

Summary
Transient Receptor Potential (TRP) channels are polymodally regulated ion channels. TRPV4 (vanilloid 4) is sensitized by PIP2 and desensitized by Syndapin3/PACSIN3 which bind to the structurally uncharacterized TRPV4 N-terminus. We determined the NMR structure of the Syndapin3/PACSIN3 SH3 domain in complex with the TRPV4 N-terminal proline rich region (PRR), which binds as a class I poly-proline II (PPII) helix. This PPII conformation is broken by a conserved proline in a cis conformation. Beyond the PPII, we find that the proximal TRPV4 N-terminus is unstructured, a feature conserved across species thus explaining the difficulties in resolving it in previous structural studies. Syndapin/PACSIN SH3 domain binding leads to rigidification of both the PRR and the adjacent PIP2-binding site. We determined the affinities of the TRPV4 N-terminus for PACSIN1, 2 and 3 SH3 domains and PIP2 and deduce a hierarchical interaction network where Syndapin/PACSIN binding influences the PIP2-binding site but not vice versa.
Keywords: TRP channel, TRPV4, PACSIN, Syndapin, PIP2, cis proline, NMR, SH3 domain, class I, proline rich region
eTOC Blurb:
Goretzki et al determined the molecular interaction between the ion channel TRPV4 and its regulatory partners, PIP2 and Syndapin/PACSIN1–3. The NMR and CD spectroscopic approach provides an atomistic view of the unstructured TRPV4 N-terminus and builds a framework for a better understanding of the intricate polymodality of TRPV4.
Introduction
Transient Receptor Potential (TRP) channels are cation-selective ion channels involved in various physiological functions, including nociception and temperature sensation (Castillo et al., 2018; Moore et al., 2018). Recently, our understanding of these complex membrane proteins made a huge leap forward through numerous high-resolution cryo-electron microscopy (cryoEM) structures of near-full length TRP channels, beginning with TRP vanilloid 1 (TRPV1) (Cao et al., 2013; Liao et al., 2013). Structures of near full-length ion channels are complemented nicely by structures from various soluble TRP channel domains (Hellmich and Gaudet, 2014b). However, the molecular insights into the functional interactions of TRP channels with lipids or regulatory proteins are still sparse.
TRPV4 is widely expressed in numerous tissues (White et al., 2016). Its physiological functions include regulation of osteoclast and chondrocyte development or cell volume regulation (Liedtke and Friedman, 2003; Muramatsu et al., 2007; Masuyama et al., 2008) as well as temperature sensation (Güler et al., 2002) and mutations can lead to severe channelopathies (Nilius and Voets, 2013). As all other members of the TRPV subfamily, TRPV4 contains a cytoplasmic ankyrin repeat domain (ARD) with six repeats preceding the first transmembrane helix. The ARD is preceded by an N-terminal region which varies in length and sequence between the different TRPV channels. In structures of TRPV1, V2 and V4, this proximal N-terminus is either unresolved or has been deleted in the constructs used for structure determination (Cao et al., 2013; Liao et al., 2013; Huynh et al., 2016; Zubcevic et al., 2016; Deng et al., 2018). In TRPV5 and V6 it forms an α-helix that folds back onto the ARD (Hughes et al., 2018; McGoldrick et al., 2018).
The ~130 amino acid TRPV4 N-terminal region harbors interaction sites for functional regulators, including both sensitizers and desensitizers of channel function. While structures of the TRPV4 ARD in isolation and in context with the transmembrane domain show an α-helical fold (Landouré et al., 2010; Inada et al., 2012; Deng et al., 2018), the structure of the TRPV4 N-terminal region preceding the ARD is currently unknown. Here, a ++W++ motif (+: lysine/arginine, W: tryptophan) termed the PIP2 binding domain (PBD), is essential for channel sensitization by PI(4,5)P2 (Garcia-Elias et al., 2013). Mutagenesis of the basic residues to alanine (++W++ → AAWAA) led to loss of TRPV4 activation through heat and osmotic stimuli (Garcia-Elias et al., 2013). Between the PBD and the ARD, TRPV4 contains a proline rich region (PRR) that interacts with the C-terminal Src homology 3 (SH3) domain of PACSINs (Protein kinase C and casein kinase substrate in neurons, also known as Syndapins) (Cuajungco et al., 2006). This SH3 domain is connected to an F-BAR (Fer-CIP4 homology Bin/Amphiphysin/Rvs) domain which interacts with membranes to sense or induce curvature and tubulation (Wang et al., 2009; Bai et al., 2012). Despite their name, only PACSIN1 is a neurospecific protein, while PACSIN2 is ubiquitously expressed and PACSIN3 is predominantly localized in the skeletal muscle and heart (Modregger et al., 2000) although it also co-localizes with TRPV4 in kidney tubule membranes (Cuajungco et al., 2006). All three PACSIN SH3 domains can interact with the PRR in the TRPV4 N-terminus, but only PACSIN3 desensitizes TRPV4 to heat stimuli (Cuajungco et al., 2006; D’hoedt et al., 2008). Additionally, mutations in the PACSIN3 SH3 domain or the TRPV4-PRR that decreased TRPV4/PACSIN3 interaction also led to a reduced plasma membrane presentation of TRPV4. This effect was not observed for PACSIN1 and 2 (Cuajungco et al., 2006). Likewise, no regulatory role for PACSIN1 and 2 on TRPV4 channel activity is known. Overall, a detailed molecular view on the interactions between TRPV4, PIP2 and PACSINs is currently missing.
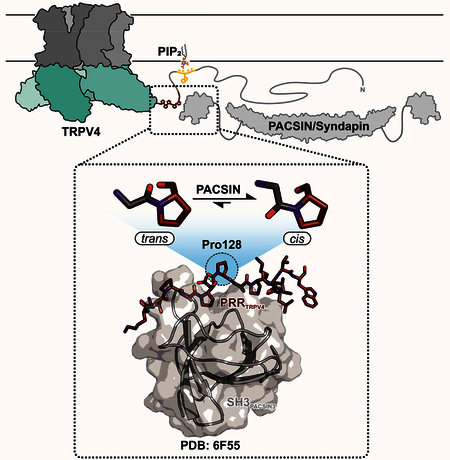
There are currently only a handful of structures showing the interaction of a TRP(V) channel with a protein regulator (Lau et al., 2012; Gao et al., 2016; Bokhovchuk et al., 2018). Structures of TRPV1 or TRPP2 have provided insights into how PIP2 interacts with the transmembrane domain of TRP channels (Gao et al., 2016; Wilkes et al., 2017) and densities attributed to lipids were identified in the Xenopus tropicalis TRPV4 pore (Deng et al., 2018), but the PIP2 interaction site responsible for TRPV4 channel sensitization is in the structurally unexplored proximal N-terminus (Garcia-Elias et al., 2013). The interaction of TRP(V) channels with lipid and protein partners in the N- and C-termini often take place within regions deleted or unresolved in cryoEM or X-ray crystallography structures of near full-length channels. This is unsurprising, because flexible regions and those predicted to be unstructured and dynamic are often deleted to develop stable constructs for high-resolution structure determination (Hellmich and Gaudet, 2014a). Indeed, although included in the crystallization construct, the PRR was not resolved in the recent frog TRPV4 structures (Deng et al., 2018). This highlights the need for complementary approaches to study ligand interactions with TRP channels. Here we use solution nuclear magnetic resonance (NMR) spectroscopy, a powerful tool to analyze dynamic proteins and their interactions even when weak and transient (Hellmich and Glaubitz, 2009; Hacker et al., 2015; Liu et al., 2016), to determine the structural details of the TRPV4 PRR interaction with the PACSIN1, 2, and 3 SH3 domains and of PIP2 with the TRPV4 N-terminal PIP2 binding domain. Importantly, we observe a binding mode we termed “skipped class I motif” of the channel prolinerich region with the PACSIN SH3 domain, characterized by a highly conserved TRPV4 proline in a cis conformation. SH3 binding affects both the TRPV4-PRR and the PBD and concomitantly rigidifies the PRR and PIP2 binding site. In contrast, PIP2 binding does not affect the channel PRR. Our results thus provide structural and mechanistic insights into the interplay of two important functional modulators of the TRPV4 channel.
Results
The proximal TRPV4 N-terminus is unstructured
The TRPV4 N-terminus contains important binding sites for PIP2 and PACSIN1, 2 and 3 (Cuajungco et al., 2006; D’hoedt et al., 2008). To elucidate the TRPV4 N-terminus structure, we expressed and purified several human and chicken TRPV4 N-terminal constructs: (i) the entire ~130 N-terminal residues with the ARD (N-ARD, 1–383); (ii) the ARD preceded by the PRR ( PRR-ARD, 121–383) and (iii) the isolated ARD (ARD, 134–383) (Fig. 1A, B). As expected, and as previously observed (Landouré et al., 2010; Inada et al., 2012), circular dichroism (CD) spectroscopy shows that the TRPV4 ARD is predominantly α-helical. With increasing length of the N-terminus, the degree of unstructured contributions increases, indicating that the regions preceding the ARD are largely unstructured. The comparison with the respective N-terminal constructs from chicken and human TRPV4 confirms that this architecture is observed across species (Fig. 1, S1).
