

Abstract
Basal transcription factors have never been considered as a priority target in the field of drug discovery. However, their unparalleled roles in decoding the genetic information in response to the appropriate signal and their association with the disease progression are very well-established phenomena. Instead of considering transcription factors as such a target, in this review, we discuss about the potential of the regulatory mechanisms that control their gene expression. Based on our recent understanding about the critical roles of c-MYB at the cellular and molecular level in several types of cancers, we discuss here how MLL-fusion protein centred SEC in leukaemia, ligand-estrogen receptor (ER) complex in breast cancer (BC) and NF-κB and associated factors in colorectal cancer regulate the transcription of this gene. We further discuss plausible strategies, specific to each cancer type, to target those bona fide activators/co-activators, which control the regulation of this gene and therefore to shed fresh light in targeting the transcriptional regulation as a novel approach to the future drug discovery in cancer.
Keywords: CDK9, transcriptional regulation, P-TEFb complex
Introduction
Mammalian oncogenic transcription factor C-MYB is a member of the MYB family of proteins, encoded by the c-MYB gene, which is located in chromosome 6. The 37.85kb long DNA sequence comprises of 15 exons, capable of generating multiple splice variants which upon translation can produce series of MYB proteins with unique c-terminal domain. c-MYB transcription is regulated by an attenuation site located within the first intron and plays a pivotal role in transcription elongation. The 75 kDa MYB protein consists of a DNA binding domain at the N-terminal end that recognizes consensus PyAACG/TG sequence motif. Besides DNA binding domain, the negative regulatory domain is located at the C-terminal end and the transactivation domain, which covers the central part of the protein, plays a major role in determining the activity of this protein. A- and B-MYB, the other two members of the MYB family, share extensive sequence homology and gene transactivation activities with C-MYB. However, in spite of the structural and functional similarities, the distinct tissue specific expression of MYB family members indicated that perhaps they are involved in controlling different regulatory pathways. A-MYB is mostly expressed in highly proliferating tissues such as developing central nervous system, in germinal centre B-lymphocytes, in mammary gland ductal epithelium and in the testis (1-3). In contrast to that, the expression of B-MYB is not restricted to any tissue types and co-expressed with other MYB proteins (4,5). The expression of the founder member C-MYB, is predominantly observed in immature hematopoietic cells, colonic crypt and breast epithelial cells (6-8). The functional diversities of MYB family members also reflected in the gene depleted mouse model. A severe defect in haematopoietic system, which leads to the embryonic lethality, was observed in C-MYB null mouse. Deletion of A-MYB impaired mammary gland development and spermatogenesis whereas; B-MYB deletion demonstrated an inhibition of the blastocyst formation (3,9,10). Microarray based global gene expression profile analysis also indicated that each member of MYB family controls the expression of a distinct and specific set of genes (11), which supports the fundamental idea of the participation of MYB family members in distinct regulatory pathways. In this review, we will only focus on C-MYB, its role in cancer, the tissue specific regulatory mechanisms that control its transcription and possibilities to target those mechanisms for new therapeutic interventions.
MYB in cancers
Over the last decade, implementation of advanced molecular biology and gene manipulation technologies in both in vitro and in vivo revealed the role of c-MYB in different types of cancer. Classically, we can include leukaemia, colon, breast and adenoid cystic carcinoma in a cohort where MYB is playing a critical role in cancer initiation and maintenance. In general, it is believed that high level of MYB expression is associated with the block in cellular differentiation and continued proliferation, which leads to the oncogenicity. Overexpression of MYB is observed in all forms of acute myeloid and lymphoid leukaemia and it is associated with poor prognosis. Several studies, including siRNA-based gene silencing experiments indicated that MYB expression is essential for the proliferation and the maintenance of leukaemia (12). Most of the cases, this overexpression is a result of transcription upregulation of MYB which is either driven by a variety of oncoproteins such as MLL-fusion proteins or chromosomal translocations where MYB is placed in the vicinity of other hyperactive regulatory sequences (13). The role of MYB in the maintenance of haematopoietic lineages was not clear until recently, when Sandberg et al. showed that MYB and one of its major coactivators, p300, is required to control the proliferation and differentiation of haematopoietic stem and progenitor cells (14). Published data from our laboratory also established that MYB-p300 interaction plays a very critical role in the initiation of leukaemia (15).
MYB is essential for the development normal colon, which was established by the fact that MYB depleted embryos developed with major defect in epithelial organization of colon (16). Human colon comprises of several millions of the crypt, the functional unit of the colon, which has stem cell niche formed by stem cells surrounded by mesenchymal cells. MYB expression observed to be highest at the base of the crypt which is the functional compartment of stem cells. Tissue specific MYB knockdown in mice indicated that MYB is required for crypt integrity, normal differentiation, and steady state proliferation (17). Therefore, it could be anticipated that MYB may contribute to the self-renewal property of those stem cells. Analysis of tumours from colorectal carcinoma patients showed a consistent co-relation between high expression of MYB and BCL-xL expression indicating that co-expression of both proteins is required for the tumorigenesis (18). In fact, a high level MYB expression, associated with poor prognosis, is frequently observed in colorectal cancer (19).
Approximately, 70% of human BCs are ER-α positive (ER+ve) and expression of MYB is observed in almost all ER+ve breast tumours. High level of MYB expression also observed in 30% of hereditary BRCA1 mutant BCs (20). It is not clear how BRCA1 mutation affects MYB expression, but several studies indicated BRCA1 as MYB target gene. Several putative MYB binding sites were identified in the BRCA1 promoter (21) and a global chip-on-chip analysis for MYB binding sites on the promoter also identified BRAC1 as a target gene (22). On the other hand, most of the BRCA1 tumours are estrogen negative where MYB expression is suppressed. Therefore, it is possible that mutant BRCA1 forms an alternative transcription complex, which could activate the expression of other oncogenes to maintain the proliferation of tumorigenic cells. Our studies on cell culture, xenograft model and in mammary tissue specific knockdown experiments indicated that MYB is essential for the proliferation of ER+ve BC cell lines and breast tumour formation (23-25). In ER+ve BC cells, expression of MYB is regulated by ligand bound ER-α at the transcriptional level operated within intron 1. Interestingly, a steady state expression of MYB was also observed before and after treatment in BC patients resistance to estrogen synthesis inhibitor aromatase. This unchanged level of MYB expression may have a co-relation with the resistance cases (unpublished).
