

Abstract
Fully π-conjugated polycyclic hydrocarbons with antiaromatic character have attracted research attention because of their unique properties such as narrow energy gaps, and thus should find application as optical and electronic materials. Although antiaromatic 16π-electron frameworks can be constructed by the incorporation of multiple seven-membered rings in a fused fashion to install methylenecycloheptatriene (heptafulvene) segments, the development of corresponding benzo[1,2:4,5]di[7]annulene (BDA)-containing π-conjugated systems remains challenging due to the difficulty of their molecular design and synthesis. In this study, we develop an unprecedented chemical structure of cycloheptatriene-bis-annelated indenofluorene, which possesses both BDA and indenofluorene frameworks in a fused fashion. Physical measurements and X-ray analyses, along with theoretical calculations, indicated that antiaromaticity appeared in the BDA framework. By using the conjugated polycyclic hydrocarbon possessing both seven-membered and five-membered rings, this study provides fundamental insight into the strong antiaromatic nature of heptafulvene-based BDA framework.
Introduction
Increased interest has emerged in fully π-conjugated polycyclic hydrocarbons, which possess electronic properties desirable for potential applications as semiconducting materials in organic thin-film electronics1–7. While the majority of these systems are based on fused [4n + 2] π-electron aromatic molecules such as pentacene, the utilization of a [4n] π-electron antiaromatic character has emerged as a useful strategy to create new molecules with unusual electronic structures8–11. Since it is well known that the representative 8π- and 12π-electron frameworks pentalene and indacene (ID), respectively, are unstable due to the high antiaromatic character, the incorporation of methylenecyclopentadiene (fulvene) segments into an acene system has become a rational molecular design to construct [4n] π molecules. Indeed, dibenzopentalene12–28 and indenofluorene (IF)29–37 derivatives have been intensively developed in recent years (Fig. 1)38,39. These fulvene units can accept an electron to form stable aromatic cyclopentadienide anions and, thus, polycyclic fully conjugated hydrocarbons containing five-membered rings have intrinsically electron-accepting characteristics31. An alternative approach to access antiaromatic frameworks relies on the use of methylenecycloheptatriene (heptafulvene) segments to form tricyclic 16π-electron benzo[1, 2:4, 5]di[7]annulene (BDA), whereas pristine heptalene exhibits nonaromatic character due to its twisted non-planar geometry40. Although several polycyclic hydrocarbons containing a BDA framework have been synthesized41–47, the development of BDA-based antiaromatic compounds is still limited due to the difficulty of (1) the molecular design to incorporate heptafulvene units into the π-conjugated systems and (2) the synthetic method for the construction of seven-membered rings41,42,44–46. In contrast to the fulvene unit, the formation of cycloheptatrienyl cation by one-electron oxidation of heptafulvene leads to aromatic stabilization. Thus, the thin-film of 1 showed hole-transporting characteristics in organic field-effect transistor (OFET) devices44–46. However, irrespective of these electronically complemental properties, fundamental studies to directly investigate the antiaromatic character between the ID and BDA frameworks have not been carried out so far. Therefore, we sought to construct an unprecedented π-conjugated polycyclic hydrocarbons by introducing multiple five-membered and seven-membered rings together in a conjugated molecule. Based on this strategy, we combined ID and BDA, resulting in the molecular framework of dicyclohepta[cd,ij]-s-indacene (DCHI), and synthesized the benzene-fused cycloheptatriene-bis-annelated indenofluorene 2 (Fig. 1). Herein, we describe the synthesis, structure, properties, and OFET characteristics of 2.
Figure 1.

Chemical structures of representative antiaromatic compounds and target compound 2.
Results and Discussion
Although the formation of the DCHI framework by photochemical reaction of o-diethynylbenzene derivatives was previously inferred48, it was disproven based on theoretical calculations of the spectroscopic data49 and reconsidered50. Therefore, the synthesis of this framework was heretofore unknown. With these precedents in mind, we used an olefin metathesis reaction as a key step to construct the seven-membered ring framework. The synthetic route to 2 starting from compound 3 is shown in Fig. 2. The Suzuki coupling reaction between 3 and allylboronic acid pinacol ester in the presence of [Pd(allyl)Cl]2 afforded di-allylated compound 4, which was reacted with allylmagnesium bromide to give the tetra-allylated compound 5. The construction of seven-membered rings was achieved by olefin metathesis using the second generation Grubbs’ catalyst to give key intermediate 6 in 95% yield51. We here report that compounds 5 and 6 were isolated as a mixture of diastereomers. Then, treatment of 6 with the Burgess reagent provided dehydrated compound 752, and subsequent oxidation with 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ) produced the target compound 2 in 50% yield. Note that the presence of the hexyl group is essential to impart enough solubility of 2 to enable characterization by NMR and other spectroscopic measurements. Detailed synthetic procedures and characterization data of the new compounds are summarized in the Methods section. The NMR spectra of 2 are shown in Supplementary Figs 1 and 2. As shown in Supplementary Fig. 2, the 1H NMR spectrum of 2 in CDCl3 showed a downfield shift of the benzene protons by 0.42–0.62 ppm as compared to the indenofluorene derivative IF(Me)-TA (structure shown in Supplementary Fig. 2), indicating the increased contribution of aromatic character in the benzene ring for 2. The proton signals in the seven-membered ring were observed in the upfield region between 5.95 and 7.29 ppm. Since the 1H NMR signals for the seven-membered ring of the antiaromatic BDA framework are reported to appear in a similar δ range of 4.88–6.30 ppm41, contribution of the antiaromatic character in the seven-membered ring of 2 is expected.
Figure 2.

Synthetic route of 2.
Thermogravimetric analysis (TGA) of 2 showed a 5% weight-loss temperature of 443 °C under a nitrogen atmosphere (Supplementary Fig. 3(a)). The differential scanning calorimetry (DSC) profile of 2 showed endothermic and exothermic peaks at 234 and 250 °C, respectively, during the first heating process (Supplementary Fig. 3(b)). On the other hand, no clear peaks were detected during the second heating process, and an insoluble black solid was observed after the DSC measurements. Given that only a slight weight loss was seen in TGA up to 443 °C, we considered that 2 melted at 234 °C, and intermolecular reactions occurred in the melting state to give unidentifiable insoluble products.
