

ABSTRACT
Mycobacteriophages are viruses that infect mycobacterial hosts. A large number of mycobacteriophages have been isolated and genomically characterized, providing insights into viral diversity and evolution, as well as fueling development of tools for mycobacterial genetics. Mycobacteriophages have intimate relationships with their hosts and provide insights into the genetics and physiology of the mycobacteria and tools for potential clinical applications such as drug development, diagnosis, vaccines, and potentially therapy.
INTRODUCTION
Because of the importance of mycobacterial diseases such as tuberculosis and leprosy, mycobacteriophages have long been studied, with the first ones being isolated in the 1940s (1–3). At that time, it was recognized that bacteriophages display particular profiles of specificity for their bacterial hosts, nearly always distinguishing between bacteria in different genera and sometimes distinguishing between strains of the same bacterial species (4–7). Mycobacteriophages thus presented a plausible means of identifying (i.e., typing) clinical isolates by scoring for phage sensitivity profiles, with the prospects of obtaining data considerably faster than standard protocols that required extensive growth of the host. Mycobacterium tuberculosis—the causative agent of human tuberculosis—has a remarkably slow growth rate (24-hour doubling time), so phage typing can speed up diagnosis by several weeks. This general concept of exploiting the relatively rapid propagation of mycobacteriophages has been a common theme in their subsequent development (8–10).
Although the important role of M. tuberculosis in human health was known from the early studies by Koch, our knowledge of the organism was limited for many years because of the inability to genetically transform it with the uptake of DNA (11). An important breakthrough in the late 1980s occurred when it was demonstrated that phage DNA could be taken up by mycobacterial spheroplasts, followed by conversion into infectious units and plaque formation (12). The approach illustrated three additional aspects of working with the mycobacteria and their phages. First, Jacobs and colleagues used Mycobacterium smegmatis as a surrogate host, because of its much faster growth rate (∼3-hour doubling time), coupled with mycobacteriophages which had been shown to infect both M. smegmatis and M. tuberculosis. Second, the construction of shuttle phasmids, which replicate as a large plasmid in Escherichia coli and as a phage in mycobacteria, facilitated the introduction of foreign DNA, because the shuttle phasmids could be manipulated readily in E. coli and then used as vectors in M. smegmatis and M. tuberculosis. Shuttle phasmids have found extensive uses for introduction of reporter genes, transposons, and allelic exchange substrates into a variety of mycobacterial hosts (11). Lastly, shuttle phasmids built with temperate phages enable stable introduction of gene cassettes that can be used to evaluate expression and activities of foreign genes such as those for antibiotic resistance (13).
Mycobacteriophages entered the genomics era in the early 1990s (14), opening up a variety of new tools that can be applied to studying the mycobacteria (15). As sequenced mycobacteriophage genomes emerged over the next 25 years, the extent of their considerable genetic diversity became apparent, culminating in a current collection of over 9,600 individual phages, of which 1,500 have been completely sequenced (16). Because all of these (with the exception of DS6A) infect a single bacterial strain (M. smegmatis mc2155), they represent a unique perspective on viral diversity and bacteriophage evolution. These also encompass more than 150,000 genes and together provide a tremendous resource for advancing our understanding and control of their mycobacterial hosts.
In this article I will review our current understanding of mycobacteriophages, their roles in mycobacterial genetics, and the insights gained from their diversity. A number of other reviews of mycobacteriophages are available (8, 9, 15, 17–26) that provide a variety of perspectives, including a historical view of this field. Here, I will focus primarily on recent developments that have not been extensively covered in the earlier reviews, the most recent of which was 4 years ago (27).
GENERAL MYCOBACTERIOPHAGE FEATURES
Current Genomic Scope
As noted above, the total number of mycobacteriophages isolated is nearing 10,000, and over 1,500 have been completely sequenced. Further details about how these were isolated and characterized are provided below, but for the most part we will constrain the discussion here to the subset of genomes that are available in the public databases, totaling 899 genomes. As should be apparent from the discrepancies in these numbers, at the time of writing there are almost 700 genomes that have been sequenced but are not yet fully annotated and submitted to GenBank. The complete and up-to-date list of sequenced phages and their current status is available at the PhagesDB (http://phagesdb.org) and Phamerator (http://phamerator.org) databases (16). It should be noted that the annotation, quality control review, revision, and GenBank submission represents a current bottleneck in the pipeline and is relatively time-consuming compared to sequence determination. Nonetheless, the dataset of 899 genomes offers a representative view of the overall diversity, which does not change substantially if the larger dataset is considered.