Adenoid cystic carcinoma is a very slow growing rare type of cancer which often metastasizes to lungs, followed by bone and liver. Published data of Persson et al. indicated that due to t(6;9) chromosomal translocation, MYB very often from a fusion protein with NFIB, which a transcription factor. As a result of this translocation, the last exon (exon 15) of MYB is deleted along with the 3'-UTR region, which has shown to be responsible for the microRNA-based regulation of transcription. High expression MYB-NFIB fusion proteins can alter several MYB target gene expression related to apoptosis, cell proliferation/differentiation (26).
A switch between proliferation and differentiation in metazoan is maintained by the programmed activity of a group of transcription factors [reviewed, Gonda et al. (27)]. Expressions of oncogenic transcription factor over the physiological levels reshuffle the whole cellular machinery and modulate them to follow the oncogene driven pathways. In all the types of cancers discussed above, a sustained and high level of MYB expression is strongly coupled with the maintenance of proliferation of tumorigenic cells and enforced a block in the differentiation. On the other hand, either in leukaemia or in the solid tumour, MYB downregulation alone adversely affected their growth and proliferation. This phenomenon of dependence of cancer cells on oncogene for their survival was defined as “oncogene addiction” which proposed that inhibition of either the synthesis or the activity of that particular oncogene could prevent neoplastic transformation or could selectively inhibit the proliferation of malignant cells (28). Therefore, targeting the oncogene MYB in “so called MYB dependent cancers” could open a new window of a novel therapeutic approach.
Targeting MYB
Transcription factors are not considered as high priority targets in the field of drug discovery mainly because of the fact that inactivation of those factors can block several essential and associated cellular activities and signal transduction pathways, which could generate severe off target effects. However, based on our recent understanding, transcription factors could be targeted in a few major ways: (I) targeting the regulatory network or a complex(s) they form with single or multiple proteins; (II) targeting the regulatory mechanism that specifically control the synthesis of this factor; (III) targeting epigenetic factors such as histone deacetylases or histone methyltransferases which are considered as extended part of the transcription complex and (IV) using short interfering RNA (siRNA) to downregulate the expression of a specific transcription factor (27). However, these approaches, mentioned above, are not free of limitations. For example, in majority of the cases, the regulatory network or complex(s) associated with the transcription factor is very complicated due to the involvement of multiple proteins with diverse array of functions. On the other hand, the mechanisms that control the synthesis of the transcription factor are often not very well defined or almost impossible to target due to the complexity of the process at multiple levels. The problem of targeting the associated epigenetic factors is that the cancer cells get multiple epigenetic aberrations and often develop resistance to the epigenetic drugs (29). The major limitations of siRNAs are: (I) poor cellular uptake; (II) off-target effects; (III) poor stability under physiological conditions (30).
Given that, MYB is playing very important role in several forms of cancers, a significant amount of data has been accumulated over the last decade to understand the regulatory mechanisms that control its synthesis or the network where it functions in association with other major protein co-factors. One of the fundamental aspects of MYB expression in mammalian cells is the diversity of the regulatory mechanisms that control its expression. Several published data indicated that such tissue specific variation of the regulatory mechanism could be a major bottleneck to derive a universal model to block the transcription of MYB in various mammalian tissues. In the following sections, we will discuss transcription regulation of MYB in leukaemia, breast and colon cancer and plausible way to target MYB in different cancers. We would like to exclude adenoid cystic carcinoma because our understanding of the regulatory mechanism that controls the MYB-NFIB is at a very rudimentary stage.
Leukaemia
Acute myeloid leukemia (AML) is a heterogeneous disease and perhaps one of the most well characterized cancers. Several analyses at the molecular and genetic level indicated chromosomal rearrangements are the major contributors to the initiation of AML. Several of such rearrangements have been reported recently which produce fusion proteins such a as AML1-ETO, MLL-AF9, MLL-ENL and found to be the drivers of transformation and maintenance of several forms of leukaemia. MLL rearranged leukaemias are considered as the most aggressive form of leukaemia where the MLL1 protein, which has histone H3 lysine 4 methyl transferase activity, form fusion proteins with several other fusion partners due to reciprocal chromosomal translocation. Till to date, more than 60 MLL fusion partners have been reported and most of them, so far characterized, are act as a transcription factor and aberrantly regulate gene expression besides maintaining H3K79 (methylation of the lysine 79 residue of the globular domain of histone H3 protein) methyl transferase activity. Interestingly, recent studies indicated that MYB is a direct downstream target MLL-fusion protein. It has been shown that MLL-AF9 fusion protein binds strongly in the promoter proximal region of MYB and activates the transcription like other canonical MLL-fusion protein target genes such as HOXA9 (12). However, the molecular architecture of the transcription complex and the mechanism by which it activates MYB transcription remain elusive.
