

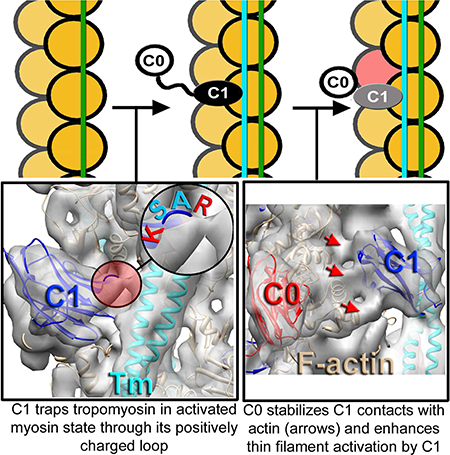
SUMMARY
Muscle contraction relies on interaction between myosin-based thick filaments and actin-based thin filaments. Myosin binding protein-C (MyBP-C) is a key regulator of actomyosin interactions. Recent studies established that the N’-terminal domains (NTDs) of MyBP-C can either activate or inhibit thin filaments, but the mechanism of their collective action is poorly understood. Cardiac MyBP-C (cMyBP-C) harbors an extra NTD which is absent in skeletal isoforms of MyBP-C and its role in regulation of cardiac contraction is unknown. Here we show that the first two domains of human cMyPB-C (i.e., C0 and C1) cooperate to activate the thin filament. We demonstrate that C1 interacts with tropomyosin via a positively charged loop and that this interaction, stabilized by the C0 domain, is required for thin filament activation by cMyBP-C. Our data reveal a mechanism by which cMyBP-C can modulate cardiac contraction and demonstrate a function of the C0 domain.
Keywords: cardiac muscle, thin filament, myosin binding protein-C, cryo electron microscopy
Graphical Abstract
INTRODUCTION
Muscle contraction is based on interactions between the thick filament, which is comprised primarily of myosin and its accessory proteins, including myosin binding protein-C MyBP-C, and the thin filament (TF), which is comprised of tropomyosin (Tm), the troponin complex (Tn), and filamentous actin (F-actin). Under relaxing conditions (low Ca2+) the Tn complex constrains Tm over the myosin binding sites, whereas under activating conditions (high Ca2+) Tm is displaced and myosin binding sites are partially exposed. Strongly bound (rigor) myosin cross-bridges are required to fully activate the TFs (Spudich et al., 1972;McKillop et al., 1993;Vibert et al., 1997). In cardiac muscle the role of strongly bound (rigor) cross-bridges is apparently diminished since ~70% activation of the cardiac TF (cTF) is achieved by binding of Ca2+ to the Tn complex alone (Houmeida et al., 2010;Heeley et al., 2006;Risi et al., 2017). Myosin binding protein-C (MyBP-C) is a striated muscle protein that directly links thick and thin filaments in muscle sarcomeres (Luther et al., 2011) and is present in three isoforms - slow-skeletal, fast-skeletal, and cardiac (ssMyBP-C, fsMyBP-C, and cMyBP-C, respectively). In vitro motility assays demonstrated that ssMyBP-C and cMyBP-C activate TF sliding at low Ca2+, while fsMyBP-C does not, suggesting that MyBP-C isoforms may be tuned to meet the needs of specific muscle types (Lin et al., 2018). Skeletal MyBP-C isoforms contain seven immunoglobulin(Ig)-like domains, three fibronectin(Fn)-like domains, and a regulatory M-domain between the C1 and C2 Ig-domains. Importantly, cMyBP-C has an additional cardiac-specific Ig-domain (e.g. C0) at its N-terminus connected to C1 with a proline/alanine-rich linker (PA-linker) (Fig. 1A) which can bind myosin regulatory light chain (Ratti et al., 2011) or actin (Orlova et al., 2011). Electron microscopy (EM) studies of TFs decorated with NTDs of cMyBP-C demonstrated the ability of NTDs to activate the TF by repositioning Tm on the surface of the TF (Mun et al., 2014;Harris et al., 2016). We recently showed that C1 binds to the native cardiac TF (cTF) in a highly polymorphic fashion and that C1 can interact with the Tm cable (Harris et al., 2016). Nevertheless, the influence of C1 on Tm position in the context of the full size NTD (e.g. C0-PAL-C1-M-C2) has yet to be established. While we previously showed that C1 but not C0 of cMyBP-C can activate the cTF (Shaffer et al., 2010) by directly interacting with the Tm cable (Harris et al., 2016), C0 still appears to contribute to activation of the TF by enhancing effects of cMyBP-C NTDs (Belknap et al., 2014;Razumova et al., 2008). However, the underlying mechanism by which the cardiac specific C0 Ig-domain contributes to the modulation of cTF activation by the NTDs of cMyBP-C remains unclear. Since mutations in the MYBPC3 gene encoding cMyBP-C are one of the most common causes of hypertrophic cardiomyopathy (HCM), a disease affecting ~1:500 people (Watkins et al., 1995;Marston, 2011;Schlossarek et al., 2011), understanding the mechanism(s) by which cMyBP-C modulates cardiac contraction may be important in designing new therapeutic targets for HCM.
Figure 1.

3D reconstructions and corresponding frequencies of structural classes revealed in the C0C1 decorated TFs. (A) cMyBP-C is composed of 8 Immunoglobulin (Ig)-like and 3 fibronectin (Fn)-like domains numbered C0 through C10 starting consecutively from the N-terminus to the C-terminus of the molecule. C0 (blue) and C1 (red) domains are connected by a flexible proline-alanine (PAL) linker, while the M-domain which links C1 and C2 contains functionally significant phosphorylation sites (PS). (B) Effects of C0 (blue curve), C1 (red curve) and tandem C0-C1 (black curve) on TF-dependent myosin-S1 ATPase activity at pCa>8. (C-K) 3D reconstructions of structural classes of C0C1-decorated TFs show all the modes of C0 (C-E) and C1 (F-I) binding to the cTF observed earlier (Harris et al., 2016) along with the two structural classes where C0 and C1 bind simultaneously to the cTF (J and K). High resolution structure of C0 (pdb: 2K1M) is shown in blue (C0-F1 mode), red (C0-F2 mode), or green (C0-S mode). The high resolution structure of the C1 (pdb: 2V6H) is shown in black (C1-F1 mode), yellow (C1-F2 mode), brown (C1-F3 mode), or cyan (C1-S mode). Actin and Tm molecules are shown as tan ribbons. Reconstructions are shown as grey transparent surfaces filtered to the resolution determined for each class (Figure S1C). Interaction of C1 with Tm in the C1-S, C0-F1+C1-S, and C0-F2+C1-S are marked with black arrows. (L) Frequencies for individual classes of cTFs decorated with individual C0 and C1 domains obtained earlier (Harris et al., 2016) are compared with those calculated for the C0C1-decorated TFs. Frequencies of classes where C1 interacts with Tm are marked in red.
Here we used cryo-electron microscopy, biochemical kinetics and mutant protein expression to show that C0 and C1, the first two Ig-like domains of cMyPB-C, when linked together by the PA-linker, cooperate to activate the cTF. We show that the interaction of a positively charged loop of C1 (residues 215–218) with Tm is a prerequisite for the activation of the cTF by the ensemble NTD of cMyBP-C. We reveal that R215 and K218 of the loop form an interface between C1 and Tm, because charge reversal mutations (R215E and K218E) inhibit cTF activation by the entire NTD of cMyPB-C. Moreover, we demonstrate that the cardiac-specific C0 Ig-domain enhances the activating effects of C1 on the cTF by securing C1 in a structural mode where it locks Tm in the same azimuthal position as in the presence of rigor S1. We conclude that C1 is the major determinant of cTF activation by cMyBP-C and that C0 is a potent enhancer of the C1-dependent TF activation. Our data reveal one of the molecular mechanisms by which cMyBP-C can influence cardiac contraction or relaxation through activation of the cTF.
RESULTS
C0 enhances the binding of C1 to the cTF in a mode where C1 tethers actin and tropomyosin.
