

Abstract
Pseudomonas aeruginosa, an opportunistic pathogen, produces redox-active pigments called phenazines. Pyocyanin (PYO, the blue phenazine) plays an important role during biofilm development. Paradoxically, PYO auto-poisoning can stimulate cell death and release of extracellular DNA (eDNA), yet PYO can also promote survival within biofilms when cells are oxidant-limited. Here we identify the environmental and physiological conditions in planktonic culture that promote PYO-mediated cell death. We demonstrate that PYO auto-poisoning is enhanced when cells are starved for carbon. In the presence of PYO, cells activate a set of genes involved in energy-dependent defenses, including: (i) the oxidative stress response, (ii) RND efflux systems and (iii) iron-sulfur cluster biogenesis factors. P. aeruginosa can avoid PYO poisoning when reduced carbon is available, but blockage of adenosine triphosphate (ATP) synthesis either through carbon limitation or direct inhibition of the F0F1-ATP synthase triggers death and eDNA release. Finally, even though PYO is toxic to the majority of the population when cells are nutrient limited, a subset of cells is intrinsically PYO resistant. The effect of PYO on the producer population thus appears to be dynamic, playing dramatically different yet predictable roles throughout distinct stages of growth, helping rationalize its multifaceted contributions to biofilm development.
Graphical Abstract
The redox active pigment, pyocyanin (PYO), made by Pseudomonas aeruginosa, is a double-edged sword, whose physiological impact changes dramatically yet predictably. Under nutrient-replete conditions, PYO is produced and can help cells achieve redox homeostasis in the absence of alternative electron acceptors. However, when cells deplete carbon or other nutrients, PYO becomes toxic and poisons the majority of the population. A small, PYO-insensitive population persists and multiplies when nutrients again become available.

Introduction
In nature, bacteria rarely grow alone and mostly live in microbial communities called biofilms. The term “biofilm” refers to aggregates of microorganisms embedded in a self-produced, extracellular matrix (Flemming et al., 2016). Biofilms are important in many contexts, not the least of which is human health, as diverse pathogens become significantly more drug-tolerant when they adopt a biofilm lifestyle rather than a free-growing state (Olsen, 2015). Among the different bacteria that can form biofilms, Pseudomonas aeruginosa is one of the most notorious: a globally-significant, opportunistic pathogen that contributes to many types of infections (Govan and Deretic, 1996).
The biofilm extracellular matrix comprises diverse extracellular polymeric substances (EPS), including polysaccharides, proteins, lipids and extracellular DNA (eDNA) (Flemming, 2016). Extracellular DNA is an important building component of the matrix and has become a target in biofilm-control approaches (Okshevsky et al., 2015). The importance of eDNA during biofilm formation has been established for several bacterial species, as recently reviewed by Ibáñez de Aldecoa et al. (2017). In the P. aeruginosa biofilm matrix, eDNA is the most abundant polymer, often accounting for more than 50% of the extracellular matrix; its removal can significantly disrupt biofilm structure (Whitchurch et al., 2002; Matsukawa and Greenberg, 2004; Allesen-Holm et al., 2006). It is well known that most of the eDNA important for biofilm formation in P. aeruginosa biofilms comes from lysis of a subpopulation of the cells (Okshevsky and Meyer, 2013). A few different mechanisms for cell lysis have been described for P. aeruginosa, including activation of prophage endolysins that disrupt the cell wall and release DNA and other cytoplasmic contents to the environment (D’Argenio et al., 2002; Webb et al., 2003; Turnbull et al., 2016). This type of eDNA release is stimulated under stressful conditions. Auto-poisoning through production of the small molecule 2-n-heptyl-4-hydroxyquinoline-N-oxide (HQNO) also leads to eDNA release and has a direct impact on biofilm formation (Hazan et al., 2016). We use the terms “auto-poisoning”, “cell death” and “cell lysis” as follows: auto-poisoning is the toxic effect caused by a molecule that eventually can kill the producing cells; after auto-poisoning, different cell death processes can be activated, which may or may not lead to cell lysis (breaking down of the cell membrane) and eDNA release (Rice and Bayles, 2008; Bayles, 2014).
In addition to being a model biofilm-forming organism, P. aeruginosa is also known for its production of colorful, redox-active molecules called phenazines—dibenzo annulated pyrazines, containing two nitrogen atoms in the center ring that mediate proton-coupled electron transfer (Laursen and Nielsen, 2004; Mavrodi et al., 2006; Price-Whelan et al., 2006). Over the last decade, a variety of beneficial physiological functions have been attributed to phenazines, for example, cell signaling, electron shuttling to diverse oxidants and promotion of survival under electron acceptor limitation (Dietrich et al., 2006; Wang et al., 2010; Wang et al., 2011; Glasser et al., 2014), with different functions attributed to different phenazines. One of the best studied phenazines is pyocyanin (PYO), which has medical importance and has long been considered a virulence factor that is toxic to host cells (Lau et al., 2004). This toxicity is mainly due to its capacity to quickly reduce molecular oxygen, thereby generating superoxide, which by enzymatic or abiotic dismutation generates hydrogen peroxide (Muller, 2002; Rada and Leto, 2013).
Even though PYO production can enhance biofilm formation in P. aeruginosa (Ramos et al., 2010; Das et al., 2015), at which stages of biofilm development and precisely how it acts, are not fully understood. Previous studies suggest that PYO might mediate biofilm development in at least three different, complementary ways: (i) causing auto-poisoning and eDNA release during early stages of biofilm formation, mostly due to generation of oxidative stress (Das and Manefield, 2012; Costa et al., 2017), which we expand upon in this study; (ii) indirectly modulating levels of the second messenger bis-(3′,5′)-cyclic-dimeric-guanosine (c-di-GMP) and EPS production (Okegbe et al., 2017); and (iii) stimulating expansion of biofilms/aggregates under anoxic/hypoxic conditions, where PYO can function as an alternative electron acceptor and alleviate reducing stress (Dietrich et al., 2013; Costa et al., 2017; Jo et al., 2017). In all cases, PYO-modulated effects—be they beneficial or deleterious, at the level of the single cell—seem to be tightly linked to environmental and physiological conditions. Accordingly, we set out to determine how different conditions commonly encountered by cellular populations at distinct growth stages result in PYO-dependent cellular effects, focusing primarily on cell death. We demonstrate that PYO production has predictable, distinct and dramatic cellular consequences, according to the physiological state of the producer.
Results
Phenazine production stimulates a morphologically-distinct type of cell death early in biofilm development
We started by imaging early-stage biofilms attached to glass, when cells settle on the surface and form microcolonies. eDNA is known to play an important structural role at this stage of biofilm development (Whitchurch et al., 2002). We performed a standard biofilm development assay and quantified phenazine auto-poisoning using the wild-type (WT) strain of P. aeruginosa PA14 (Pa PA14) that produces different phenazines, including PYO, and a ∆phz strain that cannot make phenazines (both phz1 and phz2 operons are deleted in this strain, see Dietrich et al., 2006). We grew WT and ∆phz Pa PA14 cells in 8-well chambers for 6 hours. In this assay, cells attach to the coverslip on the bottom of the chamber to initiate biofilm formation (Fig. 1A). We noticed significant amounts of cell death in these young biofilms (Fig. 1A). In addition, we found that the WT attached cells died more than ∆phz cells (Fig. 1B), consistent with the observation that phenazine production increases cell death in biofilms.
Fig. 1. Phenazine production increases cell death early in biofilm development, and cell death happens in two morphologically-distinct ways.
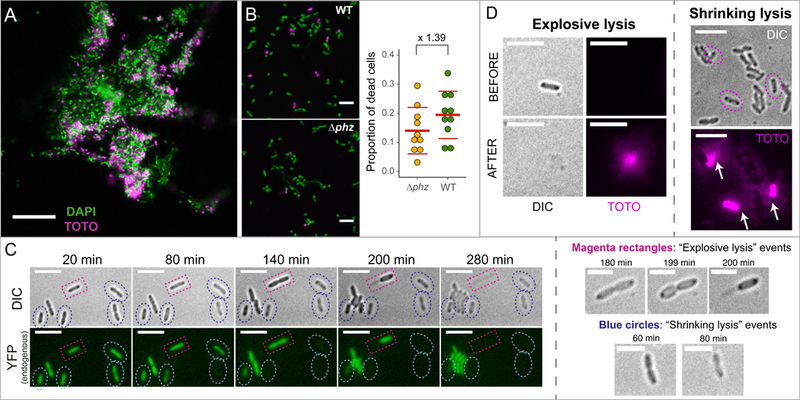
A. Picture of a 6-hour WT biofilm showing dead cells stained with TOTO-1 iodide (magenta), which does not penetrate live cells; scale bar: 20 µm. B. WT strain shows higher levels of cell death when compared to the ∆phz strain that cannot produce phenazines. Left: representative pictures of attached live (green-DAPI) and dead (magenta-TOTO) cells – scale bars: 5 µm; right: quantification of cell death for WT and ∆phz. Red lines show the average + standard deviation of the proportion of dead cells for both treatments. Ratio of cell death WT/∆phz = 1.39. C. Time-lapse of cells growing on agar pads showing the two morphologically-distinct cell death events: “shrinking” (blue circles) and “explosive” lysis (magenta rectangles); scale bars: 5 µm. Zoomed in pictures for both are shown on the right (scale bars: 3 µm). Cells were constitutively expressing yellow fluorescent protein (YFP), and fluorescence loss combined with morphology change (DIC) can be seen during cell death. D. Both explosive and shrinking lysis release DNA and other cellular contents into the environment. Explosive lysis quickly spreads eDNA extracellularly (left), but eDNA also leaks out of dead cells after shrinkage events (right: note the cloud of fluorescence [white arrows] leaking from the three dead cells [circles, DIC]). Scale bars: 5 µm. Brightness was increased for better visualization of the TOTO signal.