Due to the presence of a variety of protein binding motif at the N-terminal domain of MLL, the MLL-fusion proteins in leukemic cell recruit several proteins to from super elongation complex (SEC, Figure 1). The macromolecular SEC, comprises of more than 30 proteins, is formed by the association of MLL-fusion protein and a core protein complex WARD (WD-40 repeat protein-5, Retinoblastoma binding protein-5, Absent small homeotic 2-like protein and Dumpy30). However, besides those core group of proteins, the SEC very often associates with two other proteins, DOT1L, a histone H3 lysine 9 methyl transferase (H3K9); BRD4, a double bromo domain protein recognizes transcriptionally active chromatin with histone acetylation marks and a protein complex P-TEFb, a heterodimer of CyclinT1 and CDK9 required for transcription elongation. Such distinct but essential functional activities of DOT1L, BRD4 and P-TEFb complex have drawn major attention recently to develop a new therapeutics to control the aggressive MLL-fusion protein leukemia (31-33). A recently developed very specific DOT1L inhibitor, EPZ-5676 has been shown to be very effective in leukemia both in vitro and in vivo (34). Similarly, two BRD4 inhibitors JQ1 and iBET and CDK9 inhibitors have been shown to be very effective in preventing the proliferation of leukaemia (35,36). However, recent reports of identification and characterization of BRD4 inhibitor resistance leukaemia further indicated that a single drug treatment is not sufficient for complete remission (37,38). On the other hand, the evidences of MLL-fusion protein driven MYB activation hypothesized further that this pathway could activate a series of molecular event which leads to the initiation and later maintenance of leukemia. After activating the MYB transcription, MLL-fusion (MLL-ENL) protein forms a complex with MYB through Menin, a protein encoded by MEN1 contributes significantly in leukemic transformation by maintaining the MLL-mediated H3K4 methylation. MLL-ENL-Menin-MYB complex plays a very important role in leukaemia because MYB lacking Menin interaction domain suppressed MLL-ENL mediated transformation (39). Unpublished data from our laboratory indicated that ectopic expression of MLL-ENL and-AF9 can also activate MYB-reporter construct (40) and this activation required functionally active CDK9. Similarly, CDK9 inhibitor treatment downregulated MYB transcription and prevented the proliferation of cell lines expressing MLL-fusion protein such as MOM13, THP1. Since MYB is an important component of this MLL-Menin-MYB autoregulatory loop and being a direct target of MLL-fusion proteins, targeting MYB in leukaemia could be a potential alternative therapeutic model. Our attempt to target MYB-p300 interaction by using small peptides is currently underway. On the other hand, our preliminary experiments in ectopically MLL-AF9 expressed mouse cell line indicated that transcription is regulated by the proximal pausing mechanism at the first exon close to the transcription site which is not the known pausing site located at 1.7 kb downstream from the transcription start site. Therefore, it is possible that transcription is regulated by different pausing site in leukemic cell lines and functionally active CDK9 is required to drive this transcription by phosphorylating ser-2 residue of RNApolII. A combination of CDK9 and BRD4 inhibitors seems more effective because the latter recruits CDK9 in the SEC and in our preliminary double drug experiments to block the activity of both CDK9 and BRD4 at the same time, appeared several folds more effective to prevent proliferation of MOM13 than using any one of the drugs alone (unpublished).
Figure 1.
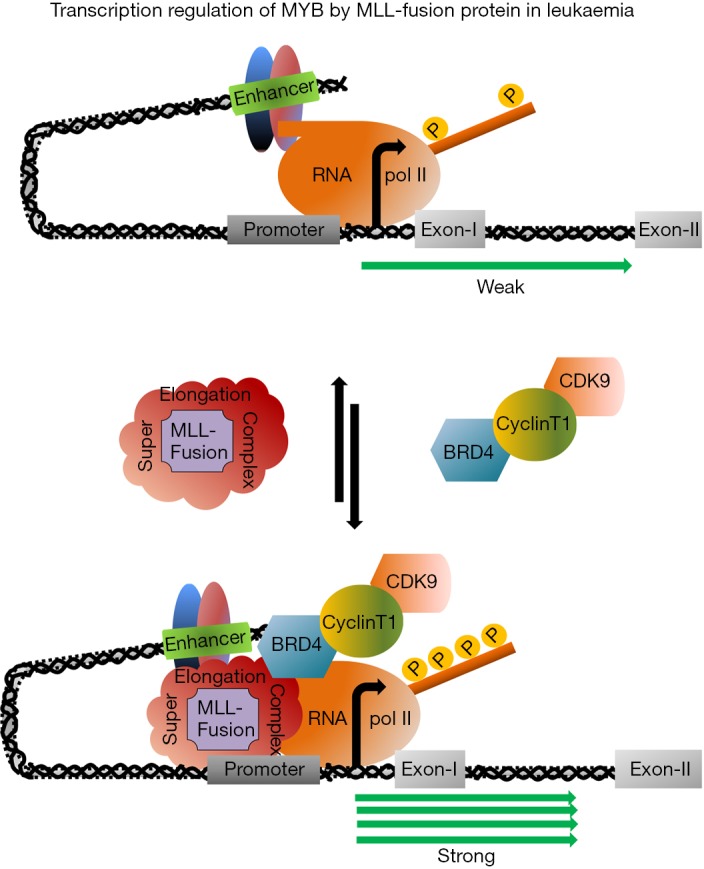
MLL-fusion protein driven transcription of c-MYB. The transcriptionally active super elongation complex is formed when MLL-fusion proteins recruits several proteins along with the core protein complex WARD. Th process initiates with the modulation of the chromosomal three-dimensional structure that brings the enhancer close to the proximal region of the promoter followed by the accumulation of the MLL-fusion protein and other co-factors to form the SEC. Phosphorylation of the CTD of PolII, the penultimate stage of this process, is triggered by the recruitment of the P-TEFb complex by BRD4. CTD, C-terminal domain; SEC, super elongation complex.