It is also worth noting that there is a growing collection of genomically sequenced phages that infect other bacterial hosts within the phylum Actinobacteria. This includes substantial numbers of phages of Arthrobacter, Corynebacterium, Gordonia, Microbacterium, Propionibacterium, Rhodococcus, and Streptomyces (16). These will not be discussed in detail here but are especially relevant in considering the processes by which bacteriophages evolve.
Phage Discovery and Genomics as a Platform for Advancing Science Education
Isolation of bacteriophages using a simple plaque assay has been performed successfully for the past 100 years (28). However, genomic analyses of these phages have only been available since about the early 1990s (14), and DNA sequencing technology has advanced rapidly since then. The coupling of phage isolation and genomic analysis facilitates defining viral diversity and provides insights into understanding their evolution (29). The finding that there are so many different phage genomes underpins the use of phage isolation and genomics as a powerful platform for integrating our missions in science education with those in phage exploration (30).
The Phage Hunters Integrating Research and Education (PHIRE) program was developed at the University of Pittsburgh in 2002 with the intent of using phage discovery and genomics to introduce high school and undergraduate students to authentic scientific research (31). The PHIRE program provides strong evidence not only that students can readily successfully engage in research, but that it is inclusive and does not require either prerequisite understanding of bacteriophages or technical skills in microbiology (30, 32). Since its inception, students in the PHIRE program have isolated over 300 individual mycobacteriophages, about one-half of which have been sequenced. However, PHIRE is essentially a local program mostly confined to the Pittsburgh region where our research laboratory is located.
The demonstration that this discovery-rich platform is well suited to scientific novices provided the rationale for broader dissemination and expansion to a nationwide program organized as a course-based research experience for first-year undergraduate students. In 2008, in collaboration with the Science Education Alliance (SEA) program at the Howard Hughes Medical Institute (HHMI), we developed the Science Education Alliance Phage Hunters Advancing Genomics and Evolutionary Science (SEA-PHAGES) program (33). SEA-PHAGES is an example of a general model for transforming science education—an inclusive research education community—in which a program infrastructure supports inclusion of different types of institutions and students from all types of backgrounds (Fig. 1). In its first year (2008) there were 12 participating institutions, but additional schools have joined SEA-PHAGES each year, and currently there are over 120 member schools, with more than 4,500 student researchers each year (33, 34). The SEA-PHAGES experience strongly promotes student engagement in studying science, with project ownership and networking being prominent contributing elements (34). It has also had a major impact on our understanding of bacteriophages, and SEA-PHAGES students and faculty have isolated a total of 9,600 actinobacteriophages, of which 1,880 have been sequenced. Of these, 8,800 are mycobacteriophages, of which over 1,200 have been sequenced (16).
FIGURE 1.

Organization and structure of the SEA-PHAGES program. SEA-PHAGES program administrators (yellow box, top) oversee support components critical to program implementation (green box, upper middle). The typical two-term course structure (red box, lower middle) includes phage isolation through comparative genomics; additional characterization includes electron microscopy, PCR/restriction analysis, and lysogeny assays (red ovals). Sequence and annotation quality control is shared with SEA-PHAGES faculty teams (purple box, bottom right). Reproduced with permission from reference 34.
In general, we have adopted a nonsystematic scheme for naming these newly isolated phages. The reasons are three-fold. First, the genomes are clearly constructed as architectural mosaics, with individual genomes composed of specific assemblages of individual genes (35) (see below). It is thus unclear how to use a systematic naming system that reflects the genomic constitution of the phages. Second, naming a newly discovered phage is interesting and stimulating for PHIRE and SEA-PHAGES students and provides an opportunity for them to define their discoveries and their contributions, expanding the sense of project ownership that is important to the impact of these programs (34). Third, from a purely practical perspective, it is much easier to remember names such as Corndog, Rosebush, Patience, Daenerys, RedRock, Phayonce, or Snazzy than some tedious series of meaningless letters and numbers. Science doesn’t have to be dull and boring all the time.
