 38 These structures are consistent with an alternating-access model in which ATP binding and hydrolysis drive the conformational change via NBD dimerization. In keeping with biochemistry, substrate transport by P-gp is often accompanied with stimulated ATP hydrolysis while the basal rate of ATPase hydrolysis is low in the absence of substrate. For P-gp, however, no outward-facing structures have been determined by X-ray crystallography to date. An ATPase-catalytically deficient mutant of mouse P-gp has been co-crystallized with ATP, but showed an inward facing conformation similar to other apo structures (Figure 2).33 Divergent from consensus views of general ABC transporter mechanisms, a recent study using double electron-electron resonance spectroscopy suggests that P-gp only attains the outward-facing state upon ATP hydrolysis.39 The difficulty of capturing this outward facing state under steady-state ATP hydrolysis conditions was indicated in previous single particle analyses of P-gp conformations by electron microscopy (EM) imaging.40 Thus the outward-facing P-gp may exist as a transient high-energy state during the transport cycle. Of note, the outward-facing P-gp were indeed enriched in the presence of substrates that stimulate the basal rate of ATP hydrolysis, as well as by the combination of ADP and vanadate which represents an artificially stabilized nucleotide-bound (post-ATP hydrolysis) condition.39, 40 However, in another interesting EM study of human P-gp with bound antibody fragment (UIC2, which binds to the extracellular region of human P-gp), the transporter displayed a significant proportion (> 50% of particles analyzed) of NBD-closed conformations even in the absence of bound nucleotide.41 Of note, there has been some controversy in the field as to whether the NBD-open, inward-facing P-gp crystal structures are physiologically relevant, as this conformation could arise from crystallographic constraints, the use of detergents, or the absence of nucleotides and transport substrates.42, 43 It can be argued that the two NBDs in P-gp, or likely in any ABC transporter, should always have nucleotide bound since the cellular concentration of ATP (~3–5 mM) far exceeds the binding constant. Despite remaining controversies and lack of a clear picture of the entire conformational pathway of P-gp, the majority of structural evidence presently suggests that the inward facing state likely represents a high affinity drug-binding conformation, which is highly relevant for the modeling of inhibitor and drug binding.
Polyspecific drug binding sites of P-gp
A significant aspect of P-gp structural studies is to disclose how this single transporter is able to recognize a wide variety of structurally and chemically diverse substrates. In early work using photoaffinity labeling, cysteine-scanning mutagenesis, and derivatization with thiol-reactive reagents, the drug binding sites of P-gp appeared to reside within the membrane-embedded region at the interface between the two TM halves.44, 45 Multiple substrate binding sites in P-gp have been documented based on drug binding and competition studies, for example the H site for binding to Hoechst-33342 and colchicine, the R site for rhodamine-123 and anthracyclines, and the P site for prazosin and progesterone.46–48 These functional characterizations establish neither exact nor relative locations of binding for these substrates. In this regard, the successful co-crystallization of P-gp with several cyclic peptide ligands revealed unprecedented insights into the polyspecific drug-binding sites.29, 34
The first compounds to be precisely localized in the drug-binding region surrounded by the TM helix bundles of inward-facing P-gp were a pair of cyclic peptide enantiomers (tris-(R)-valineselenazole: QZ59-RRR; tris-(S)-valineselenazole: QZ59-SSS), which by definition possess the same chemical composition but opposite amino acid chirality (Figure 3).29 These cyclic peptides were derived from marine natural products49–52 displaying MDR reversal activities, with several features implemented to facilitate their co-crystallization within the flexible drug binding region of P-gp, including extreme structural rigidity, and a homotrimeric structural symmetry.53 Also, labeling of the peptides with three symmetrically distributed selenium atoms as X-ray anomalous scattering markers was particularly helpful to the positioning and modeling of these molecules in the P-gp structures solved at moderate resolutions. The co-crystal structures revealed one molecule of QZ59-RRR roughly at the center of an internal cavity of the drug-binding region. Interestingly, the enantiomer QZ59-SSS bound two sites per P-gp molecule, which are distinct from but overlapped with the binding site of QZ59-RRR. These structures essentially established that P-gp can distinguish between the ligand stereoisomers with distinct binding sites, orientations, and stoichiometry.
Figure 3.

Cyclic peptide mimetics co-crystallized with P-gp. Homotrimeric cyclic peptides were designed based on natural products (e.g. Dendroamide A) and labeled with selenium atoms to facilitate co-crystallization and subsequent structural determinations. Shown on right are the two sets of co-crystal structures, resolved in different inward-facing conformations, including with the enantiomeric QZ59-RRR and QZ59-SSS (top), and with four cyclic peptides integrating systematic side chain variations (bottom). Distinct binding sites were revealed in P-gp for each ligand (rendered in sticks with the same color schemes as for the respective 2D structures) and are further described in Figure 4.
Based on the rationale from the first structural view of P-gp ligand binding, a detailed structure activity relationship (SAR) for cyclic peptide QZ59-SSS was undertaken to determine the impact of ligand side chain variation on P-gp interactions.34 Homotrimers were designed with identical cyclic peptide backbone and systematic increases in size and hydrophobicity of the side chain groups to generate the four compounds QZ-Ala, QZ-Val (QZ59-SSS), QZ-Leu and QZ-Phe (Figure 3). All these cyclic peptides inhibit substrate transport and sensitize cancer cells,34 likely through competition for P-gp binding by substrates or anticancer drugs. Measurements of P-gp’s ATPase activity revealed that the smaller and less hydrophobic QZ-Ala conferred the highest degree of stimulation followed by QZ-Val, while QZ-Leu and QZ-Phe had less effect. Consistent with these substrate-like interactions, P-gp mediated mild resistance to QZ-Ala in cells.34
Additional novel structural insights into P-gp’s ligand binding sites were gained by the newer set of P-gp co-crystal structures solved with all four cyclic peptides (Figure 4). These structures were determined at improved resolutions (3.6–3.8 Å vs 4.5 Å) and in a more open conformation than the first set of co-crystal structures with QZ59-RRR and QZ59-SSS.29, 34 Within the TMD-enclosed binding region, the smaller ligands QZ-Ala and QZ-Val shared an upper and a lower binding site, whereas the larger and more hydrophobic ligands QZ-Leu and QZ-Phe shared a different upper binding site, with QZ-Phe also binding to a second, unique lower site (Figure 4B and 4C). A remarkable finding was that P-gp binding of the substrate-like ATPase stimulators QZ-Ala and QZ-Val induces a large conformational change, a kink in helix TM4 (Figure 4B). This conformational change in TM4 is structurally linked to a short intracellular helix, which couples the movement between NBD2 and the associated TM helices. This structural insight provides a clue to the mechanism of how substrate binding in the TMD region in P-gp can stimulate ATP hydrolysis at the ATP binding sites ~40 Å away. Of note, a recent cryo-EM structure of MRP1 shows that substrate binding induces a narrower separation distance between the NBDs compared to the substrate-free structure.54 It is possible that the unchanged spacing of the two NBDs in QZ-Ala or QZ-Val-bound P-gp co-crystal structures relative to the apo structure is restricted by crystallographic constraints.
Figure 4.

Polyspecific drug binding sites of P-gp. (A) Drug binding residues identified near the binding sites of QZ-Ala, QZ-Val, QZ-Leu and QZ-Phe. Aromatic residues are shown in red sticks and non-aromatics in cyan. (B) QZ-Ala (green) and QZ-Val (blue) share similar binding sites and their binding induces a conformational change in the TM4 region compared to the apo structure (grey). (C) Location of the binding sites for QZ-Leu (wheat) and QZ-Phe (red). For clarity, only the N-terminal half of P-gp (pale green) cocrystal structures was superimposed with the apo structure in (B) and (C).