Figure 1: Structural analysis of the TRPV4 N-terminus.

(A) Schematic overview of constructs with varying N-terminus length. The α-helical Ankyrin repeat domain (ARD, cyan) is preceded by a ~130 amino acid stretch harboring the PIP2 binding domain (PBD, yellow) and the proline rich region (PRR, red) important for PACSIN SH3 domain interaction. (B) Coomassie-stained SDS-PAGE of purified chicken TRPV4 N-terminal constructs. (C) CD spectra of the entire N-terminus (N-ARD; residues 1–383), the ankyrin repeat domain with the PRR (PRR-ARD; 122–383), and the isolated Ankyrin repeat domain (ARD; 134–383). (See Fig. S1 for CD studies of human TRPV4 N-terminal constructs).
The TRPV4 N-terminal proline rich region and the PIP2 binding domain, regions important for the interaction with PACSIN proteins and PIP2, were further investigated by CD spectroscopy. Neither the isolated PRR (residues 121–134) nor the PRR with the PBD (PBD-PRR, residues 105–134) contain α-helices or β-sheets (Fig. S2). Solely based on the CD spectra, it is however difficult to estimate whether a polyproline helix (PPII) might already be present in the isolated peptides, as spectra of unfolded proteins and polyproline helices are similar (Afonin et al., 2017). The absence of α-helices or β-sheets in the TRPV4-PBD-PRR region is also supported by the secondary structure analysis based on the PBD-PRR NMR backbone assignment. Here, the chemical shift-based prediction of the torsion angles indicates that a polyproline helix contribution may be present (Fig. S3).
Since TRPV4-PBD-PRR includes a lipid-binding site, and the interaction with membrane mimetics can lead to the induction of structural elements (Roccatano et al., 2002), this was probed through the addition of SDS or liposomes but also did not lead to the formation of secondary structure (Fig. S2). In TRPV5 and V6, the N-terminal region preceding the ARD (in an analogous position to the TRPV4-PRR) forms an α-helix that folds onto the ARD (Hughes et al., 2018; McGoldrick et al., 2018). To test the propensity of this region to adopt α-helical conformations, both the TRPV4-PRR and the corresponding TRPV6 N-terminal peptides were titrated with trifluoroethanol (TFE), a solvent which leads to enhanced secondary structure formation through promoting H-bonds (Roccatano et al., 2002). Addition of TFE led to α-helical secondary structure formation for the TRPV6 N-terminus, but not for TRPV4-PRR (Fig. S2). Importantly, polyproline helices are not stabilized by H-bonds and thus no effect of TFE on such a structure would be expected.
The TRPV4 proline rich region constitutes the minimal PACSIN3 SH3 binding site
Previous pulldown and mutagenesis studies identified the TRPV4-PRR as the interaction site for the PACSIN3 SH3 domain in the context of the full-length TRPV4 channel (Cuajungco et al., 2006), but a molecular view of the interaction of TRPV4 with PACSIN3 is currently missing. The unstructured nature of the TRPV4 N-terminus makes X-ray and cryo-EM approaches difficult while NMR spectroscopy is well suited to investigate unstructured or flexible proteins and their interaction partners. To identify the minimal region of the TRPV4 N-terminus that can bind to the PACSIN3 SH3 domain without loss of affinity, we performed an NMR backbone assignment of the 13C, 15N-labeled chicken PACSIN3 SH3 domain (Fig. 2A). Different TRPV4 N-terminal constructs were then titrated to the 15N-PACSIN3 SH3 (Fig. 2B-D). For the 30 kDa PRR-ARD construct, the observed chemical shift changes indicated complex formation with the PACSIN3 SH3 domain, but the size of the complex led to unfavorable tumbling behavior and thus severe line broadening (Fig. 2B). Addition of the isolated PRR led to the same chemical shift changes in the PACSIN3 SH3 domain as observed for the PRR-ARD but without peak broadening (Fig. 2C). We further tested the PBD-PRR peptide, containing the PIP2 binding site and again observed the same chemical shift changes within the spectra of the 15N-PACSIN3 SH3 domain as for the isolated PRR. Of note, the interaction of the PBD-PRR with the PACSIN3 SH3 domain were accompanied by slight peak broadening in the final titration points (Fig. 2D, see below). However, there are no differences in the affinities of the TRPV4-PRR and the PBD-PRR for the PACSIN3 SH3 domain (KD = 68.6±5.7 and 74.2±11.8 μM, respectively). As a control, the reverse experiment using the 15N-TRPV4-PBD-PRR as a reporter titrated with unlabeled PACSIN3 SH3 also yielded KD values in the same range (Fig. S3, Table S1).
Figure 2: Identification of the minimal TRPV4/PACSIN3 interaction sites.

(A) 1H, 15N-HSQC NMR spectrum of PACSIN3 SH3 domain with complete backbone assignments. (B, C, D) Zoom in on peak of residue Y387 in the presence of increasing concentrations of TRPV4 N-terminal parts to show representative chemical shift changes: (B) addition of PRR-ARD (122–383) leads to chemical shift changes and line broadening due to unfavorable tumbling behavior of the 30 kDa complex; addition of (C) PRR (121–134) and (D) PBD-PRR (105–134) leads to well resolved chemical shift changes that are identical for all binding partners. Cartoon of ARD is based on (PDB: 3JXI), cartoon of PACSIN3 SH3 domain based on our NMR structure (PDB: 6F55). All interaction partners drawn to scale.
The TRPV4 proline rich region binds the PACSIN3 SH3 domain in a class I orientation
Typically, polyproline SH3 domain ligands form a left-handed helix (PPII) that can interact with SH3 domains in two different N- to C-terminal orientations, class I and class II. This orientation can normally be predicted from the position of a positively charged residue flanking the proline rich stretch which will bind the specificity pocket of the SH3 domain (Saksela and Permi, 2012). However, in the case of TRPV4, the proline rich region is bracketed by a lysine residue on both sides of the PRR (Fig. 3B). It is thus not possible to predict the binding mode based on sequence alone. To obtain a high-resolution view of the interaction of TRPV4 with a desensitizing protein, we determined the solution NMR structure of the chicken PACSIN3 SH3 domain in complex with the TRPV4-PRR (Fig. 3A, S4, Table 1). A chicken TRPV4-PRR with a single substitution (V131I) was used for structure determination. Human TRPV4 carries an isoleucine at this position and the valine to isoleucine substitution made side chain assignments for the PRR for structure determination significantly easier. However, titration with the native sequence (containing V131) showed that the fingerprint 1H, 15N-HSQC spectra of the complex are virtually identical for both peptide variants, i.e. the direction and intensity of chemical shift changes are the same as for the isoleucine substitution.
Figure 3: Structural basis of TRPV4 interaction with PACSIN3 SH3.

(A) Solution NMR structure of TRPV4 proline rich region in complex with PACSIN3 SH3 ( see also Table 1, Fig. S4). (B) TRPV4-PRR binds to PACSIN3 SH3 domain in a class I binding mode, with N-terminal K122 interacting with the SH3 specificity pocket. P126 dips into the first proline pocket as expected for a canonical class I binding mode, but due to the cis conformation of P128, P130 rather than P129 interacts with the second proline pocket (see also Fig. S4). PACSIN1 and 2 SH3 domains share identical binding behavior (Fig. S3). (C) P128 in the TRPV4-PRR bound to the SH3 domain adopts a cis conformation while all other proline residues are in the trans conformation. (D) In the cis conformation, the proline Cδ carbon is oriented in the same direction as the backbone carboxylate of the preceding amino acid (ω=0°), in a trans conformation, they point into opposite directions (ω=180°). The cis/trans isomerization has implications for the orientation of the N- and C-terminus and thus on the position of the adjacent amino acids. (E) 1H, 13C-HSQC of site specifically 13C-P128 labeled free (unbound) PRR shows that while P128 strongly prefers trans population in the absence of a binding partner, it has a small, detectable cis population. (F) 1D projections of the trans (blue) and cis Hγ/Cγ peaks (red) show relative populations of isomers. In the apo state, the trans state is dominant while upon addition of PACSIN1, 2 and 3 SH3 domains, the cis conformation population increases.
