

Summary
Nucleic acids can fold into well-defined 3D structures that help determine their function. Knowing precise nucleic acid structures can also be used for the design of nucleic acid-based therapeutics. However, locations of hydrogen atoms, which are key players of nucleic acid function are normally not determined with X-ray crystallography. Accurate determination of hydrogen atom positions can provide indispensable information on protonation states, hydrogen bonding, and water architecture in nucleic acids. Here, we used neutron crystallography in combination with X-ray diffraction to obtain joint X-ray/neutron structures at both room- and cryo-temperatures of a self-complimentary A-DNA oligonucleotide d[GTGG(CSe)CAC]2 containing 2’-SeCH3 modification on Cyt5 (CSe) at pH 5.6. We directly observed protonation of a backbone phosphate oxygen of Ade7 at room temperature. The proton is replaced with hydrated Mg2+ upon cooling the crystal to 100K, indicating that metal binding is favoured at low temperature, whereas proton binding is dominant at room temperature.
Keywords: A-form DNA, neutron crystallography, protonation, hydrogen bonding, metal binding, selenium modification
eTOC
Vandavasi et al. used joint X-ray/neutron macromolecular crystallography to follow structural changes in an A-DNA oligonucleotide induced by temperature. The study visualized hydrogen atom positions, revealing protonation of a backbone phosphate oxygen at room temperature and the proton replacement with hydrated magnesium ion upon cooling the crystal to 100K.
Graphical Abstract
INTRODUCTION
Nucleic acids encode life by storing and transferring genetic information in all living organisms including viruses. DNAs and RNAs of various length can fold into well-defined 3D structures, as many proteins do, with the information about their biological function implicitly woven into their sequences as well as structures (Leslie et al., 1980; Hoogstraten and Sumita, 2007; Dickerson et al., 1982). Knowing the precise 3-dimensional structures of nucleic acids can offer more information on the molecular mechanisms of various diseases, including genetic disorders, viral infections, cancers, etc (Park et al., 2011). The structural knowledge can also be used in the development of nucleic acidbased therapeutics, such as antisense oligonucleotides and aptamers (Sharma et al., 2014a; Zhou and Rossi, 2014; Sharma et al., 2014b).
Over the past several decades, the structure determination of biomacromolecules, including nucleic acids, has gone through several revolutionary changes due to advancements in X-ray crystallographic technologies, such as developments in synchrotron radiation, automation of crystal growth and data collection, and detectors. It is estimated that the number of functional nucleic acids (including noncoding RNAs) and nucleic acid-protein complexes is much larger than that of proteins. However, the rate of protein structure determination has dramatically outpaced that of nucleic acids, with >120,000 X-ray structures of proteins versus just ~3,000 of nucleic acids deposited in the Protein Data Bank (Berman et al., 2000, www.rcsb.org). Such tremendous disparity in the number of available 3D structures of proteins and nucleic acids can be attributed to the differences in their natural ability to crystallize and to the difficulties in preparation of chemically and structurally homogenous nucleic acid samples. The presence of negatively charged phosphate backbone on the surface, chemical and structural inhomogeneity, and the structural plasticity of nucleic acids make their crystal packing much more challenging (Dock-Bregeo et al., 1999; Ke and Doudna, 2004; Mooers, 2009; Choi and Majima, 2011).
X-ray crystallography has been the gold standard of structural biology. However, other techniques, such as NMR, cryo-EM and neutron crystallography, are rapidly advancing. Neutrons have the advantage over X-rays in that they scatter off atomic nuclei instead of electron clouds, and the neutron scattering power of an atom does not depend on its atomic number. Thus, the lightest atom, hydrogen (H), can be directly observed, usually as its heavier isotope deuterium (D), in neutron structures at resolutions as low as 2.5–2.6 Å (Blakeley, 2009; Banco et al., 2016; Gerlits et al., 2017). In contrast, observing H atoms in X-ray structures requires data collected to ultra-high resolutions of 1.0 Å or better; although, even at such high resolutions most interesting and functionally important H atoms are often still not detected by X-ray crystallography (Gardberg et al., 2010). In addition, neutrons used in neutron crystallographic experiments have wavelengths in the range of 1–5 Å; these ‘cold’ neutrons do not cause direct radiation damage to macromolecular crystals, in contrast to X-rays with the same wavelengths, producing radiation damage-free biomacromolecular structures. Further, the benign nature of cold neutrons allows neutron crystallographic data to be collected equally well at room and cryo temperatures, allowing functional observations to be made at room temperature.