Breast cancer
Among the five major subtypes of BC, the survival rate estrogen positive (ER+ve) BC is 10% better than ER-ve and triple negative BCs. In general, patients respond very well against systematic adjuvant endocrine therapy, which includes: (I) ovarian suppression; (II) selective ER modulators (SERMs) and down-regulators, and (III) aromatase inhibitors (AIs). Tamoxifen, though has been considered as the most widely used SERM, has adverse effect on other tissues and replaced by fulvestrant, which is a downregulator of ERs. Inhibition of aromatase system with third generation AIs improve patient survival with advance BC. However, around 30% patients develop resistance after receiving aromatase treatment for a short period of time and those patients respond partially against SERM treatment and in advance stage, eventually develop resistance to the therapy (41-44). In ER+ve BC, MYB expression is regulated by a transcription pausing mechanism, which is operated at 1.7 kb downstream form the transcription start site. Analysis of 72 bp DNA sequence followed by a polyT tract, which plays the central role of this attenuation, indicated that it can form a secondary stem-loop structure upon transcription and perhaps act as a docking site for those positive and negative regulatory factors to control the transcription. A genome wide ER-α binding experiments in MCF-7 cells identified single ER binding site close to the upstream of the pausing sequence. Our subsequent studies demonstrated that in the absence of ligand bound ERs, transcription continued till to the pausing site. However, in the presence of estrogen, ligand bound ER-α occupies the designated upstream binding site and recruits P-TEFb complex. This ER-α-P-TEFb tripartite complex phosphorylates the C-terminal domain ser-2 residue of RNA polII to drive the transcription till to the end of the gene (23,40) (Figure 2). We tested several CDK9 inhibitors such as flavopiridol, AT7519 (45,46) and BE-09-LN5 (47) and observed all of them can effectively downregulate MYB expression several folds when MCF-7 cells were exposed to those drugs for few hours and this downregulation was associated with reimposed pausing of transcription at the attenuation site. Interestingly, we observed that CDK9 inhibitors selectively inhibit proliferation of ER+veMYB+ve but not ER−veMYB−ve cells. Further investigation revealed that CDK9 inhibitors can selectively induce apoptosis in a set of ER+veMYB+ve cells such as MCF-7, T47D and ZR-75 cells but not ER−veMYB−ve cell lines such as MDA-MB-231, MDA-MB-468 and BT-10 cells when exposed for 48 to 72 h at 10–25 nM concentration. More interestingly, ectopically expressed MYB alone is sufficient to protect the MCF-7 cells form this apoptotic induction (48). Further analysis to understand CDK9 inhibitor mediated apoptosis revealed that level of BCL-2, a known MYB target; MCL-1 a known CDK9 target gene and BCL-xL expression was downregulated upon inhibitor treatment. Therefore, we concluded that by using CDK9 inhibitors we can inhibit MYB-Bcl2 axis as well major anti-apoptotic proteins MCL-1 and Bcl-xL to trigger the apoptosis in ER+ve BC cells (48). This CDK9 dependence of ER+veMYB+ve BC cells seems not very unlikely because Wang et al., (49) recently showed that proliferation of aggressive triple negative BC cells such as MDA-MB-468, BT459 absolutely depend on the functionally active CDK7. By using CRISPR/CAS9 gene editing system, they also showed that ER+ve cell line ZR-75, completely depends on CDK9 but not CDK7. Therefore, our data suggest that CDK9 inhibitors can be a potential therapeutic agent to treat estrogen positive BCs. Perhaps another advantage of using CDK9 inhibitors in ER+ve BC is to control aromatase resistant BC. Comprehensive genetic analyses on ER+ve metastatic breast tumours and endocrine therapy resistance patient derived xenografts identified mutations in the ligand binding domain of ER. Several such mutant receptors (D538G, Y537S, Y537N and Y537C) have been characterized further for their transcriptional activity. Those mutant receptors have shown to activate known ER target genes even much higher level in the absence or presence of estrogen in comparison to the wild type receptors and partially sensitive to the AIs. Our preliminary experiments indicated that unlike wild type receptors, ectopically expressed mutant receptors, D538G, Y537S and Y537N activated MYB-reporter in the absence of estrogen and required functionally active CDK9. Our published data indicated that ER-α-P-TEFb interaction is independent of the presence of estrogen (40). Therefore, it is possible that mutant receptors can interact and recruit the P-TEFb complex to activate MYB. We hypothesize that CDK9 inhibitor treatment can be an alternative therapeutic approach to control mutant receptor driven breast tumours by targeting MYB and other anti-apoptotic genes.
Figure 2.
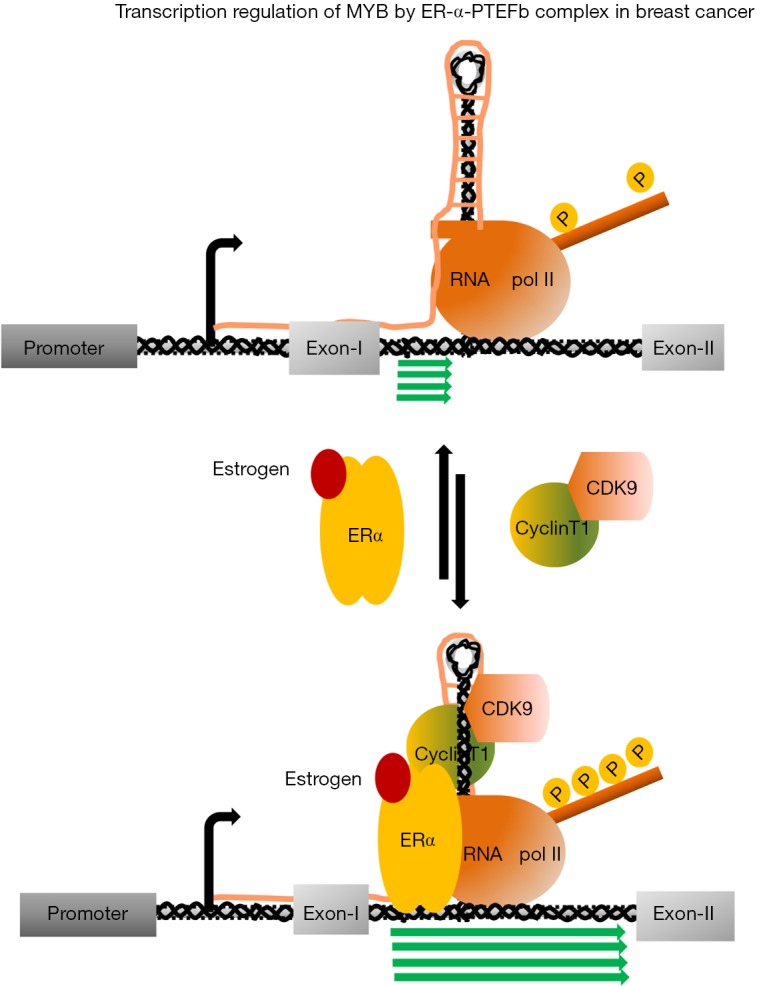
Regulation of transcription of C-MYB in estrogen positive breast cancer. In the absence of estrogen transcription pauses at the 1.7 kb downstream of the transcription start site and generates short transcripts and a stem-loop structure followed by a polyT trail upon transcription (upper panel). In the presence of estrogen, the ER’s binds to the upstream of the 72 bp regulatory elements and form a regulatory complex includes P-TEFb. Phosphorylation of the ser-2 residues at the CTD of RNA PolII drives transcription past the pausing site till to the end of the gene. CTD, C-terminal domain; ER, estrogen receptor.