Molecular basis of P-gp-mediated MDR
It is evident that P-gp possesses a large and versatile drug-binding region encompassing multiple and overlapping binding sites, which can accommodate smaller or larger molecules or several molecules simultaneously. Recently described mouse P-gp structures also correlate the opening and closing of the two halves of P-gp with rotation and translation of individual helices in the TM domains, thereby causing continuous change in surface topology.33 P-gp substrates or inhibitors are typically lipid-soluble and amphipathic molecules.55 Consistent with this observation, the drug-binding region of Pgp is made up mostly of hydrophobic and aromatic residues that bind substrates via hydrophobic and van der Waals interactions (Figure 4A). We note that there are a few polar side chains (e.g. Gln343, Gln721, Gln942, Gln986, and Ser 975; residue numbers referring to mouse P-gp) in addition to the aromatic tyrosine and tryptophan residues also facing the drug binding pocket in the inward-facing P-gp structures, and these polar residues may mediate additional H-bond interactions with certain ligands. Additionally, the drug binding residues are mostly conserved between mouse and human P-gp, so that the conclusions drawn from the current mouse model can for the most part be extended to human P-gp, though with some caution in light of a few minor differences in drug binding between human and mouse P-gp.56, 57
P-gp has been proposed to function as a “hydrophobic vacuum cleaner” by extruding drugs from the inner leaflet of plasma membranes.58 An additional binding site for QZ-Val was also observed in the co-crystal structure on the surface of P-gp facing the inner leaflet of the membrane bilayer and just above the elbow helix EH2 (Figure 3).34 This site lies close to the predicted membrane-water interface and was proposed to represent a substrate entry point. The structural data lends strong credence to the proposal of an initial lower-affinity “ON-site” for ligands near the inner leaflet of the membrane preceding the higher-affinity “ON-site(s)’ within the central binding cavity.59, 60
Overall, a molecular understanding of how P-gp mediates MDR has started to emerge from the recent structural determinations, although P-gp structures in complex with actual anticancer drugs have not yet been determined, and a detailed drug transport pathway has yet to be fully elucidated in the context of a dynamic conformational landscape for P-gp. The observed binding positions of QZ cyclic peptides in P-gp appear to correlate with the structure and activity of ligands, and this structure-activity correlation also needs to be scrutinized with a greater number of drugs and drug-bound structures.
Development of P-gp inhibitors and impeding challenges
There has been significant interest in the past in the development and discovery of small molecule P-gp inhibitors or modulators, with a hope to overcome P-gp-mediated MDR in cancer treatment. Figure 5 shows a number of such compounds that have been tested in clinical trials in conjunction with prescribed anticancer drugs. However, excitement has largely dissipated due to limited success with these compounds. The reasons for the failures of these clinical trials are complex and not completely clear, despite quite extensive discussion and analyses of these results in the literature.61, 62 Here we comment on some limiting factors related to available inhibitors. One obvious challenge arises from their toxicity to normal tissues, owing to the wide distribution of P-gp and its role in the protection of normal cells and tissues. To minimize the undesired toxicity of P-gp modulators against normal tissues, targeted delivery of these drugs to cancer cells may be developed. A second major concern with existing P-gp inhibitors is their generally low potency and specificity. Early generation P-gp inhibitors were not specifically developed to inhibit MDR and they exert other pharmacological activities. For example, verapamil, one of the first P-gp inhibitors tested in clinical trials, is a calcium channel blocker and its P-gp binding affinity is only about 10 μM.63 Cyclosporine A, another P-gp inhibitor with low μM affinity that was used in early clinical trials, is also prescribed as an immunosuppressant.64 The low affinity of these inhibitors necessitated the use of high doses, resulting in serious side effects and toxicity. In addition, co-administration of these MDR inhibitors elevated plasma concentrations of anticancer drugs by interfering with their metabolism and excretion. Relatively newer MDR drugs such as tariquidar and zosuquidar, were optimized for increased potency (EC50 < 100 nM) and higher specificities toward P-gp.65, 66 Some of these new compounds show little to no pharmacokinetic interactions with current anticancer drugs. However, contradicting data exist on whether tariquidar is an ATPase stimulator or inhibitor and whether it also acts as a substrate of P-gp.67, 68 Additionally, tariquidar is reported as a substrate and inhibitor of BCRP.69 Overall, the potency and specificity of known P-gp inhibitors are low compared to most drugs in clinical use.
Figure 5.

Examples of three generations of P-gp inhibitors.
From a structural perspective (Figures 2 and 4), P-gp does not have a well-defined drug-binding pocket as compared to many receptors or enzymes, making it intrinsically challenging to design highly potent P-gp-specific inhibitors. That identical or different ligands occupy proximal binding sites within P-gp may be utilized as a basis for fragment-based drug design to create potent compounds. Some bivalent inhibitors have been designed in literature to exploit the multiple binding sites of P-gp, showing greatly increased potency compared to monomers.70, 71 For P-gp inhibition, another related question is whether particular drug binding conformations of P-gp can be targeted by high-affinity inhibitors, an area which remains to be explored and requires additional Pgp conformations and drug binding modes/sites to be revealed. Most inhibitors block Pgp efflux likely by high-affinity binding to the open inward-facing conformations. However, a recent cross-linking study has shown that tariquidar inhibits P-gp transition to the open state during the catalytic cycle.72 As MDR remains a major obstacle in cancer treatment even with the advent of new therapies, the discovery of new and effective P-gp modulators that will overcome the drawbacks of current compounds may still be of great value despite these significant challenges.
Overcoming P-gp-mediated MDR in cancer chemotherapy - alternative strategies
As no drugs directly targeting or inhibiting P-gp have been brought to clinical use, alternative approaches have been extensively explored to overcome MDR in cancer treatment. In this section we will review two strategies that remain the major focus in today’s drug discovery: one is the development of new or modified anticancer drugs, and the other the use of delivery systems to evade transporter-mediated efflux. Other approaches, including antisense strategies (e.g. antisense oligonucleotides, ribozymes, and small interfering RNAs) that down regulate ABCB1 gene expression at the mRNA level, or transcriptional regulators that inhibit the transcriptional activation of the ABCB1 gene or by exploitation of the collateral sensitivity of MDR cells to certain drugs,14, 73 are relatively less explored, and a discussion of these strategies can be found in the provided references.
Designing new drugs or optimizing existing drugs to evade P-gp-mediated efflux
Development of anticancer drugs that are not or poor substrates of P-gp and therefore that are less susceptible to efflux by P-gp overexpressing tumor cells is an important strategy to improve the drug efficacy. This can be achieved either by identification of new lead compounds or the chemical modification of existing anticancer drugs. Notable successes have been reported where strategic modifications of known anticancer drugs or natural products significantly reduced P-gp-mediated efflux (Figure 6).74 A class of naturally occurring microtubule-targeting compounds, the epothilones, with structure and mechanism of action similar to taxanes, are shown to be poor substrates for P-gp.75
Figure 6.

Chemical modifications of several microtubule-targeting anticancer drugs to reduce P-gp-medicated efflux. The positions of chemical modifications on Epothilone B (A), paclitaxel (B), and vinblastine (C) are indicated in small shadowed boxes. Decreased susceptibility to P-gp efflux after modifications in (B) and (C) is indicated by the lower IC50 values against various drug-resistant, P-gp overexpressing cell lines.
Interestingly, structural modification at specific sites lead to altered substrate affinity that allows them to bypass resistance associated with P-gp overexpression.75–77 The use of Ixabepilone (also known as azaepothilone B and BMS-247550) (Figure 6A), a semisynthetic analogue of epothilone B, in combination with capecitabine, showed effectiveness in the treatment of metastatic breast cancer, where known anthracycline and taxane chemotherapeutics failed.78 Ixabepilone showed high cytotoxicity, comparable to epothilone B, with IC50 of 2.60 and 0.41 nM, respectively, against the drug-sensitive cell line HCT116 (Figure 6A).79 Furthermore, the IC50 values for Ixabepilone were shown in a relatively narrow range of 1.4–24.5 nM against a panel of over 20 tumor cell lines including the sensitive and P-gp overexpressing resistant cells.80
Semisynthetic chemical modifications of the framework of taxanes have given rise to several compounds with significantly improved activity against paclitaxel and docetaxel-resistant tumor cells. Promising examples include cabazitaxel and ortataxel (IDN5109) (Figure 6B), with the former received FDA approval for patients with hormone-refractory metastatic prostate cancer showing significant overall survival and higher response rate,81–83 and the latter currently in phase II clinical trials for the treatment of several solid tumors84. Both taxane derivatives significantly decreased susceptibility to Pgp efflux, giving about 7–14 (for cabazitaxel)85 and 30–50 (ortataxel)84 fold of increase in activity relative to paclitaxel, against various P-gp overexpressing tumor cell lines. Of note, several modified taxanes, including ortataxel,84 SB-T-1213 and SB-T-1250,86 have been studied not only as potent anticancer agents but also shown as good P-gp inhibitors. These compounds are less susceptible to P-gp-mediated efflux possibly by a novel mechanism of self-inhibition.