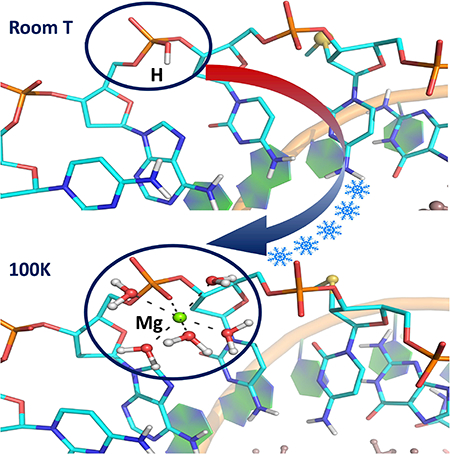
Herein we report the joint X-ray/neutron (XN) structures of the self-complimentary A-DNA octamer d[GTGG(CSe)CAC]2 (1), exchanged with D2O, obtained at room temperature (1_RT structure at 2.0 Å resolution) and at 100K (1_LT structure at 1.9 Å resolution) are examined in detail. This study was enabled by selenium (Se) derivatization at 2’-position of the ribose in Cyt5 nucleotide (CSe), making it possible to collect neutron crystallographic data from radically smaller crystals of 0.2–0.4 mm3 in volume than previously required sizes of several cubic millimetres. By introducing the 2’SeCH3 group, crystallization of the oligonucleotide was enhanced, allowing us to obtain well-diffracting crystals suitable for neutron diffraction. Chemical modification of nucleic acids with Se was pioneered and developed in the lab of Dr. Zhen Huang (Lin et al., 2011). It was demonstrated that introduction of Se to nucleobases and the ribose sugar ring significantly improved crystal growth, diffraction quality of oligonucleotide crystals and helped with phasing X-ray crystallographic data (Jiang et al., 2007; Salon et al., 2008; Salon et al., 2010). The most striking observation in the current neutron crystallographic study was the detection of the backbone protonation at RP-oxygen of Ade7 phosphate in 1_RT, crystallized without organic polycations at pH of 5.6. Moreover, when the crystal was flash-frozen in liquid nitrogen, this backbone phosphate protonation was replaced with Mg2+ ion coordination in 1_LT, with the metal cation having proper octahedral coordination by five D2O water molecules and the RP-oxygen (Figure 1). It appears that the metal coordination of a phosphate in the A-DNA is very labile, with most Mg(D2O)62+ ions being disordered in the crystal at room temperature. Conversely, metal coordination may correspond to the global energy minimum on the internal potential energy hypersurface of the A-DNA, while at room temperature the oligonucleotide adopts a configuration that corresponds to a local energy minimum, in which the backbone phosphate of Ade7 is protonated, similar to what was previously suggested in studies of proteins (Gerlits et al., 2017; Kovalevsky et al., 2018).
Figure 1.

Structure of d(GTGGCSeCAC)2 at room (A) and cryo temperatures (B). Arrows point to the Ade7 phosphate backbone, which is protonated in 1_RT and coordinated by Mg2+ in 1_LT.
RESULTS
The octameric self-complimentary oligonucleotide d[GTGG(CSe)CAC]2 (1), crystallizes in a tetragonal unit cell (P43212), with one DNA oligomer in the asymmetric unit and the double helix generated through the crystallographic 2-fold axis, similar to the PDB-deposited structure 4FP6 (unpublished). Crystals of 1 suitable for X-ray and neutron crystallography grew in organic polycation-free conditions, containing Mg2+ as the sole source of the DNA counter-ion. The neutron structures of 1, obtained at 2.0 Å and 1.9 Å resolutions at room (RT) and cryo (LT) temperatures, respectively, were refined jointly with the 1.56 Å (RT) and 1.65 Å (LT) X-ray crystallographic data to produce joint X-ray/neutron (XN) structures 1_RT and 1_LT (Tables S1 and S2). All exchangeable H atoms were observed as deuterium (D), and we were able to model 29 and 39 heavy water (D2O) molecules in 1_RT and 1_LT, respectively.
1 adopts the A-DNA form in the crystal, having C3’-endo ribose sugar ring puckering, a narrow deep major groove and a wide shallow minor groove (Figure 1). Conversely, in B-DNA the sugar rings prefer C2’-endo puckering, whereas the geometric characteristics of the grooves are opposite to those in the A-DNA. The B-DNA’s major groove is wide, and the minor groove is narrow. As usually observed in A-DNA structures (Wahl and Sundaralingam, 1997), the deep major groove is hydrated to a higher extent than the shallow minor groove, as illustrated in Figure S1, with several water molecules hydrogen-bonded to the backbone phosphates. There are only 8 ordered D2O molecules observed in the minor groove close to the oligonucleotide’s 3’- and 5’-termini in each 1_RT and 1_LT structures. These water molecules are conserved, but move slightly, when the crystal is flashfrozen in liquid nitrogen. By comparison, the major groove contains at least 30 water molecules in each structure. In addition, the major groove harbours two Mg(D2O)2+ complexes positioned close to the middle of the oligonucleotide between the nucleobases of Gua4 and Cyt6. Each magnesium ion interacts through the outersphere contacts with the nucleobases of Gua3 and Gua4, with hydrogen bond distances of 2.6–2.9 Å. Metal ions stabilize the multiple negative charges on the DNA backbone phosphates but are rarely seen in crystal structures (Wahl and Sundaralingam, 1997). The 2’-SeCH3 modification of Cyt5 is clearly beneficial for crystal growth, as the SeCH3 group forms intermolecular van der Waals contacts with the 5’-Gua1 nucleotide, which help create tight packing of the DNA molecules in the crystal (Figure S2). We did not observe H/D exchange of C8-H proton on any of the three guanine nucleobases in 1_RT or 1_LT, although significant exchange of this proton with D in Gua was previously documented in the neutron structure of hexameric Z-DNA (Chatake et al., 2005). In addition, unlike in the previous neutron structure of the decameric A-DNA (Leal et al., 2010), we did not observe protonation of N7 in guanine or N3 in adenine nucleobases in our current XN structures.