Since the indenofluorene derivatives include a p-quinodimethane core, it is known that these molecules show biradical character originating from the aromatization of the central benzene ring53. Thus, the singlet biradical characters (y) of 2(Me) and IF were estimated using the occupation numbers of the spin-unrestricted Hartree–Fock natural orbitals. As a result, 2(Me) displayed a moderate singlet biradical character (y = 0.49), which is larger than that of IF (y = 0.30). Since the calculated spin density for 2(Me) shows the largest amplitude at the fused carbons between the five- and seven-membered rings and is distributed to the seven-membered ring (Fig. 3), the biradicaloid electronic structure is thought to be stabilized by the spin delocalization at the seven-membered rings. The theoretically estimated singlet-triplet energy gap (ΔES-T ≡ ET − ES) of 2(Me) at the UB3LYP/6-31 G(d,p) level is a large positive value of 92.5 kJ mol−1, indicating that the biradical structure of 2 has an exclusively singlet nature, which is consistent with the observation of sharp 1H NMR signals (Supplementary Fig. 1).
Figure 3.
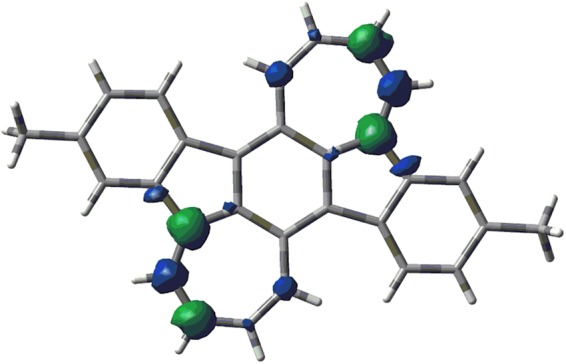
Spin density distribution of 2(Me) calculated at the UB3LYP/6-31 G(d) level.
The electrochemical behavior of 2 was investigated by cyclic voltammetry (CV) and differential pulse voltammetry (DPV) measurements in CH2Cl2 containing 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) as a supporting electrolyte. All potentials were calibrated against a ferrocene/ferrocenium (Fc/Fc+) couple as the standard. As shown in Fig. 4, the CV of 2 revealed three oxidation and two reduction processes, and the redox potentials were determined from DPV. From the first oxidation potential (Eox1) and first reduction potential (Ered1) and the assumption that the energy level of Fc/Fc+ is −4.8 eV below the vacuum level54–56, the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels (EHOMO and ELUMO) of 2 were estimated to be −4.69 and −3.46 eV, respectively. Based on these values, the HOMO-LUMO energy gap of 2 is calculated to be 1.23 eV. Interestingly, these EHOMO and ELUMO values are significantly different from those of IF(Me)-TA (EHOMO = −5.84 eV and ELUMO = −3.99 eV)30. In order to understand the origin of this phenomenon, density functional theory (DFT) calculations of 2(H), IF, and BDA at the B3LYP/6-311 + G(d, p) level were performed. As shown in Fig. 5, the theoretically estimated EHOMO and ELUMO values of 2(H) and IF qualitatively mimic the experimental values. Although the HOMO and LUMO orbitals of 2(H) are delocalized over the π-conjugated backbones, both the EHOMO and ELUMO values of 2(H) are between those of BDA and IF and closer to those of BDA. These results indicate that the electronic structure of the 2(H) molecule is formed from a hybrid of those of BDA and IF, and that the contribution of the electronic character of BDA seems to be dominant.
Figure 4.

CV (top) and DPV (bottom) of 2 in CH2Cl2 containing 0.1 M TBAPF6.
Figure 5.

Energy levels and molecular orbitals of BDA (left), 2(H) (center), and IF (right) calculated at B3LYP/6-311 + G(d,p) level.
To investigate the photophysical properties, a UV-vis-NIR absorption measurement of 2 in a CH2Cl2 solution was performed. As shown in Fig. 6, the absorption spectrum of 2 includes three intense bands with absorption maxima (λmax) at 279 (ε = 55000 M−1 cm−1), 498 (36000 M−1 cm−1), and 692 (26000 M−1 cm−1) nm and weak bands at 866 and 997 nm. A time-dependent (TD)-DFT calculation at the B3LYP/6-31 G (d,p) level indicated that these intense bands were mainly assigned to HOMO − 1 to LUMO + 1, HOMO to LUMO + 2, and HOMO to LUMO transitions, respectively (see the Electronic Supplementary Information). Considering that 2(Me) possesses a moderate singlet biradical character (Fig. 3), the weak bands in the NIR region are assignable to the double excitation state of an open biradicaloid electronic structure57,58. Compared to the solution spectra, a new broad band was observed in the thin-films. This phenomenon is attributed to the intermolecular electronic interactions of π−π stacked backbones, which is favorable for carrier transport in thin-film devices (discussed later).
Figure 6.
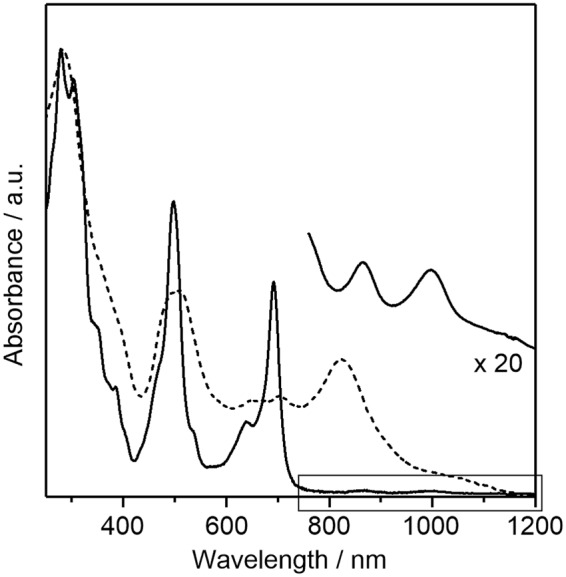
UV-vis-NIR absorption spectra of 2 in CH2Cl2 (solid line) and thin-films (dashed line). The magnified spectrum in CH2Cl2 between 800 and 1200 nm is also shown.