Extensive research has been carried out on the modification of vinca alkaloids as well.87, 88 Vinflunine (Figure 6C), a derivative of vinblastine which contains a fluorinated modification at the C20’ position, was shown to have decreased susceptibility to P-gp-mediated efflux compared to vincristine and vinorelbine by factors of 2.5 to 13-fold, with lower in vitro neurotoxicity and enhanced bioavailability.89 Vinflunine was approved for the second-line treatment of urothelial cancer by EMA in 2009, and remains under investigation in several clinical trials for different cancer treatment.90 As also demonstrated by Boger et al., this C20’ position of vinblastine represents a site with great potential for functionalization leading to substantial enhancement in potency with simultaneous decrease in P-gp binding and transport.91 The C20’ isoindoline urea derivative (1) was first shown to display a reduced differential in activity against the vinblastine-sensitive HCT116 and vinblastine-resistant HCT116/VM46 cell lines of 1020 fold (vs ca. 100-fold for vinblastine). Further modifications of the isoindoline moiety on 2 afforded ultrapotent compound 2 (IC50 values of 50–75 pM and 830–880 pM against HCT116 and HCT116/VM46 cells, respectively) (Figure 6C), which gave a stunning ~700 fold increase in activity relative to vinblastine against the same P-g overexpressing HCT116/VM46 cell line.92 A more recent and detailed study of several vinblastine C20’ aryl amide derivatives (such as compound 3, identified from the screening of 180 compounds) have confirmed little or no P-gp transport by directly assaying the P-gp transport and membrane permeability using Caco-2 cells. Compound 3 was >10-fold more potent than vinblastine against the sensitive cell lines (IC50 = 400–500 pM), and >300-fold more active against the resistant HCT116 cell line (IC50 = 1.8 nM).93
Given the structural complexity seen in many natural product-derived anticancer drugs, it is difficult to predict the exact position and/or groups to optimize these drug molecules in order to suppress P-gp-mediated efflux while maintaining drug potency. As P-gp typically recognizes hydrophobic compounds for export,55 adding polarity to existing drug entities is often perceived as a feasible strategy to overcome P-gp-medicated efflux. However, this empirical rule is not manifested in the modifications exemplified in Figure 6, specifically with the added structural complexity and hydrophobicity in the vinblastine modifications. Thus, a fundamental question still remains as to how P-gp reacts upon different chemical structural modifications on transport substrates. Nevertheless, extensive modifications on several classical anticancer drugs such as taxanes and vinca alkaloids, as illustrated in above examples through decades of research in many laboratories, have provided valuable information with regards to the optimal sites for further modification and for detailed SAR studies to elude P-gp efflux.
Complexing or conjugating with drug delivery agents to counteract the activity of P-gp
Complexation or attachment of drugs to a delivery agent has offered the possibility to outcompete P-gp mediated efflux by enhancing drug uptake or potentially modifying the mechanism of cellular entry. Various nano-sized carriers have been exploited in drug formulations,94, 95 including polymeric conjugates or nanoparticles, lipid-based carriers such as liposomes and micelles, dendrimers, carbon nanotubes, and gold nanoparticles (Figure 7A).96–98 Some of these nanocarriers have been investigated to address challenges associated with P-gp-mediated MDR within cell lines or in mouse tumor models.99–102 Among them, liposomes have received the most attention in clinical applications, with the first nano-drug, doxorubicin, approved by FDA in 1995. Many other liposome-based drugs are currently in use and in clinical development.103 Polymer nanoparticles have also been widely studied. For example, SP1049C is a P-gp targeting non-ionic block-copolymer formulation containing doxorubicin as the cytotoxic payload.104 SP1049C has been shown to circumvent P-gp-mediated doxorubicin resistance in a mouse model of leukemia,105 and the formulation with doxorubicin has gone through phase II clinical trials against advanced esophageal cancer.106 Abraxane, designed to address insolubility problems associated with paclitaxel, is an albumin-bound paclitaxel nanoparticle formulation.107 It was approved by the FDA as a second-line treatment for metastatic breast cancer.108 Other drug-polymer conjugates with cytotoxic drugs have also been extensively used to overcome MDR in highly resistant cells in vitro and in vivo; these include drugs like doxorubicin, camptothecin, paclitaxel, and platinate with polymers like N-(2-hydroxylpropyl)methacrylamide (HPMA), poly-L-glutamic acid, poly(ethylene glycol) (PEG), and dextran.94, 109 Opaxio (formerly Xyotax: PG-TXL-(poly (L-glutamic acid-paclitaxel) has advanced to Phase III clinical trials as the first polymer-drug conjugate for the treatment of breast, ovarian, lung and other types of cancers.110 The rationale behind the use of polymer-drug conjugates lies in the improved aqueous solubility of the chemotherapeutic drug by conjugation to a water-soluble polymer, which could help bypass P-gp by an endocytic internalization pathway.111
Figure 7.

Schematic representation of drug delivery systems to combat P-gp transport. Anticancer drugs are complexed or conjugated with these delivery agents and internalized for intracellular processing to release the free drugs into the cytoplasm, reducing their uptake from the plasma membrane and export by P-gp. (A) Nanoparticle carriers; (B) Guanidinium-rich molecular transporters; and (C) Antibody-drug conjugates (ADCs).
The best examples of covalent delivery systems include the use of cell-penetrating molecular transporters and antibody-drug conjugates (ADCs). The former strategy, pioneered by Wender et al., employs a cationic, guanidinium-rich molecular scaffold to tag the conjugated drug so as to drastically alter drug recognition by P-gp (Figure 7B).112, 113 Several versions of these guanidinium-rich molecular transporters have been devised, based on the scaffolds of peptides, peptoids, oligocarbamates, dendrimers, oligocarbonates, carbohydrates, or oligophosphoesters.114, 115 These small molecule transporter drug conjugates are highly water-soluble, show high cellular uptake, and are not substrates for P-gp-mediated efflux.112 The uptake of these molecular transporters was proposed to be the attribute of the number and the spatial array of the quanidinium groups, which associate with anionic lipids to form reversed-micelle-like complexes.113 Release of free drug from these conjugates can be controlled by linker design and target cell characteristics. The efficacy of this strategy was demonstrated with taxol-octaarginine conjugates to overcome resistance in vitro, in vivo and ex vivo in malignant ascites.116, 117 Taxol-octaarginine conjugates were shown to be very effective (4–100 fold over unconjugated taxol) in a variety of ovarian cancer cell lines including taxol-sensitive and taxol-resistant variants. Other examples include conjugates with doxorubicin and methotrexate.118, 119
Antibody-drug conjugates (ADCs) are designed with a goal of targeted drug delivery together with reduced systemic toxicity.120 Upon binding to tumor cell surface antigens, ADCs are internalized by receptor-mediated endocytosis, which leads to drug release inside the target cell (Figure 7C). In developing ADCs, much attention has been focused on the development of highly potent cytotoxic payloads, the linker, and the antibody components as well as conjugation technologies. Similar to other types of drug delivery systems, the use of ADC for drug internalization also reduces P-gp-mediated efflux. However, cytosolic drug release by these various delivery systems may not completely evade P-gp-mediated efflux, since the drugs can repartition into the plasma membrane. Additionally, inward facing structures suggest that drug substrates may enter P-gp from the cytosol as well as from the plasma membrane (Figure 2). As a matter of fact, sensitivity of the cytotoxic drugs to P-gp-mediated efflux still plays a crucial role in limiting efficiency of ADCs. Mylotarg121 (gemtuzumab ozogamicin) was the first ADC to gain FDA approval but later withdrawn due to lack of improvement in overall patient survival. In vitro cytotoxicity assays in AML cell lines showed that P-gp expression altered the potency of the cytotoxic payload (Calicheamicin), and potency of the drug was restored by the use of a P-gp inhibitor.122, 123
Another example related to MDR mechanisms involves AVE9633, which comprises an anti-CD33 antibody linked through a disulfide bond to the maytansine analog DM4. In vitro data demonstrated that P-gp activity was a critical factor in resistance against AVE9633 and DM4 cytotoxicity in leukemia cell lines124 Kovtun et al. have shown that modification of maytansine DM1 in an ADC with a hydrophilic oligo(ethylene oxide) linker, which is retained after intracellular processing, converts DM1 to a poor P-gp substrate, and this modification markedly improves the therapeutic index of the ADC in eradicating P-gp-expressing human xenograft tumors.125 Extensive chemical modifications have also been conducted on the potent cytotoxic drug monomethyl auristatin E (MMAE),126 which is used in many ADCs including the FDA-approved brentuximab vedotin (BV) for treatment of refractory Hodgkin lymphoma and systemic anaplastic large cell lymphoma. MMAE is susceptible to P-gp-mediated efflux, and structural optimization of its ADCs to help the drug escape efflux may thus improve the therapeutic index.127, 128
Future Perspectives and Concluding Remarks
Despite extensive efforts, MDR is as much of a problem in cancer treatment now as it ever was, even with the advent of new therapies. The strategy of using multiple anticancer drugs is helpful but cancer cells have proven to be able to adapt and develop MDR. Given the significant and abundant clinical data supporting the role of P-gp in cancer MDR, development of selective P-gp inhibitors with further assessment of safety and efficacy remains relevant. In addition to P-gp, studies toward the inhibition of other MDR transporters such as BCRP and MRP1 are also important, as these transporters may confer resistance to the same or different sets of drugs. It is thus important to analyze the cause of clinical MDR and develop tailored therapeutics to target these transporters.