Unexpectedly, we detected protonation of the RP-oxygen of Ade7 phosphate in the room temperature XN structure 1_RT (Figure 2). A strong peak is observed in the difference FO-FC neutron scattering density length map located at a distance about 1 Å from the RP-oxygen. The density peak was interpreted as D, because no extra electron density was seen near this oxygen. The D atom occupancy refined to 67%; thus, the RP-oxygen atom is ⅔ protonated. The RP-oxygen is weakly hydrogen bonded with a water molecule that bridges the phosphates of Ade6 and Cyt6, with O-D⋅⋅⋅O distances of 2.3–2.4 Å. Such phosphate-bridging water molecules are common in A-DNA structures.
Figure 2.

(A) 2FO-FC neutron scattering length density map (light green mesh) for Cyt6, Ade7 and Cyt8 nucleotides in 1_RT, contoured at 1.2 σ level, showing protonation of RP-oxygen of the Ade7 backbone phosphate. Experimentally determined D atom positions are shown as dark grey spheres, while the calculated positions of non-exchangeable H atoms are light grey. The omit difference FO-FC neutron scattering length density map (red mesh) is contoured at 3 σ level. Hydrogen bonds are shown as blue dashed lines, and distances are in Å. (B) 2FO-FC electron density map for the same nucleotides contoured at 2.0 σ level. H and D atoms are not shown.
When the crystal of oligonucleotide 1 was flash-frozen in liquid nitrogen and the XN structure 1_LT determined, to our surprise, we have found that the Ade7 backbone phosphate is no longer protonated. Instead, Mg2+ ion hydrated with five D2O molecules binds to the RP-oxygen, completing its octahedral coordination sphere (Figure 3). The metal and water molecules are clearly visible in the neutron scattering length density and electron density maps, while there is no indication of a D atom presence near the backbone phosphate. Thus, the D atom bound to RP-oxygen of Ade7 in 1_RT is replaced with metal coordination at cryo temperature in 1_LT. The water molecule that acted as a bridge between Ade7 and Cyt6 phosphates in 1_RT is pushed away from Ade7 by the incoming metal complex (W6 in Figure 3), losing its hydrogen bond with the former but keeping the hydrogen bond with Cyt6 in 1_LT.
Figure 3.

(A) 2FO-FC neutron scattering length density map (grey mesh) for Cyt6, Ade7 and Cyt8 nucleotides in 1_LT, contoured at 1.0 σ level. D atoms are coloured light green, while the non-exchangeable H atoms are grey. Metal coordination is shown as black solid lines, and hydrogen bonds are blue dashed lines. Distances are in Å. (B) 2FO-FC electron density map for the same nucleotides contoured at 2.0 σ level. Densities for nucleobases are omitted for clarity, and H and D atoms are not shown.
DISCUSSION
Neutron structures of nucleic acids have been studied mainly by fibre diffraction (Fuller et al., 2004). There are only four neutron crystallographic structures of DNA reported in the literature (Chatake et al., 2005; Leal et al., 2010; Arai et al., 2005; Fenn et al., 2011), while there are still no neutron structures of RNA. Well-diffracting crystals of Z-DNA oligonucleotides can usually be grown, and the 1.4 and 1.6 Å resolution neutron structures have been published (Arai et al., 2005; Fenn et al., 2011). On the contrary, much lower 2.4 and 3 Å resolution neutron structures of A- and B-form DNA oligonucleotides have been obtained from crystals of several cubic millimetres in volume. The 2’SeCH3 modification introduced on the ribose sugar ring of Cyt5 in A-DNA oligonucleotide 1 resulted in high diffraction quality crystals and made it possible to obtain high resolution neutron diffraction data – 2.0 Å at room temperature and 1.9 Å at 100K – from crystals that are about an order of magnitude smaller in volume. In addition, we were able to investigate the effect of temperature on the crystal structure and protonation states in DNA, via neutrons.
Neutrons are the perfect probe to visualize D atoms in biological macromolecules. With the neutron scattering power of D being as good as that of C, N, and O, positions of virtually all D atoms can be determined in a neutron structure, whereas X-rays often cannot provide this information even at ultra-high resolutions, especially for water molecules (Brzezinski et al., 2011). In the room temperature XN structure 1_RT obtained using a crystal grown at pH of 5.6, we unequivocally observed protonation of the RP oxygen atom of the Ade7 backbone phosphate (Figure 2). The D atom on the phosphate is not involved in any hydrogen bonding interactions and the O-D group is rotated inward, facing the adenine nucleobase. It is interesting to note that unusual protonation of guanine at N7 nitrogen and protonation of adenine nucleobase were demonstrated in the neutron structure of another A-DNA oligonucleotide (Leal et al., 2010). Protonated and positively charged cytosine and adenine are well-known and have been implicated in affecting polymerase fidelity and ribozyme general acid-base catalysis (Wilcox et al., 2011; Wilcox and Bevilacqua, 2013a, 2013b). However, protonation of either the backbone phosphate or the guanine nucleobase is unexpected, because the pKa values of these protonated functional groups must be much lower than 5. Perhaps, the Aconformation of DNA might induce the increase in the intrinsic basicity of some nucleobases and the phosphate backbone, shifting their intrinsic pKa up. In addition, hydrogen ion concentration in the vicinity of DNA may be significantly higher than in the bulk solution, as suggested by theoretical calculations (Lamm and Pack, 1990; Jayaram et al., 1989). Moreover, protonation of the phosphate moiety has been previously shown for uracil and thymine residues (Wu et al., 2017), and the DNA alkylating antitumor agents are believed to be activated by the DNA backbone phosphate protonating the drug’s carbonyl to generate a reactive carbocation (Hurley and Needham-VanDevanter, 1986; Warpehoski and Harper, 1994; Warpehoski and Harper, 1995; Thompson et al., 1995; Boger et al., 1996).