Recently we showed that the first two domains of cMyBP-C (e.g. C0 and C1) exhibit different patterns of binding on the surface of the native cTF so that C1 but not C0 can activate the cTF (Harris et al., 2016). However, steady-state ATPase measurements under low Ca2+ (relaxing) conditions showed that despite the inability of C0 to activate the cTF by itself (Figure 1B, blue curve), when linked to C1 in the C0C1 (e.g. C0-PA-C1) fragment it significantly enhanced activation of the cTF (Fig. 1B, black curve, compare with activation of by C1 alone, red curve). To understand how the two domains work in concert to activate cTF we examined frozen hydrated cTFs decorated with the C0C1 fragment of cMyBP-C (Figure S1A). Segments of decorated cTFs were sorted into structural classes using models previously obtained for the individual C0 and C1 domains (Harris et al., 2016) as references (see Methods section), and each class was independently reconstructed. Importantly, to exclude any bias from the model-based classification, each structural class was iteratively reconstructed (Egelman, 2000) starting from either a solid cylinder or from a 3D reconstruction of another structural class (Figure S1B). To further prove that model bias cannot be introduced at the classification step and that the resultant structural classes cannot mimic any structural feature introduced into a reference model, we used images extracted from four structural classes and repeated the sorting for those images using an additional unrelated “bogus” model (Figure S2A-E). However, a small number of images that yielded the best correlation with the “bogus” model failed to replicate the structural features of the bogus reference (Figure S2D and E).
Our success in isolating particular structural modes of binding of the C0C1 fragment of cMyBP-C strongly suggests that both C0 and C1 domains bind to the TF with high cooperativity. To validate it we analyzed the propagation of modes of binding of C0C1 along the individual cTFs (Figure S2F). Our data show that there is a substantial cooperativity in the modes of binding of C0C1 to the cTF.
The 3D reconstructions of the nine structural classes that converged to meaningful solutions (Figure 1C-K) were low-pass filtered to the resolution determined for each class using Fourier Shell Correlation (FSC) approach (Figure S1C). Classes possessing individual C0 or C1 bound to the front of F-actin (Figure 1 C-D, F-H) or C0 bound to the side of F-actin (Figure 1E) scored a resolution of ~13 – 14 Å (Figure S1C). Notably, classes that yielded better resolution (~9 – 11 Å, Figure S1C) had C1 te thering actin and the Tm cable together (Figure 1I-K, black arrows) which suggests that in this mode of binding C1 stabilizes the structure of the complex. Also, a highly cooperative fashion of C1-S mode in binding to the cTF (Figure S2F, red boxes) may lead to more homogeneous structural classes which yield higher resolution. Within the C0C1-decorated cTFs we found all the modes of binding of C0 (Figure 1C-E) and C1 (Figure 1F-I) domains previously reported for the cTFs decorated with individual C0 or C1 domains (Harris et al., 2016). C0 was thus found to interact with the front of F-actin in two structural modes C0-F1 (Figure 1C, blue ribbons) and C0-F2 (Figure 1D, red ribbons) related by ~120º clockwise rotation (Harris et al., 2016). Also, C0 was found bound to the side of actin subdomain 1 (SD1) (Figure 1E, green ribbons). C1 was seen in three positions at the front of F-actin, C1-F1, C1-F2 and C1-F3 (Figure 1F-H black, yellow and brown ribbons, respectively). These 3 positions were originally predicted to be conformations of the same mode of binding of C1 to F-actin (Harris et al., 2016). A fourth position was also found where C1 bound to the side of F-actin (Figure 1I, cyan ribbons) where it tethers actin and Tm (Figure 1I, black arrow). Lastly, we found two novel modes of binding where both C0 and C1 domains were simultaneously bound to each actin subunit (Figure 1 J and K). In these two new modes C0 was bound to the front of SD1 of actin in either C0-F1 (Figure 1J) or C0-F2 (Figure 1K) mode, while C1 was bound in the C1-S mode tethering F-actin with the Tm cable (Figure 1J and K, black arrows).
We compared the frequencies of the structural modes found for cTFs decorated with the individual (e.g. C0 or C1) domains (Harris et al., 2016) to those observed for the C0C1-decorated cTFs (Figure 1L). As expected, C0 and C1 competed for binding to the front and side sites of the actin protomer and, therefore, the frequencies for the C0-F1, C0-F2, C1-F1, C1-F2, C1-F3 and C0-S structural classes were reduced in C0C1-decorated set compared to cTF decorated with the individual C0 or C1 domains (Figure 1L). In contrast to that, the side mode of binding of C1 (C1-S) was enhanced from 14% in C1-decorated cTFs (C1-S mode) to 36% in C0C1-decorated cTFs, because C1-S, C0-F1+C1-S, and C0-F2+C1-S modes all had C1 in the C1-S position (Figure 1L, frequencies marked in red). Because C1 was shown to trap Tm in the activated state in the C1-S mode of binding (Harris et al., 2016), our data suggest that the enhanced frequency of the C1-S mode in the C0C1-decorated cTFs (Figure 1L) explains enhanced activation of the cTF by the C0C1 fragment versus C1 alone (Figure 1B).
C1 tethering of actin subdomain 1 with tropomyosin is required for the cTF activation by the complete NTD (C0-PA-C1-M-C2) of cMyBP-C.
Recently, we determined the positions of the Tm cable on the surface of frozen hydrated cTF at relaxing (low Ca2+) and activating (high Ca2+) conditions (Risi et al., 2017). To evaluate the extent of activation of the cTF by the C1 domain of cMyBP-C we compared the position of Tm in the C1-S structural mode, where C1 tethers actin with the Tm cable (Figure 1I, black arrow), with the position of Tm in either the Ca2+-activated state (Risi et al., 2017) or in the actin-tropomyosin-myosin complex (von der Ecken et al., 2016) (Figures 2A and B, respectively). Our data suggests that when C1 cross-links actin with the Tm cable, it activates the cTF to the same extent as rigor S1. Previously, it had been suggested that the NTD of cMyBP-C activates the TF to the same extent as high Ca2+ (Mun et al., 2016;Mun et al., 2014). However, it is possible that the negative staining protocol used in those studies altered the position of Tm in the complexes of TFs with cMyBP-C NTDs as observed for actin-tropomyosin complex (von der Ecken et al., 2015) and native cTFs (Risi et al., 2017).
Figure 2.

C1 binding to the Tm cable traps it in the myosin rather than Ca2+-induced c-open structural state. (A) Atomic model of the cardiac open (c-open) structural state of the cardiac TF (Protein Data Bank ID code 5NOJ) (Risi et al., 2017) was rigidly docked into the 3D reconstruction of the C1-S structural class to show that Tm (magenta ribbons) does not fit into the portion of the electron density map that corresponds to the Tm cable. (B) The model of the myosin-Tpm-F-actin complex (von der Ecken et al., 2016) (Protein Data Bank ID code 5JLH) docked into the 3D reconstruction of the C1-S structural class without any perturbations unambiguously shows that Tm (cyan ribbons) position in the 3D reconstruction of the C1-S class corresponds to the myosin state of the TF. 3D reconstructions are shown as grey transparent surfaces. Actin molecules are shown as tan ribbons, while rigor bound myosin-S1 head is shown as black ribbons.