Suppressing drug efflux is an important goal in many drug development programs. Chemically modifying or redesigning an anticancer drug to completely bypass P-gp is challenging, and there are no clear rules, as P-gp is able to recognize diverse structures that permeate cellular membranes. At the same time, modifications of an anticancer drug without diminution of drug potency presents another significant challenge. In comparison, drug delivery systems offer seemingly innumerable possibilities and provide the potential for safer and high-dose delivery of anticancer drugs, while using noninvasive tracking techniques that are target specific. In these cases, controlling drug release inside the cells could be key since the drug residence and accumulation are counter-balanced by the efflux via membrane transporters.
The recently solved X-ray structures of P-gp in complex with several ligands have provided a molecular basis of how P-gp mediates MDR. The presence of overlapping sites inside the large and flexible binding pocket lined with aromatic residues explains the extraordinary array of chemical structures that interact with Pgp. Despite the progress, many profound questions pertinent to the mechanism of P-gp and drug interactions remain to be addressed. So far the X-ray structures of P-gp have been captured only in inward-facing states, at limited resolutions of above 3.3 Å and with very few solved in complex with cyclopeptide inhibitors. Controversies remain about the range of P-gp conformations, and a complete pathway of drug transport coupled to such conformational changes is not known. No P-gp structure with an actual drug bound has been determined to date, but would presumably guide efforts to chemically alter the drug so as to evade efflux. How P-gp distinguishes a substrate for transport vs. an inhibitor is also highly relevant to drug design. Addressing all these questions should be among the next steps for the community effort to study P-gp and drug interactions.
Compared to P-gp, structural and mechanistic studies on other MDR transporters are quite limited.54, 129 Future structural biology studies along with chemistry and biochemical characterizations on all these MDR transporters are crucial not just for understanding the biology, but potentially also for structure-based drug design. The field of membrane protein structural biology has seen a boost over the last few years due to technological advances in modern X-ray crystallography and cryo-EM. Expedited progress in high-resolution structural determinations of these MDR pumps and their drug complexes should be anticipated.
Acknowledgments
We are grateful to our collaborators, Drs. G. Chang, A. Ward, I. Urbatsch, B. Carragher, and C. Potter for P-gp structural studies. This work was supported by NIH grant RO1 GM118594.
Abbreviations Used
- MDR
multidrug resistance
- P-gp
P-glycoprotein
- ABC
ATP-binding cassette
- MRP1
MDR-associated protein 1
- BCRP
breast cancer resistance protein
- TM
transmembrane
- TMD
transmembrane domain
- NBD
nucleotide binding domain
- EM
electron microscopy
- ADC
antibody-drug conjugate
Biographies:
Deepali Waghray obtained her Ph.D. degree in organic chemistry from Katholieke Universiteit Leuven (KU Leuven), Belgium. From 2009 to 2013 she worked in the laboratory of Professor Wim Dehaen on the design, synthesis and optical property studies of hetera[n]helicenes. During a short postdoctoral experience from 2014–2015 she worked on the design and synthesis of shape persistent macrocycles, synthesis of imine-linked COFs for the formation of extended 2D networks and characterization of 2D-networks using Scanning Tunneling Microscopy (STM) in the laboratory of Professor Steven De Feyter. She moved to USA in 2016 and joined the laboratory of Professor Qinghai Zhang at The Scripps Research Institute (TSRI) as a Research Associate in 2017, and her current project centers on studies of P-glycoprotein and drug interactions.
Qinghai Zhang received his Ph.D. degree in organic chemistry in 2001 under the supervision of Professor Xiyan Lu from Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences. From 2001 to 2004, he conducted postdoctoral research with Professor Jeffery Kelly at TSRI, while studying the mechanism of protein amyloidosis using chemical, biochemical, and biophysical approaches. He is now an Associate Professor in the Department of Integrative Structural and Computational Biology at TSRI. His research interests focus on chemical and structural biology of membrane proteins aiming at drug development and mechanistic studies.
Footnotes
The authors declare no competing financial interest.
References:
- 1.Holohan C; Van Schaeybroeck S; Longley DB; Johnston PG Cancer drug resistance: an evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [DOI] [PubMed] [Google Scholar]
- 2.Szakacs G; Paterson JK; Ludwig JA; Booth-Genthe C; Gottesman MM Targeting multidrug resistance in cancer. Nat. Rev. Drug. Discov 2006, 5, 219–234. [DOI] [PubMed] [Google Scholar]
- 3.Ozpolat B Resistance to Differentiation Therapy In Drug Resistance in Cancer Cells, Siddik ZH; Mehta K, Eds. Springer US: New York, NY, 2009; pp233–255. [Google Scholar]
- 4.Fan D; Kim S-J; Langley RL; Fidler IJ Metastasis and Drug Resistance In Drug Resistance in Cancer Cells, Siddik ZH; Mehta K, Eds. Springer US: New York, NY, 2009; pp21–52. [Google Scholar]
- 5.Swanton C Intratumor heterogeneity: evolution through space and time. Cancer Res 2012, 72, 4875–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dexter DL; Leith JT Tumor heterogeneity and drug resistance. J. Clin. Oncol 1986, 4, 244–257. [DOI] [PubMed] [Google Scholar]
- 7.Gillet JP; Gottesman MM Mechanisms of multidrug resistance in cancer. Methods. Mol. Biol 2010, 596, 47–76. [DOI] [PubMed] [Google Scholar]
- 8.Gottesman MM; Fojo T; Bates SE Multidrug resistance in cancer: role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [DOI] [PubMed] [Google Scholar]
- 9.Holland IB; Cole SP; Kuchler K; Higgins CF ABC proteins: from bacteria to man London: Academic Press: 2003. [Google Scholar]
- 10.Gottesman MM; Ling V The molecular basis of multidrug resistance in cancer: the early years of P-glycoprotein research. FEBS Lett 2006, 580, 998–1009. [DOI] [PubMed] [Google Scholar]
- 11.Cole SP; Bhardwaj G; Gerlach JH; Mackie JE; Grant CE; Almquist K; Stewart AJ; Kurz EU; Duncan AM; Deeley RG Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science 1992, 258, 1650–1654. [DOI] [PubMed] [Google Scholar]
- 12.Doyle LA; Yang W; Abruzzo LV; Krogmann T; Gao Y; Rishi AK; Ross D A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc. Natl. Acad. Sci. U. S. A 1998, 95, 15665–15670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alvarez M; Paull K; Monks A; Hose C; Lee JS; Weinstein J; Grever M; Bates S; Fojo T Generation of a drug resistance profile by quantitation of mdr-1/P-glycoprotein in the cell lines of the National Cancer Institute Anticancer Drug Screen. J. Clin. Invest 1995, 95, 2205–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Szakacs G; Annereau JP; Lababidi S; Shankavaram U; Arciello A; Bussey KJ; Reinhold W; Guo Y; Kruh GD; Reimers M; Weinstein JN; Gottesman MM Predicting drug sensitivity and resistance: profiling ABC transporter genes in cancer cells. Cancer Cell 2004, 6, 129–137. [DOI] [PubMed] [Google Scholar]