Next, we visualized cell death at the single-cell level. Because previous studies suggested that PYO production can cause eDNA release (Das and Manefield, 2012), we used this phenazine for these experiments. Cells in mid-exponential phase were washed and re-suspended on top of agarose pads containing 100 µM PYO and imaged using fluorescence microscopy. When looking at the cells, we observed at least two morphologically-distinct death events that we refer to as “explosive” and “shrinking lysis” (Fig. 1C). Explosive cell lysis in P. aeruginosa occurs in different strains (including PA14, PAO1, PAK, PA103 and others), stimulating eDNA release and biofilm formation (Turnbull et al., 2016). It is caused by the activation of a prophage endolysin (lys) present in the R- and F-pyocin gene cluster in the P. aeruginosa genome, and is activated under stressful conditions (Turnbull et al., 2016). We observed a very similar phenomenon happening during our experiments (Fig. 1C). Yet we also noticed a different cell death phenotype, “shrinking lysis”, where cells contract in size at the moment of death (Fig. 1C); in contrast to explosive lysis, these cells remain visible under the microscope after death (Fig. 1C). Phenazine production increases shrinking lysis during early biofilm development (Fig. 1B); to our knowledge, this cell death phenotype has not been previously characterized. It was previously shown that after explosive lysis, all the cytoplasmic contents (including DNA) are expelled to the environment (Fig. 1D-left and (Turnbull et al., 2016). To test if shrinking lysis events can also release eDNA in the medium, we used TOTO-1 iodide, a nucleic acid stain that does not penetrate living cells (Okshevsky and Meyer, 2014). We observed that eDNA slowly leaks out of shrunken cells, indicating that shrinking lysis provides an alternate route for eDNA release at an early stage in biofilm development (Fig. 1D). Together, these data show that phenazine production stimulates auto-poisoning in Pa PA14 biofilms, enhancing eDNA release to the environment by triggering a morphologically-distinct type of cell death.
PYO increases cell death in liquid cultures under stationary phase conditions
Heterogeneity during biofilm development and low efficiency of eDNA quantification in biofilm assays limit the range of conditions we can readily control to study phenazine-mediated eDNA release. Therefore, we switched to experiments using planktonic cultures. First, we checked whether the enhanced cell death we saw in our early stage biofilm experiments could be recapitulated under planktonic growth conditions. We measured the number of colony forming units (CFU) from cultures of WT and ∆phz strains over the course of batch growth (Fig. 2A). By the first time point (18h), the WT cultures had already produced considerable amounts of phenazines in the medium, particularly the blue phenazine PYO (see tubes in Fig. 2A). Standard incubator shaking (250 rpm) was sufficient to maintain PYO in its oxidized state (blue color). WT cultures consistently showed lower CFU recovery than ∆phz cultures, and this difference increased over time. Adding physiologically relevant amounts of exogenous PYO (100 µM) to the ∆phz culture recapitulated poisoning levels present in WT (Fig. 2A).
Fig. 2. Pyocyanin production causes auto-poisoning and cell death in Pa PA14.
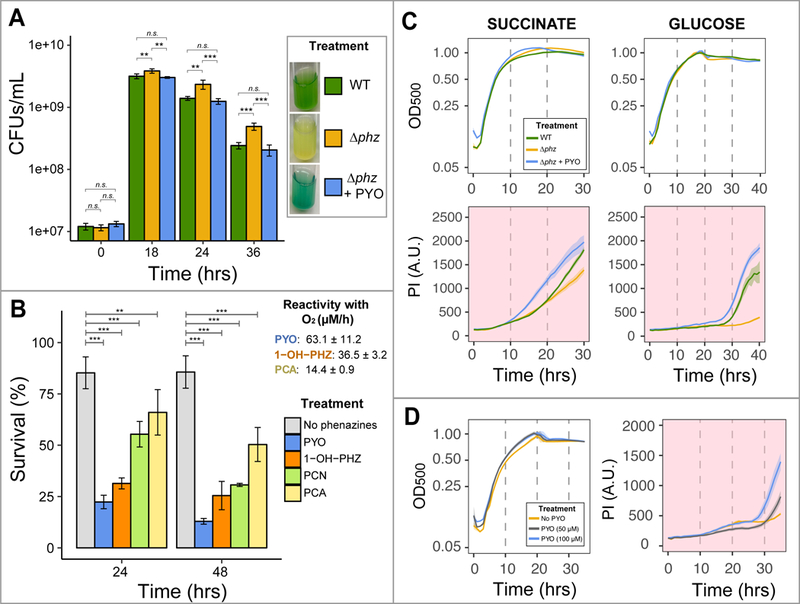
A. Viability assay shows a faster decrease of CFUs for the WT producing phenazines compared to the ∆phz strain; addition of exogenous PYO restores WT poisoning levels. Culture medium used: succinate minimal medium (SMM – see Experimental Procedures), with 40 mM sodium succinate. B. Phenazines induce a range of toxicity, with PYO being the most lethal. O2-reactivity constants previously measured for three of the four used phenazines (Wang and Newman, 2008) are also given, showing a correlation between O2 reactivity and toxicity. For A-B, we performed One-way ANOVA (p < 0.05) with Tukey’s HSD multiple-comparison test ([n.s. = p > 0.05 - not a statistically significant difference]; [* = p < 0.05]; [** = p < 0.01]; [*** = p < 0.001]). For p-values of all comparisons between phenazines, see Table S2. Bar graphs represent averages of four replicates and error bars represent standard deviation (SD). C. High-throughput plate reader assay confirms higher levels of auto-poisoning due to PYO production. Top: optical density at 500nm (OD500) measurements for growth curve; bottom: propidium iodide (PI) fluorescence, always presented with background colored in light red, indicating cell death. Minimal medium with different carbon sources were used and showed similar trends, with PYO increasing poisoning (sodium succinate concentration: 40 mM; glucose concentration: 10 mM). D. PYO poisoning is concentration dependent (glucose concentration: 10 mM). Plotted lines represent averages of six replicates and shaded areas are SD.
In addition to PYO, P. aeruginosa strains produce and secrete other phenazines, including phenazine-1-carboxylic acid (PCA), phenazine-1-carboxamide (PCN) and 1-hydroxyphenazine (1-OH-PHZ) (Price-Whelan et al., 2006). Because different phenazines can affect cells in different ways (Sakhtah et al., 2016), we compared the toxicity of these four phenazines. ∆phz cells were re-suspended in phosphate buffer with a given phenazine, and survival was measured after 24 and 48 hours. PYO is the most toxic phenazine (Fig. 2B), followed by 1-OH-PHZ, PCN and PCA. Perhaps not surprisingly, phenazine toxicity under oxic conditions correlates with their oxygen reactivity: the faster a phenazine reduces oxygen abiotically, the higher its toxicity (Fig. 2B, (Wang and Newman, 2008)). Because (i) Pa PA14 produces significant amounts of PYO, (ii) PYO causes the highest toxicity, and (iii) PYO production is known to be involved in eDNA release (Das and Manefield, 2012), we focused specifically on PYO-mediated cell death and eDNA release for the remainder of our experiments.
Though CFU counting can reveal the absolute number of viable cells in a culture for a given time point, this method is not conducive to sampling multiple conditions with high temporal resolution. To overcome this limitation, we developed a high-throughput approach using a plate reader assay to quantify cell growth and death simultaneously. Growth was measured by reading optical density (OD500); at the same time, the fluorescence of propidium iodide (PI) dye added to cultures served as a proxy for cell death. Similar to TOTO-1 iodide (used in the biofilm experiments), PI cannot penetrate live cells and thus has been used as a cell death marker (Okshevsky and Meyer, 2014). Because the TOTO fluorescence spectrum overlaps with the background fluorescence of Pseudomonas cultures, PI was a better dye for this purpose (see Experimental Procedures). By measuring OD500 and PI fluorescence together, we could track the dynamics of auto-poisoning through PYO production at fine temporal resolution.
Confirming results from the CFU experiment, WT Pa PA14 consistently had a higher cell death signal when compared to the ∆phz mutant (Fig. 2C). In addition, exogenously adding PYO to ∆phz increased the death signal (Fig. 2C). The PYO-triggered cell death phenomenon is concentration dependent, with higher concentrations of PYO promoting more cell death (Fig. 2D). The increased cell death of WT in comparison to ∆phz is evident regardless of the carbon-source (e.g. succinate or glucose – Fig. 2C). The PI signal increase correlated with cells entering stationary phase (Fig. 2C). This was expected for the WT strain, because PYO production is quorum-sensing regulated (Sakhtah et al., 2013). However, PYO was exogenously added to ∆phz cultures at the beginning of the experiment. Therefore, even when cells had 100 µM PYO present in their environment prior to exponential growth, cell death was triggered only after cells had entered stationary phase (Fig. 2C). This suggests the existence of specific physiological/environmental conditions during stationary phase that cause PYO-mediated cell death.