To evaluate the role of C1 interactions with Tm we next analyzed the interface between C1 and Tm using a ~9 Å resolution map of th e C1-S structural mode (Figure 1I). We found a bridge of density connecting the upper loop of C1 with the Tm cable (Figure 3A, red circle). The four amino acid loop, comprised of Arg-Ala-Ser-Lys (RASK) residues, was repeatedly positioned next to negatively charged patches along the Tm cable (Figure 3B), suggesting that the interactions between R215 and K218 residues of C1 with Asp and Glu residues of Tm were forming the bridge of density observed between C1 and Tm (Figure 3A, red circle). Amino acid alignment of the NTDs of cMyBP-C from different species showed that both R215 and K218 are highly conserved between species (Figure S3A, red arrows). To verify the importance of these residues for the interaction of C1 with Tm we introduced charge reversing mutations (e.g., R215E/K218E) into the C1 loop and decorated cTF with the mutant C1 fragment. Image analysis of frozen hydrated cTFs decorated with the mutant C1(R215E/K218E) showed the same structural modes of binding of the mutant C1 to the cTF observed for wild-type C1 (Figure S3B). However, C1 and Tm interactions in the C1-S mode were absent in C1(R215E/K218E) (Figure 3D, red arrows). Importantly, the Tm cable was found in the c-closed structural state (Figure 3D, green ribbons) which is an average position of Tm cable at relaxing (low Ca2+) conditions (Risi et al., 2017) instead of in the myosin state (Figure 3C, cyan ribbons). The differences between C1-S structural modes obtained for the wild-type and C1(R215E/K218E) are illustrated in Movie S1. To confirm results from structural studies we next measured cTF-dependent steady-state ATP hydrolysis by myosin-S1 at low Ca2+ for both wild-type (Figure 3E, red curve) and mutant C1(R215E/K218E) (Figure 3E, green curve). Unlike the wild-type C1, C1(R215E/K218E) was unable to activate cTFs at low Ca2+. To evaluate whether the C1 loop interaction with the Tm cable is involved in the activation of the cTF by the entire NTD (e.g. C0-PA-C1-M-C2) we introduced the C1(R215E/K218E) mutation into the full length NTD and measured its ability to activate the cTF at low Ca2+ conditions (Figure 3F). Comparison of cTF-dependent activation of steady-state ATP hydrolysis by myosin-S1 for wild-type (Figure 3F, magenta curve) and mutant C1(R215E/K218E) (Figure 3F, green curve) NTDs revealed that the R215E/K218E mutation completely inhibited cTF activation by the NTD of cMyBP-C at low Ca2+. Steady-state ATP hydrolysis assays at intermediate (pCa=5.5) and high (pCa=4) Ca2+ also showed that R215E/K218E mutation significantly reduced cTF activation by the cMyBP-C NTD at pCa=5, but effects at maximal Ca2+ activation (pCa=4) were modest (Figure S3C). To further test the hypothesis that interactions of the positively charged residues in the 215–218 loop of C1 with Tm are important for the activating effects of C1, we enhanced the positive charge of the loop by replacing A216 and S217 with R216 and K217, respectively. The positive charge mutant, C1(A216K/S218K), significantly enhanced cMyBP-C NTD-dependent activation of the TF at low and intermediate Ca2+ levels (Figure 3F and S3C, cyan curve). These data provide strong support for the idea that ionic charge interactions between Tm and R215/K218 residues of C1 are essential for the activation of the cTF by the NTD of cMyBP-C at low and intermediate submaximal Ca2+ activation.
Figure 3.

R215 and K218 amino acid residues of C1 maintain its interaction with Tm which is required for the TF activation by the cMyBP-C NTD. (A) A 9 Å 3D reconstruction of the C1-S class shows a bridge of density (red circle) between C1 (blue ribbons) and Tm (cyan ribbons). Insert shows that C1 loop involved in the interaction is comprised of two positively charged (red) and two neutral (cyan) residues. (B) Residues R215 and K218 (red spheres) of C1 loop are located in proximity to negatively charged residues of Tm (yellow spheres). Tm model is from (Behrmann et al., 2012). (C-D) In the 3D reconstruction of the wild type C1 in the C1-S mode there is a bridge of density (black arrows) between C1 (C, blue ribbons) and Tm in the myosin state (C, cyan ribbons). It is absent in the 3D reconstruction of the C1-S class of the C1(R215E/K218E)-decorated TFs (D, red arrows) where Tm is in the c-closed position (D, green ribbons). (E) Effects of C1 (red curve), and C1(R215E/K218E) (green curve) on TF-dependent myosin-S1 ATPase activity at pCa>8. (F) Effects of C0C1MC2 (magenta curve), C0C1(R215E/K218E)MC2 (green curve), and C0C1(A216R/S217K)MC2 on TF-dependent myosin-S1 ATPase activity at pCa>8.
C0 enhances interaction of C1 with F-actin when the two domains are in tandem.
To evaluate how C0 influences the ability of C1 to activate the cTF we compared 3D reconstructions of the C1-S mode when both C0 and C1 simultaneously interact with actin subunits (e.g. C0-F1+C1-S and C0-F2+C1-S). We compared the interface between C1 and SD1 of actin in the C1-S, C0-F1+C1-S and C0-F2+C1-S modes (Figure 4A-C). In the C1-S mode, C1 makes one prominent contact with SD1 of actin (Figure 4A), while in the C0-F1+C1-S mode a second contact is observed between C1 and the actin subunit (Figure 4B). Importantly, in the C0-F2+C1-S mode we found three prominent bridges of density between C1 and actin (Figure 4C), which suggests that when bound in the C0-F2 mode, C0 reinforces C1 interactions with actin. To validate the difference in connectivity between the SD1 of actin and C1 observed in the C1-S, C0-F1+C1- and C0-F2+C1S classes, we reconstructed these classes starting from a 3D reconstruction obtained from a different class (Figure S1B) and monitored the morphing of the interface between the SD1 of actin and C1 (Figure S1B, red arrows). The convergence in the number of contacts between F-actin and C1 for the C1-S, C0-F1+C1- and C0-F2+C1S classes strongly suggests that C0 reinforces C1 interactions with F-actin.
Figure 4.

Effects of C0 on the interaction of C1 with F-actin. Comparison of the contacts between actin and C1 in the reconstructions of C1-S (A), C0-F1+C1-S (B) and C0-F2+C1S (C) shows that in the absence of bound C0, C1 has one contact with actin (A), while binding of C0 in the F1 and F2 models increases the number of contacts to two (B) and three (C), respectively.
C0 and C1 need to be linked through the PA-linker to activate the cTF.
We have already shown that C0 and C1 bind to multiple overlapping sites on the surface of F-actin (Harris et al., 2016). Therefore, it is possible that C0 and C1 could compete with each other in a random fashion so that the chances of having C1 bound to the side (C1-S) or C0 bound to the front (C0-F1 or C0-F2) on the same actin molecule will be low. However, it is also possible that when the two domains are linked by the PA-linker, binding of C0 to the front (C0-F1 and C0-F2) or binding of C1 to the side (C1-S) promotes the adjacent domain to occupy the nearest site on the same actin molecule. To evaluate whether the two domains need to be linked by the PA-linker to cooperatively activate the cTF we used steady-state ATP hydrolysis assay to compare C0C1 in tandem with equimolar mixtures of C0-PA and C1 (Figure 5A). As expected, the mixture of C0-PA and C1 did not activate the cTF to the same extent as C0 and C1 when linked by the PA-linker. Moreover, when the two domains were not linked together, the C0-PA fragment reduced C1-dependent activation of the cTF, suggesting that competition between C0 and C1 (C0-S and C1-S, respectively) for binding to the side of F-actin displaces C1 from its activating C1-S mode.
Figure 5.

C0 and C1 Ig-domains of cMyBP-C work in tandem to activate the cTF. (A) Effects of C0-PA-C1 (black curve), C1 (red curve), and equimolar mixture of C0-PA and C1 on TF-dependent myosin-S1 ATPase activity at pCa>8. (B) The model of activation of the cTF by C0-PA-C1 fragment of cMyBP-C. At relaxing conditions Tm is in the inactive (c-closed and c-blocked) states (Risi et al., 2017) (green line). Upon C1 binding in the C1-S mode the Tm cable is azimuthally shifted to its myosin (cyan line) state (Figure 2B) by means of interaction with the R215 and K218 residues of C1 (Figure 3). Bound C1 enforces interaction of C0 with the front of the same actin molecule which enhances the C1 interaction with F-actin (Fig. 4A-C). Actin molecules are shown in yellow. C0 Ig-domain is shown as white circle, C1 domain bound in the C1-S mode is black oval, while when activated by the C0 domain is marked in grey.