Metal ions are believed to have essential biological roles in nucleic acid folding and enzymatic reactions (Aoki and Murayama, 2012; Sigel and Sigel, 2013). Metal ion interactions with nucleic acids are important for counterbalancing the high concentration of charged phosphate groups in DNA and RNA, but usually only a handful of metal ions are sufficiently ordered to be observed in crystal structures (Whal and Sundaralingam, 1997; Leonarski et al., 2017). In 1_RT, two Mg2+ ions are observed as hexahydrated Mg(D2O)62+ complexes bound in the major groove (Figure 1). Magnesium ions interact through the outersphere contacts with the nucleobases of Gua3 and Gua4, which is typical for A-DNA oligonucleotide structures (Aoki and Murayama, 2012; Robinson et al., 2000). Thus, Mg2+ ions and the protonated Ade7 backbone phosphates provide six positive charges to balance the fourteen negative charges present in the double helix of oligonucleotide 1, with the rest of charge balancing presumably coming from the metal ions that are disordered in the structure. Surprisingly, in the low-temperature XN structure 1_LT, the Ade7 backbone phosphate protonation is replaced with metal coordination. We detected no D atom near the Rp oxygen in the nuclear density map in 1_LT. Instead, there are clear evidences in the electron and nuclear density maps for a pentahydrated magnesium ion, Mg(D2O)52+, bound to this oxygen using its sixth available coordination site (Figure 3). Such inner sphere binding of Mg2+ ions to the backbone phosphate is also commonly observed in low-temperature A-DNA oligonucleotide crystal structures (Aoki and Murayama, 2012). This observation suggests that, while the DNA duplex retains the original two hydrated Mg2+ ions, one Mg(D2O)62+ complex (for each DNA strand) comes from the bulk and coordinates to the backbone phosphate oxygen, when the temperature is lowered. It is not unreasonable to suggest that, during this process, the sixth D2O from the hexahydrated magnesium accepts the proton from the phosphate to become a hydronium ion (D3O+) that dissociates from the metal, allowing the latter to bind to the RP-oxygen. It is also apparent that the Rp oxygen of Ade7 has a higher affinity to cations than other atoms. At room temperature, this oxygen is protonated, while the Mg-phosphate interaction is very labile. On the contrary, at low temperature Mg(D2O)52+ becomes ordered and coordinated to the backbone phosphate. This metal coordination may therefore correspond to the global energy minimum on the internal potential energy hypersurface of the ADNA, with hydrated magnesium binding being favoured by the enthalpy component of the system’s free energy but not by the entropy. Hence, magnesium coordination to the Ade7 phosphate oxygen is only observed at 100K when thermal motions are essentially frozen, and the entropy is low. The phosphate protonation could thus correspond to a local energy minimum that is achievable and mostly occupied by the molecules at room temperature due to increased thermal motion.
In summary, we have demonstrated that a backbone phosphate oxygen in an A-DNA nucleotide crystal can be protonated. The protonation state can be altered, and the proton can be replaced with a coordinated metal cation, when the temperature is lowered to 100K. Our results might suggest that, for a more complete understanding of nucleic acid structure and function, it may be necessary to obtain DNA and RNA structures at both room and low temperatures. Moreover, we have shown that the Se modification of the ribose sugar at the 2’-position in d[GTGG(CSe)CAC]2 allowed us to obtain high-resolution neutron diffraction data from DNA crystals that are an order of magnitude smaller in volume than those required previously. This work suggests that future studies of the DNA and RNA structure and function using neutron crystallography may be generally possible, including mechanistic studies of ribozymes, DNAzymes and riboswitches.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Andrey Kovalevsky (kovalevskyay@ornl.gov).
METHOD DETAILS
Synthesis of d[GTGG(CSe)CAC]2.