Databases
The large sizes of both the SEA-PHAGES program and the growing phage collections require data management that is accomplished with three interconnected databases with front-end web interfaces. One is available at http://seaphages.org and contains information about the SEA-PHAGES program, including institutional participation, contact information, protocols, forums, news events, etc. The second is at http://phagesdb.org (16) and contains all of the information associated with individual phages, including genome sequences, GPS location coordinates, BLAST functions, and many other resources. It is a place where information on newly isolated phages is deposited and where data on all of the phages is available.
The third is the Phamerator database at http://phamerator.org. The database was designed as a means of organizing mycobacteriophage-encoded genes into “phamilies,” or sequence-related groups, together with the nucleotide sequence data (36). This information can then be used for comparative genomics, such as displaying sets of genome maps in which the genes are colored according to their phamily membership and pairwise nucleotide sequence similarity can be displayed. The Phamerator database includes some genome-associated information that is not present in the other databases, and a web browser interface provides facile database usage. It liaises with both phagesdb.org and GenBank to help maintain data consistency and is also used by other programs such as Starterator and the Phage Evidence Collection and Annotation Network (PECAAN). As such, Phamerator plays a central role in the annotation pipeline, from genome sequence to GenBank submission (Fig. 1). Phamerator databases containing different numbers of genomes can be downloaded and “frozen” so that they can be archived and used for publication preparation.
Mycobacteriophage Morphologies
All of the mycobacteriophages isolated to date are tailed phages with double-stranded DNA (dsDNA) genomes; no single-stranded DNA (ssDNA) or RNA phages have been found. Three families of such Caudovirales have been described for other bacterial hosts (Siphoviridae, Myoviridae, and Podoviridae) but only siphoviral and myoviral morphotypes are present among the mycobacteriophages (Fig. 2). Because such a large number have been morphologically examined, it seems unlikely that mycobacteriophages with podoviral morphologies exist. Interestingly, phages with podoviral morphologies have been described for other actinobacterial hosts, including Arthrobacter and Streptomyces (37, 38), suggesting that the absence of podoviral mycobacteriophages is due to a physical blockage presented by mycobacterial cell walls, rather than being evolutionarily inaccessible.
FIGURE 2.

Mycobacteriophage viral morphotypes. Mycobacteriophages generally fall into two morphological families, the Siphoviridae with long flexible tails (e.g., Phaedrus and Catdawg) and the Myoviridae with contractile tails (e.g., Rizal). Most of the siphoviral phages have isometric heads (e.g., Phaedrus), but some (e.g., Catdawg) have prolate heads. Scale marker is 100 nm.
The mycobacteriophage myoviruses, which have contractile tails, are all confined to a single genomic type, with phages grouped within cluster C (Fig. 2; see below). All of the other mycobacteriophages are siphoviruses, with long flexible noncontractile tails, and these span considerable genetic diversity (Fig. 2). Most mycobacteriophages have isometric capsids (i.e., like a soccer ball) with sizes varying from a diameter of about 40 nm to about 80 nm, correlating with their genome sizes. However, some have prolate heads with the length:width ratios varying from about 2.5:1 to about 4:1 (Fig. 2). Tail length also varies considerably, ranging from about 135 nm to more than 350 nm.
MYCOBACTERIOPHAGE GENOMIC DIVERSITY
Clusters, Subclusters, and Singletons
When the first few mycobacteriophage genome sequences were determined in the 1990s, it became apparent that they do not simply represent variants of a common sequence type (14, 35, 39–41). As the number of sequenced genomes grew, it became clear that these could be grouped according to their overall nucleotide sequence similarity. These groups are referred to as “clusters,” and in general, phages within a cluster share evident DNA sequence similarity to each other that spans more than 50% of their genome lengths; phages in different clusters share little if any DNA sequence similarity (42). When 30 genome sequences were available (42), these could be assembled into six clusters (clusters A to F); nine phages were not closely related to the others and are referred to as singletons. Within 4 years, the number of genomes had doubled, and these 60 phages were grouped into 9 clusters (clusters A to I), and five singletons (43). It also became clear that within some clusters there are boundaries justifying a separation into subclusters, and average nucleotide identity values reflect these divisions. Thus, for the 9 clusters formed with 60 sequenced mycobacteriophages, 5 could be divided into subclusters. For example, cluster A contained a total of 13 phages at that time, 8 of which are in subcluster A1, and 5 are in subcluster A2 (43). As noted at the time (43), the nature of the relationships within clusters is not uniform, such that the apparent divisions revolve around different ANI values for different clusters. A similar pattern has been reported for phages of the Enterobacteriaceae (44).