- 15.Leonard GD; Fojo T; Bates SE The role of ABC transporters in clinical practice. Oncologist 2003, 8, 411–424. [DOI] [PubMed] [Google Scholar]
- 16.Ambudkar SV; Dey S; Hrycyna CA; Ramachandra M; Pastan I; Gottesman MM Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu. Rev. Pharmacol. Toxicol 1999, 39, 361–398. [DOI] [PubMed] [Google Scholar]
- 17.Sharom FJ The P-glycoprotein multidrug transporter. Essays Biochem 2011, 50, 161–178. [DOI] [PubMed] [Google Scholar]
- 18.Eckford PD; Sharom FJ ABC efflux pump-based resistance to chemotherapy drugs. Chem. Rev 2009, 109, 2989–3011. [DOI] [PubMed] [Google Scholar]
- 19.Darby RA; Callaghan R; McMahon RM P-glycoprotein inhibition: the past, the present and the future. Curr. Drug. Metab 2011, 12, 722–731. [DOI] [PubMed] [Google Scholar]
- 20.Amiri-Kordestani L; Basseville A; Kurdziel K; Fojo AT; Bates SE Targeting MDR in breast and lung cancer: discriminating its potential importance from the failure of drug resistance reversal studies. Drug Resist. Updat 2012, 15, 50–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schinkel AH; Smit JJ; van Tellingen O; Beijnen JH; Wagenaar E; van Deemter L; Mol CA; van der Valk MA; Robanus-Maandag EC; te Riele HP; et al. Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell 1994, 77, 491–502. [DOI] [PubMed] [Google Scholar]
- 22.Fojo AT; Ueda K; Slamon DJ; Poplack DG; Gottesman MM; Pastan I Expression of a multidrug-resistance gene in human tumors and tissues. Proc. Natl. Acad. Sci. U. S. A 1987, 84, 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cordon-Cardo C; O’Brien JP; Casals D; Rittman-Grauer L; Biedler JL; Melamed MR; Bertino JR Multidrug-resistance gene (P-glycoprotein) is expressed by endothelial cells at blood-brain barrier sites. Proc. Natl. Acad. Sci. U. S. A 1989, 86, 695–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Noonan KE; Beck C; Holzmayer TA; Chin JE; Wunder JS; Andrulis IL; Gazdar AF; Willman CL; Griffith B; Von Hoff DD; et al. Quantitative analysis of MDR1 (multidrug resistance) gene expression in human tumors by polymerase chain reaction. Proc. Natl. Acad. Sci. U. S. A 1990, 87, 7160–7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Tellingen O; Buckle T; Jonker JW; van der Valk MA; Beijnen JH P-glycoprotein and Mrp1 collectively protect the bone marrow from vincristine-induced toxicity in vivo. Br. J. Cancer 2003, 89, 1776–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sparreboom A; van Asperen J; Mayer U; Schinkel AH; Smit JW; Meijer K; Borst P; Nooijen WJ; Beijnen JH; van Tellingen O Limited oral bioavailability and active epithelial excretion of paclitaxel (Taxol) caused by P-glycoprotein in the intestine. Proc. Natl. Acad. Sci. U. S. A 1997, 94, 2031–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Borst P; Schinkel AH P-glycoprotein ABCB1: a major player in drug handling by mammals. J. Clin. Invest 2013, 123, 4131–4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.International Transporter C; Giacomini KM; Huang SM; Tweedie DJ; Benet LZ; Brouwer KL; Chu X; Dahlin A; Evers R; Fischer V; Hillgren KM; Hoffmaster KA; Ishikawa T; Keppler D; Kim RB; Lee CA; Niemi M; Polli JW; Sugiyama Y; Swaan PW; Ware JA; Wright SH; Yee SW; Zamek-Gliszczynski MJ; Zhang L Membrane transporters in drug development. Nat. Rev. Drug. Discov 2010, 9, 215–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aller SG; Yu J; Ward A; Weng Y; Chittaboina S; Zhuo R; Harrell PM; Trinh YT; Zhang Q; Urbatsch IL; Chang G Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 2009,323, 1718–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bai J; Swartz DJ; Protasevich II; Brouillette CG; Harrell PM; Hildebrandt E; Gasser B; Mattanovich D; Ward A; Chang G; Urbatsch IL A gene optimization strategy that enhances production of fully functional P-glycoprotein in Pichia pastoris. PLoS One 2011, 6, e22577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ward AB; Szewczyk P; Grimard V; Lee CW; Martinez L; Doshi R; Caya A; Villaluz M; Pardon E; Cregger C; Swartz DJ; Falson PG; Urbatsch IL; Govaerts C; Steyaert J; Chang G Structures of P-glycoprotein reveal its conformational flexibility and an epitope on the nucleotide-binding domain. Proc Natl. Acad. Sci. U. S. A 2013, 110, 13386–13391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jin MS; Oldham ML; Zhang Q; Chen J Crystal structure of the multidrug transporter P-glycoprotein from Caenorhabditis elegans. Nature 2012, 490, 566569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Esser L; Zhou F; Pluchino KM; Shiloach J; Ma J; Tang WK; Gutierrez C; Zhang A; Shukla S; Madigan JP; Zhou T; Kwong PD; Ambudkar SV; Gottesman MM; Xia D Structures of the Multidrug Transporter P-glycoprotein Reveal Asymmetric ATP Binding and the Mechanism of Polyspecificity. J. Biol. Chem 2017, 292, 446–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Szewczyk P; Tao H; McGrath AP; Villaluz M; Rees SD; Lee SC; Doshi R; Urbatsch IL; Zhang Q; Chang G Snapshots of ligand entry, malleable binding and induced helical movement in P-glycoprotein. Acta Crystallogr. D Biol. Crystallogr 2015, 71, 732–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li J; Jaimes KF; Aller SG Refined structures of mouse P-glycoprotein. Protein Sci 2014, 23, 34–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Katayama K; Noguchi K; Sugimoto Y Regulations of P-Glycoprotein/ABCB1/MDR1 in Human Cancer Cells. New J. Sci 2014, Article ID 476974: 1–10. [Google Scholar]
- 37.Ward A; Reyes CL; Yu J; Roth CB; Chang G Flexibility in the ABC transporter MsbA: Alternating access with a twist. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 19005–19010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dawson RJ; Locher KP Structure of a bacterial multidrug ABC transporter. Nature 2006, 443, 180–185. [DOI] [PubMed] [Google Scholar]
- 39.Verhalen B; Dastvan R; Thangapandian S; Peskova Y; Koteiche HA; Nakamoto RK; Tajkhorshid E; McHaourab HS Energy transduction and alternating access of the mammalian ABC transporter P-glycoprotein. Nature 2017, 543, 738–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moeller A; Lee SC; Tao H; Speir JA; Chang G; Urbatsch IL; Potter CS; Carragher B; Zhang Q Distinct conformational spectrum of homologous multidrug ABC transporters. Structure 2015, 23, 450–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frank GA; Shukla S; Rao P; Borgnia MJ; Bartesaghi A; Merk A; Mobin A; Esser L; Earl LA; Gottesman MM; Xia D; Ambudkar SV; Subramaniam S Cryo-EM Analysis of the Conformational Landscape of Human P-glycoprotein (ABCB1) During its Catalytic Cycle. Mol. Pharmacol 2016, 90, 35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gottesman MM; Ambudkar SV; Xia D Structure of a multidrug transporter. Nat. Biotechnol 2009, 27, 546–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jones PM; George AM A reciprocating twin-channel model for ABC transporters. Q. Rev. Biophys 2014, 47, 189–220. [DOI] [PubMed] [Google Scholar]