Nutrient depletion triggers PYO-mediated cell death
Intrigued by the fact that PYO-triggered cell death only occurred during stationary phase, we hypothesized that a lack of specific nutrients might drive this phenotype. Accordingly, we used our plate reader assay to test the effect of removing each of the main nutritional elements (carbon, nitrogen, phosphorus and sulfur) from the medium on cells in the presence and absence of PYO. Cells were grown to mid-exponential phase, washed, and re-suspended at high cell density in medium lacking each of these nutrients. The absence of nitrogen did not seem to impact the cells with respect to PYO sensitivity (Fig. 3A and Fig. S1). Therefore, we used nitrogen removal as a way to limit growth (cells stopped growing ~5h after the experiment started), enabling us to test the importance of the other nutrients systematically (see Experimental Procedures). Removal of carbon, phosphorus or sulfur significantly stimulated PYO toxicity (Fig. 3B-E), indicating that these nutrients might support PYO defense mechanisms under oxic conditions. To confirm that cells were indeed dying, we performed viability tests with CFU as a read out; this gave us confidence that the observed PI fluorescence increase could be interpreted as cell death (Fig. S2). The absence of PYO always resulted in lower levels of cell death (Fig. 3).
Fig. 3. Nutrient depletion triggers PYO-mediated cell death and PYO toxicity is coupled to the available reducing power.

A-E. Survival assays under different nutritional conditions. In all experiments, nitrogen was removed to minimize growth (see Experimental Procedures). Starvation for carbon, phosphorus and sulfur causes increased PYO sensitivity. F. Carbon starvation in stationary phase results in increased PYO-mediated cell death under normal growth conditions. Cell-death signal increase correlates with carbon limitation, but higher levels of glucose (> 40 mM) inhibit cell death. Concentrations for all other major nutrients (N, P, S) were the standard used for our minimal medium (see Experimental Procedures). Plotted lines represent averages of six replicates and shaded areas are SD.
Even though phosphorus and sulfur seem to be very important for PYO defense, cells usually need significantly lower levels of these nutrients compared to carbon, with S/C and P/C intracellular ratios in bacteria ranging from 0.02 to 0.08 and 0.04 to 0.09, respectively (Fagerbakke et al., 1996). Therefore, it is likely that carbon depletion would occur first, leading us to speculate that its depletion would be the central driver of the observed stationary phase PYO-stimulated cell death. To test this hypothesis, we grew ∆phz cells under different concentrations of carbon (glucose) in the absence or presence of PYO to track poisoning temporally. A clear correlation between glucose availability and cell death in the presence of PYO can be seen in Fig. 3F: lower levels of glucose resulted in increased cell death in a dose-dependent manner. Giving cells 4 mM glucose stimulated earlier PYO poisoning compared to when cells had 10 mM of glucose available. When given 20 mM glucose, cells only started dying after ~ 45 hours, close to the end of the experiment. Further, increasing levels of glucose to 40 mM was sufficient to completely inhibit PYO-mediated cell death (Fig. 3F). In the absence of PYO, cell death was attenuated (Fig. S3), indicating that under these conditions, PYO was primarily responsible for cell death. Finally, we confirmed that glucose in the medium was completely consumed before PYO-mediated cell death started by measuring bulk glucose levels in cultures at different time points (Fig. S4). These data indicate that PYO’s toxic effects on P. aeruginosa under oxic conditions are mitigated by available carbon, the source of reducing power.
PYO also poisons cells under anoxic conditions
The most commonly ascribed mechanism for PYO toxicity in P. aeruginosa is oxidative stress (Muller, 2002; Das and Manefield, 2012; Rada and Leto, 2013). Reduced PYO can partially reduce O2 to superoxide (O2-), which by enzymatic or abiotic dismutation generates hydrogen peroxide (H2O2). H2O2 can be toxic to cells mainly by participating in the Fenton reaction (step 1: Fe2+ + H2O2 → [FeO]2+ + H2O; step 2: [FeO]2+ + H+ → Fe3+ + HO•; Imlay, 2013) which generates hydroxyl radical (HO•). However, in addition to triggering oxidative stress, PYO can also be toxic under anoxic conditions (Barakat et al., 2014). Accordingly, we sought to determine whether limiting carbon utilization would also impact PYO sensitivity under anoxic conditions using the same survival approach described above. This was done with a plate reader in an anoxic chamber (see Experimental Procedures). In this experiment, no alternative electron acceptor was available (i.e. no O2 or NO3) and cells should not be able to oxidize the carbon. In addition, there was no extracellular oxidizing potential (e.g. electrode) to re-oxidize PYO in the medium, preventing the anaerobic survival effect caused by the phenazines cycling under anaerobic conditions (Wang et al., 2010; Glasser et al., 2014). If energy generation from oxidation of reduced carbon is necessary for PYO defense, incubation of cells with PYO in the absence of an electron acceptor should cause toxicity. In agreement with this hypothesis, we observed that PYO increased cell death anaerobically, even when the medium was carbon-replete (Fig. 4A). We also confirmed that PYO present in the medium was fully reduced by two complementary approaches: (i) measuring absorbance at 310 and (ii) measuring fluorescence (excitation: 360/40, emission: 460/40). Oxidized PYO strongly absorbs at 310 nm (Wang and Newman, 2008) and if reduction happens, OD310 absorbance drops. In parallel, only the reduced form of the phenazine is fluorescent, leading to an increase in fluorescence emission at ~500 nm if PYO is reduced (Wang and Newman, 2008). As expected, PYO reduction happened very quickly and after one hour PYO was completely reduced (Fig. 4B-C). No detectable re-oxidation occurred during the experiment. These data reveal that reduced PYO itself can be harmful and cause poisoning in P. aeruginosa cultures under anoxic conditions.
Fig.4. PYO also poisons cells under anoxic conditions.

A. Survival experiments as in Fig. 3A, but performed under anoxic conditions also reveal PYO toxicity. Available carbon cannot be used by cells due to the absence of an electron acceptor. B. PYO is completely reduced by cells at the beginning of the experiment, as measured by both decrease in OD310 absorbance (left) as well as increase in fluorescence (right). Note that PYO stays oxidized (no fluorescence) in the absence of cells. Fluorescence levels reached maximum values before the first hour of the experiment. Plotted lines represent averages of six replicates and shaded areas are SD.
ATP synthesis is required to avoid PYO poisoning
Because (i) under oxic conditions PYO-mediated cell death happens when cells are carbon-limited and (ii) PYO is still toxic under anoxic conditions when cells are carbon replete (but cannot catabolize carbon), we hypothesized that to avoid PYO poisoning, cells must be able to synthesize ATP to power cellular processes involved in PYO resistance. To begin to test this hypothesis, we measured bulk ATP levels in cultures incubated with high and low amounts of carbon under oxic conditions. We found that, in the presence of PYO, ATP levels substantially decreased when cells were carbon-limited (Fig. 5A-left-top). Confirming our plate reader results, a significant fraction of the population died (Fig. 5A-left-bottom). No substantial decrease in ATP levels or viability happened to the control without PYO. On the other hand, for cells incubated with high levels of glucose, the presence of PYO did not elicit a drop in bulk ATP levels nor a survival decrease (Fig. 5A-right). These data indicate that there is a consistent correlation between ATP depletion and PYO poisoning, with subsequent cell death.
Fig. 5. Reduced carbon and ATP synthesis are required to avoid poisoning caused by PYO.
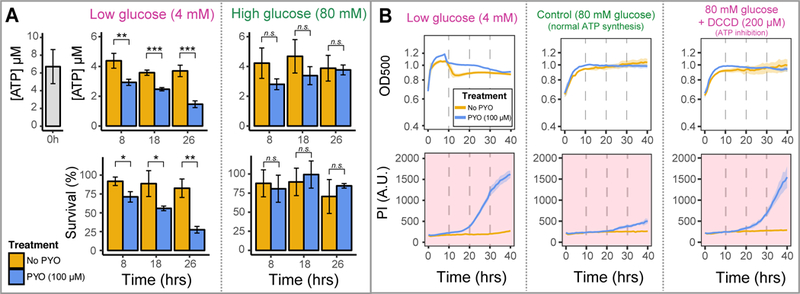
A. ATP levels decrease if cells are in the presence of PYO under low carbon (glucose) concentrations (left-top); at the same time, a survival drop can be detected, indicating poisoning and cell death (left-bottom). However, no decrease in ATP levels (right-top) or survival (right-bottom) is detected if cells high levels of glucose are available. Differences were assessed using an unpaired t-test ([n.s. = p > 0.05 - not a statistically significant difference]; [* = p < 0.05]; [** = p < 0.01]; [*** = p < 0.001]). Bar graphs represent averages of three replicates and error bars are SD. B. Inhibition of ATP synthesis using the F0F1-ATPase inhibitor DCCD (right) recapitulates the amount of cell death observed when cells are incubated under low carbon concentrations (left). Significantly lower cell death is seen when cells can metabolize the available and abundant reduced carbon (center). Cells survive well in all treatments when PYO is not present (beige lines). Plotted lines represent averages of six replicates and shaded areas are SD.