DISCUSSION
Recent studies established that the N’-terminal domains (NTDs) of MyBP-C (e.g. C0, PA, C1, M and C2) can bind to and modulate activation state of the cardiac TF (cTF) (Razumova et al., 2008;Razumova et al., 2006;Herron et al., 2006;Mun et al., 2014). However, the contribution of individual domains of MyBP-C to cTF activation and the role of the cardiac specific C0 domain remain poorly understood. Recently, we have shown that despite being structural homologs, C0 and C1 domains of human cMyBP-C exhibit different patterns of binding on the surface of F-actin and, importantly, that C1 but not C0 can activate the cTF (Harris et al., 2016). These data provided important evidence that the NTDs of cMyBP-C are not equivalent in their functional characteristics, but still left open questions of how adjacent domains and linkers work together to activate the cTF. That is, despite our evidence that C1 can trap Tm in its activated state, it remained unclear if C1 is the only activator of the cTF and whether its ability to trap Tm is necessary for cTF activation when all four NTDs are present. Here, our mutagenesis experiments demonstrate that tethering of actin to the Tm cable by C1 in the C1-S mode is required to activate the cTF even when all NTDs of cMyBP-C are present (Figure 3 and S3C). We further demonstrate that the interaction of C1 with Tm is electrostatic and involves a 4 amino acid loop (RASK residues 215–218) (Figure 3B). This C1 loop may therefore also be a reasonable target for targeted drug design because modulation of charge on the loop modulates cMyPB-C-dependent cTF activation (Figure 3F and S3C) and thus could modulate cardiac contraction/relaxation. Consistent with this, A177H and A216T mutations identified in hypertrophic cardiomyopathy patients are predicted to cause charge redistributions in the 215–218 loop (Gajendrarao et al., 2013).
We show here that the cardiac specific C0 domain of cMyBP-C, despite being unable to activate the cTF on its own, potentiates the ability of C1 to activate the cTF (Figure 1B and 5A). These data demonstrate that C0 communicates with C1 through the PAL upon binding to the cTF. Image analysis of frozen hydrated cTF decorated with the C0C1 fragment of cMyBP-C reveals that when the two domains are present in tandem (joined by the PAL), the frequency of the C1-S mode is significantly enhanced (Figure 1L) consistent with reinforcement of C1-S interactions with F-actin by C0 (Figure 4A-C).
We suggest the following model for how C0 facilitates cTF activation by C1 (Figure 5B): under relaxing conditions Tm is in the inactive (c-closed and c-blocked) states (Risi et al., 2017) (Figure 5B, green line). Upon C1 binding to the cTF (in the C1-S mode) the Tm cable is azimuthally shifted to its myosin (cyan line) state (Figure 2B) by means of interaction with the R215 and K218 loop residues of C1 with Tm (Figure 3). Because C0 is linked to C1, it is well positioned to occupy the front of the same actin molecule which enforces interaction of C1 with the SD1 of actin (Figure 5B, denoted in pink) and lock C1 in the C1-S structural mode crucial for the TF activation by the NTD of cMyBP-C. Alternatively, the binding of C0 to the front of the actin subunit may also increase the likelihood of C1 interaction with the side of the filament in the C1-S mode.
The cooperative binding of C0 and C1, the first two Ig-domains of cMyBP-C, indirectly implicates the importance of the PA-linker for their cooperative effects in tandem on activation of the cTF. Our data consistently shows that if C0 and C1 are not connected with the PA-linker, their combined effect on the activation of the cTF is smaller than the effect of C1 alone (Figure 5A). The PA-linker length has been shown to correlate with the heart rate in mammals (Shaffer et al., 2009), suggesting that it may work as an extended spacer that modulates timing of interactions of flanking domains with the TF (Colson et al., 2016). The absence of an observable density that can be attributed to the PA-linker in our 3D reconstructions of C0C1-decorated cTFs also provides indirect support for a role for the PA-linker as a spatial regulator which controls positions of NTDs of cMyBP-C on the surface of the cTF without itself having tight interactions with the cTF.
The C0 domain is absent in skeletal isoforms of MyBP-C. It has been proposed that the absence of C0 in skeletal muscle can be explained by redundancy in the interaction of the first two Ig-domains of MyBP-C with the actin filament (Orlova et al., 2011). However, arguing against this possibility are our observations that the absence of C0 reduces the efficacy of cMyBP-C NTDs to activate the cTF (Belknap et al., 2014). Taken together, our data demonstrate that C0 and C1 are not functionally redundant but instead work cooperatively to activate the cTF.
The primary difficulty in defining molecular mechanisms for how cMyBP-C affects force in the myocardium until now has been that cMyBP-C binds to both thick (Gruen et al., 1999;Kampourakis et al., 2014;Nag et al., 2017) and thin (Shaffer et al., 2009;Belknap et al., 2014;Harris et al., 2016) filament proteins and it has thus been challenging to discriminate individual effects of cMyBP-C on either system. The problem is further compounded when considering that both Ca2+ and phosphorylation regulate cMyBP-C function and its interactions with the two filament systems (Stelzer et al., 2006;Stelzer et al., 2007). Phosphorylation of cMyBP-C disrupts its binding to myosin relaxed heads, thereby increasing the number of myosin heads available for binding to cTFs (Kensler et al., 2017). On the other hand, phosphorylation reduces the affinity of the NTDs of cMyBP-C to the TF at low Ca2+, presumably because of phosphorylation induces closure of the NTDs (Previs et al., 2016;Colson et al., 2016). However, at elevated Ca2+ phosphorylated NTDs of cMyBP-C adopt an open active conformation (Previs et al., 2016). It is conceivable that phosphorylation-induced closing of NTDs prevents C0 and C1 from binding to the cTF at low Ca2+, whereas binding to the cTF can still occur at elevated levels of Ca2+ even if the NTDs are phosphorylated because of Ca2+-induced extension of the NTDs. If so, phosphorylated cMyBP-C may accelerate the rate of force development as Ca2+ increases during a twitch by accelerating the rate of cooperative cross-bridge recruitment by broadening the activation zones around sites of Ca2+-bound troponin (Moss et al., 2015). This hypothesis is consistent with the ability of C1 to activate cTFs to the same extent as rigor-S1 (Figure 2B), thus enhancing the extent of propagation of the activating effect along the thin filament in a cooperative fashion. At the same time, effects of Ca2+ and phosphorylation on NTD binding to the cTF could also speed relaxation because phosphorylation would be expected to induce the closed configuration of the NTDs and thus promote their unbinding from the cTF as Ca2+ falls back to diastolic levels.