The molecular structure of 2 was unambiguously determined using X-ray crystallographic analysis of crystals grown by the slow evaporation of hexane/CHCl3 solutions. As shown in Fig. 7(a), the π-conjugated framework of compound 2 holds a nearly planar structure: the deviations of carbon atoms constituting the BDA core from the mean plane of this core are less than 0.04 Å, and the dihedral angles between the mean planes of central BDA and the outer benzene rings are 4.9°, as shown in Fig. 7(a). The structure of 1 was reported to show clear bond alternation in the p-quinodimethane core due to the increased contribution from resonance form 1a (structure shown in Supplementary Fig. 4), which in turn is attributed to the presence of the outer fused benzene rings44. On the other hand, the extent of the bond alternation for the BDA framework in 2 was rather small, with C-C bond lengths varying within the range of 1.37–1.44 Å (Fig. 7(b)). To further assess the degree of bond alternation, we calculated the harmonic oscillator model of aromaticity (HOMA) values59 based on the reported X-ray information. As shown in Supplementary Fig. 5, the HOMA value of the BDA framework in 1 was determined to be 0.31. In contrast, the HOMA value of the BDA framework in 2 was calculated to be as high as 0.81, supporting the small degree of bond alternation and resulting delocalized electronic structure. A similar trend was also observed for the antiaromatic ID framework: the absence of fused benzene rings in compound A leads to an increase in the HOMA value (Supplementary Fig. 5). These results indicate that pristine antiaromatic frameworks show large HOMA values; thus, the BDA framework in 2 is expected to show antiaromatic character. Interestingly, this HOMA value is larger than that of the indenofluorene framework in 2 (0.56), indicating that the contribution of 16π (BDA) and 6π × 2 (two outer benzene rings) is larger than that of 20π (indenofluorene) in the electronic structure of 2. To investigate the electronic contribution of the π electrons in 2, we performed an electron localization function (ELF) estimation60. The ELF isosurface plot (Fig. 7(c)) showed that the π electron pairs are uniformly distributed over the entire molecule. This result clearly indicates that the π-conjugation is delocalized over the entire π conjugated framework in 2, despite the different HOMA values of BDA and IF frameworks.
Figure 7.

(a) ORTEP drawing of 2 for top view and side view; (b) bond lengths for the crystal structure (left) and HOMA values for BDA and IF frameworks. (c) Isosurface plot of 2 at an isosurface level of 0.73.
Based on this molecular structure, we estimated the aromaticity of the π-conjugated framework of 2(H) by the nucleus independent chemical shift (NICS). The results of the NICS(1.7)πzz-XY-scans61 are summarized in Fig. 8. The NICS(1.7)πzz values of 2(H), calculated at the GIAO-B3LYP/6-311 + G(d) level, are δ = +12.40, +6.19, −9.48, and −18.90 ppm for the rings A, B, C, and D, respectively. The positive values of rings A and B indicate that the BDA framework has an antiaromatic character. On the other hand, the indenofluorene core composed of rings C and D in 2(H) exhibits an aromatic character, which is in contrast with the result of pristine indenofluorene IF, in which the five-membered ring shows an antiaromatic character33. Since framework 2(H) contains both five- and seven-membered rings, this calculated aromatic character of the C ring is considered to result from increased electron density owing to electrical polarization between these rings. This was confirmed by the calculation of the electrostatic potential of 2(H), which was similar to that calculated for benz[a]azulene, as shown in Supplementary Fig. 6. These results are in agreement with the aforementioned findings based on the HOMA values, indicating the large electronic contribution of the BDA framework. To further support the antiaromaticity of the BDA framework in 2, we conducted anisotropy of the current-induced density (ACID) analysis62. Compound 2 showed continuous paratropic ring currents in the BDA framework (Fig. 9). On the other hand, diatropic ring currents were seen in the C5-C6 frameworks. This result clearly indicates that 2 has local antiaromaticity in the BDA framework and local aromaticity in the C5-C6 frameworks, consistent with the results of NICS calculation.
Figure 8.

(a) NICS(1.7)πzz-X-scans for 2(H) (black) and IF (red) and (b) NICS(1.7)πzz-Y-scans for 2(H).
Figure 9.

ACID-derived induced ring current map for 2.
In the molecular packing diagram, 2 takes a herringbone π-stacked motif with minimum intermolecular π–π distances of 3.42 Å (Supplementary Fig. 7(a)). On the basis of the calculation by the Amsterdam Density Functional (ADF) program at the PW91/TZP level, the transfer integrals for hole transport (tHOMO) and electron transport (tLUMO) between adjoining molecules were estimated. As summarized in Supplementary Fig. 7(b), 2 showed large tHOMO and tLUMO of 91.4 and 111.6 meV, respectively, between facial-stacked molecules, and, thus, the construction of charge-carrier transporting pathways is expected along the stacking direction.
Reflecting the high thermal stability of 2, the thin-films for OFET measurements could be prepared by vacuum deposition onto hexamethyldisilazane (HMDS)-modified Si/SiO2 substrates. The atomic force microscopy (AFM) image of this film exhibited interconnected micrometer-sized grains (Supplementary Fig. 8(a)). X-ray diffraction (XRD) of the thin-film showed clear diffractions, indicating the formation of crystalline structures in thin-films. According to the X-ray crystal structure (Supplementary Fig. 7(a)), the peak at 2θ = 5.2° can be indexed as a (001) diffraction peak with a d spacing of 17.0 Å, implying that the molecules are aligned with the crystal c-axis perpendicular to the SiO2 surface. To evaluate the charge-transport characteristics of the thin-films, OFET devices with bottom-gate bottom-contact configuration were fabricated. As shown in Fig. 10, this device showed hole-transporting characteristics with a field-effect hole mobility of 3.0 × 10−5 cm2 V−1 s−1 with a current on/off ratio of 105. On the other hand, electron-transporting behavior was not observed for 2. This p-type response is explained by the high-lying HOMO energy level of 2. Importantly, taking the intrinsic electron-transporting behavior of the indenofluorene chromophore into consideration, embedding the BDA framework significantly influences the type of charge carriers. This trend is in good agreement with the estimated antiaromaticity of the BDA framework.