The 899 mycobacteriophages considered here assemble into a total of 28 clusters together with 5 singletons (Table 1). These clusters are designated clusters A to Z and clusters AB and AC (note that cluster AA is constituted with genomes that are yet to be submitted to GenBank and are thus excluded from the current list). Twelve of these clusters (A, B, C, D, F, G, H, I, K, L, M, and P) are currently divided into subclusters (Table 1). The largest of the clusters is cluster A with 322 phages, and this is split into 18 subclusters (A1 to A14 and A16 to A19; note that A15 is made up solely of Gordonia phages). The largest of the clusters that is not subdivided is cluster E, with 52 members (Table 1). These all share substantial nucleotide sequence similarity but are far from identical and differ with numerous small insertions/deletions/substitutions relative to each other, often constrained to a single gene or a small group of genes. Although it typically requires two or more members to constitute a cluster, three of the clusters in the dataset discussed here (X, Y, AC) contain only a single member, and the other members of these are sequenced but are not yet publicly available in GenBank (Table 1).
TABLE 1.
Mycobacteriophage clusters and subclusters
| Cluster | No. of Phages | No. of Subclustersa | Avg genome length | Avg G+C% |
|---|---|---|---|---|
| A | 318 | 18 | 51,656 | 63.3 |
| B | 147 | 6 | 68,705 | 67.2 |
| C | 49 | 2 | 155,858 | 64.7 |
| D | 10 | 2 | 64,965 | 59.4 |
| E | 52 | 75,446 | 63.0 | |
| F | 84 | 3 | 57,427 | 61.5 |
| G | 28 | 4 | 42,429 | 66.9 |
| H | 6 | 2 | 69,292 | 57.3 |
| I | 5 | 2 | 51,129 | 66.3 |
| J | 17 | 109,962 | 61.0 | |
| K | 71 | 6 | 60,000 | 66.8 |
| L | 27 | 3 | 74,978 | 59.0 |
| M | 7 | 2 | 81,539 | 61.2 |
| N | 14 | 43,200 | 66.2 | |
| O | 9 | 70,805 | 65.4 | |
| P | 17 | 2 | 47,901 | 67.0 |
| Q | 6 | 53,753 | 67.5 | |
| R | 5 | 71,424 | 56.0 | |
| S | 3 | 64,989 | 63.4 | |
| T | 5 | 42,847 | 66.2 | |
| U | 2 | 69,942 | 50.4 | |
| V | 2 | 78,262 | 56.8 | |
| W | 3 | 60,820 | 67.5 | |
| X | 1 | 90,460 | 56.8 | |
| Y | 1 | 77,832 | 67.3 | |
| Z | 2 | 50,806 | 66.0 | |
| AB | 2 | 47,976 | 58.7 | |
| AC | 1 | 68,869 | 49.1 | |
| Dorib | N/A | 64,613 | 66.0 | |
| Kumaob | N/A | 70,373 | 62.1 | |
| MooMoob | N/A | 55,178 | 62.0 | |
| DS6Ab | N/A | 60,588 | 68.4 | |
| Sparkyb | N/A | 63,334 | 65.2 |
The numbers of subclusters are shown but are not applicable (N/A) for singleton phages.
Singleton phages with no close relatives. Phage names are provided.
Cluster assignments simplify the analyses and descriptions of phage genomes, but clusters represent groupings of convenience rather than hard biological boundaries, consistent with them having mosaic architectures (45). As more phage genomes are sequenced, the boundaries between different clusters and subclusters have become more obscure. Thus, these divisions likely reflect unequal sampling of phages from an underlying continual spectrum of diversity, of which not all parts are equally prevalent in the biosphere (45). Thus, while the vast majority of mycobacteriophages within a cluster do indeed share nucleotide sequence similarity spanning greater than 50% of the genome lengths, some phages have genomes that fall only just below that, complicating their cluster assignment. This has become especially apparent in the comparative genomic analyses of Gordonia phages (46, 47). As a consequence, for those genomes that have partially shared sequences where nucleotide-based assignments are difficult, it is recommended that the encoded genes be first grouped into phamilies using Phamerator and the proportions of shared genes be calculated (http://phagesdb.org). Phages sharing more than 35% of their genes should be grouped into the same cluster, and those with fewer than 35% should be in different clusters (46).