Colon cancer
Like BC cells, proliferation of colorectal cancer cells (CRC) is also tightly regulated with MYB expression. Downregulation of MYB expression was observed when the human CRC line LIM 1215 was treated with differentiation inducing reagent sodium butyrate (50). The mechanism, by which MYB drives the CRC cell proliferation, was not known for a long period. Very recently, Cheasley et al. showed that MYB regulates one of the important cell cycle regulatory genes CyclinE1 by binding to the distal promoter element of this gene (51). MYB binding in association with the transcription factor E2F-1 and TCF4 is required to activate the promoter to continue the expression of CyclinE1. Like BC cells, nuclear run on analysis in LIM 12151 and CaCo-2 indicated that the transcription is regulated at intron 1. Analysis of the DNA sequence of the 72 bp pausing site followed by ployT tract of several colon tumours and cell lines, identified a significant variability. The length of the polyT tract is shorter in CRC cell line (average size is 12–15 nucleotides) in comparison to the control (average size is 19–21 nucleotides) and this shortening of polyT tract also reflected in 70% CRC of patient samples. On the other hand, it was noticed that patients, with normal length of polyT tract, were accompanied by either disruption with additional nucleotide insertion or acquired mutations in the stem-loop region. The introduction of those point mutations in the stem-loop region and reducing the length of polyT site in MYB-reporter construct increased the level of transcription several folds which corroborates well with the high level of MYB expression in CRCs (52). Altogether, above evidences clearly indicated that the integrity of the three-dimensional structure of this attenuation site, is very important for the regulation of MYB transcription. It was discussed earlier, in case of BC that P-TEFb complex is recruited on this site by ER-α. Therefore, it was hypothesized in CRC, that tissue specific transcription factors and associated co-factors which include P-TEFb complex, may be recruited on this site to drive the transcription. Recent studies of Pereira et al. (53) showed that the p50 subunit of NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) binds to the nascent stem-loop RNA synthesized by transcription of the 72 bp regulatory region. NF-κB p50 subunit forms a heterodimer with p65 subunit of NF-κB which is required to drive the transcription. However, it was not clear whether this NF-κB binding to the stem-loop RNA is very critical to overcome the transcription pausing (Figure 3). Therefore, to establish this hypothesis, it became explicit to get further evidence that RNA polII C-terminal domain (CTD) phosphorylating protein kinase complex, such as P-TEFb is recruited at this docking site. By using NF-κB inhibitor BAY-11-7082 and CDK9 inhibitor DRB, Pereira et al. further demonstrated that both are required for the transcription of MYB in CRC Cells. This observation provides a strong indication of the involvement of CDK9 to initiate transcription form the pausing site. The role of NF-kB in transcription elongation was reported almost a decade ago in a study to understand the role of this protein in the regulation of pro-inflammatory gene IL-8 and ICAM-1 (54). Subsequently, it was shown that P-TEFb is an integral part of this regulatory process and interacts with NF-κB to activate IL-8 transcription (55,56). In case of MYB activation in CRC, perhaps NF-κB is playing the same role like BRD4 in leukaemia and ER-α in BC cells. This NF-κB mediated regulation of MYB in CRC may have a strong correlation with observed sensitivity of CRC to the inhibitors of this protein. Several studies have established the contribution of NF-κB signalling pathway in the initiation and progression of the CRC. Constitutive expression of NF-κB has been reported in more than 66% of CRC cell lines and 50% of CRCs which is associated with resistance to chemotherapy, continued proliferation and angiogenesis (57-59). A strong growth inhibitory effect in combination with apoptosis was observed when human colon carcinoma cell line SW480 and HT29 cells were treated with NF-κB inhibitor BAY11-7082 that blocks TNF-α induced phosphorylation of IκB-α (60,61). In a cell line-based xenograft model, BAY11-7082 was shown to be very effective to reduce the tumour volume, indicating that inhibition of NF-κB has the potential to be an alternative therapeutic intervention in CRC (60,62,63). However, targeting multifunctional protein NF-κB alone may not be very fruitful because it might generate irrecoverable side effect. Targeting transcriptional regulation of MYB where NF-κB is participating with P-TEFb complex can be an alternative therapeutic model for CRCs. Combination of optimal dosages of NF-κB as well as P-TEFb inhibitory drugs may be more effective than either of the drugs alone.
Figure 3.