Figure 10.
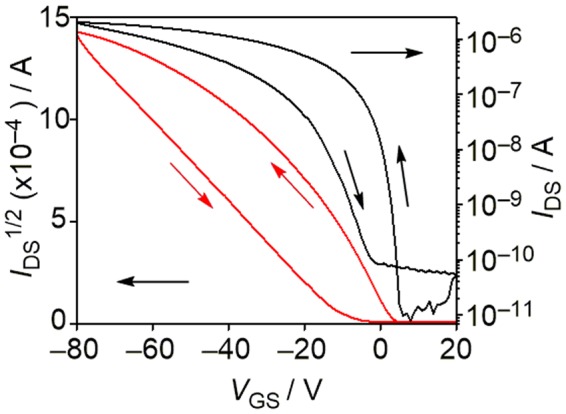
OFET transfer characteristics of thin-film based on 2. IDS and VGS denote source-drain current and gate voltage, respectively.
Summary
In conclusion, in order to directly investigate the antiaromatic character between the ID and BDA frameworks, we successfully synthesized new polycyclic hydrocarbon 2, which contains a fused dicyclohepta[cd,ij]-s-indacene framework in the molecule. Electrochemical and photophysical measurements revealed that 2 has a relatively high HOMO energy level and a narrow HOMO-LUMO energy gap, and these properties mainly come from the BDA framework. Investigation of the X-ray crystal structure and theoretical calculation indicates that the contribution of antiaromatic character for the BDA framework is dominant in the molecule. These results clearly demonstrate that the unique character of 2 originated from the heptafulvene-based BDA framework, and we can conclude that the development of new antiaromatic compounds possessing the DCHI or BDA frameworks will pave the way to the fundamental understanding of antiaromaticity. Considering that 2 shows hole-transporting characteristics, fine-tuning of molecular design would aid the development of high-performance electronic materials. Further investigation on the development of such compounds to reveal the structure-property-semiconducting performance relationship is currently underway in our group.
Methods
General Information
Column chromatography was performed on silica gel. KANTO Chemical silica gel 60N (40–50 μm). Thin-layer Chromatography (TLC) plates were visualized with UV light. Preparative gel-permeation chromatography (GPC) was performed on a Japan Analytical LC-918 equipped with JAI-GEL 1H/2H. 1H and 13C NMR spectra were recorded on a JEOL JNM-ECS400 or JEOL JNM-ECA600 spectrometer in CDCl3 with tetramethylsilane (TMS) as an internal standard. Data are reported as follows: chemical shift in ppm (δ), multiplicity (s = singlet, d = doublet, t = triple, m = multiplet, br = broad), coupling constant (Hz), and integration. UV-vis-NIR spectra were recorded on a Shimadzu UV-3600 spectrophotometer. All spectra were obtained in spectrograde solvents. TGA and DSC were performed under nitrogen at a heating rate of 10 °C min−1 with a Shimadzu TGA-50 and a Shimadzu DSC-60, respectively. Cyclic voltammetry was carried out on a BAS CV-620C voltammetric analyzer using a platinum disk as the working electrode, platinum wire as the counter electrode, and Ag/AgNO3 as the reference electrode at a scan rate of 100 mV s−1. High-resolution mass spectrum (HRMS) was obtained atmospheric pressure chemical ionization (APCI) or electrospray ionization (ESI) methods using a Thermo scientific LTQ Orbitrap XL. Elemental analyses were performed on PerkinElmer LS-50B by the elemental analysis section of the Comprehensive Analysis Center (CAC) of ISIR, Osaka University. The surface structures of the deposited organic film were observed by atomic force microscopy (Shimadzu, SPM9600), and the film crystallinity was evaluated by an X-ray diffractometer (Rigaku, SmartLab). X-ray diffraction patterns were obtained using Bragg-Brentano geometry with CuKα radiation as an X-ray source with an acceleration voltage of 45 kV and a beam current of 200 mA. The scanning mode was set to 2θ–θ scans between 2°–30° with scanning steps of 0.01°.
Synthetic information
Unless stated otherwise, all reagents were purchased from commercial sources and used without purification. Synthetic procedure of 3 was shown in Supplementary Fig. 9, and the corresponding characterization data was summarized in the Electronic Supplementary Information.
Synthesis of 4
Compound 3 (200 mg, 0.329 mmol), allylboronic acid pinacol ester (0.240 mL, 1.32 mmol), allylpalladium(II) chloride dimer (24.0 mg, 0.0658 mmol), SPhos (108 mg, 0.263 mmol), and K3PO4 (350 mg, 1.65 mmol), 1,4-dioxance (7.0 mL) and water (1.9 mL) were added to a reaction vial. The vial was purged with N2. After stirring for 4 h at 40 °C, water was poured into the reaction mixture. The resultant mixture was extracted with ethyl acetate (EtOAc) and the combined organic layer was washed with brine. After drying with MgSO4, the solvent was removed under reduced pressure and the residue was purified by column chromatography on silica gel (hexane: CHCl3 = 2: 1) to give 4 (113 mg, 65%) as a magenta solid. 1H NMR (400 MHz, CDCl3, TMS): δ 0.89 (t, J = 6.9 Hz, 6 H), 1.27–1.38 (m, 12 H), 1.56–1.65 (m, 4 H), 2.66 (t, J = 7.6 Hz, 4 H), 4.24 (d, J = 5.0 Hz, 4 H), 5.08–5.13 (m, 4 H), 6.05–6.16 (m, 2 H), 7.30 (dd, J = 7.8, 1.8 Hz, 2 H), 7.47 (d, J = 1.8 Hz, 2 H), 7.57 (d, J = 7.8 Hz, 2 H); 13C NMR (150 MHz, CDCl3): δ 14.1, 22.6, 28.9, 30.3, 31.1, 31.7, 35.6, 116.4, 123.6, 124.0, 133.9, 134.3, 134.9, 134.9, 136.1, 140.2, 144.1, 145.5; HRMS (m/z): [M + H]+ calcd. for C38H43O2, 531.3258; found, 531.3248.