Allocation of Cluster Designations
When only a relatively modest number of mycobacteriophages were sequenced and the first few clusters were designated, their naming was simple, using ordered alphabetical names (cluster A, cluster B, etc.). Three events prompted further consideration of this naming system. First, as the numbers of clusters grew, the possibility arose that new clusters would be designated even after cluster Z was named. How should these be designated? Second, as our attention shifted to actinobacterial hosts in other genera, how should these be termed? Third, phages isolated on different host genera might group into the same cluster either frequently or very rarely. How should this be accommodated?
We decided to implement a scheme in which blocks of letters are set aside for clusters of phages isolated on particular hosts (Table 2). In brief, the alphabetical series is extended beyond clusters A to Z into two-letter (e.g., AA, AB, AC, etc.) and then three-letter (AAA, AAB, AAC, etc.) names as needed, and a block is assigned to each host. For example, phages isolated on mycobacteria have a reserved block of clusters A to Z and AA to AJ. Currently, only clusters up to AC are actually used, so there are seven unoccupied clusters. The size of the initial block (40 for mycobacteriophages; Table 2) is mostly a guess based on an estimate of how many phages may be isolated for that host and their predicted diversity. For example, a relatively small block is set aside for Cutibacterium (including the formerly named Propionibacterium acnes) phages, because the phages of Cutibacterium acnes are very similar to each other and are all in a single cluster (48). If a block of cluster allocations becomes fully used, then additional blocks can be added, even though they are likely to be nonsequential. For example, phages of Arthrobacter were initially assigned clusters AK to AZ, but these were quickly used, and an additional block of FA to FZ was added (Table 2). Similarly, Gordonia phages were found to be quite diverse, and the initial allocation of clusters CQ to CZ was quickly populated. However, this could be easily extended to clusters DA to DZ, because these had not been previously assigned (Table 2), preserving a series of consecutive letters.
TABLE 2.
Allocation of cluster designations
| Host phylum | Host genus | Designations | Number |
|---|---|---|---|
| Actinobacteria | Mycobacterium | A–Z, AA–AJ | 40 |
| Arthrobacter | AK–AZ, FA–FZ | 42 | |
| Streptomyces | BA–BT | 20 | |
| Propionibacterium | BU–BZ | 6 | |
| Rhodococcus | CA–CP | 14 | |
| Gordonia | CQ–DZ | 36 | |
| Microbacterium | EA–EM | 13 | |
| Corynebacterium | EN–EZ | 13 |
This scheme works in large part because most of the phages isolated on a host of a different genus form new clusters from those previously described. However, we do not expect a strict correlation between the genus of the host and the phage cluster designation. An excellent example is provided by cluster A. Cluster A is highly populated (Table 1) with individual phages, by far the majority of which were isolated on M. smegmatis. However, some cluster A phages were isolated on Gordonia strains, all of which constitute subcluster A15; it is noteworthy, however, that in spite of the genomic relationships, subcluster A15 phages do not infect M. smegmatis, and other cluster A phages that have been tested do not infect Gordonia terrae. Likewise, we note that several Gordonia phages described previously also infect Nocardia strains (49). In general, we predict that the more closely related the bacterial hosts are phylogenetically, the more likely it is that their phages may be grouped into the same clusters.
We have constrained these cluster allocations to phages of hosts in the phylum Actinobacteria. However, this could plausibly be extended to phages of hosts in other phyla using a similar scheme and designations, using a prefix to denote the phylum in circumstances where confusion might arise.