Table 1:
Structural statistics for the solution NMR structure of PACSIN3 SH3 domain in complex with TRPV4-PRR
| Conformational restricting restraints | |
| Total NOE distance restraints | 948 |
| intraresidue |i = j| | 98 |
| sequential |i - j| = 1 | 230 |
| medium-range 1 < |i - j| < 5 | 143 |
| long-range |i - j| ≥ 5 | 477 |
| Dihedral angle restraints (Talos-N) | 108 |
| No. of restraint per residue | 13.5 |
| No. of long-range restraints per residue | 6.1 |
| Residual restraint violationsa | |
| Average no. of distance violations | |
| per structure | |
| 0.1–0.2 Å | 3.8 |
| 0.2–0.5 Å | 0.3 |
| >0.5 Å | 1.5 |
| Average no. of dihedral angle violations | |
| per structure | |
| 1–10° | 7.8 |
| >10° | 0 |
| Model quality (ordered residues)a | |
| RMSD backbone atoms (Å) | 0.4 |
| RMSD heavy atoms (Å) | 0.9 |
| RMSD bond lengths (Å) | 0.012 |
| RMSD bond angles (°) | 2.3 |
| MolProbity Ramachandran statisticsa | |
| Most favored regions | 93.5% |
| Allowed regions | 5.5% |
| Disallowed regions | 1.1% |
| Global quality scores (raw/Z score)a | |
| Verify 3D | 0.23/−3.69 |
| Prosall | 0.17/−1.99 |
| PROCHECK (ϕ-ψ) | −0.76/−2.68 |
| PROCHECK (all) | −0.89/−5.26 |
| MolProity clash score | 4.9/0.68 |
| Model contents | |
| Ordered residue ranges (HetNOE > 0.6) | 6–62,103–111 |
| Total no. of residues | 82 |
| BMRB accession number | 34211 |
| PDBID code | 6F55 |
calculated using PSVS 1.5 for using ordered residues (HetNOE > 0.6) (Bhattacharya et al., 2007).
Average distance violations were calculated using the sum over r−6
The structure of the PACSIN3 SH3 domain contains the typical SH3 domain five-stranded β-sheet core and a long loop that acts as the “lid” for recognition and binding of the TRPV4 proline rich region. The TRPV4-PRR binds between the PACSIN3 SH3 Src and RT-loops in a class I binding mode (Fig. 3A, B), with K122 interacting with the conserved glutamate in the RT loop constituting the specificity pocket (Saksela and Permi, 2012) and P126 integrating into the first proline binding pocket (Fig. S4A, B). In a canonical class I binding orientation, P129 would be predicted to occupy the second proline pocket (Fig. 3B, S4C). However, because P128 adopts a cis conformation for the peptide bond and introduces a kink in the peptide, this is sterically not possible and instead it is P130 that interacts with the second proline pocket (Fig. S4) in a “skipped class I motif”.
PACSIN1, 2 and 3 SH3 domains interact similarly with the TRPV4 proline rich region
Besides PACSIN3, PACSIN1 and 2 can also interact with TRPV4, but to date no similar functional consequences for channel sensitization and localization have been described (Cuajungco et al., 2006). To investigate whether all PACSINs nonetheless share a similar binding interface, we obtained NMR backbone assignments of chicken PACSIN1 and PACSIN2 SH3 domains (Fig. S3). The NMR secondary structure analysis in combination with CD spectroscopy of all three SH3 domains showed the expected β-sheet structure (Fig. S5A, B). Using 15N-labeled PACSIN1 and 2 SH3 domains as reporters, we performed chemical shift perturbation experiments to map the TRPV4-PRR interacting residues. All three PACSIN SH3 domains share the same binding interface with the TRPV4-PRR. The affinities of the interactions between TRPV4-PRR and PACSIN1 SH3 (KD = 51.6±7.6 μM) or PACSIN2 SH3 (12.7±2.4 μM) are in the same range as the PACSIN3 SH3 domain (68.6±5.7 μM) (Fig. S3, Table S1).
TRPV4 Pro128 binds to PACSIN SH3 domains in a cis conformation and breaks the polyproline helix
In the structure of the TRPV4-PRR-bound PACSIN3 SH3 domain, residues T121 to N127 in the TRPV4-PRR adopt a classical Polyproline type II (PPII) left-handed helix. The PACSIN SH3 domain’s specificity and first proline pocket are occupied as expected for a peptide interacting in a class I orientation. However, at residue P128, the classical PPII is broken since the peptide bond between N127 and P128 adopts a cis conformation, introducing a kink in the bound peptide and thus leading to the observed “skipped Class I” binding motif. P128 is followed by two consecutive proline residues (P129, P130), both of which feature trans peptide bonds. This three-proline stretch is conserved in TRPV4 across species (Fig. S1).
While the P128 peptide bond is in a cis conformation in the TRPV4-PRR/PACSIN3 SH3 complex structure, all other proline residues are in the thermodynamically more stable trans peptide bond conformation (Fig. 3C, D). To measure whether P128 is also in a cis conformation when not bound to a target protein, 1H, 13C-HSQC experiments on site-specifically 13C-P128 labeled PRR were carried out (Fig. 3E). Here, two sets of signals with different intensities are observed for the P128 spin system. The chemical shifts for the Hβ/Cβ and the Hγ/Cγ groups show that the P128 peptide bond adopts both the trans and the cis conformation (Schubert et al., 2002), but that in the free PRR, the trans conformer is strongly preferred (see intensities of the 1D projections for the cis and trans Hγ/Cγ peaks) (Fig. 3F). The SH3 domains of PACSIN1, 2 and 3 form complexes with the PRR which are in the fast exchange regime on the NMR time scale. Thus, for the labelled PRR in the presence of an SH3 domain only averaged signals between the free and the bound state are observable. Nonetheless, upon addition of any of the three SH3 domains from PACSIN1, 2 and 3 the overall cis population for P128 increases proportionally to the amount of PRR/SH3 domain complex in the sample (Fig. 3F). Thus, in agreement with the PACSIN3 SH3 domain/PRR complex structure, the three SH3 domains bind the PRR with P128 in the cis conformation.
A P128 to alanine substitution (also in combination with a P129L mutation) was previously shown to abrogate the interaction of TRPV4 with PACSINs in co-immunoprecipitation assays (Cuajungco et al., 2006). In our system, neither the introduction of the single P128A mutation in the TRPV4-PRR, nor the double mutant, P128A/P129L, fully abolished the interaction with the PACSIN3 SH3 domain, but in both cases, the affinity was weakened (Fig. S6, Table S1).
PACSIN3 SH3 domain binding influences PIP2 interacting residues in the TRPV4 N-terminus
The TRPV4-PRR is close to the PIP2 binding domain (PBD) necessary for channel sensitization. Mutagenesis studies identified a cluster of four basic amino acids (107KRWRR111) to be essential for PIP2 interaction of TRPV4 (Garcia-Elias et al., 2013). However, these mutagenesis studies do not necessarily delineate the complete binding site as residues responsible for weak interactions may be missed.
To identify the residues in the TRPV4 N-terminus that sense either PACSIN3 SH3 or PIP2 binding, we determined a backbone NMR resonance assignment of the 15N,13C-TRPV4-PBD-PRR (Fig. S3) and performed chemical shift perturbation experiments with either unlabeled PACSIN3 SH3 domain or the water soluble lipid diC8-PI(4,5)P2 (Fig. 4A-G). As expected, PACSIN3 SH3 domain binding to the 15N-TRPV4-PBD-PRR induces chemical shift changes for residues in the PRR, particularly K122, G123, A125 and N127 (Fig. 4A, red line). Chemical shift perturbations are also observed for residues in the linker between the PBD and PRR (e.g. V120 and R112) and even within the PBD (e.g. W109). For the 15N-labeled PACSIN3 SH3 domain, we observe chemical shift differences between PRR and PBD-PRR interactions on the backside of the RT loop, specifically residues E408 and following (Fig. S4, S6). Of note, only addition of wildtype sequence PBDKRWRR-PRR, but neither PBDAAWAA-PRR (with a mutated PIP2 binding site) nor isolated PRR led to line broadening in the 1H, 15N-HSQC spectrum of 15N-labeled PACSIN3 SH3 (Fig. 2). We thus also tested the effects of PACSIN3 SH3 binding to 15N-labeled PBDAAWAA-PRR (Fig. 4A, light grey line). Intriguingly, while the chemical shift changes on the PRR residues are identical compared to PBDKRWRR-PRR (Fig. 4A, red line), the chemical shift changes of the PBD-PRR linker residues are smaller and PBDAAWAA residues are unperturbed by the SH3 domain. This demonstrates that while the interaction between the PACSIN3 SH3 domain and the TRPV4 N-terminus is dominated by the PRR, the presence of the PACSIN3 SH3 domain is also sensed in the PIP2 binding domain.
Figure 4: Interactions between TRPV4 N-terminus, PACSIN3 SH3 domain and PIP2.

(A) Chemical shift differences of TRPV4-PBD-PRR (bottom) and TRPV4-PBDAAWAA-PRR (top) free and bound states with PACSIN3 SH3 (red and light grey traces) and PIP2 (yellow and dark grey traces). PIP2 only leads to chemical shift changes in the previously determined PIP2 binding domain (Garcia-Elias et al., 2013), while PACSIN3 SH3 domain has effects on both the PRR and the PBD. For PBDAAWAA-PRR, mostly the same residues are affected by PACSIN3 SH3 binding, while PIP2 binding leads to line broadening in the linker region (empty boxes) and only very slight chemical shifts in the PBD. (B) {1H},15N-HetNOE values of TRPV4-PBD-PRR backbone amides. Lower values are indicative of higher flexibility. (C) Position of the three phosphate groups, P1, P4 and P5 in PI(4,5)P2. (D) 31P titration curves of PIP2 P4 and P5 with PBD-PRR (blue), PBDAAWAA-PRR (green) and PRR (red) derived from 31P chemical shift changes: (E-G) 31P NMR spectra of PIP2 phosphate groups with (E) PBD-PRR; (F) PBDAAWAA-PRR and (G) PRR. (H) Effect of adding PIP2 and PBD-PRR to 15N-PACSIN3 SH3 domain in different orders:Peak corresponding to PACSIN3 SH3 Y387 is shown in the 2D 1H, 15N-spectrum and as a slice through the nitrogen dimension to illustrate line width (left, grey)Upon addition of PIP2 to the SH3 domain, no chemical shift differences nor line broadening is observed, thus showing that lipid and SH3 domain do not interact (bottom, orange). In contrast, addition of PBD-PRR to 15N-PACSIN3 SH3 shows a chemical shift and concomitant line broadening indicative of interaction between SH3 domain and peptide (top, blue). In a second step, the respective missing binding partner (PIP2 in upper and PBD-PRR in lower path) is added. In both cases, identical final chemical shift positions and broad line widths are observed (right) showing that PACSIN3 SH3, PIP2 and TRPV4-PBD-PRR form the same tripartite complex regardless of order of binding partner addition.