Synthesis of 5
To a solution of 4 (190 mg, 0.358 mmol) in THF (23 mL), 1.0 M allylmagnesium bromide solution in Et2O (0.788 mL, 0.788 mmol) was added at −78 °C. After stirring for 1 h at −78 °C, the reaction was quenched by pouring saturated NH4Cl aqueous solution. The combined organic was extracted with EtOAc and washed with water. After drying over with MgSO4, the solvent was removed under reduced pressure to give 5 (220 mg, quant.) as an orange solid. 1H NMR (600 MHz, CDCl3, TMS): δ 0.89 (t, J = 6.9 Hz, 6 H), 1.27–1.38 (m, 12 H), 1.59–1.68 (m, 4 H), 2.19–2.23 (m, 2 H), 2.61–2.69 (m, 4 H), 2.92–3.07 (m, 4 H), 3.95–4.01 (m, 1 H), 4.08–4.14 (m, 1 H), 4.30–4.36 (m, 1 H), 4.42–4.47 (m, 1 H), 4.65–4.77 (m, 4 H), 4.86–4.96 (m, 2 H), 5.06–5.16 (m, 4 H), 6.14–6.23 (m, 2 H), 7.11–7.15 (m, 2 H), 7.32 (s, 2 H), 7.54–7.60 (m, 2 H); 13C NMR (150 MHz, CDCl3): δ 14.1, 22.6, 28.9, 29.0, 31.5, 31.8, 31.9, 35.9, 36.0, 43.7, 43.8, 82.7, 116.1, 116.2, 117.9, 118.0, 122.8, 123.0, 123.4, 123.5, 128.8, 128.9, 129.5, 129.9, 132.5, 132.6, 136.0, 136.2, 136.4, 138.7, 138.8, 142.1, 147.3, 147.5, 149.8; HRMS (m/z): [M + Na]+ calcd. for C44H54O2Na, 637.4016; found, 637.4014.
Synthesis of 6
Grubbs catalyst (22.4 mg, 0.0264 mmol) was added to a solution of 5 (203 mg, 0.330 mmol) in CH2Cl2 (8.0 mL), and the reaction was refluxed. After stirring for 2 h, the reaction mixture was filtrated through pad of celite. The solvent was removed under reduced pressure, and the residue was purified by column chromatography on silica gel (CHCl3) to give 6 (175 mg, 95%) as a brown solid. 1H NMR (600 MHz, CDCl3, TMS): δ 0.89 (t, J = 6.5 Hz, 6 H), 1.28–1.40 (m, 12 H), 1.62–1.69 (m, 4 H), 2.30–2.40 (m, 4 H), 2.66 (t, J = 7.6 Hz, 4 H), 2.98–3.04 (m, 2 H), 4.09–4.21 (m, 4 H), 5.68–5.74 (m, 2 H), 5.89–6.00 (m, 2 H), 7.16–7.19 (m, 2 H), 7.34–7.37 (m, 2 H), 7.73–7.81 (m, 2 H); 13C NMR (150 MHz, CDCl3): δ 14.1, 22.6, 28.4, 28.9, 29.1, 31.5, 31.7, 35.9, 38.8, 38.9, 80.9, 81.0, 123.0, 123.1, 123.2, 125.5, 125.9, 126.1, 126.4, 129.1, 131.1, 131.4, 135.0, 135.1, 136.7, 136.8, 142.4, 142.5, 148.5, 148.9, 150.8, 150.9; HRMS (m/z): [M + Na]+ calcd. for C40H46O2Na, 581.3390; found, 581.3392.
Synthesis of 7
Burgess reagent (180 mg, 0.754 mmol) was added to a solution of 6 (175 mg, 0.314 mmol) in THF (7.0 mL). After stirring for 1 h at 65 °C, the reaction was quenched by pouring water. The combined organic was extracted with EtOAc and washed with water. After drying over with MgSO4, the solvent was removed under reduced pressure and the residue was purified by column chromatography on silica gel (hexane: CHCl3 = 1: 1) to give 7 (110 mg, 67%) as a brown solid. 1H NMR (400 MHz, CDCl3, TMS): δ 0.90 (t, J = 6.9 Hz, 6 H), 1.28–1.42 (m, 12 H), 1.64–1.74 (m, 4 H), 2.70 (t, J = 7.3 Hz, 4 H), 3.76 (d, J = 7.3 Hz, 4 H), 5.95–6.04 (m, 2 H), 6.46–6.53 (m, 2 H), 7.14–7.19 (m, 4 H), 7.59 (d, J = 1.4 Hz, 2 H), 7.98 (d, J = 7.8 Hz, 2 H); 13C NMR (150 MHz, CDCl3): δ 14.1, 22.7, 29.0, 29.4, 31.6, 31.8, 36.1, 120.1, 122.3, 122.8, 122.9, 123.8, 127.4, 127.9, 133.3, 137.3, 140.9, 141.2, 141.3, 141.9; HRMS (m/z): [M + H]+ calcd. for C40H43, 523.3359; found, 523.3353.
Synthesis of 2
To a solution of 7 (22 mg, 0.042 mmol) in CH2Cl2 (3.2 mL), a solution of DDQ (9.6 mg, 0.042 mmol) in CH2Cl2 (1.0 mL) was added slowly at −40 °C. After stirring for 20 min, the reaction was allowed to warm to ambient temperature. The solvent was removed under reduced pressure and the residue was washed with acetone to give 2 (11 mg, 50%) as a dark green solid. Recrystallization from hexane/CHCl3 gave a pure compound. mp: 233–235 °C; 1H NMR (400 MHz, CDCl3, TMS): δ 0.89 (t, J = 6.9 Hz, 6 H), 1.27–1.38 (m, 12 H), 1.64–1.72 (m, 4 H), 2.75 (t, J = 7.6 Hz, 4 H), 5.92–5.97 (m, 2 H), 6.46–6.51 (m, 2 H), 7.02 (d, J = 11.5 Hz, 2 H), 7.28–7.34 (m, 4 H), 7.66 (s, 2 H), 7.80 (d, J = 8.7 Hz, 2 H); 13C NMR (175 MHz, CDCl3): δ 14.2, 22.6, 29.1, 31.8, 31.9, 36.2, 119.5, 120.7, 123.6, 126.0, 128.8, 129.0, 133.0, 134.4, 134.6, 136.5, 138.0, 140.4, 149.1, 150.6; UV/vis (CH2Cl2): λmax (ε [M−1 cm−1]) = 279 (55000), 304 (51000), 498 (36000), 692 (26000); HRMS (m/z): [M + H]+ calcd. for C40H41, 521.3203; found, 521.3196.