Mycobacteriophage Gene Phamilies
The central questions in deciphering these mycobacteriophage genomes are how are they architecturally structured and how have they evolved? To simplify the comparative genomic analyses, we can take the total gene set, compare each gene to all other genes pairwise at the amino acid sequence level, and place similar genes into groups that we call phamilies (43), often abbreviated to phams. This is a different strategy to using a hidden Markov model, in part in an attempt to accommodate the complexity arising from the substantial number of modular genes. When the dataset was small, phamilies were constructed using BlastP and ClustalW, using cutoff values of 10−50 (E value) and 32.5% (amino acid identity), respectively (36). These values were set to capture distantly related homologues into the same phamily but to minimize false-positive inclusion of nonhomologues in phamilies (36). As the dataset expanded, the use of BlastP and ClustalW became computationally prohibitive, and they were substituted with a kmer-based method for phamily assembly (45). Different phams are identified by numbers (pham 1, pham 2, pham 3, etc.), and we attempt to maintain pham designations as additional phages are added to the databases. The program Phamerator uses k-clust to sort genes into phamilies and displays genome maps that include pham designations and other genome and gene information (36). The web-based version of Phamerator is available at http://phamerator.org, which uses a default database(Actino_Draft) that contains information on all sequenced phages of actinobacterial hosts. The Actino_Draft database is constantly updated as new phages are isolated and their genomes sequenced, and as such contains genomes of several different status classes including “draft” (autoannotated) annotations, “final” annotations that have been manually reviewed and revised, and “Gbk,” whose annotation history is unknown.
A Phamerator database (Mycobacteriophage_899) was constructed for the 899 phages discussed here, which contains only genomes that are available in GenBank. This database contains 96,979 genes, which are sorted into 6,046 phamilies. The average number of genes per phamily is 16.03, and approximately 25% (1,506) contain only a single gene and are designated as “orphams.” The largest phamily has 337 gene members.
MYCOBACTERIOPHAGE GENOME ORGANIZATION AND EXPRESSION
Genome Annotation and Gene Naming
When phage genomes are sequenced within the PHIRE or SEA-PHAGES program, they can be autoannotated simply with Glimmer (50) and GeneMark (51) using the program DNA Master (http://cobamide2.bio.pitt.edu/). Although these programs are quite good at identifying coding regions and predicting translational start sites, they do this incorrectly for about 10% of the genes. To obtain as well-supported an annotation as possible, each of the predicted genes is manually inspected, and putative functional assignments are made. The genomes are also inspected for missing genes and for tRNA genes and other features. All annotated genomes are reviewed through a quality control process prior to submission to GenBank (Fig. 1).
To simplify the gene naming process, each open reading frame or RNA gene is assigned a gene name as a number, sequentially starting with 1 at the left end of the genome and proceeding rightward. It is important to note that gene names (e.g., 1, 2, 3, etc.) and their gene products (e.g., gp1, gp2, gp3, etc.) have no functional meaning. Thus, for example, both phage Corndog and phage Rosebush have a gene 20, each coding for a protein, gp20, but Corndog gp20 and Rosebush gp20 are unrelated at the sequence level and are not functionally related.
Genome Organization
Although there is great sequence diversity among the mycobacteriophages, there are several common themes arising in their overall organization and genomic architecture. An example is shown in Fig. 3. First, there is a common organization of virion structural genes in all of the phages with siphoviral morphologies, which includes all of the clusters and singletons, with the exception of cluster C. The genomes are typically presented such that these structural genes are at the left end of the genome and transcribed rightward (Fig. 3). The gene order is typically terminase (small and large subunit genes), portal, capsid maturation protease, scaffold, capsid, tail capping and head-tail connectors, major tail subunit, tail assembly chaperones (typically expressed via a programmed translational frameshift), tape measure protein, and minor tail genes (of which there may be 4 to 10) (Fig. 3).
FIGURE 3.

Genome organization of mycobacteriophage Taheera. The genome of phage Taheera (cluster G) is represented as a scale bar (major intervals: 1 kbp) with predicted genes shown as boxes either above (rightward transcribed) or below (leftward transcribed). Gene numbers are shown within each box, and the phamily designation is shown either above or below with the number of phamily members shown in parentheses. Putative gene functions are indicated.
In some genomes, usually those that are relatively short (40 to 45 kbp), the 20 to 25 closely packed virion structure and assembly genes span about 22 to 23 kbp, occupying ∼50% of the whole genome (Fig. 3). In such phages, the terminase small subunit gene is usually very close to the physical left end of the genome (in phage Taheera, the translation start site of the small terminase subunit genes is predicted to be at coordinate 47). In other genomes, the gene order may be conserved, but the virion structure and assembly genes are interspersed with nonstructural genes. In phage Omega and its relatives (52), the virion structure and assembly genes span over 35 kbp, and the inserted genes include a glycosyl transferase, an O-methyl transferase, and an IS110-like transposon. In phage Marvin and its relatives two of the minor tail protein genes are atypically located near the right end of the genome, separated by about 20 kbp from the other structural genes (53).