To further test the necessity of ATP synthesis to avoid PYO poisoning, we exposed cells to N,N-dicyclohexylcarbodiimide (DCCD), a chemical agent that blocks the F0F1-ATP synthase and inhibits ATP synthesis (Sebald et al., 1980), and measured their survival under different conditions. We predicted that by inhibiting ATP synthesis, DCCD would increase PYO toxicity even when abundant levels of glucose were available. Indeed, addition of DCCD caused cells to die in the presence of PYO even under high glucose concentrations, indicating that a lack of ATP sensitizes cells to PYO (Fig. 5B-right). Inhibiting ATP synthesis had a similar effect to limiting cells for carbon (Fig. 5B-left), but when ATP synthesis was not inhibited, PYO poisoning was minimal (Fig. 5B-center). Together, these data indicate that ATP synthesis is necessary for cells to prevent PYO toxicity.
Energy-dependent defense mechanisms are induced when cells are exposed to PYO
Having observed a strong correlation between nutritional conditions (particularly carbon metabolism and ATP synthesis) and PYO toxicity, we sought to explore whether PYO induces particular defense mechanisms that require reduced carbon and ATP synthesis. We performed survival assays under conditions where cells tolerate PYO well (i.e. oxic, nutrient-replete). After exposing cells to PYO for 5 hours, we measured the transcriptional response of genes encoding different categories of energy-dependent defense systems. We separated the tested genes into four main categories: (i) oxidative stress response, (ii) efflux systems, (iii) SOS response and (iv) iron-sulfur (Fe-S) clusters biogenesis (Fig. 6A).
Fig. 6. PYO induces energy-dependent defense mechanisms.
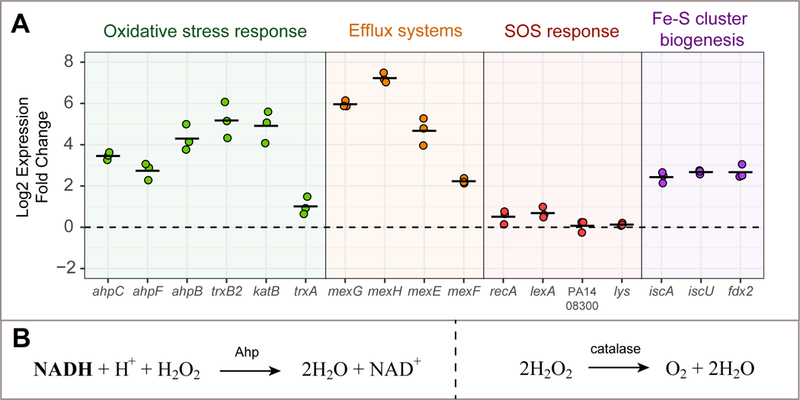
A. After cells are exposed to PYO under optimal nutritional conditions (i.e. auto-poisoning does not happen), genes involved in the oxidative stress response, RND efflux systems and Fe-S clusters biogenesis are induced. The SOS response is not up-regulated under these conditions, and genes directly related to pyocin clusters/explosive lysis (PA14_08300 and lys) are not induced. Black lines show averages of three replicates. B. Reactions performed by Ahps and catalases when detoxifying H2O2 (Imlay, 2013).
A strong induction of oxidative stress-related genes was observed after cells were exposed to PYO. These genes included alkyl hydroperoxide reductases (Ahps: ahpC, ahpF, ahpB), a thioredoxin reductase (trxB2) and a catalase (katB) (Fig. 6A). These genes are regulated by the transcription factor OxyR and induced by H2O2 (Ochsner et al., 2000; Hishinuma et al., 2008; Panmanee and Hassett, 2009). A thioredoxin gene (trxA) was also mildly upregulated (Fig. 6). The catalytic activity of Ahps depends upon reducing power provided by reduced electron carriers such as nicotinamide adenine dinucleotide (phosphate) (NAD(P)H) to perform the reduction of H2O2 to water (Fig. 6B, (Imlay, 2013)). Catalases on the other hand, reduce H2O2 to water and O2 in a NAD(P)H-independent manner and do not require stoichiometric reductants (Fig. 6B). That a strong oxidative stress response is part of the PYO defense mechanism under oxic conditions is not surprising (Hassan and Fridovich, 1980; Hassett et al., 1992; Rada and Leto, 2013). In addition, at least part of the oxidative stress response is dependent upon reducing power.
Besides the oxidative stress response, we suspected other cellular processes would be upregulated by PYO and important for defense against it. For example, energy-dependent RND efflux systems such a mexGHI-opmD are induced by PYO and involved in phenazine transport (Dietrich et al., 2006; Sakhtah et al., 2016). We tested two genes in this operon (mexG and mexH) and both of them were strongly induced by PYO in our survival experiments (Fig. 6A). In addition, we tested another efflux pump system, the mexEF-oprN operon, which is a multidrug efflux system in P. aeruginosa (Köhler et al., 1997); its genes were also transcribed more in the presence of PYO (Fig. 6A). We were also curious whether PYO exposure would lead to DNA damage and a subsequent SOS response. Yet the SOS-related genes recA and lexA were only mildly induced (Fig. 6A). Moreover, we did not detect PYO-upregulation of the endolysin lys, known to be responsible for the explosive lysis phenotype in P. aeruginosa (Turnbull et al., 2016) (Fig. 6A). This suggests that the SOS response and ensuing lys-mediated explosive lysis are minimized when cells have sufficient nutrients to power other defense mechanisms.
Finally, we were intrigued that sulfur seemed to significantly affect PYO resistance (Fig. 3D). Fe-S clusters are ubiquitous prosthetic groups that allow proteins to perform several functions, including electron transfer and disulfide reduction (Johnson et al., 2005). PYO stimulates generation of ROS (O2- and H2O2), and these species can damage Fe-S clusters (Imlay, 2006). However, phenazines can also directly oxidize Fe-S clusters in proteins (Gu and Imlay, 2011), which could potentially result in damage. Because (i) Fe-S cluster biogenesis machinery might participate in the replacement/repair of damaged Fe-S clusters (Djaman et al., 2004), and (ii) the Fe-S cluster biogenesis regulator IscR contributes to resistance to oxidants (Romsang et al., 2014), we tested whether PYO induces genes involved in Fe-S cluster biogenesis. In P. aeruginosa, the isc operon (iron-sulfur-cluster formation) is the system for Fe-S cluster biogenesis (Romsang et al., 2014; Lee et al., 2015). We measured the induction of three different genes in this operon (iscA, iscU, fdx2) and found that all three genes were upregulated in the presence of PYO (Fig. 6A).
A subpopulation of cells resists PYO poisoning even under nutrient-limited conditions
Together, our previous experiments indicated that continuous ATP synthesis through oxidation of reduced carbon helps cells avoid PYO auto-poisoning during the transition from a growth to non-growth state. However, these experiments assessed the bulk response of populations, leading us to wonder whether a subset of the population might respond differently. When exposing cells to PYO under stressful conditions (i.e. oxic, no nutrients), the majority of the population dies (1–3 days after exposure), but a small percentage of the population survives (usually around 5–15% for cells pre-grown in succinate, Fig. 2B). We were interested in understanding what enables this subset of cells to resist PYO, and hypothesized that resistance could occur by two distinct means: (i) the surviving cells are PYO resistant because they are in a distinct physiological state (i.e. “persister-like”) due to population heterogeneity; or (ii) they could be surviving based on the nutrients released when the majority of the population dies.
To differentiate between these hypotheses, we performed the experiment shown in Fig. 7. Growing cells were exposed to PYO in the absence of nutrients, causing massive cell death (~5% survival, Fig. 7A). Surviving cells were washed and re-suspended in the same buffer medium (oxic, no nutrients) with or without PYO. If cells were relying on nutrients released from the dead cells to power defense mechanisms against PYO, poisoning and cell death should be high after the wash. However, this was not the case: re-exposure of survivors did not result in any decrease in viability (Fig. 7B). This result suggested that the subpopulation of surviving cells was in a persister-like state (Van den Bergh et al., 2017) that does not require nutrients to survive PYO stress. When we re-grew the survivor cells in fresh medium and re-exposed the population to PYO, the majority of the population was re-sensitized to PYO, with only a small subpopulation again surviving (Fig. 7C). Such population dynamics indicate that PYO sensitivity is cyclic, dependent upon the environmental conditions.
Fig. 7. A subpopulation of cells can resist PYO even under nutrient-limited conditions.

The experimental protocol is shown on the left with results of survival after exposure to PYO on the right. Survival was quantified by plating and CFU counting. Differences were assessed using an unpaired t-test. Bar graphs represent the average of three replicates and error bars are SD.
Discussion
Over the last decade, a variety of physiological functions for phenazines have been identified (Dietrich et al., 2006; Price-Whelan et al., 2007; Dietrich et al., 2008; Wang et al., 2010; Wang et al., 2011; Dietrich et al., 2013; Glasser et al., 2014; Muller and Merrett, 2014; Okegbe et al., 2017; Glasser et al., 2017; Lin et al., 2018). Phenazines benefit their producers when they are experiencing reducing stress (i.e. cells are electron donor replete but electron acceptor limited). Under these conditions, phenazines can serve as alternative electron acceptors, allowing cells to maintain redox homeostasis, anoxic energy generation, and survive in regions of biofilms where oxygen is limiting (Price-Whelan et al., 2007; Wang et al., 2010; Glasser et al., 2014; Jo et al., 2017). Even aerobically-grown nutrient replete planktonic cultures achieve significantly greater cell yields when grown in the presence of PYO compared to its absence (Fig. S5), possibly due to oxygen limitation in high-density cultures. Despite these positive effects, phenazines are more commonly considered to be toxic compounds that kill competitors and damage host cells, and can even poison their producers (Lau et al., 2004; Voggu et al., 2006; Rada et al., 2008; Gibson et al., 2009; Das and Manefield, 2012; Wang et al., 2016). These opposing functions are striking and paradoxical. The objective of this work was to gain a better understanding of how PYO’s “double edged sword” effect impacts P. aeruginosa under particular conditions.