Electronic supplementary material
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research (B) (16H04191) and Innovative Areas (JP25110004) and “Dynamic Alliance for Open Innovation Bridging Human, Environmental and Materials” from The Ministry of Education, Culture, Sports, Science and Technology, Japan and ACT-C programs from the Japan Science and Technology Agency Japan. Thanks are extended to the Comprehensive Analysis Center (CAC), ISIR, for assistance in obtaining elemental analyses and HRMS. We acknowledge to Prof. H. Shinokubo at Nagoya University and Prof. Y. Tobe and Dr. T. Ohto at Osaka University for helpful discussion. We also acknowledge to Ms. Y. Okumura at ISIR for technichal support of calculation.
Author Contributions
K.Y. contributed all the experiments. N.T. contributed X-ray analyses. F.K. contributed experimental supports for OFETs. Y.I. and Y.A. designed this study.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Yutaka Ie, Email: yutakaie@sanken.osaka-u.ac.jp.
Yoshio Aso, Email: aso@sanken.osaka-u.ac.jp.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-35839-w.
References
- 1.Bendikov M, Wudl F, Perepichka DF. Tetrathiafulvalenes, oligoacenenes, and their buckminsterfullerene derivatives: the brick and mortar of organic electronics. Chem. Rev. 2004;104:4891–4945. doi: 10.1021/cr030666m. [DOI] [PubMed] [Google Scholar]
- 2.Anthony JE. Functionalized acenes and heteroacenes for organic electronics. Chem. Rev. 2006;106:5028–5048. doi: 10.1021/cr050966z. [DOI] [PubMed] [Google Scholar]
- 3.Anthony JE. The larger acenes: versatile organic semiconductors. Angew. Chem. Int. Ed. 2008;47:452–483. doi: 10.1002/anie.200604045. [DOI] [PubMed] [Google Scholar]
- 4.Zade SS, Bendikov M. Heptacene and beyond: the longest characterized acenes. Angew. Chem. Int. Ed. 2010;49:4012–4015. doi: 10.1002/anie.200906002. [DOI] [PubMed] [Google Scholar]
- 5.Li J, Zhang Q. Mono- and oligocyclic aromatic ynes and diynes as building blocks to approach larger acenes, heteroacenes, and twistacenes. Synlett. 2013;24:686–696. doi: 10.1055/s-0032-1318157. [DOI] [Google Scholar]
- 6.Watanabe M, Chen K-Y, Chang YJ, Chow T. Acenes generated from precursors and their semiconducting properties. Acc. Chem. Res. 2013;46:1606–1615. doi: 10.1021/ar400002y. [DOI] [PubMed] [Google Scholar]
- 7.Ye Q, Chi C. Recent highlights and perspectives on acene based molecules and materials. Chem. Mater. 2014;26:4046–4056. doi: 10.1021/cm501536p. [DOI] [Google Scholar]
- 8.Randić M. Aromaticity of polycyclic conjugated hydrocarbons. Chem. Rev. 2003;103:3449–3605. doi: 10.1021/cr9903656. [DOI] [PubMed] [Google Scholar]
- 9.Kertesz M, Choi CH, Yang S. Conjugated polymers and aromaticity. Chem. Rev. 2005;105:3448–3481. doi: 10.1021/cr990357p. [DOI] [PubMed] [Google Scholar]
- 10.Hopf H. Pentalenes−from highly reactive antiaromatics to substrates for material science. Angew. Chem. Int. Ed. 2013;52:12224–12226. doi: 10.1002/anie.201307162. [DOI] [PubMed] [Google Scholar]
- 11.Breslow R. Novel aromatic and antiaromatic systems. Chem. Rec. 2014;14:1174–1182. doi: 10.1002/tcr.201402070. [DOI] [PubMed] [Google Scholar]
- 12.Blood, C. T. & Linstead, R. P. Fused carbon rings. Part XXI. Dibenzopentalene. J. Chem. Soc. 2263–2268 (1952).
- 13.Chuen CC, Fenton SW. Dibenzopentalene. J. Org. Chem. 1958;23:1538–1539. doi: 10.1021/jo01104a037. [DOI] [Google Scholar]
- 14.Brown R, Eastwood F, Wong N. The ethyne-ethylidene rearrangement: formation of indeno[2,1-a]indene and fluoranthene on flash vacuum pyrolysis of 1,4-diphenylbutadiyne. Tetrahedron Lett. 1993;34:3607–3608. doi: 10.1016/S0040-4039(00)73648-3. [DOI] [Google Scholar]
- 15.Preda DV, Scott LT. Phenyl migrations in dehydroaromatic compounds. A new mechanistic link between alternant and nonalternant hydrocarbons at high temperatures. Org. Lett. 2000;2:1489–1492. doi: 10.1021/ol005849z. [DOI] [PubMed] [Google Scholar]
- 16.Kendall JK, Shechter H. Intramolecular behaviors of anthryldicarbenic systems: dibenzo[b,f]pentalene and 1h,5h-dicyclobuta[de,kl]anthracene. J. Org. Chem. 2001;66:6643–6649. doi: 10.1021/jo010404p. [DOI] [PubMed] [Google Scholar]
- 17.Babu G, Orita A, Otera J. Facile carbolithiation of bent alkyne without catalyst. Tandem route to dibenzo[b,f]pentalenes from dibenzocyclooctadiyne. Chem. Lett. 2008;37:1296–1297. doi: 10.1246/cl.2008.1296. [DOI] [Google Scholar]
- 18.Kawase T, et al. An extremely simple dibenzopentalene synthesis from 2-bromo-1-ethynylbenzenes using nickel(0) complexes: construction of its derivatives with various functionalities. Chem. Eur. J. 2009;15:2653–2661. doi: 10.1002/chem.200802471. [DOI] [PubMed] [Google Scholar]