Syntheses of Se-modified nucleotides and the DNA oligonucleotides incorporating SeCH3 substituent at the 2’ position of the sugar moiety have been described previously (Lin et al., 2011; Jiang et al., 2007; Salon et al., 2008; Salon et al., 2010). The GTGG(CSe)CAC was synthesized by following the standard solid-phase synthesis and using 5-benzylmercaptotetrazole (5-BMT) as the coupling reagent. The coupling yield using 5-BMT activator was higher than that using tetrazole. The synthesis was conducted on an ABI model 394 synthesizer using standard β-cyanoethylphosphoramidite solid-phase synthesis protocol with mild I2 oxidation conditions (20 mM, 20 seconds). The solid-phase coupling yield of the GTGG(CSe)CAC using the 2’-Se-C phosphoramidite was higher than 99%. The synthesized oligonucleotide was cleaved from the beads by incubation with concentrated ammonia for 11 hr at 60oC. After filtration or centrifugation to remove the beads, ammonia was evaporated by speed vacuum. The crude residue was re-dissolved in water (500 μL) and the pH adjusted to 7. The reverse phase HPLC analysis of the crude DNA synthesized using 2’-Se-C phosphoramidite was performed. 50% aqueous acetonitrile, 50 mM triethylammonium acetate (TEAAc) pH 7.1 was used as buffer B, while a 25 mM TEAAc buffer (pH 7.1) as buffer A. The oligonucleotide was first purified with the DMTr group on. The purification was run from 0% → 20% B in 30 minutes (10 mL/min). Detrytilation was performed by incubating the oligonucleotide at pH 4.0 and at 40oC for 1 hour, followed by quenching with an aqueous solution of triethylamine (from a 10X stock solution) and by extracting with petroleum ether to remove the DMTr-OH residue. Desalting was carried out to remove the salts from the GTGG(CSe)CAC.
Crystallization.
Solution of the purified d[GTGG(CSe)CAC] oligonucleotide was first heated to 90°C for 1 min, and then allowed to cool slowly to room temperature (20°C) to give d[GTGG(CSe)CAC]2, as the sequence is self-complimentary. The crystallization conditions were screened using the nucleic acid mini screen, Natrix, and Natrix 2 supplied by Hampton Research (Aliso Viejo, CA, USA) using 0.20.5 mM oligonucleotide dissolved in water. The best crystals shaped as square bipyramids were obtained in conditions #3 and #16 of the Natrix screen at 18°C. To increase the size of the crystals the crystallization conditions were then optimized by changing the temperature, concentrations of the DNA and precipitant, and DNA-to-reservoir solution ratio. Crystals as large as 0.2–0.4 mm3 were obtained using the reservoir solution with 0.1M Mg(OAc)2, 30% MPD and 0.1M MES (pH = 5.6). The crystallization drops were set by mixing 1 mM d[GTGG(CSe)CAC]2 with the reservoir solution at the 2:1 ratio. The crystals were grown by vapor diffusion using Hampton Research sandwich box setup and siliconized 9-well plates. Initially, the sandwich boxes were placed in a temperature-controlled incubator set at 30°C. Crystals were allowed to grow over the period of one month, and then the temperature was slowly reduced to 18°C over the following two months. Crystals for room- and cryo-temperature crystallographic experiments were taken from the same crystallization set-up and were mounted in quartz capillaries with the reservoir solution plugs made with perdeuterated MPD and D2O. The labile H atoms were allowed to exchange with D by vapor for at least 4 weeks before starting crystallographic data collection. The crystal for the cryo-temperature diffraction experiment was mounted on a Molecular Dimensions litholoop and frozen in liquid nitrogen after being soaked in the deuterated mother liquor containing 30% perdeuterated MPD for cryo-protection and in order to reduce background in the neutron diffraction images caused by CH hydrogen atoms present in the MPD molecule.
X-ray and neutron data collection.
Room- and cryo-temperature X-ray data were collected on an in-house Rigaku HomeFlux system equipped with the R-AXIS IV++ image plate detector, the Rigaku MicroMax-007 HF Cu rotatinganode generator, Osmic VariMax HR optics, and the Oxford CryoStream operating at 100K. Quasi-Laue neutron data to 2.0 Å resolution (room temperature) and to 1.9 Å resolution (100K) were collected from the 0.4 and 0.2 mm3 DNA crystals, respectively. The DNA crystal diffraction quality was tested, and preliminary data were collected at room temperature on the IMAGINE (Meilleur et al., 2013) instrument located at the High Flux Isotope Reactor (Oak Ridge National Laboratory). The diffraction quality and the preliminary data were considered worthy of the full data collection. The complete quasi-Laue neutron diffraction datasets were collected at room temperature and at 100K using the LADI-III beamline at the ILL, Grenoble, France (Blakeley et al., 2010). During the neutron diffraction experiment, the crystal was held stationary at different ϕ settings for each exposure. In total, 22 diffraction images were collected (with an exposure time of 2 hours per image) from 3 different crystal orientations for the room-temperature experiment. 25 diffraction images (6 hours per image) were collected for a crystal at 100K.
X-ray and neutron data processing.
X-ray diffraction data indexing, integration and scaling were performed using HKL-3000 (Minor et al., 2006). Starting phases were calculated from PDB entry 4FP6 with waters removed. Refinement was done using the SHELX package (Sheldrick and Schneider, 1997; Sheldrick, 2008; Sheldrick, 2015), with 5% of the unique reflections selected randomly for Rfree (Brünger, 1992), and alternated with manual refitting of the model in Coot (Emsley et al., 2010). The neutron data from LADI-III were processed using the Daresbury Laboratory LAUE suite program LAUEGEN modified to account for the cylindrical geometry of the detector (Campbell, 1995; Campbell et al., 1998). The program LSCALE was used to determine the wavelength-normalization curve using the intensities of symmetry-equivalent reflections measured at different wavelengths (Artz et al., 1999). No explicit absorption corrections were applied. These data were then merged in SCALA (Weiss, 2001). A summary of the experimental and refinement statistics is given in Tables S1 and S2.
Joint XN structure refinement.