STAR Methods
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial strains | ||
| Escherichia coli M15 [pREP4] (K12) (NaIS, StrS, RifS, Thi–, Lac–, Ara+, Gal+, Mtl–, F–, RecA+, Uvr+, Lon+) | Qiagen | Cat. #34210 |
| Chemicals, Tissues, Peptides, and Recombinant Proteins | ||
| Luria-Bertani powder | Fisher | Cat. #BP9723 |
| Bacto Agar | DIFCO | Cat. #214050 |
| Kanamycin | Sigma | Cat. #K4000 |
| Carbenicillin disodium salt | Alfa Aesar | Cat. #J61949 |
| Magnesium sulfate hexahydrate | Baker | Cat. #1–2500 |
| Potassium chloride | Sigma | Cat. #P4504 |
| Isopropyl-β-D-thiogalactopyranoside (IPTG) | RPI Corp. | Cat. #I-56000 |
| Nickel-nitrilotriacetic acid (NiNTA) agarose | Qiagen | Cat. #30210 |
| Tris(hydroxymethyl)aminomethane | Sigma | Cat. #T1503 |
| Sodium chloride | Sigma | Cat. #S9625 |
| Imidazole | Sigma | Cat. #I2399 |
| Bacterial Protein Extraction Reagent (B-per) | Thermo | Cat. #78248 |
| HALT protease inhibitor | Thermo | Cat. #78439 |
| Pepstatin A | Santa Cruz Biotech | Cat. #SC-45036A |
| Dimethylsulfoxide (DMSO) | Sigma | Cat. #D2650 |
| Phenylmethylsulfonyl fluoride (PMSF) | Sigma | Cat. #P7626 |
| E-64 protease inhibitor | Fisher | Cat. #501010687 |
| 2-mercaptoethanol | Bio-Rad | Cat. #161–0710 |
| cOmplete Mini protease inhibitor tablets – EDTA free | Sigma | Cat. #11836170001 |
| Lysozyme | Sigma | Cat. #L6876 |
| DNase I recombinant | Sigma | Cat. #04536282001 |
| Triton X-100 | Fisher | Cat. #BP151 |
| Sodium phosphate dibasic | Fisher | Cat. #S374 |
| Pig hearts | Pel Freez | Cat. #59416–1 |
| Recombinant DNA | ||
| pQE-2 vector | Qiagen | Cat. #32932 |
| Zero-Blunt vector kit | Invitrogen | Cat. #K2750 |
| pQE-2 vector | Qiagen | Cat. #32932 |
| Zero-Blunt vector kit | Invitrogen | Cat. #K2750 |
| Deposited Data | ||
| NMR structure of domain cC0 of cardiac myosin binding protein C | (Ratti et al., 2011) | PDB ID 2K1M |
| Crystal structure of the C1 domain of cardiac myosin binding protein-C | Govada et al., 2008) | PDB ID 2V6H |
| Pseudo-atomic model of the myosin-Tpm-F-actin complex | (Behrmann et al., 2012) | PDB ID 4A7F |
| Pseudo-atomic model of the myosin-Tpm-F-actin complex | (von der Ecken et al., 2016) | PDB ID 5JLH |
| Pseudo-atomic model of the cardiac open (c-open) structural state of the cardiac TF | (Risi et al., 2017) | PDB ID 5NOJ |
| C1 bound to the TF in C1-S mode | This paper | EMD-4346, PDB ID 6G2T |
| C0C1 bound to the TF in the C0-F1+C1-S mode | This paper | EMD-7780, PDB ID 6CXI |
| C0C1 bound to the TF in the C0-F2+C1-S mode | This paper | EMD-7781, PDB ID 6CXJ |
| Software and Algorithms | ||
| SPIDER | (Frank et al., 1981) | https://spider.wadsworth.org |
| CTFFIND3 | (Mindell et al., 2003) | http://grigoriefflab.janelia.org |
| EMAN | (Ludtke et al., 1999) | http://blake.bcm.edu/emanwiki |
| IHRSR | (Egelman, 2000) | egelman@virginia.edu |
| DireX | (Schroder et al., 2007) | https://simtk.org/home/direx |
| CNS | (Adams et al., 2010) | http://cns-online.org |
| UCSF Chimera | (Pettersen et al., 2004) | http://www.cgl.ucsf.edu/chimera |
| ExPASy | SIB Swiss Institute of Bioinformatics | https://web.expasy.org/protparam |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to, and will be fulfilled by, the lead contact, Dr. Vitold E. Galkin (galkinve@evms.edu).
Method Details
Isolation and Purification of cDNA:
N-terminal cMyBP-C proteins containing the C0, C1, C0C1 C0-PA and C0C2 domains were cloned by PCR amplification of a full-length human cMyBP-C cDNA sequence (GenBankTM accession number NM000256) using upstream and downstream primers containing restriction site sequences for NdeI and HindIII, respectively. The resulting PCR products were cloned into the Zero-Blunt vector (Invitrogen #K2750) and gel-purified following NdeI/HindIII digestion. For expression in bacteria, gel-purified inserts were subcloned into the pQE-2 expression vector (Qiagen #32932) in frame with a His6-tag at the N terminus of the clone sequence to facilitate purification. Mutant constructs C1(R66E/K69E), C0C2(R215E/K218E) and C0C2(A216R/S217K) were generated by site directed mutagenesis by Mutagenex Inc.(Suwanee, GA, USA). The mutations were verified by sequencing the full coding sequence of the DNA.
Protein Expression in E. coli:
A 100 μl aliquot of competent M15[pREP4] cells (Qiagen #34210 – no longer commercially available) was thawed on ice. 2 μg cDNA was added and flicked to mix. The mixture was left on ice for 20 minutes followed by heat shocking at 42ºC for 90 seconds. 500 μl of pre-warmed sterile Psi broth (10 ml LB broth without antibiotics, 4 mM MgSO4, 10 mM KCl) was added to the cell/DNA mixture. This was incubated at 37ºC and 230 rpm for 90 minutes. Aliquots of 10 and 25 μl were plated on LB Agar (1.5% (w/v) agar) containing 25 μg/ml kanamycin (in water) and 100 μg/ml carbenicillin (in 20% (w/v) ethanol (LB+K+C) followed by overnight incubation at 37ºC. Several 5 ml LB+K+C were inoculated with single colonies and incubated by shaking at 230 rpm for 16 hours at 37ºC. Following overnight incubation, the cells were pelleted by centrifugation at 3500 rpm for 10 minutes. The supernatants were removed and the pellets were resuspended with 5 ml of fresh LB+K+C. Each resuspended 5 ml culture was transferred to a 2 liter baffled flask containing 500 ml of pre-warmed LB+K+C. The flasks were incubated at 37ºC/200 rpm until the OD595 = 0.5 – 0.7. IPTG induction was done by using 1 mM IPTG. Then the temperature was quickly reduced to 30ºC and the cultures were incubated for 6 hours at 170 rpm. The cells from each culture were collected by centrifugation at 5500×g for 15 minutes. The supernatants were discarded and the pellets were resuspended in 10 ml of cold LB+K+C and transferred to 50 ml conical tubes. The transferred pellets were collected by centrifugation at 3,000×g for 10 minutes at 4OC. The supernatants were discarded and the pellets were stored at −20ºC.
Protein Purification:
A 5 ml nickel-nitrilotriacetic acid (NiNTA: Qiagen #30210) was prepared for use by placing 10 ml of a 50% slurry of resin in a disposable column. This was rinsed with 5 bed volumes of water before being equilibrated with 10 bed volumes of 2x Lysis Buffer (50 mM TRIS, 400 mM NaCl, 30 mM imidazole (pH 7.5)). The buffer was drained from the resin and the column was left at 4º C until needed. A frozen cell pellet containing the desired expressed cMyBP-C was thawed on ice. The pellet was completely resuspended in cold Extraction Buffer (15 ml of Bacterial Protein Extraction Reagent (BPER), 100 μl HALT EDTA-free protease cocktail, 30 μl 1 mg/ml pepstatin-A in DMSO, 30 μl 100 mM PMSF, 30 μl 10 mM E64 protease inhibitor, 2.1 μl 2-mercaptoethanol and 1 complete Mini protease inhibitor tablet. The resuspended pellet was transferred to a 50 ml conical centrifuge tube. 30 mg of lysozyme was added to the mixture and mixed gently. This was placed on a rotator at 50 rpm for 15 minutes at 4ºC. After 15 minutes, 75 μl of 10,000 units/ml DNase (final concentration of 50 units per ml) was slowly added to the suspension. The tube was returned to the rotator for another 15 minutes. The suspension was diluted with an equal volume (15 ml) of 2X Lysis Buffer (50 mM TRIS (pH 7.5), 400 mM NaCl, 30 mM imidazole) before centrifugation at 25,000×g for 30 minutes at 4ºC. The supernatant was used to resuspend the equilibrated NiNTA resin. The resuspended resin/lysate was placed in a 50 ml conical and placed on a rotator at 4ºC/50 rpm for 75 minutes. The resuspended resin was removed from the rotator and poured into a 30 ml disposable column and allowed to drain completely. 5 ml aliquots of NiNTA Wash Buffer (25 mM TRIS (pH 7.5), 200 mM NaCl, 30 mM imidazole, 1 mM 2-mercaptoethanol, 0.1 mM PMSF, 1 μg/μl pepstatin) were added and fractions were collected. Washing continued until the OD280 of the eluted fractions was less than 0.04. The column was then eluted by adding 1 ml aliquots of NiNTA Elution Buffer (25 mM TRIS (pH 7.5), 200 mM NaCl, 250 mM imidazole, 1 mM 2-mercaptoethanol, 0.1 mM PMSF, 1 μg/μl pepstatin) until the OD280 returned to baseline. The fractions eluted from the NiNTA column were run on a 15% polyacrylamide gel and appropriate fractions were pooled based on the presence of the desired protein and purity. Extinction coefficients and molecular weights for each construct were determined from their amino acid sequences using the ExPASy web tool. Concentrations were determined from the net OD280 after accounting for light scattering:
The pooled fractions were concentrated and the buffer was exchanged using a 15 ml Millipore spin concentrating unit with a 10,000 MWCO. The final cMyPB-C samples were in 0.2 M potassium acetate, 10 mM MOPS, 3 mM MgCl2 (pH 7.0) and were drip frozen and stored in liquid nitrogen until needed.