- 19.Levi ZU, Tilley TD. Versatile synthesis of pentalene derivatives via the Pd-catalyzed homocoupling of haloenynes. J. Am. Chem. Soc. 2009;131:2796–2797. doi: 10.1021/ja809930f. [DOI] [PubMed] [Google Scholar]
- 20.Kawase T, et al. Dinaphthopentalenes: pentalene derivatives for organic thin-film transistors. Angew. Chem. Int. Ed. 2010;49:7728–7732. doi: 10.1002/anie.201003609. [DOI] [PubMed] [Google Scholar]
- 21.Takahashi K, Ito S, Shintani R, Nozaki K. Selective synthesis of unsymmetric dibenzo[a,e]pentalenes by a rhodium-catalysed stitching reaction. Chem. Sci. 2017;8:101–107. doi: 10.1039/C6SC04560J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Willner I, Rabinovitz M. 1,9-Dimethyldibenzo[b,f]pentalene dication and dianion. New 14π and 18π aromatic systems. J. Am. Chem. Soc. 1978;100:337–338. doi: 10.1021/ja00469a085. [DOI] [Google Scholar]
- 23.Willner I, Becker JY, Rabinovitz M. Manifestation of dual aromaticity in doubly charged annelated pentalenes. J. Am. Chem. Soc. 1979;101:395–401. doi: 10.1021/ja00496a020. [DOI] [Google Scholar]
- 24.Saito M, Nakamura M, Tajima T, Yoshioka M. Reduction of phenyl silyl acetylenes with lithium: unexpected formation of a dilithium dibenzopentalenide. Angew. Chem. Int. Ed. 2007;46:1504–1507. doi: 10.1002/anie.200604067. [DOI] [PubMed] [Google Scholar]
- 25.Saito M, Nakamura M, Tajima T. New reactions of a dibenzo[a,e]pentalene. Chem. Eur. J. 2008;14:6062–6068. doi: 10.1002/chem.200800451. [DOI] [PubMed] [Google Scholar]
- 26.Kuwabara T, Ishimura K, Sasamori T, Tokitoh N, Saito M. Facile synthesis of dibenzopentalene dianions and their application as new π-extended ligands. Chem. Eur. J. 2014;20:7571–7575. doi: 10.1002/chem.201402304. [DOI] [PubMed] [Google Scholar]
- 27.Oshima H, Fukazawa A, Yamaguchi S. Facile synthesis of polycyclic pentalenes with enhanced hückel antiaromaticity. Angew. Chem. Int. Ed. 2017;56:3270–3274. doi: 10.1002/anie.201611344. [DOI] [PubMed] [Google Scholar]
- 28.Konishi A, et al. Synthesis and characterization of dibenzo[a,f]pentalene: harmonization of the antiaromatic and singlet biradical character. J. Am. Chem. Soc. 2017;139:15284–15287. doi: 10.1021/jacs.7b05709. [DOI] [PubMed] [Google Scholar]
- 29.Chase DT, Rose BD, McClintock SP, Zakharov LN, Haley MM. Indeno[1,2-b]fluorenes: fully conjugated antiaromatic analogues of acenes. Angew. Chem. Int. Ed. 2011;50:1127–1130. doi: 10.1002/anie.201006312. [DOI] [PubMed] [Google Scholar]
- 30.Chase DT, et al. Electron-accepting 6,12-diethynylindeno[1,2-b]fluorenes: synthesis, crystal structures, and photophysical properties. Angew. Chem. Int. Ed. 2011;50:11103–11106. doi: 10.1002/anie.201104797. [DOI] [PubMed] [Google Scholar]
- 31.Chase DT, et al. 6,12-Diarylindeno[1,2-b]fluorenes: syntheses, photophysics, and ambipolar OFETs. J. Am. Chem. Soc. 2012;134:10349–10352. doi: 10.1021/ja303402p. [DOI] [PubMed] [Google Scholar]
- 32.Young BS, et al. Synthesis and properties of fully-conjugated indacenedithiophenes. Chem. Sci. 2014;5:1008–1014. doi: 10.1039/C3SC53181C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frederickson CK, Zakharov LN, Haley MM. Modulating paratropicity strength in diareno-fused antiaromatics. J. Am. Chem. Soc. 2016;138:16827–16838. doi: 10.1021/jacs.6b11397. [DOI] [PubMed] [Google Scholar]
- 34.Dressler JJ, et al. M. M. Synthesis of the unknown indeno[1,2-a]fluorene regioisomer: crystallographic characterization of its dianion. Angew. Chem. Int. Ed. 2017;56:15363–15367. doi: 10.1002/anie.201709282. [DOI] [PubMed] [Google Scholar]
- 35.Shimizu A, Tobe Y. Indeno[2,1-a]fluorene: an air-stable ortho-quinodimethane derivative. Angew. Chem. Int. Ed. 2011;50:6906–6910. doi: 10.1002/anie.201101950. [DOI] [PubMed] [Google Scholar]
- 36.Shimizu A, et al. Indeno[2,1-b]fluorene: a 20-π-electron hydrocarbon with very low-energy light absorption. Angew. Chem. Int. Ed. 2013;52:6076–6079. doi: 10.1002/anie.201302091. [DOI] [PubMed] [Google Scholar]
- 37.Nishida J-I, Tsukaguchi S, Yamashita Y. Synthesis, crystal structures, and properties of 6,12-diaryl-substituted indeno[1,2- b]fluorenes. Chem. Eur. J. 2012;18:8964–8970. doi: 10.1002/chem.201200591. [DOI] [PubMed] [Google Scholar]
- 38.Tobe, Y. Non-alternant non-benzenoid aromatic compounds: past, present, and future. Chem. Rec. 15, 86–96 (2015). [DOI] [PubMed]
- 39.Frederickson CK, Rose BD, Haley MM. Explorations of the indenofluorenes and expanded quinoidal analogues. Acc. Chem. Res. 2017;50:977–987. doi: 10.1021/acs.accounts.7b00004. [DOI] [PubMed] [Google Scholar]
- 40.Oth JFM, Müllen K, Königshofen H, Wassen J, Vogel E. The dianion of heptalene. Helv. Chim. Acta. 1974;57:2387–2398. doi: 10.1002/hlca.19740570811. [DOI] [Google Scholar]