Both room- and cryo-temperature joint XN structures of d[GTGG(CSe)CAC]2 were determined using nCNS (Adams et al., 2009). The structures were solved by molecular replacement with PDB 4FP6 as a starting model utilizing CCP4 (Winn et al., 2011). Initial rigid-body refinement was followed by several cycles of positional, atomic displacement parameter (B factor), and D occupancy refinement. Between each cycle the structures were examined, and water molecule orientations were built based on the omit FO-FC difference neutron scattering length density map, and with meaningful hydrogen bonding interactions in mind. The 2FO-FC and FO-FC neutron scattering length density maps were then checked to determine the correct orientation of hydroxyl groups. No protonation of the nucleic bases was observed, whereas in the room-temperature structure the phosphate backbone of Ade7 was observed protonated at the RP oxygen. All water molecules were refined as D2O. Initially, water oxygen atoms were positioned according to their electron density peaks, and then were shifted slightly in accordance with the neutron scattering density maps. Labile H positions in d[GTGG(CSe)CAC]2 were modeled as D and then the occupancies of D atoms were allowed to refine within the range of −0.56 to 1.00 (the scattering length of H is −0.56 times the scattering length of D). Before depositing the final structure to the Protein Data Bank (PDB), a script was run that converts a record for the coordinate of D atom into two records corresponding to an H and a D atom partially occupying the same site, both with positive partial occupancies that add up to unity.
QUANTIFICATION AND STATISTICAL ANALYSIS
No statistical approaches were used in this manuscript.
DATA AND SOFTWARE AVAILABILITY
Atomic coordinates and structure factors for the reported crystal structures 1_RT and 1_LT have been deposited with the Protein Data bank under accession numbers 6D4L and 6D54, respectively.
Supplementary Material
Highlights.
Joint X-ray/neutron structures of A-form DNA
Backbone phosphate of Ade7 is protonated at room temperature
Metal ion replaces proton on backbone phosphate at low temperature
Neutron structures show importance of structural analyses at different temperatures
Acknowledgements
This research at ORNL’s High Flux Isotope Reactor (IMAGINE beamline) was sponsored by the Scientific User Facilities Division, Office of Basic Energy Sciences, U.S. Department of Energy. The authors thank Institut Laue Langevin (beamline LADI-III) for awarded neutron beamtime. Notice: This manuscript has been authored by UT-Battelle LLC under DOE Contract No. DE-AC05–00OR22725. This work was supported by the ORNL Laboratory Directed Research and Development grant to A.K. and by NIH (R01GM095881 and R42ES026935) to Z.H.; and the US Department of Energy’s (DOE) Office of Basic Energy Sciences.
Footnotes
Declaration of Interests
The authors declare that they have no conflicts of interest with the contents of this article.
SUPPLEMENTAL INFORMATION
Supplemental Information includes the data collection and refinement statistics tables, and figures and can be found with this article online at:
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Mustyakimov M, Afonine PV & Langan P (2009). Generalized X-ray and neutron crystallographic analysis: more accurate and complete structures for biological macromolecules. Acta Crystallogr. D Biol. Crystallogr 65, 567–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki K and Murayama K (2012) Nucleic acid-metal ion interactions in the solid state In Sigel A, Sigel H, Sigel RKO (eds.) Interplay between metal ions and nucleic acids, Metal ions in Life Sciences, Springer Science, vol. 10, pp. 43–102. [DOI] [PubMed] [Google Scholar]
- Arai S, Chatake T, Ohhara T, Kurihara K, Tanaka I, Suzuki N, Fujimoto Z, Mizuno H and Niimura N (2005) Nucleic Acids Res, 33, 3017–3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arzt S, Campbell JW, Harding MM, Hao Q & Helliwell JR (1999). LSCALE—the new normalization, scaling and absorption correction program in the Daresbury Laue software suite. Journal of Applied Crystallography. 32, 554–562. [Google Scholar]
- Banco MT, Mishra V, Ostermann A, Schrader TE, Evans GB, Kovalevsky A and Ronning DR (2016) Neutron structures of the Helicobacter pylori 5’-methylthioadenosine nucleosidase highlight proton sharing and protonation states. Proc. Natl. Acad. Sci. U.S.A, 113, 13756–13761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN and Bourne PE (2000) The Protein Data Bank. Nucl. Acids Res, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakeley MP (2009) Neutron macromolecular crystallography. Cryst. Rev, 15, 157–218. [Google Scholar]
- Blakeley MP, Teixeira SCM, Petit-Haertlein I, Hazemann I, Mitschler A, Haertlein M, Howard E & Podjarny AD (2010). Neutron macromolecular crystallography with LADI-III. Acta Crystallogr. D Biol. Crystallogr 66, 1198–1205. [DOI] [PubMed] [Google Scholar]