An overlap of the PACSIN3 SH3 binding site with the PBD is further supported by {1H},15N-hetNOE (heteronuclear Overhauser Effect) measurements of the 15N-PBD-PRR in the absence or presence of the PACSIN3 SH3 domain (Fig. 4B). Here, the dynamics of the PBD-PRR backbone is measured as fluctuations of the N-H amide bond vector on the ps-ns timescale (Farrow et al., 2002). In general, low values represent highly mobile residues, while higher values represent increasing rigidification. As expected for an unstructured peptide, the PBD-PRR peptide shows overall high flexibility in the absence of the PACSIN3 SH3 domain, with very high mobility in the C-terminus. Upon binding of PACSIN3 SH3, the entire peptide becomes less flexible. In agreement with the complex structure, the effects are most pronounced in the PRR, particularly for K122, G123, A125 and N127. In the complex, G123, A125 and N127 contact the SH3 domain Src loop, with the highly conserved A125 wedged between Q412 and W414 of the PACSIN3 SH3. The backbone carbonyl of G123 hydrogen bonds to the indole amide of W414 in the SH3 domain, as evidenced by the very strong chemical shift differences for NHε of W414 between the bound and unbound states of all PACSIN SH3 domains (Fig. S3, S4). Surprisingly, residues in the PBD rigidify upon PACSIN3 SH3 binding as indicated by the significantly increased {1H},15N-hetNOE values for the amides of R108, W109, R110 and R111. Taken together, the chemical shift differences upon binding of the SH3 domain and the changes in the {1H},15N-hetNOE values show that the PBD is involved in transient interactions with the SH3 domain.
PIP2 binding does not influence PACSIN3 SH3 interacting residues in TRPV4 N-terminus
In contrast to the effects of the PACSIN3 SH3 domain, when PIP2 is added, it exclusively affects residues in and around the previously identified PIP2 binding domain (107KRWRR111) in the PBD-PRR (Garcia-Elias et al., 2013), as indicated by strong shifts and peak broadening of the corresponding NMR resonances (Fig. 4A, yellow line). However, while mutating the four positively charged residues was sufficient to abolish TRPV4 sensitization and eliminate PIP2-dependent protection in limited proteolysis experiments (Garcia-Elias et al., 2013), residues beyond the ++W++ motif do participate in lipid binding as residues in the PBD-PRR linker (e.g. R112, V113, V114) experience strong chemical shift changes. In contrast, in the PBDAAWAA-PRR, the PIP2 interaction is essentially abolished (Fig. 4A, dark grey line).
We also probed the TRPV4 N-terminus/PIP2 interaction via 31P NMR spectroscopy (Fig. 4C-G). 31P NMR allows to study protein-lipid interactions from the lipid’s point of view and, since 31P has 100% natural abundance, no elaborate isotope labeling schemes are required (Ullrich et al., 2011). PI(4,5)P2 gives rise to three peaks in the 31P spectrum corresponding to the three phosphate groups at the 1, 4, and 5 positions of the lipid headgroup (Fig. 4E-G). PIP2 was titrated with PBDKRWRR-PRR or the mutant PBDAAWAA-PRR. While the PBDKRWRR-PRR peptide interacts with PIP2 with a KD in the μM range (139.8±1.9 μM), the PBDAAWAA-PRR affinity is more than one order of magnitude lower (2.7±0.1 mM) (Fig. 4D). The different affinities of PIP2 are further reflected by the different line widths during the titration experiments. The interaction of PIP2 with PBD-PRR is characterized by broad (intermediate exchange) compared to sharper (fast exchange) resonances in the case of PBDAAWAA-PRR (Fig. 4E, F). In agreement with the finding that the PIP2 interaction is dominated by the PBD and surrounding residues (Fig. 4A), titration of the isolated PRR showed essentially no interaction (Fig. 4G). The data thus show that PIP2 binding is sensed only by residues in and around the TRPV4-PBD but not in the proline rich region.
PACSIN3 SH3 and PIP2 can form a tripartite complex with TRPV4 N-terminus
The chemical shift perturbation experiments of 15N-PBD-PRR titrated with PIP2 or the PACSIN3 SH3 domain show that the PIP2 binding site extends beyond the region previously inferred from mutagenesis (Garcia-Elias et al., 2013), but that PIP2 binding has no effects on residues within the PRR. In contrast, PACSIN3 SH3 domain binding is dominated by the PRR, but affects residues both in the PRR and the PBD. Considering this overlap, we assessed whether binding of PACSIN3 SH3 and PIP2 are mutually exclusive. 15N-PACSIN3 SH3 was titrated with PBD-PRR followed by PIP2 addition and vice versa and the chemical shifts followed in 1H, 15N-HSQC spectra. The SH3 domain alone does not interact with PIP2 (Fig. 4H, lower branch, yellow spectra) as indicated by the absence of chemical shift changes upon addition of PIP2 to the isolated SH3 domain. Addition of PBD-PRR to the PACSIN3 SH3 domain (Fig. 4H, upper branch, blue spectra) leads to a shift change and line broadening for residues in the contact site thus indicating binding (also see Fig. 2C). In a second step, the respective second ligand (PBD-PRR to PACSIN3 SH3 + PIP2 or PIP2 to PACSIN3 SH3 + PBD-PRR) was added. Regardless of the order of addition, the final peak position and peak widths at the titration endpoints are identical and show the same increased line broadening (Fig. 4H right). This indicates that the order of addition does not matter and in both cases, the formation of a tripartite PBD-PRR/SH3/PIP2 complex is observed.
Discussion
TRP channels are regulated by intricate interactions with proteins and lipids, and TRPV4 is a prime example for this, but structural and dynamic information on such interactions is sparse. Our structure of the PACSIN3 SH3/TRPV4-PRR complex confirmed the TRPV4-PRR as the minimal interaction region for the PACSIN3 SH3 domain. TRPV4 interacts with PACSIN3 in a class I SH3 domain binding mode, with an unusual arrangement enabled by a cis peptide bond at P128 leading to a “skipped class I” binding mode. Using an isolated PBD-PRR peptide we showed that PIP2 and the PACSIN3 SH3 domain can interact simultaneously with the TRPV4 N-terminus. Therefore, the opposing effects of the PIP2 and PACSIN3 regulatory factors on TRPV4 activity are not due to a simple competitive binding interaction. Instead, we propose that the preferential binding of the PACSIN3 SH3 domain to the cis P128 PRR configuration sequesters the N-terminal PIP2 binding domain (PBD) away from the PIP2-containing cellular membrane (Fig. 5), as detailed below.
Figure 5: Mechanistic model of the interaction of the TRPV4-PRR with PACSIN1–3 SH3 domains.

For TRPV4 to readily react to stimuli, the channel PBD will normally be PIP2 bound while P128 in the PRR exists mostly in the trans conformation. Because the trans/cis conformations are in exchange, PACSIN SH3 domain binding can select and stabilize P128 in the cis conformation in a “skipped class I” binding mode. This leads to TRPV4-PBD-PRR rigidification and presumably a relative reorientation of the TRPV4 N-terminus to the plasma membrane. While PIP2 and PACSIN SH3 can form a tertiary complex in vitro, in a cellular setting the TRPV4 N-terminus reorientation and rigidification may lead to release of PIP2 and subsequent channel desensitization.