- 44.Loo TW; Clarke DM Determining the structure and mechanism of the human multidrug resistance P-glycoprotein using cysteine-scanning mutagenesis and thiol-modification techniques. Biochim. Biophys. Acta 1999, 1461, 315–325. [DOI] [PubMed] [Google Scholar]
- 45.Loo TW; Clarke DM The transmembrane domains of the human multidrug resistance P-glycoprotein are sufficient to mediate drug binding and trafficking to the cell surface. J. Biol. Chem 1999, 274, 24759–24765. [DOI] [PubMed] [Google Scholar]
- 46.Shapiro AB; Ling V Positively cooperative sites for drug transport by P-glycoprotein with distinct drug specificities. Eur. J. Biochem 1997, 250, 130–137. [DOI] [PubMed] [Google Scholar]
- 47.Shapiro AB; Fox K; Lam P; Ling V Stimulation of P-glycoprotein-mediated drug transport by prazosin and progesterone. Evidence for a third drug-binding site. Eur. J. Biochem 1999, 259, 841–850. [DOI] [PubMed] [Google Scholar]
- 48.Martin C; Berridge G; Higgins CF; Mistry P; Charlton P; Callaghan R Communication between multiple drug binding sites on P-glycoprotein. Mol. Pharmacol 2000, 58, 624–632. [DOI] [PubMed] [Google Scholar]
- 49.Ogino J; Moore RE; Patterson GM; Smith CD Dendroamides, new cyclic hexapeptides from a blue-green alga. Multidrug-resistance reversing activity of dendroamide A. J. Nat. Prod 1996, 59, 581–586. [DOI] [PubMed] [Google Scholar]
- 50.Banker R; Carmeli S Tenuecyclamides A-D, cyclic hexapeptides from the cyanobacterium Nostoc spongiaeforme var. tenue. J. Nat. Prod 1998, 61, 1248–1251. [DOI] [PubMed] [Google Scholar]
- 51.Fu X; Do T; Schmitz FJ; Andrusevich V; Engel MH New cyclic peptides from the ascidian Lissoclinum patella. J. Nat. Prod 1998, 61, 1547–1551. [DOI] [PubMed] [Google Scholar]
- 52.Perez LJ; Faulkner DJ Bistratamides E-J, modified cyclic hexapeptides from the Philippines ascidian Lissoclinum bistratum. J. Nat. Prod 2003, 66, 247–250. [DOI] [PubMed] [Google Scholar]
- 53.Tao H; Weng Y; Zhuo R; Chang G; Urbatsch IL; Zhang Q Design and synthesis of Selenazole-containing peptides for cocrystallization with P-glycoprotein. Chembiochem 2011, 12, 868–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Johnson ZL; Chen J Structural Basis of Substrate Recognition by the Multidrug Resistance Protein MRP1. Cell 2017, 168, 1075–1085 e1079. [DOI] [PubMed] [Google Scholar]
- 55.Sharom FJ Complex Interplay between the P-Glycoprotein Multidrug Efflux Pump and the Membrane: Its Role in Modulating Protein Function. Front. Oncol 2014, 4, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pluchino KM; Hall MD; Moen JK; Chufan EE; Fetsch PA; Shukla S; Gill DR; Hyde SC; Xia D; Ambudkar SV; Gottesman MM Human-Mouse Chimeras with Normal Expression and Function Reveal That Major Domain Swapping Is Tolerated by P-Glycoprotein (ABCB1). Biochemistry 2016, 55, 10101023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim IW; Booth-Genthe C; Ambudkar SV Relationship between drugs and functional activity of various mammalian P-glycoproteins (ABCB1). Mini. Rev. Med. Chem 2008, 8, 193–200. [DOI] [PubMed] [Google Scholar]
- 58.Gottesman MM; Pastan I Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu. Rev. Biochem 1993, 62, 385–427. [DOI] [PubMed] [Google Scholar]
- 59.Dey S; Ramachandra M; Pastan I; Gottesman MM; Ambudkar SV Evidence for two nonidentical drug-interaction sites in the human P-glycoprotein. Proc. Natl. Acad. Sci. U. S. A 1997, 94, 10594–10599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Al-Shawi MK; Omote H The remarkable transport mechanism of P-glycoprotein: a multidrug transporter. J. Bioenerg. Biomembr 2005, 37, 489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shaffer BC; Gillet JP; Patel C; Baer MR; Bates SE; Gottesman MM Drug resistance: still a daunting challenge to the successful treatment of AML. Drug Resist. Updat 2012, 15, 62–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dean M; Fojo T; Bates S Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [DOI] [PubMed] [Google Scholar]
- 63.Hollt V; Kouba M; Dietel M; Vogt G Stereoisomers of calcium antagonists which differ markedly in their potencies as calcium blockers are equally effective in modulating drug transport by P-glycoprotein. Biochem. Pharmacol 1992, 43, 2601–2608. [DOI] [PubMed] [Google Scholar]
- 64.List AF; Kopecky KJ; Willman CL; Head DR; Persons DL; Slovak ML; Dorr R; Karanes C; Hynes HE; Doroshow JH; Shurafa M; Appelbaum FR Benefit of cyclosporine modulation of drug resistance in patients with poor-risk acute myeloid leukemia: a Southwest Oncology Group study. Blood 2001, 98, 32123220. [DOI] [PubMed] [Google Scholar]
- 65.Slate DL; Bruno NA; Casey SM; Zutshi N; Garvin LJ; Wu H; Pfister JR RS-33295–198: a novel, potent modulator of P-glycoprotein-mediated multidrug resistance. Anticancer Res 1995, 15, 811–814. [PubMed] [Google Scholar]
- 66.Martin C; Berridge G; Mistry P; Higgins C; Charlton P; Callaghan R The molecular interaction of the high affinity reversal agent XR9576 with P-glycoprotein. Br. J. Pharmacol 1999, 128, 403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Loo TW; Bartlett MC; Detty MR; Clarke DM The ATPase activity of the P-glycoprotein drug pump is highly activated when the N-terminal and central regions of the nucleotide-binding domains are linked closely together. J. Biol. Chem 2012, 287, 26806–26816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Weidner LD; Fung KL; Kannan P; Moen JK; Kumar JS; Mulder J; Innis RB; Gottesman MM; Hall MD Tariquidar Is an Inhibitor and Not a Substrate of Human and Mouse P-glycoprotein. Drug Metab. Dispos 2016, 44, 275282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kannan P; Telu S; Shukla S; Ambudkar SV; Pike VW; Halldin C; Gottesman MM; Innis RB; Hall MD The “specific” P-glycoprotein inhibitor Tariquidar is also a substrate and an inhibitor for breast cancer resistance protein (BCRP/ABCG2). ACS Chem. Neurosci 2011, 2, 82–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Emmert D; Campos CR; Ward D; Lu P; Namanja HA; Bohn K; Miller DS; Sharom FJ; Chmielewski J; Hrycyna CA Reversible dimers of the atypical antipsychotic quetiapine inhibit p-glycoprotein-mediated efflux in vitro with increased binding affinity and in situ at the blood-brain barrier. ACS Chem. Neurosci 2014, 5, 305–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bohn K; Lange A; Chmielewski J; Hrycyna CA Dual Modulation of Human P-Glycoprotein and ABCG2 with Prodrug Dimers of the Atypical Antipsychotic Agent Paliperidone in a Model of the Blood-Brain Barrier. Mol. Pharmacol 2017, 14, 11071119. [DOI] [PubMed] [Google Scholar]
- 72.Loo TW; Clarke DM Tariquidar inhibits P-glycoprotein drug efflux but activates ATPase activity by blocking transition to an open conformation. Biochem. Pharmacol 2014, 92, 558–566. [DOI] [PubMed] [Google Scholar]
- 73.Abbasi M; Lavasanifar A; Uludag H Recent attempts at RNAi-mediated P-glycoprotein downregulation for reversal of multidrug resistance in cancer. Med. Res. Rev 2013, 33, 33–53. [DOI] [PubMed] [Google Scholar]
- 74.Nobili S; Landini I; Mazzei T; Mini E Overcoming tumor multidrug resistance using drugs able to evade P-glycoprotein or to exploit its expression. Med. Res. Rev 2012, 32, 1220–1262. [DOI] [PubMed] [Google Scholar]