Native porcine cardiac thin filaments purification:
Native porcine cardiac thin filaments were prepared according to the procedure of Spiess et al. as modified by Matsumoto et al. Whole frozen porcine hearts were obtain from Pel Freez and stored at −80º C until needed. Approximately 100–125 grams of ventricular tissue was removed and thawed on ice. The tissue was passed through a cold meat grinder and collected in a 1 liter beaker on ice. The tissue was suspended in 400 ml of Buffer F1 (10 mM Na2HPO4, 100 mM NaCl, 5 mM MgCl2, 1 mM EGTA, 1 mM NaN3, 1 mM DTT (pH 7)) containing 1% Triton-X 100 (v/v) and homogenized for 40 seconds using a Polytron foam-reducing homogenizer. The tissue was pelleted by spinning at 15,000×g for 8 minutes. The resulting supernatant was discarded and the homogenization step was repeated once more with Buffer F1 + Triton-X and an additional three times using Buffer F1 without detergent. The thin filaments were extracted from the washed myofibrils by resuspending and homogenizing twice in 180 ml of Buffer F1 + 5 mM ATP followed by spinning at 15,000×g for 8 minutes. The supernatants were combined and centrifuged at 186,000×g for 15 minutes. The supernatants were transferred to fresh tubes and centrifuged for 2.5 hours at 186,000×g. The pelleted crude thin filaments were gently resuspended using a tissue homogenizer to approximately 120 ml in Dialysis Buffer (20 mM TRIS, 100 mM NaCl, 5 mM MgCl2, 1 mM EGTA, 1 mM NaN3, 1 mM DTT (pH 7.0)) and dialyzed overnight against the same. The following morning, the solution was adjusted to 20 mM TRIS, 200 mM NaCl, 2 mM ATP, 2.5 mM MgCl2, 1 mM EGTA, 1 mM DTT, 1 mM NaN3 (pH 7.0) by the addition of an equal volume of ATP Release Adjustment Buffer (20 mM TRIS, 300 mM NaCl, 1 mM EGTA, 1 mM DTT, 1 mM NaN3, 4 mM ATP (pH 7.0)). The solution was stirred on ice for 10 minutes before spinning at 186,000×g for 15 minutes, discarding the pellets and spinning for another 2.5 hours at the same speed. The pellets were resuspended with Dialysis Buffer as described previously to approximately 90 ml and dialyzed overnight. The thin filaments were again diluted with an equal volume of ATP Release Adjustment Buffer, stirred on ice for 10 minutes and centrifuged as above. The final thin filament pellets were resuspended to approximately 10–15 ml to provide a concentration of 150–200 μM. The concentration was determined after accounting for light scattering (OD280 – (OD320 × 1.5)) using an extinction coefficient of 0.765 mg−1/ml−1 and a molecular weight of 63,000.
Kinetic measurements:
cTF activation of steady-state ATP hydrolysis was measured by a colorimetric assay in 50 mM KAc, 10 mM MOPS, 3 mM MgCl2, 1 mM ATP, pH 7.0 in presence of 0.1 μM A1-S1, 5 μM TF and indicated concentrations of cMyBP-C and Ca2+ levels.
Cryo EM sample preparation:
[2 μM] of TFs in 50 mM KAc, 10 mM MOPS, 3 mM MgCl2, 2 mM EGTA (pCa>8), pH 7.0 were incubated with human C0C1 or C1R215E/K218E constructs [15– 20 μM] for 1–2 min and applied to lacey carbon 300 mesh copper grids (Ted Pella), blotted with Whatman #1 filter paper for 3 sec and vitrified in a Vitrobot Mark IV (FEI,Inc.).
Image analysis:
Samples were imaged with a Titan Krios at 300 keV using the Falcon II direct electron detector. The images were dose-fractionated into seven frames. Two frames out of seven where used and represented a dose of ~20 electrons/Å 2. The SPIDER software package (Frank et al., 1981) was used for image processing, except the CTFFIND3 software (Mindell et al., 2003) was used to determine the defocus values in the micrographs. EMAN package (Ludtke et al., 1999) was used to extract filament images from micrographs. Fourier Shell Correlation was used to estimate the resolution (Supplemental Figure S1C).
3D-reconstruction of C0C1-decorated TFs:
We used 6,080 micrographs possessing a defocus range from 0.5 to 3.5 μm at a raster of 1.05 Å per pixel. Initial correcti on for the contrast transfer function (CTF) was made by multiplying each image by its theoretical CTF. From these CTF- corrected images, 354,161 overlapping segments (460 pixels long) were extracted. Nineteen model volumes were created: pure F-actin (1), F-actin with Tm in the “myosin” state (von der Ecken et al., 2016), F-actin with Tm in the “myosin” state decorated with C0 (pdb: 2K1M) in the “front” and “side” posit ions observed earlier (Harris et al., 2016) (e.g. C0-F1, C0-F2, C0-S), F-actin with Tm in the “myosin” state decorated with C1 (pdb: 2V6H) in the “front” and “side” positions observed earlier (Harris et al., 2016) (e.g. C1-F1, C1-F2, C1-F3, C1-S), F-actin with Tm in the “myosin” state decorated with both C0 and C1 Ig-domains (e.g. C0-F1+C1-S, C0-F2+C1-S, C1-F1+C0-S, C1-F2+C0-S, C1-F3+C0-S), and F-actin with Tm in the “myosin” state decorated with either two C0 or two C1 Ig-domains per actin subunit (e. g. C0-F1+C0-S, C0-F2+C0-S, C1-F1+C1-S, C1-F1+C1-S, C1-F3+C1-S). These volumes were scaled to 4.2 Å per pixel and pr ojected onto 115 × 115 px images with an azimuthal rotational increment of 4°, generating 1,710 reference projections (19 models × 90 projections). The C0C1-decorated TF segments were down-sampled to 4.2 Å per pixel and cross-correlated with 1,710 reference projections. Segments that possessed in-plane rotation angle larger than 15º were discarded (n=243,507). Such a large portion of images that yielded large in-plan rotation angle suggests that the high C0C1 concentration (20 μM) used to prevent stripping of C0C1 from the TFs presumably reduced the SNR in the images and, hence, made images with relatively thick ice unusable due to poor SNR. Segments of images that yielded the best correlation with the pure F-actin model (n=2,222), Tmp-F-actin model (n=3,418), or having either C0 or C1 bound to two actin protomers (e. g. C0-F1+C0-S (n=8,051), C0-F2+C0-S (n=9,115), C1-F1+C1-S (n=5,890), C1-F1+C1-S (n=4,766), C1-F3+C1-S (n=4,427)) were discarded (3D reconstruction of the C1-F1+C1-S class is shown as an example in Figure S4A), while the remaining twelve structural classes were reconstructed with the IHRSR method (Egelman, 2000) starting from a solid cylinder, or a reconstruction originated from the other structural class as a starting model (Figure S1B). Segments that yielded the best correlation with C1-F1+C0-S (n=7,103), C1-F2+C0-S (n=6,049) and C1-F3+C0S (n=5,737) did not return interpretable 3D reconstructions (3D reconstruction of the C1-F1+C0-S class is shown as an example in Figure S4B), suggesting that C0 cannot bind to the side of the TF when C1 is bound to the front. These three sets were disregarded. The nine classes that demonstrated stable structural solutions were reconstructed at a raster of 2.1 Å per pixel and yielded the following helical p arameters: C0-F1 set (n=6,034) yielded a symmetry of −166.5°/27.8 Å; C0-F2 set (n=6,092) yie lded a symmetry of −166.6°/27.7 Å; C0-S set (n=5,294) yielded a symmetry of −166.6°/27.8 Å; C1-F1 set (n=7,371) yielded a symmetry of −166.6°/27.7 Å; C1-F2 set (n=5,067) yielded a sy mmetry of −166.6°/27.7 Å; C1-F3 set (n=5,110) yielded a symmetry of −166.7°/27.7 Å; C1- S (n=7,051) yielded a symmetry of - 166.6°/27.7 Å; C0-F1+C1-S (n=6,117) yielded a symm etry of −166.6°/27.5 Å; C0-F2+C1-S (n=5,830) yielded a symmetry of −166.6°/27.5 Å. Ove rall, the difference in the helical parameters between the structural classes was small and insignificant. The volumes were corrected for the CTF by using a Wiener filter assuming that the signal-to-noise ratio in the volume was very large. Negative B-factors of 500–800 were used to amplify high frequencies in the reconstruction that were dampened by the envelope function of the microscope.