We measured an affinity in the μM range for PI(4,5)P2 binding to the TRPV4-PBD, and pinpointed the binding interactions to the PBD and adjoining linker to the PRR. With approximately 1% PI(4,5)P2 in the plasma membrane (van Meer et al., 2008), it is likely almost always bound to the channel in the two-dimensionally diffusion limited context of the membrane. This means that TRPV4 will be primed for activation by heat or osmotic shock, both of which depend on PIP2 (Garcia-Elias et al., 2013). Importantly, PIP2 rarely acts alone on proteins, it often co-plays with another regulator (Hilgemann et al., 2001). In the case of TRPV4, PACSIN3 antagonizes PIP2 function and reduces sensitivity to stimuli such as heat (D’hoedt et al., 2008). Originally, we hypothesized that PACSIN3 binding may prevent PIP2 binding to the TRPV4-PBD. However, while the interaction of the PACSIN SH3 domain influences the PIP2 binding site, PIP2 does not influence the PRR (Fig. 4A). Intriguingly, the SH3 domain region affected by the TRPV4-PBD corresponds to the “RT under-groove” (Fig. S4, S6). Similarly, the PACSIN1 SH3 RT under-groove is important for dynamin PRR binding affinity: the dynamin PRR binds the SH3 domain in a canonical PPII while a neighboring stretch of amino acids affects binding affinity through interactions with the under-groove (Luo et al., 2016). In contrast, the TRPV4-PBD had no effect on affinity of the PRR for the PACSIN3 SH3 domain. This interaction may thus serve other regulatory purposes. Importantly, the interaction of PACSIN3 SH3 with the PRR is not sufficient to release bound PIP2. Instead, PACSIN3 SH3 and PIP2 can form a tripartite complex with the TRPV4 N-terminus. The regulation of TRPV4 by these two antagonistic players thus does not depend on a straight-forward competition mechanism.
The isolated PRR has some propensity to adopt a PPII fold, although we observed low hetNOE values indicating high flexibility. Consistently, this region was not resolved in the recent frog TRPV4 structure (Deng et al., 2018). The remaining ~130 residues of the TRPV4 N-terminus are unstructured. The N-terminus length and PRR are conserved in TRPV4 across species (but not other TRPV channels), and both chicken and human TRPV4 N-terminal regions behaved similarly (Fig. 1, S1). In contrast, the N-terminus preceding the ARD is α-helical in the TRPV5 and V6 structures (Hughes et al., 2018; McGoldrick et al., 2018). Consistently, a peptide corresponding to the TRPV6 N-terminal region readily adopts an α-helical fold (Fig. S2). Thus, important regulatory mechanisms specific to different TRPV channels are encoded in their unique N-terminal features.
In isolation, P128 of the PRR is predominantly in a trans conformation, thus we propose that the PIP2-bound state with trans P128 is the basal state of the channel, ready to be activated by various stimuli. When bound to the PACSIN3 SH3 domain, the N-terminus of the TRPV4-PRR adopts a canonical PPII fold up to P128. This residue features a cis peptide bond in the bound state introducing a kink in the peptide. This enables P130 to occupy the second proline pocket, thus skipping one residue (P129) compared to the typical class I binding mode (Fig. 3, S4). The binding mode also explains why the P128A and P128A/P129L mutants have the same reduction in binding affinity for PACSIN3 SH3. P128 is in a conserved three-proline stretch of low flexibility (Fig. 4B), directly preceding the ARD. We thus hypothesize that by selecting the PRR P128 cis conformation, the SH3 domain changes the relative orientation of the PRR (and thus the PBD). PRR binding by the SH3 domain is accompanied by a rigidification of PRR and PBD. This is further supported by the strong line broadening we observed upon titration of the PACSIN3 SH3 domain with the 30.5 kDa TRPV4-PRR-ARD (Fig. 2B), indicating the SH3 domain constitutes part of a larger complex rather than moving like a bead on a loose string.
A PACSIN1 structure shows its SH3 domain interacting electrostatically with the F-BAR domain, providing an elegant structural model for the intermolecular rearrangements of PACSIN upon binding to a target protein (Rao et al., 2010): The SH3 domain cleft binding to a PRR is occupied by the F-BAR domain and upon PRR interaction, the SH3 domain is released thus enabling or changing the interaction of the F-BAR domain with the membrane. The relevant residues of the SH3 and F-BAR domains are conserved between PACSIN1 and 3. Importantly, SH3 domain residues affected by TRPV4-PRR binding also coordinate the interaction with the F-BAR domain (Fig. S6). In TRPV4, PACSIN3 SH3 domain binding would lead to the channel N-terminus changing its relative orientation, which could enable the PACSIN F-BAR domain to attach to the plasma membrane.
Expressing the isolated PACSIN3 SH3 domain showed no effects on TRPV4 desensitization (Garcia-Elias et al., 2013). However the full-length proteins are both membrane-associated, TRPV4 is a homotetramer and PACSIN proteins can form homodimers through the F-BAR domain (Rao et al., 2010; Bai et al., 2012). Thus, the avidity of the TRPV4/PACSIN interaction may be quite high in a physiological system. Yet the affinity could allow transient regulatory interactions in response to changing environments, partly through cis/trans isomerization of P128. Proline cis/trans isomerization reactions have been shown to act as biological regulatory switches Sarkar et al., 2011),also in TRP channels (Shim et al., 2009).
In summary, our structure of the PACSIN3 SH3 domain in complex with the TRPV4 proline rich region lays the foundation for a structural understanding of the interaction of these two important proteins and revealed a unique SH3 domain/proline rich region binding mode. Our biophysical analyses of the interaction and potential cross-talk of PIP2 and PACSIN3 SH3 binding on the TRPV4 N-terminus provide constraints to better understand the interplay between these two regulatory factors in a physiological context. Our data suggest a model in which the opposing regulatory effects of PIP2 and PACSIN3 relies on conformational differences imparted on the TRPV4 N-terminus. Our data thus complement and enhance available functional and structural data for TRPV4 and have implications for our understanding of TRP channel regulation by lipids and proteins.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ute Hellmich (u.hellmich@uni-mainz.de).
EXPERIMENTAL MODELS
We used E. coli BL21-Gold(DE3) cells for recombinant expression of His6SUMO-tagged TRPV4 peptides, His6-tagged TRPV4 N-terminal and PACSIN SH3 constructs. The cells were cultured using standard practices in luria broth (LB) and terrific broth (TB) media.
METHOD DETAILS
Cloning, expression and purification of TRPV4 N-terminal constructs:
Human and chicken TRPV4 N-terminal constructs were cloned from cDNA with NdeI and NotI restriction sites into pET21 with a C-terminal Hexa-His-tag (Landoure et al., 2010, Inada et al., 2012). Constructs were expressed in E. coli BL21-Gold(DE3) in terrific broth medium supplemented with 0,04 % (w/v) glucose and 100 μg/mL ampicillin. Cells were grown at 37°C to an OD600 of 0.4, moved to room temperature (RT), grown to OD600 of 0.8 for induction (75 μM IPTG) and then grown overnight at 20°C (typically 16 h). After harvest, cells were stored at −80°C until further use. For purification, cells were dissolved in lysis buffer (20 mM Tris pH8, 20 mM imidazole, 300 mM NaCl, 0.1% (v/v) Triton X-100, 1 mM DTT, 1 mM benzamidine, 1 mM PMSF, lysozyme, DNAse, RNAse and protease inhibitor (Sigmafast)) and sonicated. Debris was removed by centrifugation and the supernatant applied to a NiNTA gravity flow column (Qiagen). After washing (20 mM Tris pH8, 20 mM imidazole, 300 mM NaCl), protein was eluted with 250 mM imidazole. Protein containing fractions were dialyzed overnight into low salt buffer (10 mM Tris pH7, 50 mM NaCl, 10% glycerol, 1 mM DTT, 0.5 mM PMSF), followed by an ion exchange column (ResQ, GE Healthcare). Protein containing fractions were concentrated and run on a size exclusion column (SEC; HiLoad prep grade 16/60 Superdex200, GE Healthcare) with 10 mM Tris pH7, 300 mM NaCl, 10% glycerol, 1mM DTT. All steps were carried out at 4°C. After SEC, samples were concentrated to required concentrations, flash frozen in liquid nitrogen and stored at −80°C until further use.
Expression and purification of PACSIN SH3 domains:
Gene fragments encoding chicken PACSIN1, 2 and 3 SH3 domains were bought (Genescript) and cloned into a pET11a vector with an N-terminal Hexa-Histdine tag. Constructs were expressed similarly to TRPV4 constructs except in luria broth, or for isotope labeling in minimal media supplemented with 13C-glucose and/or 15N-ammonium chloride (Eurisotope). Cells were lysed as for TRPV4 constructs and lysate applied to a NiNTA gravity flow column (Qiagen). After washing (20 mM Tris pH8, 20/50/75 mM imidazole, 300 mM NaCl), protein was eluted with 100/150/200/250 mM imidazole. Protein containing fractions were concentrated and run on SEC (HiLoad prep grade 16/60 Superdex75, GE Healthcare) with 10 mM Tris pH7, 100 mM NaCl. All steps were carried out at 4°C. After SEC, samples were concentrated to required concentrations, flash frozen in liquid nitrogen and stored at −80°C until further use.