- 75.Wartmann M; Altmann KH The biology and medicinal chemistry of epothilones. Curr. Med. Chem: Anticancer Agents 2002, 2, 123–148. [DOI] [PubMed] [Google Scholar]
- 76.Bollag DM; McQueney PA; Zhu J; Hensens O; Koupal L; Liesch J; Goetz M; Lazarides E; Woods CM Epothilones, a new class of microtubule-stabilizing agents with a taxol-like mechanism of action. Cancer Res 1995, 55, 2325–2333. [PubMed] [Google Scholar]
- 77.Altmann KH; Wartmann M; O’Reilly T Epothilones and related structures--a new class of microtubule inhibitors with potent in vivo antitumor activity. Biochim. Biophys. Acta 2000, 1470, M79–91. [DOI] [PubMed] [Google Scholar]
- 78.Rak Tkaczuk KH Ixabepilone as monotherapy or in combination with capecitabine for the treatment of advanced breast cancer. Breast Cancer (Auckl) 2011, 5, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee FY; Borzilleri R; Fairchild CR; Kamath A; Smykla R; Kramer R; Vite G Preclinical discovery of ixabepilone, a highly active antineoplastic agent. Cancer Chemother. Pharmacol 2008, 63, 157–166. [DOI] [PubMed] [Google Scholar]
- 80.Lee FY; Borzilleri R; Fairchild CR; Kim SH; Long BH; Reventos-Suarez C; Vite GD; Rose WC; Kramer RA BMS-247550: a novel epothilone analog with a mode of action similar to paclitaxel but possessing superior antitumor efficacy. Clin. Cancer Res 2001, 7, 1429–1437. [PubMed] [Google Scholar]
- 81.Kingston DG; Newman DJ Taxoids: cancer-fighting compounds from nature. Curr. Opin. Drug Discov. Devel 2007, 10, 130–144. [PubMed] [Google Scholar]
- 82.Abidi A Cabazitaxel: A novel taxane for metastatic castration-resistant prostate cancer-current implications and future prospects. J. Pharmacol. Pharmacother 2013, 4, 230–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bouchet BP; Galmarini CM Cabazitaxel, a new taxane with favorable properties. Drugs Today (Barc) 2010, 46, 735–742. [DOI] [PubMed] [Google Scholar]
- 84.Vredenburg MR; Ojima I; Veith J; Pera P; Kee K; Cabral F; Sharma A; Kanter P; Greco WR; Bernacki RJ Effects of orally active taxanes on P-glycoprotein modulation and colon and breast carcinoma drug resistance. J. Natl. Cancer Inst 2001, 93, 1234–1245. [DOI] [PubMed] [Google Scholar]
- 85.Duran GE; Wang YC; Francisco EB; Rose JC; Martinez FJ; Coller J; Brassard D; Vrignaud P; Sikic BI Mechanisms of resistance to cabazitaxel. Mol. Cancer Ther 2015, 14, 193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ferlini C; Distefano M; Pignatelli F; Lin S; Riva A; Bombardelli E; Mancuso S; Ojima I; Scambia G Antitumour activity of novel taxanes that act at the same time as cytotoxic agents and P-glycoprotein inhibitors. Br. J. Cancer 2000, 83, 1762–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sears JE; Boger DL Total synthesis of vinblastine, related natural products, and key analogues and development of inspired methodology suitable for the systematic study of their structure-function properties. Acc. Chem. Res 2015, 48, 653–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Voss ME; Ralph JM; Xie D; Manning DD; Chen X; Frank AJ; Leyhane AJ; Liu L; Stevens JM; Budde C; Surman MD; Friedrich T; Peace D; Scott IL; Wolf M; Johnson R Synthesis and SAR of vinca alkaloid analogues. Bioorg. Med. Chem. Lett 2009, 19, 1245–1249. [DOI] [PubMed] [Google Scholar]
- 89.Etievant C; Barret J-M; Kruczynski A; Perrin D; Hill BT Vinflunine (20’, 20’-difluoro-3’, 4’-dihydrovinorelbine), a novel Vinca alkaloid, which participates in P-glycoprotein (Pgp)-mediated multidrug resistance in vivo and in vitro. Inves. New Drugs 1998, 16, 3–17. [DOI] [PubMed] [Google Scholar]
- 90.Gerullis H; Wawroschek F; Kohne CH; Ecke TH Vinflunine in the treatment of advanced urothelial cancer: clinical evidence and experience. Ther. Adv. Urol 2017, 9, 28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Barker TJ; Duncan KK; Otrubova K; Boger DL Potent Vinblastine C20’ Ureas Displaying Additionally Improved Activity Against a Vinblastine-Resistant Cancer Cell Line. ACS Med. Chem. Lett 2013, 4, 985–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Carney DW; Lukesh JC 3rd; Brody DM; Brutsch MM; Boger DL Ultrapotent vinblastines in which added molecular complexity further disrupts the target tubulin dimer-dimer interface. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 96919698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lukesh JC 3rd; Carney DW; Dong H; Cross RM; Shukla V; Duncan KK; Yang S; Brody DM; Brutsch MM; Radakovic A; Boger DL Vinblastine 20’ Amides: Synthetic Analogues That Maintain or Improve Potency and Simultaneously Overcome Pgp-Derived Efflux and Resistance. J. Med. Chem 2017, 60, 7591–7604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Duncan R Polymer therapeutics as nanomedicines: new perspectives. Curr. Opin. Biotechnol 2011, 22, 492–501. [DOI] [PubMed] [Google Scholar]
- 95.Ferrari M Cancer nanotechnology: opportunities and challenges. Nat. Rev. Cancer 2005, 5, 161–171. [DOI] [PubMed] [Google Scholar]
- 96.Peer D; Karp JM; Hong S; Farokhzad OC; Margalit R; Langer R Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol 2007, 2, 751–760. [DOI] [PubMed] [Google Scholar]
- 97.Huang Y; Cole SP; Cai T; Cai YU Applications of nanoparticle drug delivery systems for the reversal of multidrug resistance in cancer. Oncol. Lett 2016, 12, 11–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shi J; Kantoff PW; Wooster R; Farokhzad OC Cancer nanomedicine: progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Goren D; Horowitz AT; Tzemach D; Tarshish M; Zalipsky S; Gabizon A Nuclear delivery of doxorubicin via folate-targeted liposomes with bypass of multidrug-resistance efflux pump. Clin. Cancer Res 2000, 6, 1949–1957. [PubMed] [Google Scholar]
- 100.Duncan R; Vicent MJ; Greco F; Nicholson RI Polymer-drug conjugates: towards a novel approach for the treatment of endrocine-related cancer. Endocr. Relat. Cancer 2005, 12 Suppl 1, S189–199. [DOI] [PubMed] [Google Scholar]
- 101.Wong HL; Rauth AM; Bendayan R; Manias JL; Ramaswamy M; Liu Z; Erhan SZ; Wu XY A new polymer-lipid hybrid nanoparticle system increases cytotoxicity of doxorubicin against multidrug-resistant human breast cancer cells. Pharm. Res 2006, 23, 1574–1585. [DOI] [PubMed] [Google Scholar]
- 102.Garcion E; Lamprecht A; Heurtault B; Paillard A; Aubert-Pouessel A; Denizot B; Menei P; Benoit JP A new generation of anticancer, drug-loaded, colloidal vectors reverses multidrug resistance in glioma and reduces tumor progression in rats. Mol. Cancer Ther 2006, 5, 1710–1722. [DOI] [PubMed] [Google Scholar]