All of the phages code for a lysis system that typically includes three components: a holin, an endolysin (lysin A), and a serine-esterase, referred to as lysin B or lysterase (see Fig. 3). The lysis cassette is located in one of two genomic positions. In most phages it is immediately downstream (to the right) of the virion structural genes (Fig. 3). However, in the cluster A phages it is positioned to the left of the structural genes, between the leftmost cos end and the terminase genes (14, 54). Within the lysis cassettes, the holin gene is sometimes difficult to assign, because of there being several genes with predicted transmembrane domains near the lysin A gene; it is plausible that several of them play some role in the regulation of lysis. The endolysin genes are constructed modularly, with individual modules shared between otherwise distantly related phages (55). The lysin B genes code for esterases that cleave the mycolic acids from the cell wall to promote efficient lysis, and at least in phage Giles the lysin B is not essential for plaque formation, although it forms smaller plaques with fewer particles (56). Some of the mycobacteriophages appear to lack a lysin B gene altogether (e.g., Rosebush), and it is unclear if other genes substitute for its function (56).
Over one-half of the clusters of mycobacteriophages appear to be temperate and contain an integration cassette (e.g., clusters A, E, F, G, I, J, K, L, M, N, O, P, Q, T, X, Y, Z; singletons DS6A, Dori, Kumao, MooMoo, and Sparky). Typically, the integration cassette is located near the center of the genome and contains an integrase gene and an attP site (see Fig. 3). Both major families of integrases—tyrosine integrases and serine-integrases—are represented, although the serine-integrases are typically found in cluster A and K genomes. Over a dozen attB sites are predicted to be used for prophage integration (22). Interestingly, some phages—predominantly members of cluster A—do not have an integration cassette and instead code for a parABS partitioning system (57, 58). In lysogeny, these phages replicate extrachromosomally at low copy number, although the replication functions have yet to be determined (58).
The mycobacteriophages have a variable number of genes that are not involved in virion structure and assembly, lysis, or integration, depending on the overall genome size. Thus, the smallest of the mycobacteriophages (e.g., clusters G, N, T) may have only 25 to 30 additional genes (Fig. 3), whereas larger genomes (e.g., cluster J) may have over 150 such genes. A few of these have predicted functions in DNA metabolism, including replication and recombination, but the vast majority are relatively small and are of unknown function. In phage Giles, it has been shown that about half of the nonstructural genes are dispensable for lytic growth (59). Little is known about the essentiality of any of the predicted DNA replication functions, including the Pol I type genes and Pol III alpha genes present in some of the phages. Many mycobacteriophages do not encode any DNA polymerase, so those that do may need them to synthesize genomes with altered chemistry or because the host polymerases are degraded or inactivated. However, this has yet to be investigated.
Mycobacteriophage Gene Expression
A general picture of mycobacteriophage gene expression is emerging, although only a rather small subset of phages has been examined. Lytic gene expression has been described for several cluster A phages including L5, D29, StarStuff, Kampy, and SWU1 (54, 60, 61); Giles (cluster Q) (59); Fruitloop (cluster F) (62); and several cluster N phages (63). In general, there appear to be two major types of transcription that occur, in early and late periods. Early gene expression occurs in the first 30 minutes, with late transcription initiating about 30 minutes after infection and continuing until lysis, typically about 180 minutes after infection. These general profiles are in agreement with patterns of phage protein synthesis detected by radiolabeling (14, 40).
Early gene transcription usually encompasses nonstructural genes in the right arm, and the virion structure and assembly genes and the lysis cassette are expressed late. In phages such as Fruitloop (Fig. 4), late transcription starts ∼5 kbp from the right genome end (in the 113-bp intergenic region between genes 91 and 92) and proceeds rightward through cos into the structural genes (62) (Fig. 4). Of the 11 right arm genes (92 to 102) at the beginning of the late operon, 4 of the gene products have predicted functions. One (gp92) is a predicted transcriptional regulator, two (gp99 and gp102) are putative glycosyltransferases, and gp100 is a putative kinase. It is plausible that several of these may be involved in postreplication DNA modification as protection against host defenses. Currently, very little is understood about the factors needed to activate late transcription in any of the mycobacteriophages. Presumably, they are encoded as early genes, but they have not been identified.
FIGURE 4.