3D-reconstruction of C1R215E/K218E-decorated TFs:
We used 1,066 micrographs possessing a defocus range from 0.5 to 3.5 μm at a raster of 1.05 Å per pixel. Initial correcti on for the contrast transfer function (CTF) was made by multiplying each image by its theoretical CTF. From these CTF- corrected images, 79,816 overlapping segments (460 pixels long) were extracted. Six model volumes were created: pure F-actin, F-actin with Tm in the c-closed state (Risi et al., 2017), F-actin with Tm in the c-closed state decorated with C1 (pdb: 2V6H) in the “front” and “side” positions observed earlie r (Harris et al., 2016) (e.g. C1-F1, C1-F2, C1-F3, C1-S). These volumes were scaled to 4.2 Å pe r pixel and projected onto 115 × 115 px images with an azimuthal rotational increment of 4°, generating 540 reference projections (6 models × 90 projections). The C1 R215E/K218E-decorated TF segments were down-sampled to 4.2 Å per pixel and cross-correlated with 540 refer ence projections. Segments that possessed in-plane rotation angle larger than 15º were discarded (n=22,764). Segments of images that yielded the best correlation with the pure F-actin model (n=25,877), Tm-F-actin model (n=10,663) were discarded. The remaining four classes (Figure S3A) were reconstructed at a raster of 2.1 Å per pixel and yi elded the following helical parameters: C1-F1 set (n=6,821) yielded a symmetry of −166,6°/27.5 Å; C1-F2 set (n=5,396) yielded a symmetry of −166,7°/27.5 Å; C1-F3 set (n=5,701) yie lded a symmetry of −166,5°/27.5 Å; C1-S (n=2,594) yielded a symmetry of −166,8°/27.6 Å.
Modeling:
The model of the Tm-F-actin complex extracted from the myosin-Tm-F-actin complex (von der Ecken et al., 2016) (PDB: 5JLH) was used as the starting model for the refinement against the Cryo-EM density maps. The program DireX (Schroder et al., 2007) was used for the model refinement. The density maps were masked around the rigid-body docked starting model with a radius of 8 Å using a smooth edge. H-bonds present in the starting structure were restrained during the refinement. The C0-F1+C1-S and C0-F2+C1-S models were refined for 100 steps against a target map filtered to 12 Å, while the Fourier interval 10–12 Å was used for cross-validation to p revent overfitting. The C1-S model was refined for 100 steps against a target map filtered to 11 Å, and the Fourier interval 9–11 Å was used for cross-validation. All DireX refinements were followed by minimization with CNS using Ramachandran restraints (Adams et al., 2010). Images were produced using UCSF Chimera (Pettersen et al., 2004).
Data and Software Availability:
The atomic coordinates and structure factors for the C1-S (EMD-4346, PDB ID 6G2T), C0-F1+C1-S (EMD-7780, PDB ID 6CXI) and C0-F2+C1S (EMD-7781, PDB ID 6CXJ) structural modes are deposited in the Protein Data Bank and Electron Microscopy Data Bank.
Supplementary Material
{kind=link}
Highlights:
C0 and C1 domains of Myosin Binding Protein-C cooperate to activate thin filament
C1 traps tropomyosin in the activated myosin state via a positively charged loop
Cardiac-specific C0 domain enhances activating effects of C1
Acknowledgments:
The EM work was conducted at the Molecular Electron Microscopy Core facility at the University of Virginia, which is supported by the School of Medicine, and NIH grants S10-RR025067 (Titan Krios) and S10-OD018149 (Falcon camera). This work was supported by AHA Grant in Aid 560851 (VEG) and R01 HL080367 (SPH).
Footnotes
Declaration of Interests: The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Spudich JA, Huxley HE, and Finch JT (1972). Regulation of skeletal muscle contraction. II. Structural studies of the interaction of the tropomyosin-troponin complex with actin. J. Mol. Biol 72, 619–632. [DOI] [PubMed] [Google Scholar]
- 2.McKillop DFA, and Geeves MA (1993). Regulation of the interaction between actin and myosin subfragment-1: evidence for three states of the thin filament. Biophys. J 65, 693–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vibert P, Craig R, and Lehman W (1997). Steric-model for activation of muscle thin filaments. J. Mol. Biol 266, 8–14. [DOI] [PubMed] [Google Scholar]
- 4.Houmeida A, Heeley DH, Belknap B, and White HD (2010). Mechanism of regulation of native cardiac muscle thin filaments by rigor cardiac myosin-S1 and calcium. J. Biol. Chem 285, 32760–32769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heeley DH, Belknap B, and White HD (2006). Maximal activation of skeletal muscle thin filaments requires both rigor myosin S1 and calcium. J. Biol. Chem 281, 668–676. [DOI] [PubMed] [Google Scholar]
- 6.Risi C, Eisner J, Belknap B, Heeley DH, White HD, Schroder GF, and Galkin VE (2017). Ca2+-induced movement of tropomyosin on native cardiac thin filaments revealed by cryoelectron microscopy. Proc. Natl. Acad. Sci. U. S. A 114, 6782–6787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luther PK, Winkler H, Taylor K, Zoghbi ME, Craig R, Padron R, Squire JM, and Liu J (2011). Direct visualization of myosin-binding protein C bridging myosin and actin filaments in intact muscle. Proc. Natl. Acad. Sci. U. S. A 108, 11423–11428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin BL, Li A, Mun JY, Previs MJ, Previs SB, Campbell SG, dos Remedios CG, Tombe PP, Craig R, Warshaw DM, and Sadayappan S (2018). Skeletal myosin binding protein-C isoforms regulate thin filament activity in a Ca(2+)-dependent manner. Sci. Rep 8, 2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ratti J, Rostkova E, Gautel M, and Pfuhl M (2011). Structure and interactions of myosin-binding protein C domain C0: cardiac-specific regulation of myosin at its neck? J. Biol. Chem 286, 12650–12658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Orlova A, Galkin VE, Jeffries CM, Egelman EH, and Trewhella J (2011). The N-terminal domains of myosin binding protein C can bind polymorphically to F-actin. J. Mol. Biol 412, 379–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mun JY, Previs MJ, Yu HY, Gulick J, Tobacman LS, Beck PS, Robbins J, Warshaw DM, and Craig R (2014). Myosin-binding protein C displaces tropomyosin to activate cardiac thin filaments and governs their speed by an independent mechanism. Proc. Natl. Acad. Sci. U. S. A 111, 2170–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harris SP, Belknap B, Van Sciver RE, White HD, and Galkin VE (2016). C0 and C1 N-terminal Ig domains of myosin binding protein C exert different effects on thin filament activation. Proc. Natl. Acad. Sci. U. S. A 113, 1558–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shaffer JF, Wong P, Bezold KL, and Harris SP (2010). Functional differences between the N-terminal domains of mouse and human myosin binding protein-C. J. Biomed. Biotechnol 2010, 789798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Belknap B, Harris SP, and White HD (2014). Modulation of thin filament activation of myosin ATP hydrolysis by N-terminal domains of cardiac myosin binding protein-C. Biochemistry 53, 6717–6724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Razumova MV, Bezold KL, Tu AY, Regnier M, and Harris SP (2008). Contribution of the myosin binding protein C motif to functional effects in permeabilized rat trabeculae. J. Gen. Physiol 132, 575–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Watkins H, Conner D, Thierfelder L, Jarcho JA, MacRae C, McKenna WJ, Maron BJ, Seidman JG, and Seidman CE (1995). Mutations in the cardiac myosin binding protein-C gene on chromosome 11 cause familial hypertrophic cardiomyopathy. Nat. Genet 11, 434–437. [DOI] [PubMed] [Google Scholar]