Cloning, Expression and purification of TRPV4 N-terminal peptides:
For a complete list of peptides used for this study, see Table S2. Chicken TRPV4 N-terminal peptides were expressed as N-terminally tagged His6-SUMO fusion proteins in E. coli BL21-Gold(DE3) from a pET11a vector. Mutation of the PIP2 binding site (107KRWRR111→107AAWAA111, termed PBDAAWAA-PRR, forward primer: GTGAAAACGCAGCCTGGGCCGCGCGTGTGGTTGAAAAACCAGTGG, reverse primer: CACACGCGCGGCCCAGGCTGCGTTTTCACCACCAATCTGTTCACG) and Pro128 and Pro129 in the PRR (termed PBD-PRR_P128A, forward primer: CCGAACGCGCCGCCAGTGCTGAAAGTG, reverse primer: ACTGGCGGCGCGTTCGGCGCCGGAC, and PBD-PRR_P128A/P129L, forward primer: GCCGAACGCGCTGCCAGTGCTGAAAGTG, reverse primer: CACTTTCAGCACTGGCAGCGCGTTCGG) were obtained by sited directed mutagenesis of PBD-PRR. For uniformly 13C,15N and 15N isotope labeled peptides, isotope supplemented M9 medium was used whereas non-labeled peptides were expressed in LB medium containing 100 μg/mL ampicillin. Cells were grown at 37°C to OD600 = 0.5 and then induced (0.5 mM final IPTG conc.) for 2 h. Cells were lysed by sonication in lysis buffer (see above). The lysate was cleared by centrifugation and the supernatant applied to a NiNTA affinity gravity flow column (Qiagen). The column was washed (20 mM imidazole pH8, 20 mM Tris pH8, 300 mM NaCl) and protein eluted with 500 mM imidazole. Then, a SEC step (HiLoad prep grade 16/60 Superdex75 column, GE Healthcare) was carried out (10 mM Tris pH7, 100 mM NaCl, 1 mM EDTA, 1 mM DTT). His6SUMO-peptides were digested with Ulp-1 protease (15:1 mol:mol) at 4 °C overnight and a reverse NiNTA affinity purification carried out to remove uncleaved peptides and His6-SUMO tag from cleaved peptides. Processed peptides then only comprised the native residues of the TRPV4 N-terminus. For concentration purposes, the peptides were dialyzed against autoclaved bidest. H2O at 4°C and then lyophilized. Residue-specific isotope labeled peptides used for NMR structure determination were purchased from jpt peptide technologies (Berlin, Germany) (Table S2).
NMR spectroscopy:
NMR measurements were carried out on Bruker 600, 700, 800, 900 and 950 MHz spectrometers equipped with cryogenic triple resonance probes. The proton chemical shifts of 13C, 15N-labeled PACSIN3 SH3 domain were externally (apo) and internally (in complex with PRR) referenced to 2.2-dimethyl-2-silapentane-5-sulfonic acid (DSS) and the heteronuclear 13C and 15N chemical shifts were indirectly referenced with the appropriate conversion factors (Markley et al., 1998). All spectra were processed using Bruker TopSpin™ 2.1 or 3.2 and analyzed using the programs CARA (Keller, 2004) and CcpNmr Analysis 2.2 (Vranken et al., 2005).
For peptide and lipid titration experiments, a standard Bruker HSQC pulse sequence was used. A 250 μM solution of 15N-PACSIN1, 2 or 3 SH3 domain in 10 mM Tris pH7, 100 mM NaCl was titrated with peptide and/or lipid (Cayman Chemical and Avanti Polar Lipids) from a concentrated stock solution. To evaluate NMR titration experiments the chemical shifts were determined using TopSpin 3.2 (Bruker). For 31P NMR measurements, PI(4,5)P2 dIC8 (Cayman Chemical) was used at 500 μM in 10 mM Tris pH7, 100 mM NaCl and titrated with peptide from a concentrated stock solution.
Backbone NMR assignments of apo PACSIN SH3 domains:
Backbone resonance assignments of 13C15N labeled PACSIN1, 2 and 3-SH3 domains in the absence of the PRR peptide were carried out using HNCO, HN(CA)CO, HNCA, HN(CO)CA and HNCACB spectra recorded with standard Bruker pulse sequences including water suppression with WATERGATE (Grzesiek and Bax, 1993). The {1H},15N-hetNOE data were recorded on a Bruker 600 MHz spectrometer in an interleaved manner with a 1H saturation period of 5 s duration on resonant or 10000 Hz off-resonant for the cross- and reference experiment, respectively. The relaxation delay was set to 3s.
Assignment of PACSIN3 SH3 in complex with PRR:
The backbones assignments from the apo PACSIN3 SH3 domain could be largely transferred to the 13C, 15N labeled chicken PACSIN3 SH3 domain in the presence of 5-fold unlabeled TRPV4-PRR peptide (V131I, see results section) in gradual titration experiments and were verified by (H)C(CO)NH (mixing time 25 ms) and H(CCO)NH (mixing time 25 ms) experiments used for additional side chain assignments. Additional 3D experiments to assign the side chains were 13C-NOESY-HSQC (mixing time 150 ms, aliphatic and aromatic carbons) and 15NNOESY-HSQC (mixing time 150 ms) experiments (Sattler, 1999). Using this approach, a nearly complete assignment of the PACSIN3 SH3 domain bound to the TRPV4-PRR peptide was obtained. The assignment of the aliphatic side chain protons and carbons was completed to 97 and 75 %, respectively. The aromatic protons and carbons were assigned with 64 %.
Assignment of the 1H resonances of the unlabeled TRPV4-PRR peptide in complex with the 13C, 15N labeled PACSIN3 SH3 domain was carried out using 13C-filtered TOCSY (mixing time 60 ms) (Ogura et al., 1996; Zwahlen et al., 1997; Breeze, 2000; Iwahara et al., 2001) and NOESY (mixing time 120 ms) (Ikura and Bax, 1992; Piotto et al., 1992; Sklenár et al., 1993) experiments. In addition, 13C or 15NHSQC and 2D 13C-NOESY-HSQC (mixing time 200 ms) spectra of selectively labeled 13C, 15N TRPV4-PRR peptides (see Table S2) in the presence of unlabeled PACSIN3 SH3 domain were used to support assignment and verify assignments for overlapping resonances. The complex of the PACSIN SH3 domain with the TRPV4-PRR peptide is in fast exchange on the NMR time scale and NMR samples used for structure determination of the complex contain the SH3 domain and the peptide in a 1:5 ratio. Thus, only a single set of signals is observed for the peptide with a population weighted average chemical shift between the bound and the free peptide which has very similar chemical shifts to the peptide in the absence of the SH3 domain.
Structure calculation of the PACSIN3 SH3 / TRPV4-PRR complex:
First, peak picking and NOE assignment of the PACSIN3 SH3 domain was performed with the ATNOS/CANDID module in UNIO (Herrmann et al., 2002) in combination with CYANA (Würz et al., 2017) using the 3D NOESY spectra listed above. Peak lists were reviewed manually and corrected in case of artifacts and possible NOEs to the TRPV4-PRR peptide. Distance restrains within the PACSIN3 SH3 domain were obtained using the automated NOE assignment and structure calculation protocol available in CYANA (Würz et al., 2017). Obvious NOEs to the peptide were assigned manually. NOEs to the peptide were essentially detected at the chemical shifts of the free peptide since the complex is in fast exchange on the NMR time scale and contains a significant excess of free peptide. In addition, 2D and 3D 13C-filtered and 13C-edited NOESY-HSQC experiments (Ogura et al., 1996; Zwahlen et al., 1997; Breeze, 2000; Iwahara et al., 2001) were used to obtain distance restrains of the 13C, 15N labeled PACSIN3 SH3 domain to the unlabeled PRR peptide. Additional intermolecular distance restraints were obtained from comparing 2D 13C-NOESY-HSQC experiments of selectively labeled 13C, 15N TRPV4-PRR peptides in the presence of unlabeled PACSIN3 SH3 domain. In total, 52 intermolecular distance restraints for structure calculation of the complex were assigned manually. Furthermore, 12 intrapeptide distance restraints of the PRR peptide were manually obtained using above described TOCSY and NOESY spectra. Backbone H, N, Cα, Cβ chemical shifts were used to calculate torsion angle restraints using TALOS-N (Shen and Bax, 2013).
For the structure calculation of the complex, the peptide sequence was connected to the C-terminus of the PACSIN3 SH3 domain sequence through a set of weightless noninteracting dummy atoms. Using the three sets of distance restraints 100 conformers were calculated with CYANA. 10 structures with the lowest target function were submitted to a restrained energy refinement with OPALp (Konradi et al., 2000) and the AMBER94 force field (Ponder and Case, 2003). Structure validation was carried out with the Protein Structure Validation Software suite 1.5 (Bhattacharya et al., 2007) restricted to residues with hetNOE values >0.6. (see Table 1)
{1H},15N-hetNOE experiments of 15N-PBD-PRR:
{1H},15N-heteronuclear nuclear Overhauser effect (hetNOE) experiments for 15N-labeled PBD-PRR were recorded using Bruker standard pulse sequences. Experiments were run in an interleaved fashion with and without proton saturation during the recovery delay. Peak integrals were obtained using Bruker TopSpin 3.2.