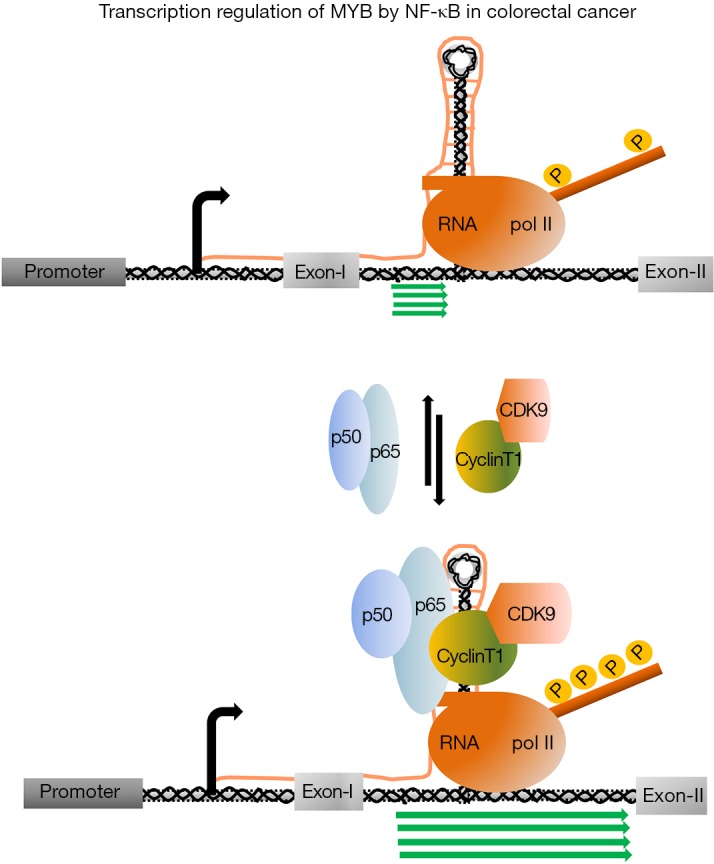
Transcriptional regulation of C-MYB in colon cancer cells. Transcription of the C-MYB depends upon the length of the polyT trail and the stem-loop structure that forms during transcription. The heterodimeric NF-κB p50 binds to the nascent stem-loop RNA forms upon transcription through its p50 subunit and recruits the P-TEFb complex by interacting with its p65 subunit. Multiprotein complex formed by NF-κB-PTEFb functions as a major driver of the transcription.
Conclusions
The hypothesis of targeting transcription regulations, in recent days, is showing a new horizon in the field of molecular approaches to drug discovery. It appeared to be more promising because of our accumulating knowledge in recent years about the associated factors involved in the regulatory process and our preliminary success to inhibit their functions. In depth studies in recent years, helped to identify and characterized several such co-factor proteins such as BRD4, DoT1L, and PTEF-b complex as possible targets in cancer treatment. Inhibitors targeting to those proteins came across very promising results in both in vitro and in vivo and some of them are currently in human clinical trials. Using c-MYB as an example, we have discussed how the expression of this oncogene is inextricably related to certain types of cancer. The unique tissue specific regulation of this gene showed not only both the diversity and the complexity of the mammalian gene transcription machinery, but at the same time, it provides opportunities to identify tissue specific factors or co-factors which could be a new potential target to control the expression of this gene. Though, this model of targeting MYB needs further validation in in vivo models, but this approach of identification of an oncogene addiction to cancer and targeting the regulatory mechanisms that control the expression of the driver oncogene, could be a unique approach for future anticancer drug discovery. Since cancer cells are intelligent enough to figure out a new survival pathway, therefore, further studies are required to investigate the risk of this approach of targeting transcription regulation in developing a resistance phenotype.
Acknowledgements
This review is written based on the data generated in the laboratory of Prof. Thomas J. Gonda, at the University of Queensland, Brisbane, Australia and in the laboratory of Prof. Robert Ramsey, Peter MacCallum Cancer Centre, Melbourne, Australia.
Footnotes
Conflicts of Interest: The author has no conflicts of interest to declare.
References
- 1.Mettus RV, Litvin J, Wali A, et al. Murine A-myb: evidence for differential splicing and tissue-specific expression. Oncogene 1994;9:3077-86. [PubMed] [Google Scholar]
- 2.Trauth K, Mutschler B, Jenkins NA, et al. Mouse A-myb encodes a trans-activator and is expressed in mitotically active cells of the developing central nervous system, adult testis and B lymphocytes. EMBO J 1994;13:5994-6005. 10.1002/j.1460-2075.1994.tb06945.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Toscani A, Mettus RV, Coupland R, et al. Arrest of spermatogenesis and defective breast development in mice lacking A-myb. Nature 1997;386:713-7. 10.1038/386713a0 [DOI] [PubMed] [Google Scholar]
- 4.Saville MK, Watson RJ. B-Myb: a key regulator of the cell cycle. Adv Cancer Res 1998;72:109-40. 10.1016/S0065-230X(08)60701-0 [DOI] [PubMed] [Google Scholar]
- 5.Sala A, Watson R. B-Myb protein in cellular proliferation, transcription control, and cancer: latest developments. J Cell Physiol 1999;179:245-50. 10.1002/(SICI)1097-4652(199906)179:3<245::AID-JCP1>3.0.CO;2-H [DOI] [PubMed] [Google Scholar]
- 6.Kastan MB, Slamon DJ, Civin CI. Expression of protooncogene c-myb in normal human hematopoietic cells. Blood 1989;73:1444-51. [PubMed] [Google Scholar]
- 7.Sitzmann J, Noben-Trauth K, Klempnauer KH. Expression of mouse c-myb during embryonic development. Oncogene 1995;11:2273-9. [PubMed] [Google Scholar]
- 8.Ess KC, Witte DP, Bascomb CP, et al. Diverse developing mouse lineages exhibit high-level c-Myb expression in immature cells and loss of expression upon differentiation. Oncogene 1999;18:1103-11. 10.1038/sj.onc.1202387 [DOI] [PubMed] [Google Scholar]
- 9.Mucenski ML, McLain K, Kier AB, et al. A functional c-myb gene is required for normal murine fetal hepatic hematopoiesis. Cell 1991;65:677-89. 10.1016/0092-8674(91)90099-K [DOI] [PubMed] [Google Scholar]
- 10.Tanaka Y, Patestos NP, Maekawa T, et al. B-myb is required for inner cell mass formation at an early stage of development. J Biol Chem 1999;274:28067-70. 10.1074/jbc.274.40.28067 [DOI] [PubMed] [Google Scholar]
- 11.Rushton JJ, Davis LM, Lei W, et al. Distinct changes in gene expression induced by A-Myb, B-Myb and c-Myb proteins. Oncogene 2003;22:308-13. 10.1038/sj.onc.1206131 [DOI] [PubMed] [Google Scholar]
- 12.Zuber J, Rappaport AR, Luo W, et al. An integrated approach to dissecting oncogene addiction implicates a Myb-coordinated self-renewal program as essential for leukemia maintenance. Genes Dev 2011;25:1628-40. 10.1101/gad.17269211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stadhouders R, Thongjuea S, Andrieu-Soler C, et al. Dynamic long-range chromatin interactions control Myb proto-oncogene transcription during erythroid development. EMBO J 2012;31:986-99. 10.1038/emboj.2011.450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sandberg ML, Sutton SE, Pletcher MT, et al. c-Myb and p300 regulate hematopoietic stem cell proliferation and differentiation. Dev Cell 2005;8:153-66. 10.1016/j.devcel.2004.12.015 [DOI] [PubMed] [Google Scholar]