- Boger DL, Han N, Tarby CM, Boyce CW, Cai H, Jin Q and Kitos PA (1996) Synthesis, chemical properties, and preliminary evaluation of substituted CBI analogs of CC-1065 and the duocarmycins incorporating the 7-cyano-1,2,9,9a-tetrahydrocyclopropa[c]benz[e]indol-4-one alkylation subunit: Hammett quantitation of the magnitude of electronic effects on functional reactivity. J. Org. Chem, 61, 4894–4912. [DOI] [PubMed] [Google Scholar]
- Brünger AT (1992). Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 355, 472–475. [DOI] [PubMed] [Google Scholar]
- Brzezinski K, Brzuszkiewicz A, Dauter M, Kubicki M, Jaskolski M and Dauter Z (2011) Hihg regularity of Z-DNA revealed by ultra high-resolution crystal structure at 0.55 Å. Nucleic Acids Res, 39, 6238–6248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JW (1995). LAUEGEN, an X-windows-based program for the processing of Laue diffraction data. Journal of Applied Crystallography. 28, 228–236. [Google Scholar]
- Campbell JW, Hao Q, Harding MM, Nguti ND & Wilkinson C (1998). LAUEGEN version 6.0 and INTLDM. Journal of Applied Crystallography. 31, 496–502. [Google Scholar]
- Chatake T, Tanaka I, Umino H, Arai S and Niimura N (2005) The hydration structure of a ZDNA hexameric duplex determined by a neutron diffraction technique. Acta Cryst, D61, 1088–1098. [DOI] [PubMed] [Google Scholar]
- Choi J and Majima T (2011) Conformational changes of non-B DNA. Chem. Soc. Rev, 40, 5893–5909. [DOI] [PubMed] [Google Scholar]
- Dickerson RE, Drew HR, Conner BN, Wing RM, Fratini AV and Kopka ML (1982) The anatomy of A-, B-, and Z-DNA. Science, 216, 475–485. [DOI] [PubMed] [Google Scholar]
- Dock-Bregeon A-C, Moras D and Giege R (1999) Nucleic acids and their complexes In Ducruix A and Giege R (eds.), Crystallization of Nucleic Acids and Proteins – A Practical Approach, Oxford University Press, Oxford, pp. 209–243. [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Crystallogr. D 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenn TD, Schnieders MJ, Mustyakimov M, Wu C, Langan P, Pande VS and Brunger AT (2011) Reintroducing electrostatics into macromolecular crystallographic refinement: application to neutron crystallography and DNA hydration. Structure, 19, 523–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller W, Forsyth T, Mahendrasingam A (2004) Water-DNA interactions as studied by X-ray and neutron fibre diffraction. Phil. Trans. R. Soc. Lond. B, 359, 1237–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardberg AS, Del Castillo AR, Weiss KL, Meilleur F, Blakeley MP and Myles DAA (2010) Unambiguous determination of H-atom positions: comparing results from neutron and high-resolution X-ray crystallography. Acta Cryst D66, 558–567. [DOI] [PubMed] [Google Scholar]
- Gerlits OO, Coates L, Woods RJ and Kovalevsky A (2017) Mannobiose binding induces changes in hydrogen bonding and protonation states of acidic residues in concanavalin A as revealed by neutron crystallography. Biochemistry, 56, 4747–4750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlits O, Keen DA, Blakeley MP, Louis JM, Weber IT and Koavelsvky A (2017) Room temperature neutron crystallography of drug resistant HIV-1 protease uncovers limitations of X-ray structural analysis at 100K. J. Med. Chem, 60, 2018–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogstraten CG and Sumita M (2007) Structure-function relationships in RNA and RNP enzymes: recent advances. Biopolymers, 87, 317–328. [DOI] [PubMed] [Google Scholar]
- Hurley LH and Needham-VanDevanter DR (1986) Covalent binding of antitumor antibiotics in the minor groove of DNA. Mechanism of action of CC-1065 and the pyrrolo(1,4)benzodiazepines. Acc. Chem. Res, 19, 230–237. [Google Scholar]
- Jayaram B, Sharp KA and Honig B (1989) The electrostatic potential of B-DNA. Biopolymers, 28, 975–993. [DOI] [PubMed] [Google Scholar]
- Jiang J, Sheng J, Carrasco N and Huang Z (2007) Selenium derivatization of nucleic acids for crystallography. Nucleic Acids Res, 35, 477–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke A and Doudna JA (2004) Crystallization of RNA and RNA-protein complexes. Methods, 34, 408–414. [DOI] [PubMed] [Google Scholar]
- Kovalevsky A, Aggarwal M, Velazquez H, Cuneo MJ, Blakeley MP, Weiss KL, Smith JC, Fisher SZ and McKenna R (2018) “To be or not to be” protonated: atomic details of human carbonic anhydrase-clinical drug complexes by neutron crystallography and simulation. Structure, 26, 383–390. [DOI] [PubMed] [Google Scholar]
- Lamm G and Pack GR (1990) Acidic domains around nucleic acids. Proc. Natl. Acad. Sci. U.S.A, 87, 9033–9036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leal RMF, Callow S, Callow P, Blakeley MP, Cardin CJ, Denny WA, Teixeira SCM, Mitchell EP and Forsyth VT (2010) Combined neutron and X-ray diffraction studies of DNA in crystals and solutions. Acta Cryst, D66, 1244–1248. [DOI] [PubMed] [Google Scholar]