CD spectroscopy:
CD measurements were carried out on a Jasco-815 CD spectrometer (Jasco, Gross-Umstadt, Germany) with 1 mm quartz cuvettes. Spectra were recorded at 20°C in a spectral range between 195–260 nm or 190–260 nm with 1 nm scanning intervals, 5 nm band-width and 50 nm/min scanning speed. All spectra were obtained from the automatic averaging of three measurements. Baseline corrections were also performed automatically. For titration experiments, TRPV4 N-terminal peptides were used in a concentration of 30 μM in bidest. H2O in the presence of TFE (2,2,2-trifluoroethanol, 0–90% (v/v)), SDS (0.5, 1.0, 2.5, 5.0 and 8.0 mM) and liposomes (0.5 and 1.0 mM). Liposomes were prepared from POPG and POPC at a molar ratio of 3:1. CD based secondary structure analysis was performed using the
QUANTIFICATION AND STATISTICAL ANALYSIS
The chemical shift differences were calculated using the following function:
| (Mulder et al., 1999). |
Affinities were determined from chemical shift changes upon peptide addition using the following function:
| (Williamson, 2013). |
Here, [P]t and [L]t are the total protein and ligand concentrations, KD is the affinity, Δδobs is the observed chemical shift difference and Δδmax is the maximum chemical shift difference in saturation.
DATA AND SOFTWARE AVAILABILITY
Coordinates and chemical shift data of the PACSIN3 SH3 domain in complex with the TRPV4-PRR described in this article have been deposited in the PDB with accession number PDB: 6F55 and in the BMRB with accession number BMRB: 34211.
Supplementary Material
Highlights.
-
-
TRPV4 proline rich region/PACSIN SH3 domain complex structure is presented
-
-
Conserved V4 proline in cis conformation leads to a “skipped class I” binding mode
-
-
PACSIN SH3 interaction affects TRPV4 PIP2 binding site but not vice versa
-
-
TRPV4 N-terminus is unstructured, a feature conserved across species
Acknowledgements
We thank Victoria D’Souza, Harvard University for generous spectrometer access during the early project stages and Hitoshi Inada, Serena Blacklow and Dorothea Winkelvoß for technical support. BG acknowledges a Max Planck Graduate Centre (MPGC) PhD fellowship, NAG a Boehringer Ingelheim travel grant and ED a TransMED – Mainz Research School of Translational Biomedicine PhD fellowship. This work was funded through the National Institutes of Health (R01 GM081340 to RG), the Carl Zeiss Foundation and the Naturwissenschaftlich-Medizinisches Forschungszentrum (NMFZ), Mainz University and University Medicine Mainz (to UAH), as well as by the Center of Biomolecular Magnetic Resonance (BMRZ) funded by the state of Hesse.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing financial interests: The authors declare no competing financial interests.
References
- Afonin S, Kubyshkin V, Mykhailiuk PK, Komarov IV, and Ulrich AS (2017). Conformational Plasticity of the Cell-Penetrating Peptide SAP As Revealed by Solid-State19F-NMR and Circular Dichroism Spectroscopies. The journal of physical chemistry. B 121, 6479–6491. [DOI] [PubMed] [Google Scholar]
- Bai X, Meng G, Luo M, and Zheng X (2012). Rigidity of wedge loop in PACSIN 3 protein is a key factor in dictating diameters of tubules. The Journal of biological chemistry 287, 22387–22396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya A, Tejero R, and Montelione GT (2007). Evaluating protein structures determined by structural genomics consortia. Proteins 66, 778–795. [DOI] [PubMed] [Google Scholar]
- Bokhovchuk FM, Bate N, Kovalevskaya NV, Goult BT, Spronk CAEM, and Vuister GW (2018). The Structural Basis of Calcium Dependent Inactivation of the Transient Receptor Potential Vanilloid 5 Channel. Biochemistry. [DOI] [PubMed] [Google Scholar]
- Breeze AL (2000). Isotope-filtered NMR methods for the study of biomolecular structure and interactions. Progress in nuclear magnetic resonance spectroscopy 36, 323–372. [Google Scholar]
- Cao E, Liao M, Cheng Y, and Julius D (2013). TRPV1 structures in distinct conformations reveal activation mechanisms. Nature 504, 113–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo K, Diaz-Franulic I, Canan J, Gonzalez-Nilo F, and Latorre R (2018). Thermally activated TRP channels. Molecular sensors for temperature detection. Physical biology 15, 21001. [DOI] [PubMed] [Google Scholar]
- Cuajungco MP, Grimm C, Oshima K, D’hoedt D, Nilius B, Mensenkamp AR, Bindels RJM, Plomann M, and Heller S (2006). PACSINs bind to the TRPV4 cation channel. PACSIN 3 modulates the subcellular localization of TRPV4. The Journal of biological chemistry 281, 18753–18762. [DOI] [PubMed] [Google Scholar]
- Deng Z, Paknejad N, Maksaev G, Sala-Rabanal M, Nichols CG, Hite RK, and Yuan P (2018). Cryo-EM and X-ray structures of TRPV4 reveal insight into ion permeation and gating mechanisms. Nature structural & molecular biology 25, 252–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’hoedt D, Owsianik G, Prenen J, Cuajungco MP, Grimm C, Heller S, Voets T, and Nilius B (2008). Stimulus-specific modulation of the cation channel TRPV4 by PACSIN 3. The Journal of biological chemistry 283, 6272–6280. [DOI] [PubMed] [Google Scholar]
- Farrow NA, Muhandiram R, Singer AU, Pascal SM, Kay CM, Gish G, Shoelson SE, Pawson T, Forman-Kay JD, and Kay LE (2002). Backbone Dynamics of a Free and a Phosphopeptide-Complexed Src Homology 2 Domain Studied by 15N NMR Relaxation. Biochemistry 33, 5984–6003. [DOI] [PubMed] [Google Scholar]
- Gao Y, Cao E, Julius D, and Cheng Y (2016). TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action. Nature 534, 347–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Elias A, Mrkonjic S, Pardo-Pastor C, Inada H, Hellmich UA, Rubio-Moscardó F, Plata C, Gaudet R, Vicente R, and Valverde MA (2013). Phosphatidylinositol-4,5-biphosphate-dependent rearrangement of TRPV4 cytosolic tails enables channel activation by physiological stimuli. Proceedings of the National Academy of Sciences of the United States of America 110, 9553–9558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzesiek S, and Bax A (1993). The importance of not saturating water in protein NMR. Application to sensitivity enhancement and NOE measurements. J. Am. Chem. Soc, 12539–12594. [Google Scholar]
- Güler AD, Lee H, Iida T, Shimizu I, Torninaga M, and Caterina MJ (2002). Heat-Evoked Activation of the Ion Channel, TRPV4. The Journal of Neuroscience, 6408–6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacker C, Christ NA, Duchardt-Ferner E, Korn S, Göbl C, Berninger L, Düsterhus S, Hellmich UA, Madl T, and Kötter P, et al. (2015). The Solution Structure of the Lantibiotic Immunity Protein NisI and Its Interactions with Nisin. The Journal of biological chemistry 290, 28869–28886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmich UA, and Gaudet R (2014a). High-resolution views of TRPV1 and their implications for the TRP channel superfamily. Handbook of experimental pharmacology 223, 991–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmich UA, and Gaudet R (2014b). Structural biology of TRP channels. Handbook of experimental pharmacology 223, 963–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmich UA, and Glaubitz C (2009). NMR and EPR studies of membrane transporters. Biological chemistry 390, 815–834. [DOI] [PubMed] [Google Scholar]
- Herrmann T, Güntert P, and Wüthrich K (2002). Protein NMR Structure Determination with Automated NOE Assignment Using the New Software CANDID and the Torsion Angle Dynamics Algorithm DYANA. Journal of molecular biology 319, 209–227. [DOI] [PubMed] [Google Scholar]
- Hilgemann DW, Feng S, and Nasuhoglu C (2001). The Complex and Intriguing Lives of PIP2 with Ion Channels and Transporters. Science’s STKE 2001, re19. [DOI] [PubMed] [Google Scholar]
- Hughes TET, Lodowski DT, Huynh KW, Yazici A, Del Rosario J, Kapoor A, Basak S, Samanta A, Han X, and Chakrapani S, et al. (2018). Structural basis of TRPV5 channel inhibition by econazole revealed by cryo-EM. Nature structural & molecular biology 25, 53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh KW, Cohen MR, Jiang J, Samanta A, Lodowski DT, Zhou ZH, and Moiseenkova-Bell VY (2016). Structure of the full-length TRPV2 channel by cryo-EM. Nature communications 7, 11130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikura M, and Bax A (1992). Isotope-filtered 2D NMR of a protein-peptide complex: study of a skeletal muscle myosin light chain kinase fragment bound to calmodulin. J. Am. Chem. Soc, 2433–2440. [Google Scholar]
- Inada H, Procko E, Sotomayor M, and Gaudet R (2012). Structural and biochemical consequences of disease-causing mutations in the ankyrin repeat domain of the human TRPV4 channel. Biochemistry 51, 6195–6206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janz JM, Sakmar TP, Min KC (2007). A Novel Interaction between Atrophin-interacting protein 4 and β-p21 activated Kinase interactive Exchange Factor is Mediated by an SH3 Domain. J. Biol. Chem 282, 28893–28903. [DOI] [PubMed] [Google Scholar]
- Keller RLJ (2004). The Computer Aided Resonance Tutorial (Goldau, Switzerland: Cantina Verlag; ). [Google Scholar]