Our central hypothesis was that PYO’s versatile redox chemistry explains its orthogonal cellular effects in a predictable fashion, depending upon the environment and physiological state of the cells. PYO is only produced at high cell density and when high levels of carbon are available because it is regulated by quorum sensing (Dietrich et al., 2006). Under these conditions, when cells are oxidant-limited, they can thrive by utilizing PYO as an electron acceptor and avoiding toxicity because they are able to power defense mechanisms. However, this benefit to the overall population can quickly change, with PYO becoming dangerous to the majority of the population as it begins to starve for certain nutrients (e.g. carbon). Even after cell death due to PYO auto-poisoning, an intrinsically PYO-tolerant subset of the population still survives, resulting in the opportunity for PYO to again become net beneficial to the population when conditions alter. It is also important to consider that even though the toxic effects of PYO can be detrimental at the level of the single cell, in the context of a biofilm, the “altruistic” death of individuals permits eDNA release that provides a structural support for the survivors. Accordingly, PYO’s nuanced “double edged sword” effect can be rationalized as net-beneficial for the producer population over its lifecycle (Fig. 8).
Fig. 8. Model of PYO’s “double-edged sword” effect throughout the P. aeruginosa life cycle.
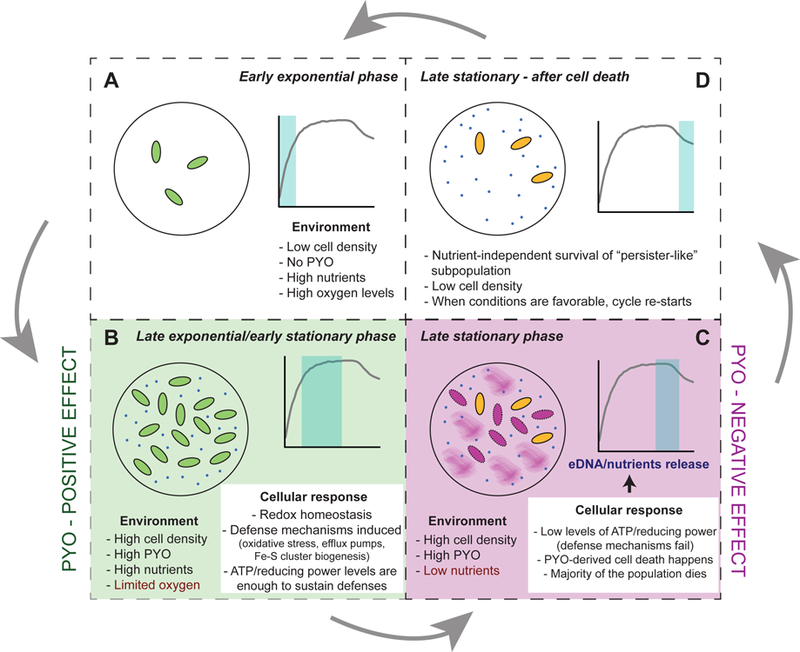
At least four different growth stages where PYO differentially affects P. aeruginosa can be distinguished and were tested by our experiments (A-D). A-B. Early stages where cells are resistant to PYO. Positive effect of PYO and the ability of cells to turn on energy-dependent mechanisms that result in survival (see Figs. 3A and F, Fig. 5 and Fig. 6). C. Negative effects of PYO (poisoning and cell death) when nutritional conditions are unfavorable to power defense mechanisms (see Fig. 1, Figs. 3B-F, Fig. 4 and Fig. 5). D. Demonstration of the existence of a “persister-like” subpopulation that is resistant to PYO (see Fig. 7).
Critical to P. aeruginosa’s tolerance of PYO is its extensive genetic arsenal to cope with the consequences of PYO’s redox activity (Hassett et al., 1992). This includes catalases, Ahps, multiple thioredoxins and thioredoxins reductases (Ochsner et al., 2000). Thioredoxins reduce disulfide bonds in thiol groups that become oxidized due to oxidative stress (Lu and Holmgren, 2014) and together with Ahps, many thioredoxin systems depend on the availability of reduced carbon provided by (NAD(P)H) (Lu and Holmgren, 2014). Because PYO is membrane permeable (Lau et al., 2004; Hall et al., 2016), its containment (by chaperones and/or efficient transport systems) is also important to minimize unwanted cellular redox reactions, such as reactivity with Fe-S clusters; RND efflux systems powered by the proton-motive force are involved in this task (Piddock, 2006; Dietrich et al., 2006). Our results showing a requirement for reducing power (carbon) and sulfur to avoid PYO toxicity (Fig. 3), together with the induction of genes involved in the oxidative stress response, efflux pump and Fe-S cluster systems in the presence of PYO (Fig. 6A), indicate that these defense systems are particularly important for PYO tolerance. Finally, though PYO can also intercalate into DNA (Hollstein and Van Gemert, 1971; Mavrodi et al., 2006; Das et al., 2015), in our experiments, cells seem to avoid extensive DNA damage (at least at the population level) when nutritional conditions are adequate to support other energy-dependent defense mechanisms (only a mild SOS response was detected). However, this does not exclude the possibility that an SOS response might be involved in auto-poisoning and lysis for a subset of the population (Turnbull et al., 2016). Future studies using single-cell tracking will allow this to be determined.
Though we performed the majority of our experiments in this report in planktonic culture for technical reasons, the implications for biofilm development are clear. Biofilms are heterogeneous and dynamic systems, and the ultimate impact of PYO on the different subpopulations would be expected to vary between beneficial and detrimental, depending upon the microenvironment present within the biofilm at different stages of development. Parameters such as nutrient concentrations, availability of electron acceptors and the respective energetic state of the cells should dictate the effect that PYO (and other phenazines) will have on different regions of the biofilm. For example, under nutrient-replete conditions, PYO production can cause beneficial expansion of biofilm aggregates under hypoxic/anoxic conditions (Costa et al., 2017). However, in regions of the biofilm where cells are starved for nutrients and PYO is present, PYO-mediated auto-poisoning would contribute to eDNA release, an important structural component of the matrix. Finally, we note that fluctuations of nutrients, which occur in open systems (DeAngelis et al., 1989; Odum et al., 1995), would be expected to significantly impact the effect of PYO. These predictions are testable, and going forward, we hope to leverage the findings reported in this work to better understand how versatile, redox-active molecules such as PYO and other phenazines can modulate biofilm development.
Experimental procedures
Biofilm and single-cell experiments
For biofilm experiments, cells of WT and ∆phz (both phz1 and phz2 operons are deleted in this strain, see Dietrich et al., 2006) strains from an overnight culture were washed and diluted (1:50) in succinate minimal medium (SMM, composition: 40 mM sodium succinate, 50 mM KH2PO4/K2HPO4[pH 7], 42.8 mM NaCl, 1 mM MgSO4, 9.35 mM NH4Cl, trace elements solution (Widdel and Pfennig, 1981)), and grown until OD500 = 0.4–0.5. Cells were centrifuged, washed once, and diluted to OD500 = 0.25, which was used as the final inoculum. Then, 250 µL of WT or ∆phz inoculum were transferred to Nunc Lab-Tek 8-well chambered coverglass (Thermo Scientific) and incubated at 37 ºC in shaking conditions (250 rpm) during six hours for biofilm development. This experiment was repeated 10 times, with two independent wells for WT and ∆phz cultures every time. After six hours of incubation, liquid was removed and the chambers were washed twice with PBS (Phosphate buffer saline – 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.2) to remove planktonic cells, leaving only the attached biofilms on the bottom of the chamber. Finally, biofilms were stained with both TOTO-1 Iodide (1 µM) and DAPI (20 µM), and chambers were imaged using confocal microscopy (Leica TCS SPE). For quantification of cell death of attached cells, 10 different fields of view per well were imaged. Images were then analyzed in Oufti (Paintdakhi et al., 2016) using the DAPI channel for total cell counting (live and dead); when checked manually, the software was able to count 80–90% of the total cells for both WT and ∆phz treatments. Dead cells were manually counted using the TOTO channel and calculation of the percentage of dead cells was done by dividing the number of dead cells/total number of cells (live and dead). The final proportions of dead cells for each of the 10 replicates for WT and ∆phz are plotted in Fig. 1B.
Single-cell experiments were performed using agarose pads (SMM with 2% agarose). Cells were grown from an overnight culture until OD500 = 0.4–0.5, spotted on top of the pads and imaged using fluorescence microscopy (Leica TCS SPE). Agarose pads contained 100 µM PYO and we used both WT and WT-YPF (constitutive YFP expression) strains for these experiments. Temperature control was set to 37 ºC. Shrinking and explosive cell lysis were monitored in both Differential Interference Contrast (DIC) and fluorescence channels under 100x magnification. For eDNA staining, we used agarose pads containing TOTO-1 Iodide (1 µM).