- 103.Sercombe L; Veerati T; Moheimani F; Wu SY; Sood AK; Hua S Advances and Challenges of Liposome Assisted Drug Delivery. Front. Pharmacol 2015, 6, 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Danson S; Ferry D; Alakhov V; Margison J; Kerr D; Jowle D; Brampton M; Halbert G; Ranson M Phase I dose escalation and pharmacokinetic study of pluronic polymer-bound doxorubicin (SP1049C) in patients with advanced cancer. Br. J. Cancer 2004, 90, 2085–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Alakhova DY; Zhao Y; Li S; Kabanov AV Effect of doxorubicin/pluronic SP1049C on tumorigenicity, aggressiveness, DNA methylation and stem cell markers in murine leukemia. PLoS One 2013, 8, e72238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Valle JW; Armstrong A; Newman C; Alakhov V; Pietrzynski G; Brewer J; Campbell S; Corrie P; Rowinsky EK; Ranson M A phase 2 study of SP1049C, doxorubicin in P-glycoprotein-targeting pluronics, in patients with advanced adenocarcinoma of the esophagus and gastroesophageal junction. Invest. New Drugs 2011, 29, 1029–1037. [DOI] [PubMed] [Google Scholar]
- 107.Damascelli B; Cantu G; Mattavelli F; Tamplenizza P; Bidoli P; Leo E; Dosio F; Cerrotta AM; Di Tolla G; Frigerio LF; Garbagnati F; Lanocita R; Marchiano A; Patelli G; Spreafico C; Ticha V; Vespro V; Zunino F Intraarterial chemotherapy with polyoxyethylated castor oil free paclitaxel, incorporated in albumin nanoparticles (ABI-007): Phase I study of patients with squamous cell carcinoma of the head and neck and anal canal: preliminary evidence of clinical activity. Cancer 2001, 92, 2592–2602. [DOI] [PubMed] [Google Scholar]
- 108.Miele E; Spinelli GP; Miele E; Tomao F; Tomao S Albumin-bound formulation of paclitaxel (Abraxane ABI-007) in the treatment of breast cancer. Int. J. Nanomedicine 2009, 4, 99–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Duncan R; Ringsdorf H; Satchi-Fainaro R Polymer therapeutics--polymers as drugs, drug and protein conjugates and gene delivery systems: past, present and future opportunities. J. Drug Target 2006, 14, 337–341. [DOI] [PubMed] [Google Scholar]
- 110.Singer JW; Shaffer S; Baker B; Bernareggi A; Stromatt S; Nienstedt D; Besman M Paclitaxel poliglumex (XYOTAX; CT-2103): an intracellularly targeted taxane. Anti-cancer Drugs 2005, 16, 243–254. [DOI] [PubMed] [Google Scholar]
- 111.Zhang L; Gu FX; Chan JM; Wang AZ; Langer RS; Farokhzad OC Nanoparticles in medicine: therapeutic applications and developments. Clin. Pharmacol. Ther 2008, 83, 761–769. [DOI] [PubMed] [Google Scholar]
- 112.Vargas JR; Stanzl EG; Teng NN; Wender PA Cell-penetrating, guanidinium-rich molecular transporters for overcoming efflux-mediated multidrug resistance. Mol. Pharm 2014, 11, 2553–2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wender PA; Mitchell DJ; Pattabiraman K; Pelkey ET; Steinman L; Rothbard JB The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: peptoid molecular transporters. Proc. Natl. Acad. Sci. U. S. A 2000, 97, 13003–13008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Stanzl EG; Trantow BM; Vargas JR; Wender PA Fifteen years of cell-penetrating, guanidinium-rich molecular transporters: basic science, research tools, and clinical applications. Acc. Chem. Res 2013, 46, 2944–2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.McKinlay CJ; Waymouth RM; Wender PA Cell-Penetrating, Guanidinium-Rich Oligophosphoesters: Effective and Versatile Molecular Transporters for Drug and Probe Delivery. J. Am. Chem. Soc 2016, 138, 3510–3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Dubikovskaya EA; Thorne SH; Pillow TH; Contag CH; Wender PA Overcoming multidrug resistance of small-molecule therapeutics through conjugation with releasable octaarginine transporters. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 12128–12133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wender PA; Galliher WC; Bhat NM; Pillow TH; Bieber MM; Teng NN Taxol-oligoarginine conjugates overcome drug resistance in-vitro in human ovarian carcinoma. Gynecol. Oncol 2012, 126, 118–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Aroui S; Brahim S; Waard MD; Kenani A Cytotoxicity, intracellular distribution and uptake of doxorubicin and doxorubicin coupled to cell-penetrating peptides in different cell lines: a comparative study. Biochem. Biophys. Res. Commun 2010, 391, 419–425. [DOI] [PubMed] [Google Scholar]
- 119.Lindgren M; Rosenthal-Aizman K; Saar K; Eiriksdottir E; Jiang Y; Sassian M; Ostlund P; Hallbrink M; Langel U Overcoming methotrexate resistance in breast cancer tumour cells by the use of a new cell-penetrating peptide. Biochem. Pharmacol 2006, 71, 416–425. [DOI] [PubMed] [Google Scholar]
- 120.Perez HL; Cardarelli PM; Deshpande S; Gangwar S; Schroeder GM; Vite GD; Borzilleri RM Antibody-drug conjugates: current status and future directions. Drug Discov. Today 2014, 19, 869–881. [DOI] [PubMed] [Google Scholar]
- 121.Linenberger ML; Hong T; Flowers D; Sievers EL; Gooley TA; Bennett JM; Berger MS; Leopold LH; Appelbaum FR; Bernstein ID Multidrug-resistance phenotype and clinical responses to gemtuzumab ozogamicin. Blood 2001, 98, 988–994. [DOI] [PubMed] [Google Scholar]
- 122.Naito K; Takeshita A; Shigeno K; Nakamura S; Fujisawa S; Shinjo K; Yoshida H; Ohnishi K; Mori M; Terakawa S; Ohno R Calicheamicin-conjugated humanized anti-CD33 monoclonal antibody (gemtuzumab zogamicin, CMA-676) shows cytocidal effect on CD33-positive leukemia cell lines, but is inactive on P-glycoprotein-expressing sublines. Leukemia 2000, 14, 1436–1443. [DOI] [PubMed] [Google Scholar]
- 123.Larson RA; Boogaerts M; Estey E; Karanes C; Stadtmauer EA; Sievers L; Mineur P; Bennett JM; Berger MS; Eten CB; Munteanu M; Loken MR; Van Dongen JJ; Bernstein ID; Appelbaum FR; Mylotarg Study G Antibody-targeted chemotherapy of older patients with acute myeloid leukemia in first relapse using Mylotarg (gemtuzumab ozogamicin). Leukemia 2002, 16, 1627–1636. [DOI] [PubMed] [Google Scholar]
- 124.Tang R; Cohen S; Perrot JY; Faussat AM; Zuany-Amorim C; Marjanovic Z; Morjani H; Fava F; Corre E; Legrand O; Marie JP P-gp activity is a critical resistance factor against AVE9633 and DM4 cytotoxicity in leukaemia cell lines, but not a major mechanism of chemoresistance in cells from acute myeloid leukaemia patients. BMC Cancer 2009, 9, 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kovtun YV; Audette CA; Mayo MF; Jones GE; Doherty H; Maloney EK; Erickson HK; Sun X; Wilhelm S; Ab O; Lai KC; Widdison WC; Kellogg B; Johnson H; Pinkas J; Lutz RJ; Singh R; Goldmacher VS; Chari RV Antibody-maytansinoid conjugates designed to bypass multidrug resistance. Cancer Res 2010, 70, 2528–2537. [DOI] [PubMed] [Google Scholar]
- 126.Maderna A; Leverett CA Recent advances in the development of new auristatins: structural modifications and application in antibody drug conjugates. Mol. Pharm 2015, 12, 1798–1812. [DOI] [PubMed] [Google Scholar]
- 127.Chen R; Hou J; Newman E; Kim Y; Donohue C; Liu X; Thomas SH; Forman SJ; Kane SE CD30 Downregulation, MMAE Resistance, and MDR1 Upregulation Are All Associated with Resistance to Brentuximab Vedotin. Mol. Cancer Ther 2015, 14, 1376–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chen RW; Hou J; Nair I; Wu J; Synold T; Kwak LW; Rosen ST; Forman SJ; Kane S; Newman E Inhibition of MDR1 Overcomes Brentuximab Vedotin Resistance in Hodgkin Lymphoma Cell Line Model and Is Synergistic with Brentuximab Vedotin in Mouse Xenograft Model. Blood 2016, 128, 752. [Google Scholar]
- 129.Taylor NMI; Manolaridis I; Jackson SM; Kowal J; Stahlberg H; Locher KP Structure of the human multidrug transporter ABCG2. Nature 2017, 546, 504–509. [DOI] [PubMed] [Google Scholar]