- Konradi R, Billeter M, and Güntert P (2000). Point-centered domain for decomposition for parallel molecular dynamics simulation. Computer Physics Communications, 139–147. [Google Scholar]
- Landouré G, Zdebik AA, Martinez TL, Burnett BG, Stanescu HC, Inada H, Shi Y, Taye AA, Kong L, and Munns CH, et al. (2010). Mutations in TRPV4 cause Charcot-Marie-Tooth disease type 2C. Nature genetics 42, 170–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau S-Y, Procko E, and Gaudet R (2012). Distinct properties of Ca2+-calmodulin binding to N- and C-terminal regulatory regions of the TRPV1 channel. The Journal of general physiology 140, 541–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao M, Cao E, Julius D, and Cheng Y (2013). Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 504, 107–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedtke W, and Friedman JM (2003). Abnormal osmotic regulation in trpv4−/− mice. Proceedings of the National Academy of Sciences of the United States of America 100, 13698–13703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Gong Z, Dong X, and Tang C (2016). Transient protein-protein interactions visualized by solution NMR. Biochimica et biophysica acta 1864, 115–122. [DOI] [PubMed] [Google Scholar]
- Luo L, Xue J, Kwan A, Gamsjaeger R, Wielens J, Kleist L. von, Cubeddu L, Guo Z, Stow JL, and Parker MW, et al. (2016). The Binding of Syndapin SH3 Domain to Dynamin Proline-rich Domain Involves Short and Long Distance Elements. The Journal of biological chemistry 291, 9411–9424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markley JL, Bax A, Arata Y, Hilbers CW, Kaptein R, Skyes BD, Wright PE, and Wüthrich K (1998). Recommendations for the presentation of NMR structures of proteins and nucleic acids. IUPAC-IUBMB-IUPAB Inter-Union Task Group on the Standardization of Data Bases of Protein and Nucleic Acid Structures Determined by NMR Spectroscopy. Journal of Biomolecular NMR, 1–23. [DOI] [PubMed] [Google Scholar]
- Masuyama R, Vriens J, Voets T, Karashima Y, Owsianik G, Vennekens R, Lieben L, Torrekens S, Moermans K, and Vanden Bosch A, et al. (2008). TRPV4-mediated calcium influx regulates terminal differentiation of osteoclasts. Cell metabolism 8, 257–265. [DOI] [PubMed] [Google Scholar]
- McGoldrick LL, Singh AK, Saotome K, Yelshanskaya MV, Twomey EC, Grassucci RA, and Sobolevsky AI (2018). Opening of the human epithelial calcium channel TRPV6. Nature 553, 233–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micsonai A, Wien F, Kernva L, Lee YH, Goto Y, Réfrégiers M, and Kardo J (2015). Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proceedings of the National Academy of Sciences of the United States of America 112, 3094–3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modregger J, Ritter B, Witter B, and Paulsson Mats and Plomann Markus (2000). All three PACSIN isoforms bind to endocytic proteins and inhibit endocytosis. Journal of Cell Science, 4511–4521. [DOI] [PubMed] [Google Scholar]
- Moore C, Gupta R, Jordt S-E, Chen Y, and Liedtke WB (2018). Regulation of Pain and Itch by TRP Channels. Neuroscience bulletin 34, 120–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder FA, Schipper D, Bott R, and Boelens R (1999). Altered flexibility in the substrate-binding site of related native and engineered high-alkaline Bacillus subtilisins. Journal of molecular biology 292, 111–123. [DOI] [PubMed] [Google Scholar]
- Muramatsu S, Wakabayashi M, Ohno T, Amano K, Ooishi R, Sugahara T, Shiojiri S, Tashiro K, Suzuki Y, and Nishimura R, et al. (2007). Functional gene screening system identified TRPV4 as a regulator of chondrogenic differentiation. The Journal of biological chemistry 282, 32158–32167. [DOI] [PubMed] [Google Scholar]
- Nilius B, and Voets T (2013). The puzzle of TRPV4 channelopathies. EMBO reports 14, 152–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura K, Terasawa H, and Inagaki F (1996). An improved double-tuned and isotope-filtered pulse scheme based on a pulsed field gradient and a wide-band inversion shaped pulse. Journal of Biomolecular NMR, 492–498. [DOI] [PubMed] [Google Scholar]
- Piotto M, Saudek V, and Sklenár V (1992). Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. Journal of Biomolecular NMR, 661–666. [DOI] [PubMed] [Google Scholar]
- Ponder JW, and Case DA (2003). Force fields for protein simulations. Adv Protein Chem, 27–85. [DOI] [PubMed] [Google Scholar]
- Rao Y, Ma Q, Vahedi-Faridi A, Sundborger A, Pechstein A, Puchkov D, Luo L, Shupliakov O, Saenger W, and Haucke V (2010). Molecular basis for SH3 domain regulation of F-BAR-mediated membrane deformation. Proceedings of the National Academy of Sciences of the United States of America 107, 8213–8218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roccatano D, Colombo G, Fioroni M, and Mark AE (2002). Mechanism by which 2,2,2-trifluoroethanolwater mixtures stabilize secondary-structure formation in peptides: A molecular dynamics study. Proceedings of the National Academy of Sciences of the United States of America, 12179–12184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saksela K, and Permi P (2012). SH3 domain ligand binding. What’s the consensus and where’s the specificity? FEBS letters 586, 2609–2614. [DOI] [PubMed] [Google Scholar]
- Sarkar P, Saleh T, Tzeng S-R, Birge RB, and Kalodimos CG (2011). Structural basis for regulation of the Crk signaling protein by a proline switch. Nature chemical biology 7, 51–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattler M (1999). Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Progress in nuclear magnetic resonance spectroscopy 34, 93–158. [Google Scholar]
- Schubert M, Labudde D, Oschkinat H, and Schmieder P (2002). A software tool for the prediction of Xaa-Pro peptide bond conformations in proteins based on 13C chemical shift statistics. Journal of Biomolecular NMR, 149–154. [DOI] [PubMed] [Google Scholar]
- Shen Y, and Bax A (2013). Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. Journal of Biomolecular NMR 56, 227–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Delaglio F, Cornilescu G, and Bax A (2009). TALOS+. A hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. Journal of Biomolecular NMR 44, 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim S, Yuan JP, Kim JY, Zeng W, Huang G, Milshteyn A, Kern D, Muallem S, Ming G. l., and Worley PF (2009). Peptidyl-prolyl isomerase FKBP52 controls chemotropic guidance of neuronal growth cones via regulation of TRPC1 channel opening. Neuron 64, 471–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh AK, Saotome K, Sobolevski A (2017). Swapping of transmembrane domains in the epithelial calciuim channel TRPV4, Sci. Rep 7, 10669–10669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J Thompson, J.D., and Higgins, D.G. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sklenár V, Piotto M, Leppik R, and Saudek V (1993). Gradient-Tailored Water Suppression for 1H-15N HSQC Experiments Optimized to Retain Full Sensitivity. Journal of Magnetic Resonance Series A, 241–245. [Google Scholar]
- Ullrich SJ, Hellmich UA, Ullrich S, and Glaubitz C (2011). Interfacial enzyme kinetics of a membrane bound kinase analyzed by real-time MAS-NMR. Nature chemical biology 7, 263–270. [DOI] [PubMed] [Google Scholar]
- van Meer G, Voelker DR, and Feigenson GW (2008). Membrane lipids. Where they are and how they behave. Nature reviews. Molecular cell biology 9, 112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, Ulrich EL, Markley JL, Ionides J, and Laue ED (2005). The CCPN data model for NMR spectroscopy. Development of a software pipeline. Proteins 59, 687–696. [DOI] [PubMed] [Google Scholar]
- Wang Q, Navarro MVAS, Peng G, Molinelli E, Goh SL, Judson BL, Rajashankar KR, and Sondermann H (2009). Molecular mechanism of membrane constriction and tubulation mediated by the F-BAR protein Pacsin/Syndapin. Proceedings of the National Academy of Sciences of the United States of America 106, 12700–12705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JPM, Cibelli M, Urban L, Nilius B, McGeown JG, and Nagy I (2016). TRPV4. Molecular Conductor of a Diverse Orchestra. Physiological reviews 96, 911–973. [DOI] [PubMed] [Google Scholar]
- Wilkes M, Madej MG, Kreuter L, Rhinow D, Heinz V, Sanctis S. de, Ruppel S, Richter RM, Joos F, and Grieben M, et al. (2017). Molecular insights into lipid-assisted Ca2+ regulation of the TRP channel Polycystin-2. Nature structural & molecular biology 24, 123–130. [DOI] [PubMed] [Google Scholar]
- Williamson MP (2013). Using chemical shift perturbation to characterise ligand binding. Progress in nuclear magnetic resonance spectroscopy 73, 1–16. [DOI] [PubMed] [Google Scholar]
- Würz JM, Kazemi S, Schmidt E, Bagaria A, and Güntert P (2017). NMR-based automated protein structure determination. Archives of biochemistry and biophysics 628, 24–32. [DOI] [PubMed] [Google Scholar]
- Zubcevic L, Herzik MA, Chung BC, Liu Z, Lander GC, and Lee S-Y (2016). Cryo-electron microscopy structure of the TRPV2 ion channel. Nature structural & molecular biology 23, 180–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.