- 15.Pattabiraman DR, McGirr C, Shakhbazov K, et al. Interaction of c-Myb with p300 is required for the induction of acute myeloid leukemia (AML) by human AML oncogenes. Blood 2014;123:2682-90. 10.1182/blood-2012-02-413187 [DOI] [PubMed] [Google Scholar]
- 16.Thompson MA, Rosenthal MA, Ellis SL, et al. c-Myb down-regulation is associated with human colon cell differentiation, apoptosis, and decreased Bcl-2 expression. Cancer Res 1998;58:5168-75. [PubMed] [Google Scholar]
- 17.Malaterre J, Carpinelli M, Ernst M, et al. c-Myb is required for progenitor cell homeostasis in colonic crypts. Proc Natl Acad Sci U S A 2007;104:3829-34. 10.1073/pnas.0610055104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Biroccio A, Benassi B, D'Agnano I, et al. c-Myb and Bcl-x overexpression predicts poor prognosis in colorectal cancer: clinical and experimental findings. Am J Pathol 2001;158:1289-99. 10.1016/S0002-9440(10)64080-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cross RS, Malaterre J, Davenport AJ, et al. Therapeutic DNA vaccination against colorectal cancer by targeting the MYB oncoprotein. Clin Transl Immunology 2015;4:e30. 10.1038/cti.2014.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kauraniemi P, Hedenfalk I, Persson K, et al. MYB oncogene amplification in hereditary BRCA1 breast cancer. Cancer Res 2000;60:5323-8. [PubMed] [Google Scholar]
- 21.DeRisi J, Penland L, Brown PO, et al. Use of a cDNA microarray to analyse gene expression patterns in human cancer. Nat Genet 1996;14:457-60. 10.1038/ng1296-457 [DOI] [PubMed] [Google Scholar]
- 22.Quintana AM, Liu F, O'Rourke JP, et al. Identification and regulation of c-Myb target genes in MCF-7 cells. BMC Cancer 2011;11:30. 10.1186/1471-2407-11-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Drabsch Y, Hugo H, Zhang R, et al. Mechanism of and requirement for estrogen-regulated MYB expression in estrogen-receptor-positive breast cancer cells. Proc Natl Acad Sci U S A 2007;104:13762-7. 10.1073/pnas.0700104104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miao RY, Drabsch Y, Cross RS, et al. MYB is essential for mammary tumorigenesis. Cancer Res 2011;71:7029-37. 10.1158/0008-5472.CAN-11-1015 [DOI] [PubMed] [Google Scholar]
- 25.Knopfova L, Benes P, Pekarcikova L, et al. c-Myb regulates matrix metalloproteinases 1/9, and cathepsin D: implications for matrix-dependent breast cancer cell invasion and metastasis. Mol Cancer 2012;11:15. 10.1186/1476-4598-11-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Persson M, Andren Y, Mark J, et al. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proc Natl Acad Sci U S A 2009;106:18740-4. 10.1073/pnas.0909114106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gonda TJ, Ramsay RG. Directly targeting transcriptional dysregulation in cancer. Nat Rev Cancer 2015;15:686-94. 10.1038/nrc4018 [DOI] [PubMed] [Google Scholar]
- 28.Torti D, Trusolino L. Oncogene addiction as a foundational rationale for targeted anti-cancer therapy: promises and perils. EMBO Mol Med 2011;3:623-36. 10.1002/emmm.201100176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kelly TK, De Carvalho DD, Jones PA. Epigenetic modifications as therapeutic targets. Nat Biotechnol 2010;28:1069-78. 10.1038/nbt.1678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Williford JM, Wu J, Ren Y, et al. Recent advances in nanoparticle-mediated siRNA delivery. Annu Rev Biomed Eng 2014;16:347-70. 10.1146/annurev-bioeng-071813-105119 [DOI] [PubMed] [Google Scholar]
- 31.Cho YW, Hong T, Hong S, et al. PTIP associates with MLL3- and MLL4-containing histone H3 lysine 4 methyltransferase complex. J Biol Chem 2007;282:20395-406. 10.1074/jbc.M701574200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dou Y, Milne TA, Ruthenburg AJ, et al. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol 2006;13:713-9. 10.1038/nsmb1128 [DOI] [PubMed] [Google Scholar]
- 33.Smith E, Lin C, Shilatifard A. The super elongation complex (SEC) and MLL in development and disease. Genes Dev 2011;25:661-72. 10.1101/gad.2015411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Daigle SR, Olhava EJ, Therkelsen CA, et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 2013;122:1017-25. 10.1182/blood-2013-04-497644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Delmore JE, Issa GC, Lemieux ME, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011;146:904-17. 10.1016/j.cell.2011.08.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garcia-Cuellar MP, Fuller E, Mathner E, et al. Efficacy of cyclin-dependent-kinase 9 inhibitors in a murine model of mixed-lineage leukemia. Leukemia 2014;28:1427-35. 10.1038/leu.2014.40 [DOI] [PubMed] [Google Scholar]