- 41.Sugihara Y, Saito J, Murata I. 2-Triphenylmethyldicyclohept[cd,g]indene: a novel cata-peri condensed nonalternant hydrocarbon. Angew. Chem. Int. Ed. 1991;30:1174–1175. doi: 10.1002/anie.199111741. [DOI] [Google Scholar]
- 42.Murata I. Novel bonding structure of some nonalternant polycyclic systems. Pure Appl. Chem. 1993;65:97–103. doi: 10.1351/pac199365010097. [DOI] [Google Scholar]
- 43.Sugihara Y, Saito J, Murata I. [14π] Electrocyclization followed by preferential [1,9]hydrogen migration in the thermolysis of 1-formyl-4-[(2,4,6-cycloheptatrienyl)(phenylsulfinyl)methyl]azulene. Bull. Chem. Soc. Jpn. 1992;65:3198–3199. doi: 10.1246/bcsj.65.3198. [DOI] [Google Scholar]
- 44.Yang X, Liu D, Miao Q. Heptagon-embedded pentacene: synthesis, structures, and thin-film transistors of dibenzo[d,d’]benzo[1,2-a:4,5-a’]dicycloheptenes. Angew. Chem. Int. Ed. 2014;53:6786–6790. doi: 10.1002/anie.201403509. [DOI] [PubMed] [Google Scholar]
- 45.Xu X, et al. Solution-processed ambipolar organic thin-film transistors by blending p- and n-type semiconductors: solid solution versus microphase separation. ACS Appl. Mater. Interfaces. 2015;7:28019–28026. doi: 10.1021/acsami.5b01172. [DOI] [PubMed] [Google Scholar]
- 46.Yang X, et al. Benzo[4,5]cyclohepta[1,2-b]fluorene: an isomeric motif for pentacene containing linearly fused five-, six- and seven-membered rings. Chem. Sci. 2016;7:6176–6181. doi: 10.1039/C6SC01795A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheung KY, Xu X, Miao Q. Aromatic saddles containing two heptagons. J. Am. Chem. Soc. 2015;137:3910–3914. doi: 10.1021/jacs.5b00403. [DOI] [PubMed] [Google Scholar]
- 48.Müller E, Sauerbier M, Heiß J. Ein beitrag zum cyclobutadienproblem. Tetrahedaron Lett. 1966;22:2473–2478. doi: 10.1016/S0040-4039(00)75678-4. [DOI] [Google Scholar]
- 49.Fischer H, Ege G. Quantenchemische berechnungen als beitrag zur konstitutionsaufklarung kondensierter azulensysteme. Chem. Ber. 1967;100:395–404. doi: 10.1002/cber.19671000205. [DOI] [Google Scholar]
- 50.Müller E, et al. Zur struktur des verdens. Liebigs Ann. Chem. 1970;735:99–112. doi: 10.1002/jlac.19707350115. [DOI] [Google Scholar]
- 51.Scholl M, Ding S, Lee CW, Grubbs RH. Synthesis and activity of a new generation of ruthenium-based olefin metathesis catalysts coordinated with 1,3-dimesityl-4,5-dihydroimidazol-2-ylidene ligands. Org. Lett. 1999;1:953–956. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- 52.Burgess E, Penton HR, Jr., Taylor EA. Thermal reactions of alkyl N-carbomethoxysulfamate esters. J. Org. Chem. 1973;38:26–31. doi: 10.1021/jo00941a006. [DOI] [Google Scholar]
- 53.Ito S, Minami T, Nakano M. Diradical character based design for singlet fission of condensed-ring systems with 4nπ electrons. J. Phys. Chem. C. 2012;116:19729–19736. doi: 10.1021/jp3072684. [DOI] [Google Scholar]
- 54.Bard, A. J. & Faulkner, L. R. Electrochemical methods-fundamentals and applications. Wiley, New York (1984).
- 55.Pommerehne J, et al. Efficient two layer LEDs on a polymer blend basis. Adv. Mater. 1995;7:551–554. doi: 10.1002/adma.19950070608. [DOI] [Google Scholar]
- 56.Cardona CM, Li W, Kaifer AE, Stockdale D, Bazan GC. Electrochemical considerations for determining absolute frontier orbital energy levels of conjugated polymers for solar cell applications. Adv. Mater. 2011;23:2367–2371. doi: 10.1002/adma.201004554. [DOI] [PubMed] [Google Scholar]
- 57.Kolc J, Downing JW, Manzara AP, Michl J. π, π-Biradicaloid hydrocarbons. The pleiadene family. II. A doubly exited state of pleiadene. J. Am. Chem. Soc. 1976;98:930–937. doi: 10.1021/ja00420a011. [DOI] [Google Scholar]
- 58.Di Motta S, Negri F, Fazzi D, Castiglioni C, Canesi EV. Biradicaloid and polyenic character of quinoidal oligothiophenes revealed by the presence of a low-lying double-exciton state. J. Phys. Chem. Lett. 2010;1:3334–3339. doi: 10.1021/jz101400d. [DOI] [Google Scholar]
- 59.Kruszewski J, Krygowski TM. Definition of aromaticity basing on the harmonic oscillator model. Tetrahedron Lett. 1972;36:3839–3942. doi: 10.1016/S0040-4039(01)94175-9. [DOI] [Google Scholar]
- 60.Poater J, Duran M, Solà M, Bernard Silvi B. Theoretical Evaluation of Electron Delocalization in Aromatic Molecules by Means of Atoms in Molecules (AIM) and Electron Localization Function (ELF) Topological Approaches. Chem. Rev. 2014;105:3911–3947. doi: 10.1021/cr030085x. [DOI] [PubMed] [Google Scholar]
- 61.Gershoni-Poranne R, Stanger A. The NICS-XY-scan: identification of local and global ring currents in multi-ring systems. Chem. Eur. J. 2014;20:5673–5688. doi: 10.1002/chem.201304307. [DOI] [PubMed] [Google Scholar]
- 62.Herges R, Geuenich D. Delocalization of Electrons in Molecules. J. Phys. Chem. A. 2001;105:3214–3220. doi: 10.1021/jp0034426. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.