Liquid culture assays and quantification of phenazine toxicity
WT and ∆phz cells were grown from overnight cultures to OD500 = 0.5 and then diluted into four independent OD500 = 0.05 cultures (replicates) using 5 mL glass tubes containing fresh SMM (with and without 100 µM PYO), where the experiment started. Cells were incubated at 37 ºC for 36h, and time-points were taken at 0, 18, 24 and 36 hours for plating and CFU counting. Plating for CFU counting was always done in lysogeny broth (LB) (Difco) plates containing 1.5% agar. PYO final concentration in WT cultures was quantified using high-performance liquid chromatography (HPLC) (Fig. S6). For all experiments shown in this paper, with the exception of the toxicity assay comparing different phenazines (see below), PYO was dissolved in 20 mM HCl, which was always added to PYO-untreated cultures as a control. The PYO used was synthesized and purified using methods previously described (Dietrich et al., 2006; Cheluvappa, 2014). Other phenazines were purchased from Princeton Biomolecular Research Inc and TCI America.
Toxicity of different phenazines was assessed by performing shock experiments where cells were shifted from growing conditions to minimal phosphate buffer (MPB, 50 mM KH2PO4/ K2HPO4[pH 7], 42.8 mM NaCl) containing 100 µM of each of the four phenazines tested: PYO, 1-OH-PHZ, PCN and PCA. Cells were grown in SMM until OD500 = 0.7–0.8, washed, and concentrated to final OD500 = 2 (which corresponds to ~2–3×109 cells) in MPB + phenazines. In this experiment, phenazines needed to be dissolved using a mutual solvent (DMSO), which was used as a negative control. Incubation was done under shaking conditions (250 rpm, 37 ºC) in 1.5 mL Eppendorf tubes containing 1 mL of culture. Cells were plated for CFU counting after 24 and 48 hours of exposure to the phenazines. Four replicates were performed for each treatment.
Plate reader assay for quantification of cell death
The plate reader assay (using a BioTek Synergy 4 plate reader) was developed to efficiently quantify cell death under a variety of environmental conditions. For growth curves presented in Fig. 1C-D, cells were grown in either SMM or glucose minimal medium (GMM: same composition as SMM, but with glucose instead of succinate as the carbon source) and propidium iodide (PI) at a concentration of 5 µM was added to the cultures. Cells were incubated in the plate reader, with shaking at 37 ºC and monitoring of OD500 and fluorescence (xenon lamp, excitation/emission = 535/617 nm). PI was used for cell death monitoring instead of TOTO-1 iodide because cultures displayed auto-fluorescence in the same wavelength as TOTO and the background auto-fluorescence was different for WT and ∆phz strains. This is likely due to different amounts of siderophore production, NAD(P)H and/or reduced phenazines present in the culture (Sullivan et al., 2011). This was not observed when using PI (Fig. S7), and therefore this dye was used in all plate reader experiments. Addition of 5 µM PI only mildly impacted cell growth (Fig. S8). For every plate reader experiment reported here, six independent wells were analyzed for each treatment (i.e. six wells with no PYO added, and six wells with 100 µM PYO added), and the averages and standard deviations for the six replicates were plotted. Each plate reader set up was repeated at least twice, with very similar results.
For survival experiments under different nutritional conditions presented in Fig. 3, ∆phz cells were grown in GMM + 100 mM MOPS until OD500 = 0.7–0.8, centrifuged, washed and re-suspended at OD500 = 2 in buffer (100 mM MOPS + 42.8 mM NaCl, pH = 7). Then, the different nutrients were added as indicated. Final concentrations for each nutrient were: carbon = 80 mM glucose; phosphorus = 50 mM KH2PO4/ K2HPO4 [pH 7]; sulfur = 1 mM MgSO4. In all experiments, trace metals solution (Widdel and Pfennig, 1981) at the same concentration as the one used for growth was added. During the survival experiments, the nitrogen source (NH4Cl) was omitted in all treatments as a way to minimize growth (OD500 still increased during the first ~5h of the experiment, but shortly afterward cells stopped growing). Removing nitrogen from the medium did not sensitize the cells to PYO (Fig. 3A). To confirm that an increase in PI fluorescence indeed meant cell death, we plated cells for CFU counting after 50 hours of incubation (Fig. S2).
Finally, to further test the importance of carbon availability to survival against PYO, cells were grown in the presence or absence of PYO in GMM under different glucose concentrations (4 mM, 10 mM, 40 mM, 80 mM). For all survival and growth experiments, OD500 measurements during inoculum preparation were performed using a Beckman Coulter DU 800 spectrophotometer. Measurements of pH using pH strips after the end of the experiments revealed no significant changes.
PYO toxicity under anaerobic conditions
To quantify poisoning and cell death caused by PYO under anaerobic conditions, ∆phz cells were grown aerobically exactly as described for the survival experiment above, but then were shifted to an anaerobic chamber, where cells were incubated for 4 hours before addition of PYO to minimize potential oxidative stress. Resazurin (1 µg/mL) was added to a control tube to monitor oxygen consumption. This control tube contained an aliquot culture derived from the experiment culture, but to avoid any potential interference of resazurin in the experiment, these cells were only used for monitoring of O2 consumption. The 4 hours incubation resulted in a colorless resazurin in the control tube (indicating consumption of trace levels of O2 in the medium, (Bacic and Smith, 2008). PYO was then added to cultures and cells were transferred to the plate reader present in the anaerobic chamber. PYO solution and 96-well plates used in this experiment had been acclimatized under anaerobic conditions for at least 3 days. Incubation conditions were the same as before, but fluorescence measurements were performed with a tungsten lamp using a red filter cube (excitation: 486/20, emission: 620/40; settings available for the anaerobic plate reader). Direct comparison of absolute fluorescence values between aerobic and anaerobic measurements are not possible due to the different plate readers used. Reduction of PYO by cells was monitored by two ways: (i) absorbance at OD310, which is intense for oxidized PYO, but weak for reduced PYO (Wang and Newman, 2008); (ii) fluorescence at ~500nm using a green filter cube (excitation: 360/40, emission: 460/40) which is intense for reduced PYO, but null for oxidized PYO (Wang and Newman, 2008).
ATP measurements and ATP synthesis inhibition
ATP measurements were performed using the BacTiter-Glo reagent (Promega) in 96-well opaque white microtiter plates as previously described (Glasser et al., 2014). In summary, aerobic survival experiments as described above were set up under high or low carbon concentrations (80 mM or 4 mM glucose, respectively). The expectation was that cells under low carbon availability would starve for carbon first, resulting in ATP depletion and cell death. After incubation, cultures were sampled at 8, 18 and 26 hours for both ATP quantification and viability through CFUs counting. For ATP measurements, 20 µL aliquot of each culture was added to 180 µL of DMSO. This solution was diluted with 800 µL of 100 mM HEPES (pH 7.5) and stored at −80ºC until analysis (no more than 2 days). Thawed samples were mixed 1:1 BacTiter-Glo reagent and luminescence was measured at 30ºC using a plate reader (BioTek Synergy 4). For determining ATP concentrations in the sample, a standard curve with known concentrations of ATP was generated.
To test for a direct connection between ATP depletion and PYO toxicity, experiments using the molecule N,N-dicyclohexylcarbodiimide (DCCD) were performed. DCCD is classical inhibitor of the F0F1-ATP synthase (Sebald et al., 1980), disrupting ATP synthesis. Aerobic survival experiments as described before were set up where cells had three different treatments: (i) low carbon (4 mM glucose) with no DCCD; (ii) high carbon (80 mM glucose) with no DCCD (i.e. normal ATP synthesis) or (iii) high carbon (80 mM glucose) + 200 µM DCCD (i.e. ATP inhibition). This concentration of the inhibitor was used because it did not seem to cause a negative impact on the negative control (No PYO treatment, Fig. 5B). OD500 and PI fluorescence were measured over a time course of 40 hours.
RNA extraction and quantitative reverse transcriptase PCR (qRT-PCR)
For RNA extraction, three independent cultures of ∆phz cells were grown in GMM and a survival experiment as described in Fig. 3A was performed, where cells were incubated under two treatments: (i) 100 µM PYO and (ii) No PYO. Under these conditions, cells were able to survive and had nutrients necessary to power defense mechanisms against PYO toxicity. For all three replicates, 750 µL of culture containing +/− PYO were split into five wells in a 96-well BRAND® microplate followed by incubation in the plate reader for 5h. Cells were then harvested from the wells, pelleted and re-suspended in 215 µL of TE buffer (30 mM Tris.Cl, 1 mM EDTA, pH 8.0) containing 15 mg/mL of lysozyme + 15 µL of proteinase K solution (20 mg/mL, Qiagen), and incubated for 8–10 min. Further lysis steps and RNA extraction were performed using an RNeasy kit (Qiagen) following the manufacturer’s instructions. For further removal of contaminant genome DNA, additional treatment of the extracted RNA with TURBO DNA-free kit (Invitrogen) was performed following the manufacturer’s instructions. Finally, cDNA was synthesized from 1 µg of total RNA using iScript cDNA Synthesis kit (Bio-Rad) according to the manufacturer’s protocol.
For qRT-PCR, iTaq Universal SYBR Green Supermix (Bio-Rad) was used. Reactions were performed in 20 µL volume containing 10 µL of the supermix, 5 µL of cDNA and 5 µL of forward and reverse primer solution (2 µM each). Primers are listed in Table S1. Reactions were run using a 7500 Fast Real-Time PCR System machine (Applied Biosystems). Standard curves for each primer pair using known concentrations of Pa PA14 genomic DNA were used to back calculate concentrations of cDNA for each gene of interest. Normalizations for a control gene were done using the house-keeping gene oprI (Babin et al., 2016). Reactions with the additional gene proC were also run as a second negative control (Heussler et al., 2015). After normalization by oprI, data is displayed as a log2 fold change in expression (Fig. 6A).