- 37.Rathert P, Roth M, Neumann T, et al. Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature 2015;525:543-7. 10.1038/nature14898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fong CY, Gilan O, Lam EY, et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature 2015;525:538-42. 10.1038/nature14888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jin S, Zhao H, Yi Y, et al. c-Myb binds MLL through menin in human leukemia cells and is an important driver of MLL-associated leukemogenesis. J Clin Invest 2010;120:593-606. 10.1172/JCI38030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mitra P, Pereira LA, Drabsch Y, et al. Estrogen receptor-alpha recruits P-TEFb to overcome transcriptional pausing in intron 1 of the MYB gene. Nucleic Acids Res 2012;40:5988-6000. 10.1093/nar/gks286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Robinson DR, Wu YM, Vats P, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet 2013;45:1446-51. 10.1038/ng.2823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Toy W, Shen Y, Won H, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet 2013;45:1439-45. 10.1038/ng.2822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Early Breast Cancer Trialists' Collaborative Group (EBCTCG) , Davies C, Godwin J, et al. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet 2011;378:771-84. 10.1016/S0140-6736(11)60993-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Palmieri C, Patten DK, Januszewski A, et al. Breast cancer: current and future endocrine therapies. Mol Cell Endocrinol 2014;382:695-723. 10.1016/j.mce.2013.08.001 [DOI] [PubMed] [Google Scholar]
- 45.Squires MS, Cooke L, Lock V, et al. AT7519, a cyclin-dependent kinase inhibitor, exerts its effects by transcriptional inhibition in leukemia cell lines and patient samples. Mol Cancer Ther 2010;9:920-8. 10.1158/1535-7163.MCT-09-1071 [DOI] [PubMed] [Google Scholar]
- 46.Squires MS, Feltell RE, Wallis NG, et al. Biological characterization of AT7519, a small-molecule inhibitor of cyclin-dependent kinases, in human tumor cell lines. Mol Cancer Ther 2009;8:324-32. 10.1158/1535-7163.MCT-08-0890 [DOI] [PubMed] [Google Scholar]
- 47.Lu H, Xue Y, Yu GK, et al. Compensatory induction of MYC expression by sustained CDK9 inhibition via a BRD4-dependent mechanism. Elife 2015;4:e06535. Erratum in: Elife 2015;4:e09993. Xue, Yuahua [corrected to Xue, Yuhua]. 10.7554/eLife.06535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mitra P, Yang RM, Sutton J, et al. CDK9 inhibitors selectively target estrogen receptor-positive breast cancer cells through combined inhibition of MYB and MCL-1 expression. Oncotarget 2016;7:9069-83. 10.18632/oncotarget.6997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Y, Zhang T, Kwiatkowski N, et al. CDK7-Dependent Transcriptional Addiction in Triple-Negative Breast Cancer. Cell 2015;163:174-86. 10.1016/j.cell.2015.08.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ramsay RG, Ciznadija D, Sicurella C, et al. Colon epithelial cell differentiation is inhibited by constitutive c-myb expression or mutant APC plus activated RAS. DNA Cell Biol 2005;24:21-9. 10.1089/dna.2005.24.21 [DOI] [PubMed] [Google Scholar]
- 51.Cheasley D, Pereira L, Sampurno S, et al. Defective Myb Function Ablates Cyclin E1 Expression and Perturbs Intestinal Carcinogenesis. Mol Cancer Res 2015;13:1185-96. 10.1158/1541-7786.MCR-15-0014 [DOI] [PubMed] [Google Scholar]
- 52.Hugo H, Cures A, Suraweera N, et al. Mutations in the MYB intron I regulatory sequence increase transcription in colon cancers. Genes Chromosomes Cancer 2006;45:1143-54. 10.1002/gcc.20378 [DOI] [PubMed] [Google Scholar]
- 53.Pereira LA, Hugo HJ, Malaterre J, et al. MYB elongation is regulated by the nucleic acid binding of NFkappaB p50 to the intronic stem-loop region. PLoS One 2015;10:e0122919. 10.1371/journal.pone.0122919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nissen RM, Yamamoto KR. The glucocorticoid receptor inhibits NFkappaB by interfering with serine-2 phosphorylation of the RNA polymerase II carboxy-terminal domain. Genes Dev 2000;14:2314-29. 10.1101/gad.827900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barboric M, Nissen RM, Kanazawa S, et al. NF-kappaB binds P-TEFb to stimulate transcriptional elongation by RNA polymerase II. Mol Cell 2001;8:327-37. 10.1016/S1097-2765(01)00314-8 [DOI] [PubMed] [Google Scholar]
- 56.Diamant G, Dikstein R. Transcriptional control by NF-kappaB: elongation in focus. Biochim Biophys Acta 2013;1829:937-45. 10.1016/j.bbagrm.2013.04.007 [DOI] [PubMed] [Google Scholar]
- 57.Sakamoto K, Maeda S, Hikiba Y, et al. Constitutive NF-kappaB activation in colorectal carcinoma plays a key role in angiogenesis, promoting tumor growth. Clin Cancer Res 2009;15:2248-58. 10.1158/1078-0432.CCR-08-1383 [DOI] [PubMed] [Google Scholar]
- 58.Voboril R, Weberova-Voborilova J. Constitutive NF-kappaB activity in colorectal cancer cells: impact on radiation-induced NF-kappaB activity, radiosensitivity, and apoptosis. Neoplasma 2006;53:518-23. [PubMed] [Google Scholar]
- 59.Lind DS, Hochwald SN, Malaty J, et al. Nuclear factor-kappa B is upregulated in colorectal cancer. Surgery 2001;130:363-9. 10.1067/msy.2001.116672 [DOI] [PubMed] [Google Scholar]
- 60.Fernandez-Majada V, Aguilera C, Villanueva A, et al. Nuclear IKK activity leads to dysregulated notch-dependent gene expression in colorectal cancer. Proc Natl Acad Sci U S A 2007;104:276-81. 10.1073/pnas.0606476104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rauert-Wunderlich H, Siegmund D, Maier E, et al. The IKK inhibitor Bay 11-7082 induces cell death independent from inhibition of activation of NFkappaB transcription factors. PLoS One 2013;8:e59292. 10.1371/journal.pone.0059292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Scaife CL, Kuang J, Wills JC, et al. Nuclear factor kappaB inhibitors induce adhesion-dependent colon cancer apoptosis: implications for metastasis. Cancer Res 2002;62:6870-8. [PubMed] [Google Scholar]
- 63.Sakamoto K, Maeda S. Targeting NF-kappaB for colorectal cancer. Expert Opin Ther Targets 2010;14:593-601. 10.1517/14728221003769903 [DOI] [PubMed] [Google Scholar]