- 17.Marston SB (2011). How do mutations in contractile proteins cause the primary familial cardiomyopathies? J. Cardiovasc. Transl. Res 4, 245–255. [DOI] [PubMed] [Google Scholar]
- 18.Schlossarek S, Mearini G, and Carrier L (2011). Cardiac myosin-binding protein C in hypertrophic cardiomyopathy: mechanisms and therapeutic opportunities. J. Mol. Cell Cardiol 50, 613–620. [DOI] [PubMed] [Google Scholar]
- 19.Egelman EH (2000). A robust algorithm for the reconstruction of helical filaments using single-particle methods. Ultramicroscopy 85, 225–234. [DOI] [PubMed] [Google Scholar]
- 20.von der Ecken.J., Heissler SM, Pathan-Chhatbar S, Manstein DJ, and Raunser S (2016). Cryo-EM structure of a human cytoplasmic actomyosin complex at near-atomic resolution. Nature 534, 724–728. [DOI] [PubMed] [Google Scholar]
- 21.Mun JY, Kensler RW, Harris SP, and Craig R (2016). The cMyBP-C HCM variant L348P enhances thin filament activation through an increased shift in tropomyosin position. J. Mol. Cell Cardiol. 91, 141–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.von der Ecken.J., Muller M, Lehman W, Manstein DJ, Penczek PA, and Raunser S (2015). Structure of the F-actin-tropomyosin complex. Nature 519, 114–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Razumova MV, Shaffer JF, Tu AY, Flint GV, Regnier M, and Harris SP (2006). Effects of the N-terminal domains of myosin binding protein-C in an in vitro motility assay: Evidence for long-lived cross-bridges. J. Biol. Chem 281, 35846–35854. [DOI] [PubMed] [Google Scholar]
- 24.Herron TJ, Rostkova E, Kunst G, Chaturvedi R, Gautel M, and Kentish JC (2006). Activation of myocardial contraction by the N-terminal domains of myosin binding protein-C. Circ. Res 98, 1290–1298. [DOI] [PubMed] [Google Scholar]
- 25.Gajendrarao P, Krishnamoorthy N, Kassem HS, Moharem-Elgamal S, Cecchi F, Olivotto I, and Yacoub MH (2013). Molecular modeling of disease causing mutations in domain C1 of cMyBP-C. PLoS. One 8, e59206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shaffer JF, and Harris SP (2009). Species-specific differences in the Pro-Ala rich region of cardiac myosin binding protein-C. J. Muscle Res. Cell Motil 30, 303–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Colson BA, Thompson AR, Espinoza-Fonseca LM, and Thomas DD (2016). Site-directed spectroscopy of cardiac myosin-binding protein C reveals effects of phosphorylation on protein structural dynamics. Proc. Natl. Acad. Sci. U. S. A 113, 3233–3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gruen M, and Gautel M (1999). Mutations in beta-myosin S2 that cause familial hypertrophic cardiomyopathy (FHC) abolish the interaction with the regulatory domain of myosin-binding protein-C. J. Mol. Biol 286, 933–949. [DOI] [PubMed] [Google Scholar]
- 29.Kampourakis T, Yan Z, Gautel M, Sun YB, and Irving M (2014). Myosin binding protein-C activates thin filaments and inhibits thick filaments in heart muscle cells. Proc. Natl. Acad. Sci. U. S. A 111, 18763–18768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nag S, Trivedi DV, Sarkar SS, Adhikari AS, Sunitha MS, Sutton S, Ruppel KM, and Spudich JA (2017). The myosin mesa and the basis of hypercontractility caused by hypertrophic cardiomyopathy mutations. Nat. Struct. Mol. Biol 24, 525–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shaffer JF, Kensler RW, and Harris SP (2009). The myosin-binding protein C motif binds to F-actin in a phosphorylation-sensitive manner. J. Biol. Chem 284, 12318–12327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stelzer JE, Patel JR, and Moss RL (2006). Protein kinase A-mediated acceleration of the stretch activation response in murine skinned myocardium is eliminated by ablation of cMyBP-C. Circ. Res 99, 884–890. [DOI] [PubMed] [Google Scholar]
- 33.Stelzer JE, Patel JR, Walker JW, and Moss RL (2007). Differential roles of cardiac myosin-binding protein C and cardiac troponin I in the myofibrillar force responses to protein kinase A phosphorylation. Circ. Res 101, 503–511. [DOI] [PubMed] [Google Scholar]
- 34.Kensler RW, Craig R, and Moss RL (2017). Phosphorylation of cardiac myosin binding protein C releases myosin heads from the surface of cardiac thick filaments. Proc. Natl. Acad. Sci. U. S. A 114, E1355–E1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Previs MJ, Mun JY, Michalek AJ, Previs SB, Gulick J, Robbins J, Warshaw DM, and Craig R (2016). Phosphorylation and calcium antagonistically tune myosin-binding protein C’s structure and function. Proc. Natl. Acad. Sci. U. S. A 113, 3239–3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moss RL, Fitzsimons DP, and Ralphe JC (2015). Cardiac MyBP-C regulates the rate and force of contraction in mammalian myocardium. Circ. Res 116, 183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ratti J, Rostkova E, Gautel M, and Pfuhl M (2011). Structure and interactions of myosin-binding protein C domain C0: cardiac-specific regulation of myosin at its neck? J. Biol. Chem 286, 12650–12658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Govada L, Carpenter L, da Fonseca PC, Helliwell JR, Rizkallah P, Flashman E, Chayen NE, Redwood C, and Squire JM (2008). Crystal structure of the C1 domain of cardiac myosin binding protein-C: implications for hypertrophic cardiomyopathy. J. Mol. Biol 378, 387–397. [DOI] [PubMed] [Google Scholar]
- 39.Frank J, Shimkin B, and Dowse H (1981). SPIDER - A modular software system for electron image processing. Ultramicroscopy 6, 343–358. [Google Scholar]
- 40.Mindell JA, and Grigorieff N (2003). Accurate determination of local defocus and specimen tilt in electron microscopy. J. Struct. Biol 142, 334–347. [DOI] [PubMed] [Google Scholar]
- 41.Ludtke SJ, Baldwin PR, and Chiu W (1999). EMAN: semiautomated software for high-resolution single-particle reconstructions. J. Struct. Biol 128, 82–97. [DOI] [PubMed] [Google Scholar]
- 42.Schroder GF, Brunger AT, and Levitt M (2007). Combining efficient conformational sampling with a deformable elastic network model facilitates structure refinement at low resolution. Structure. 15, 1630–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, and Zwart PH (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Behrmann E, Muller M, Penczek PA, Mannherz HG, Manstein DJ, and Raunser S (2012). Structure of the rigor actin-tropomyosin-myosin complex. Cell 150, 327–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.