Testing a “persister-like” phenotype upon PYO exposure
To test if the survival of a subpopulation of cells upon exposure to PYO under nutrient-limiting conditions happens (i) due to release of nutrients from dead cells or (ii) due to a “persister-like” phenotype, experiments followed the scheme presented in Fig. 7. First, a ∆phz culture was grown in SMM as described for experiments measuring phenazines toxicity, split in three different replicates, washed and shifted to MPB and exposed to 100 µM PYO for three days. The negative control did not contain PYO. Then, survivor cells were pelleted and washed twice to remove nutrients released from dead cells, followed by re-suspension in MPB and a second exposure to PYO for three days. Finally, after second exposure to PYO, survivors were grown again in SMM until OD500 = 1.0 and a third exposure to PYO (during six days) was performed to test if cells had become sensitive again. During all exposures to PYO, incubation was done under shaking conditions (250 rpm) at 37 ºC. PYO remained blue during the entire experiment, indicating that oxygenation was sufficient to maintain the phenazine in its oxidized state. Survival was assessed by plating and CFU counting.
Statistical analyses
All statistical analyses were performed using R (R Core Team, 2018). One-way ANOVA with post-hoc Tukey’s HSD test for multiple comparisons was used in Fig. 2A-B. Unpaired t-tests were used for data presented in Fig. 5A and Fig. 7. Before performing the mentioned statistical tests, normality and homogeneity of variances were assessed using the Shapiro-Wilk test and Levene’s test, respectively.
Supplementary Material
Acknowledgements
We thank all the members of the Newman lab for experimental advice and feedback on the manuscript; Megan Bergkessel was particularly generous with her time and intellectual support. We also thank Scott Saunders for helping with pyocyanin quantification. Grants to D.K.N. from the ARO (W911NF-17–1-0024) and NIH 341 (1R01AI127850–01A1) supported this research.
References
- Allesen-Holm M, Barken KB, Yang L, Klausen M, Webb JS, Kjelleberg S, et al. (2006) A characterization of DNA release in Pseudomonas aeruginosa cultures and biofilms. Mol Microbiol 59: 1114–1128. [DOI] [PubMed] [Google Scholar]
- Babin BM, Bergkessel M, Sweredoski MJ, Moradian A, Hess S, Newman DK, and Tirrell DA (2016) SutA is a bacterial transcription factor expressed during slow growth in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 113: E597–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacic MK, and Smith CJ (2008) Laboratory maintenance and cultivation of bacteroides species. Curr Protoc Microbiol Chapter 13: Unit 13C.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barakat R, Goubet I, Manon S, Berges T, and Rosenfeld E (2014) Unsuspected pyocyanin effect in yeast under anaerobiosis. Microbiologyopen 3: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayles KW (2014) Bacterial programmed cell death: making sense of a paradox. Nat Rev Microbiol 12: 63–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheluvappa R (2014) Standardized chemical synthesis of Pseudomonas aeruginosa pyocyanin. MethodsX 1: 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa KC, Glasser NR, Conway SJ, and Newman DK (2017) Pyocyanin degradation by a tautomerizing demethylase inhibits Pseudomonas aeruginosa biofilms. Science 355: 170–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Argenio DA, Calfee MW, Rainey PB, and Pesci EC (2002) Autolysis and autoaggregation in Pseudomonas aeruginosa colony morphology mutants. J Bacteriol 184: 6481–6489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das T, Kutty SK, Tavallaie R, Ibugo AI, Panchompoo J, Sehar S, et al. (2015) Phenazine virulence factor binding to extracellular DNA is important for Pseudomonas aeruginosa biofilm formation. Sci Rep 5: 8398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das T, and Manefield M (2012) Pyocyanin promotes extracellular DNA release in Pseudomonas aeruginosa. PLoS ONE 7: e46718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeAngelis DL, Mulholland PJ, Palumbo AV, Steinman AD, Huston MA, and Elwood JW (1989) Nutrient Dynamics and Food-Web Stability. Annu Rev Ecol Syst 20: 71–95. [Google Scholar]
- Dietrich LEP, Okegbe C, Price-Whelan A, Sakhtah H, Hunter RC, and Newman DK (2013) Bacterial community morphogenesis is intimately linked to the intracellular redox state. J Bacteriol 195: 1371–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich LEP, Price-Whelan A, Petersen A, Whiteley M, and Newman DK (2006) The phenazine pyocyanin is a terminal signalling factor in the quorum sensing network of Pseudomonas aeruginosa. Mol Microbiol 61: 1308–1321. [DOI] [PubMed] [Google Scholar]
- Dietrich LEP, Teal TK, Price-Whelan A, and Newman DK (2008) Redox-active antibiotics control gene expression and community behavior in divergent bacteria. Science 321: 1203–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djaman O, Outten FW, and Imlay JA (2004) Repair of oxidized iron-sulfur clusters in Escherichia coli. J Biol Chem 279: 44590–44599. [DOI] [PubMed] [Google Scholar]
- Fagerbakke KM, Heldal M, and Norland S (1996) Content of carbon, nitrogen, oxygen, sulfur and phosphorus in native aquatic and cultured bacteria. Aquat Microb Ecol 10: 15–27. [Google Scholar]
- Flemming H-C (2016) EPS-Then and Now. Microorganisms 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flemming H-C, Wingender J, Szewzyk U, Steinberg P, Rice SA, and Kjelleberg S (2016) Biofilms: an emergent form of bacterial life. Nat Rev Microbiol 14: 563–575. [DOI] [PubMed] [Google Scholar]
- Gibson J, Sood A, and Hogan DA (2009) Pseudomonas aeruginosa-Candida albicans interactions: localization and fungal toxicity of a phenazine derivative. Appl Environ Microbiol 75: 504–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasser NR, Kern SE, and Newman DK (2014) Phenazine redox cycling enhances anaerobic survival in Pseudomonas aeruginosa by facilitating generation of ATP and a proton-motive force. Mol Microbiol 92: 399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasser NR, Saunders SH, and Newman DK (2017) The colorful world of extracellular electron shuttles. Annu Rev Microbiol 71: 731–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govan JR, and Deretic V (1996) Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol Rev 60: 539–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu M, and Imlay JA (2011) The SoxRS response of Escherichia coli is directly activated by redox-cycling drugs rather than by superoxide. Mol Microbiol 79: 1136–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall S, McDermott C, Anoopkumar-Dukie S, McFarland AJ, Forbes A, Perkins AV, et al. (2016) Cellular effects of pyocyanin, a secreted virulence factor of Pseudomonas aeruginosa. Toxins (Basel) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan HM, and Fridovich I (1980) Mechanism of the antibiotic action pyocyanine. J Bacteriol 141: 156–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassett DJ, Charniga L, Bean K, Ohman DE, and Cohen MS (1992) Response of Pseudomonas aeruginosa to pyocyanin: mechanisms of resistance, antioxidant defenses, and demonstration of a manganese-cofactored superoxide dismutase. Infect Immun 60: 328–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazan R, Que YA, Maura D, Strobel B, Majcherczyk PA, Hopper LR, et al. (2016) Auto poisoning of the respiratory chain by a quorum-sensing-regulated molecule favors biofilm formation and antibiotic tolerance. Curr Biol 26: 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heussler GE, Cady KC, Koeppen K, Bhuju S, Stanton BA, and O’Toole GA (2015) Clustered regularly interspaced short palindromic repeat-dependent, biofilm-specific death of Pseudomonas aeruginosa mediated by increased expression of phage-related genes. MBio 6: e00129–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hishinuma S, Ohtsu I, Fujimura M, and Fukumori F (2008) OxyR is involved in the expression of thioredoxin reductase TrxB in Pseudomonas putida. FEMS Microbiol Lett 289: 138–145. [DOI] [PubMed] [Google Scholar]
- Hollstein U, and Van Gemert RJ (1971) Interaction of phenazines with polydeoxyribonucleotides. Biochemistry 10: 497–504. [DOI] [PubMed] [Google Scholar]
- Ibáñez de Aldecoa AL, Zafra O, and González-Pastor JE (2017) Mechanisms and regulation of extracellular DNA release and its biological roles in microbial communities. Front Microbiol 8: 1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imlay JA (2006) Iron-sulphur clusters and the problem with oxygen. Mol Microbiol 59: 1073–1082. [DOI] [PubMed] [Google Scholar]
- Imlay JA (2013) The molecular mechanisms and physiological consequences of oxidative stress: lessons from a model bacterium. Nat Rev Microbiol 11: 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo J, Cortez KL, Cornell WC, Price-Whelan A, and Dietrich LE (2017) An orphan cbb3-type cytochrome oxidase subunit supports Pseudomonas aeruginosa biofilm growth and virulence. eLife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DC, Dean DR, Smith AD, and Johnson MK (2005) Structure, function, and formation of biological iron-sulfur clusters. Annu Rev Biochem 74: 247–281. [DOI] [PubMed] [Google Scholar]