- Leonarski F, D’Ascenzo L and Auffinger P (2017) Mg2+ ions: do they bind to nucleobase nitrogens?. Nulc. Acids Res, 45, 987–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie AG, Arnott S Chandrasekaran R and Ratliff RL (1980) Polymorphism of DNA double helices. J. Mol. Biol, 143, 49–72. [DOI] [PubMed] [Google Scholar]
- Lin L, Sheng J and Huang Z (2011) Nucleic acid X-ray crystallography via direct selenium derivatization. Chem. Soc. Rev. 40, 4591–4602. [DOI] [PubMed] [Google Scholar]
- Meilleur F, Munshi P, Robertson L, Stoica AD, Crow L, Kovalevsky A, Koritsanszky T, Chakoumakos BC, Blessing R and Myles DAA (2013) The IMAGINE instrument: first neutron protein structure and new capabilities for neutron macromolecular crystallography, Acta Crystallogr. D Biol. Crystallogr 69, 2157–2160. [DOI] [PubMed] [Google Scholar]
- Minor W, Cymborowski M, Otwinowski Z & Chruszcz M (2006). HKL-3000: the integration of data reduction and structure solution--from diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr 62, 859–866. [DOI] [PubMed] [Google Scholar]
- Mooers BHM (2009) Crystallographic studies of DNA and RNA. Methods, 47, 168–176. [DOI] [PubMed] [Google Scholar]
- Park YJ, Claus R, Weichenhan D and Plass C (2011) Genome-wide epigenetic modifications in cancer. Prog. Drug. Res, 67, 25–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson H, Gao Y-G, Sanishvili R, Joachimiak A and Wang AH-J (2000) Hexahydrated magnesium ions bind in the deep major groove and at the outer mouth of A-form nucleic acid duplexes. Nucleic Acids Res, 28, 1760–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salon J, Jiang J, Sheng J, Gerlits OO and Huang Z (2008) Derivatization of DNAs with selenium at 6-position of guanine for function and crystal structure studies. Nucleic Acids Res, 36, 7009–7018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salon J, Sheng J, Gan J and Huang Z (2010) Synthesis and crystal structure of 2’-Se-modified guanosine containing DNA. J. Org. Chem, 75, 637–641. [DOI] [PubMed] [Google Scholar]
- Sharma VK, Sharma RK and Singh SK (2014a) Antisense oligonucleotides: modifications and clinical trials. Med. Chem. Comm, 5, 1454–1471. [Google Scholar]
- Sharma VK, Rungta P and Prasad AK (2014b) Nucleic acid therapeutics: basic concepts and recent developments. RSC Adv, 4, 16618–16631. [Google Scholar]
- Sheldrick GM & Schneider TR SHELXL: High-resolution refinement. Methods Enzymol. 277, 319–343 (1997). [PubMed] [Google Scholar]
- Sheldrick GM (2008) A short history of SHELX. Acta Crystallogr. A 64, 112–122. [DOI] [PubMed] [Google Scholar]
- Sheldrick GM (2015) Crystal structure refinement with SHELXL. Acta Crystallogr. C 71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigel RKO and Sigel H (2013) Metal-ion interactions with nucleic acids and their constituents In Reedijk J, Peoppelmeier K (eds) Comprehensive inorganic chemistry II, Elsevier, vol. 3, pp. 623–660. [Google Scholar]
- Thompson AS, Sun D and Hurley LH (1995) Monoalkylation and cross-linking of DNA by cyclopropapyrroloindoles entraps bent and straight forms of A-tracts. J. Am. Chem. Soc, 117, 2371–2372. [Google Scholar]
- Wahl MC and Sundaralingam (1997) Crystal structures of A-DNA duplexes. Biopolymers, 44, 45–63. [DOI] [PubMed] [Google Scholar]
- Warpehoski MA and Harper DE (1994) Acid-dependent electrophilicity of cyclopropylpyrroloindoles. Nature’s masking strategy for a potent DNA alkylator. J. Am. Chem. Soc, 116, 75737580. [Google Scholar]
- Warpehoski MA and Harper DE (1995) Enzyme-like rate acceleration in the DNA minor groove. Cyclopropylpyrroloindoles as mechanism-based inactivators of DNA. J. Am. Chem. Soc, 117, 29512952. [Google Scholar]
- Weiss MS (2001). Global indicators of X-ray data quality. Journal of Applied Crystallography. 34, 130–135. [Google Scholar]
- Wilcox JL, Ahluwalia AK and Bevilacqua PC (2011) Charged nucleobases and their potential for RNA catalysis. Acc. Chem. Res, 44, 1270–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox JL and Bevilacqua PC (2013a) A simple fluorescence method for pKa determination in RNA and DNA reveals highly shifted pKa’s. J. Am. Chem. Soc, 135, 7390–7393. [DOI] [PubMed] [Google Scholar]
- Wilcox JL and Bevilacqua PC (2013b) pKa shifting in double-stranded RNA is highly dependent upon nearest neighbours and bulge positioning. Biochemistry, 52, 7470–7476. [DOI] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A and Wilson KS (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D, 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu RR, Hamlow LA, He CC, Nei Y.-w., Berden G, Oomens J and Rodgers MT (2017) The intrinsic basicity of the phosphate backbone exceeds that of uracil and thymine residues: protonation of the phosphate moiety is preferred over the nucleobase for pdThd and pUrd. Phys. Chem. Chem. Phys, 19, 30351–30361. [DOI] [PubMed] [Google Scholar]
- Zhou J and Rossi J (2014) Cell-type-specific aptamer and aptamer-small interfering RNA conjugates for targeted human immunodeficiency virus type 1 therapy. J. Investig. Med, 62, 914–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.