- Köhler T, Michéa-Hamzehpour M, Henze U, Gotoh N, Curty LK, and Pechère JC (1997) Characterization of MexE-MexF-OprN, a positively regulated multidrug efflux system of Pseudomonas aeruginosa. Mol Microbiol 23: 345–354. [DOI] [PubMed] [Google Scholar]
- Lau GW, Hassett DJ, Ran H, and Kong F (2004) The role of pyocyanin in Pseudomonas aeruginosa infection. Trends Mol Med 10: 599–606. [DOI] [PubMed] [Google Scholar]
- Laursen JB, and Nielsen J (2004) Phenazine natural products: biosynthesis, synthetic analogues, and biological activity. Chem Rev 104: 1663–1686. [DOI] [PubMed] [Google Scholar]
- Lee SA, Gallagher LA, Thongdee M, Staudinger BJ, Lippman S, Singh PK, and Manoil C (2015) General and condition-specific essential functions of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 112: 5189–5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y-C, Sekedat MD, Cornell WC, Silva GM, Okegbe C, Price-Whelan A, et al. (2018) Phenazines regulate Nap-dependent denitrification in Pseudomonas aeruginosa biofilms. J Bacteriol 10.1128/JB.00031-18 [DOI] [PMC free article] [PubMed]
- Lu J, and Holmgren A (2014) The thioredoxin antioxidant system. Free Radic Biol Med 66: 75–87. [DOI] [PubMed] [Google Scholar]
- Matsukawa M, and Greenberg EP (2004) Putative exopolysaccharide synthesis genes influence Pseudomonas aeruginosa biofilm development. J Bacteriol 186: 4449–4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrodi DV, Blankenfeldt W, and Thomashow LS (2006) Phenazine compounds in fluorescent Pseudomonas spp. biosynthesis and regulation. Annu Rev Phytopathol 44: 417–445. [DOI] [PubMed] [Google Scholar]
- Muller M (2002) Pyocyanin induces oxidative stress in human endothelial cells and modulates the glutathione redox cycle. Free Radic Biol Med 33: 1527–1533. [DOI] [PubMed] [Google Scholar]
- Muller M, and Merrett ND (2014) Pyocyanin production by Pseudomonas aeruginosa confers resistance to ionic silver. Antimicrob Agents Chemother 58: 5492–5499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochsner UA, Vasil ML, Alsabbagh E, Parvatiyar K, and Hassett DJ (2000) Role of the Pseudomonas aeruginosa oxyR-recG operon in oxidative stress defense and DNA repair: OxyR-dependent regulation of katB-ankB, ahpB, and ahpC-ahpF. J Bacteriol 182: 4533–4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odum WE, Odum EP, and Odum HT (1995) Nature’s Pulsing Paradigm. Estuaries 18: 547. [Google Scholar]
- Okegbe C, Fields BL, Cole SJ, Beierschmitt C, Morgan CJ, Price-Whelan A, et al. (2017) Electron-shuttling antibiotics structure bacterial communities by modulating cellular levels of c-di-GMP. Proc Natl Acad Sci U S A 114: E5236–E5245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okshevsky M, and Meyer RL (2013) The role of extracellular DNA in the establishment, maintenance and perpetuation of bacterial biofilms. Crit Rev Microbiol 41: 341–352. [DOI] [PubMed] [Google Scholar]
- Okshevsky M, and Meyer RL (2014) Evaluation of fluorescent stains for visualizing extracellular DNA in biofilms. J Microbiol Methods 105: 102–104. [DOI] [PubMed] [Google Scholar]
- Okshevsky M, Regina VR, and Meyer RL (2015) Extracellular DNA as a target for biofilm control. Curr Opin Biotechnol 33: 73–80. [DOI] [PubMed] [Google Scholar]
- Olsen I (2015) Biofilm-specific antibiotic tolerance and resistance. Eur J Clin Microbiol Infect Dis 34: 877–886. [DOI] [PubMed] [Google Scholar]
- Paintdakhi A, Parry B, Campos M, Irnov I, Elf J, Surovtsev I, and Jacobs-Wagner C (2016) Oufti: an integrated software package for high-accuracy, high-throughput quantitative microscopy analysis. Mol Microbiol 99: 767–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panmanee W, and Hassett DJ (2009) Differential roles of OxyR-controlled antioxidant enzymes alkyl hydroperoxide reductase (AhpCF) and catalase (KatB) in the protection of Pseudomonas aeruginosa against hydrogen peroxide in biofilm vs. planktonic culture. FEMS Microbiol Lett 295: 238–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piddock LJV (2006) Multidrug-resistance efflux pumps - not just for resistance. Nat Rev Microbiol 4: 629–636. [DOI] [PubMed] [Google Scholar]
- Price-Whelan A, Dietrich LEP, and Newman DK (2006) Rethinking “secondary” metabolism: physiological roles for phenazine antibiotics. Nat Chem Biol 2: 71–78. [DOI] [PubMed] [Google Scholar]
- Price-Whelan A, Dietrich LEP, and Newman DK (2007) Pyocyanin alters redox homeostasis and carbon flux through central metabolic pathways in Pseudomonas aeruginosa PA14. J Bacteriol 189: 6372–6381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2018). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: URL https://www.R-project.org/. [Google Scholar]
- Rada B, Lekstrom K, Damian S, Dupuy C, and Leto TL (2008) The Pseudomonas toxin pyocyanin inhibits the dual oxidase-based antimicrobial system as it imposes oxidative stress on airway epithelial cells. J Immunol 181: 4883–4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada B, and Leto TL (2013) Pyocyanin effects on respiratory epithelium: relevance in Pseudomonas aeruginosa airway infections. Trends Microbiol 21: 73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos I, Dietrich LEP, Price-Whelan A, and Newman DK (2010) Phenazines affect biofilm formation by Pseudomonas aeruginosa in similar ways at various scales. Res Microbiol 161: 187–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice KC, and Bayles KW (2008) Molecular control of bacterial death and lysis. Microbiol Mol Biol Rev 72: 85–109, table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romsang A, Duang-Nkern J, Leesukon P, Saninjuk K, Vattanaviboon P, and Mongkolsuk S (2014) The iron-sulphur cluster biosynthesis regulator IscR contributes to iron homeostasis and resistance to oxidants in Pseudomonas aeruginosa. PLoS ONE 9: e86763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakhtah H, Koyama L, Zhang Y, Morales DK, Fields BL, Price-Whelan A, et al. (2016) The Pseudomonas aeruginosa efflux pump MexGHI-OpmD transports a natural phenazine that controls gene expression and biofilm development. Proc Natl Acad Sci U S A 113: E3538–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakhtah H, Price-Whelan A, and Dietrich LE (2013) Regulation of phenazine biosynthesis. In Microbial Phenazines: Biosynthesis, Agriculture and Health Springer, . [Google Scholar]
- Sebald W, Machleidt W, and Wachter E (1980) N,N’-dicyclohexylcarbodiimide binds specifically to a single glutamyl residue of the proteolipid subunit of the mitochondrial adenosinetriphosphatases from Neurospora crassa and Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 77: 785–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan NL, Tzeranis DS, Wang Y, So PTC, and Newman D (2011) Quantifying the dynamics of bacterial secondary metabolites by spectral multiphoton microscopy. ACS Chem Biol 6: 893–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull L, Toyofuku M, Hynen AL, Kurosawa M, Pessi G, Petty NK, et al. (2016) Explosive cell lysis as a mechanism for the biogenesis of bacterial membrane vesicles and biofilms. Nat Commun 7: 11220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Bergh B, Fauvart M, and Michiels J (2017) Formation, physiology, ecology, evolution and clinical importance of bacterial persisters. FEMS Microbiol Rev 41: 219–251. [DOI] [PubMed] [Google Scholar]
- Voggu L, Schlag S, Biswas R, Rosenstein R, Rausch C, and Götz F (2006) Microevolution of cytochrome bd oxidase in Staphylococci and its implication in resistance to respiratory toxins released by Pseudomonas. J Bacteriol 188: 8079–8086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Yu JM, Dorosky RJ, Pierson LS, and Pierson EA (2016) The phenazine 2-hydroxy-phenazine-1-carboxylic acid promotes extracellular DNA release and has broad transcriptomic consequences in Pseudomonas chlororaphis 30–84. PLoS ONE 11: e0148003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Kern SE, and Newman DK (2010) Endogenous phenazine antibiotics promote anaerobic survival of Pseudomonas aeruginosa via extracellular electron transfer. J Bacteriol 192: 365–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, and Newman DK (2008) Redox reactions of phenazine antibiotics with ferric (hydr)oxides and molecular oxygen. Environ Sci Technol 42: 2380–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wilks JC, Danhorn T, Ramos I, Croal L, and Newman DK (2011) Phenazine-1-carboxylic acid promotes bacterial biofilm development via ferrous iron acquisition. J Bacteriol 193: 3606–3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb JS, Thompson LS, James S, Charlton T, Tolker-Nielsen T, Koch B, et al. (2003) Cell death in Pseudomonas aeruginosa biofilm development. J Bacteriol 185: 4585–4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitchurch CB, Tolker-Nielsen T, Ragas PC, and Mattick JS (2002) Extracellular DNA required for bacterial biofilm formation. Science 295: 1487. [DOI] [PubMed] [Google Scholar]
- Widdel F, and Pfennig N (1981) Studies on dissimilatory sulfate-reducing bacteria that decompose fatty acids. Arch Microbiol 129: 395–400. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.