Gene expression in mycobacteriophage Fruitloop. RNA was isolated from M. smegmatis infected with phage Fruitloop at either early (30 min) or late (150 min) time points after infection. Following strand-specific RNAseq analysis, sequence reads were mapped to the Fruitloop genome (shown below). Reads mapping to the forward and reverse strands are indicated on the right. Scales for the numbers of sequence reads are from 0 to 5,000. Reproduced with permission from reference 62.
Curiously, in the cluster A phages that have been transcriptionally characterized (L5, D29, StarStuff, RedRock, Kampe, SWU1), a noncoding region near the right genome end is transcribed extremely high very early in lytic growth (54, 58, 60, 61). Several small transcripts may be made, but their functions are not known. However, they appear to be quite toxic to M. smegmatis, and even E. coli, somewhat complicating their study (54). It was previously shown that host gene expression is shut down during L5 early lytic growth (14), and a role for these early RNAs in host shutdown would be consistent with their severe toxicity.
Lysogenic gene expression has been explored in a few mycobacteriophages, primarily those in clusters A, F, Q, and N (58, 59, 62, 63). Not unexpectedly, the repressor is typically expressed, and the lytic genes are switched off. However, it is not uncommon for additional genes—typically located near the repressor—to also be expressed (see below). The cluster A phages are unusual in that they contain a large number of putative repressor binding sites (54, 64–66), spanning the entire genome. These sites are 13 to 14 bp long, asymmetric, and predominantly present in short intergenic regions. Furthermore, the sites are mostly oriented in one direction relative to the direction of transcription (64). It is proposed that at least one of these sites acts as a true operator site for regulation of the early lytic promoter Pleft (67), whereas the others, termed “stoperators,” act to promote transcription termination, primarily to prevent expression of toxic genes during lysogeny (64).
Unusual Life Cycle Regulation
Some of the mycobacteriophages employ an interesting and noncanonical system for the establishment of lysogeny that differs from the well-studied paradigms such as in coliphage lambda. Examples of integration-dependent immunity are found in mycobacteriophages in clusters G, N, and P, and the best studied is that of phage BPs (cluster G) (68). However, these systems have specific bioinformatic features that are readily recognizable in a variety of other temperate phages (46).
Perhaps the most striking feature of the integration-dependent immunity systems is that the phage attachment site (attP) is located within the coding region of the immunity repressor (Fig. 5). At first, this is seemingly a bizarre organization, because integrase-mediated site-specific recombination between the phage attP and host attB sites results in disruption of the repressor gene, even though repressor synthesis is absolutely required for maintenance of lysogeny (68). A rationalization of this emerges from a second common feature, a C-terminal tag in the virally encoded repressor gene product that promotes degradation, typically via ssrA-like proteolysis. Because of this ssrA-like tag, the virally encoded form of the repressor is rapidly turned over, and insufficient repressor is made to maintain lysogeny or confer superinfection immunity (68). Integration results in removal of the C-terminal 30 to 35 amino acids, including the ssrA-like tag, such that the prophage-encoded form of the repressor is active for maintaining lysogeny and immunity to superinfection (Fig. 5). These phages thus require integration as a prerequisite for lysogeny, and the efficiency of lysogeny is determined by the level of integration, which in turn is modulated by a tag for proteolysis at the C-terminus of integrase (68).
FIGURE 5.

Integration-dependent immunity regulation. BPs is a cluster G phage, and a central ∼4 kbp segment of the genome is shown at the top. Genes are shown as colored boxes, transcribed rightward or leftward as depicted above or below the genome, respectively. The repressor and integrase genes (33 and 32, respectively) are transcribed leftward from the Prep promoter. The attP site (black bar) is located within the repressor open reading frame and can recombine with an attB site overlapping an M. smegmatis tRNAarg gene. Establishment of lysogeny involves formation of an integrated prophage in which an active form of the repressor is expressed from near attL, and the 3′ end of the gene is near attR. The virally encoded form of the repressor contains a C-terminal ssrA-like tag that targets it for degradation, and it fails to confer immunity. Integration results in removal of the C-terminal tag such that the prophage-expressed form of the repressor is stable and confers immunity.
These features impose constraints on other aspects of immunity regulation and are accompanied by some notable consequences. First, we should note that in all of the systems studied to date, the integrases are of the tyrosine-recombinase family and use an attB site that overlaps a host tRNA gene (Fig. 5). The common core, a region of sequence identity between attP and attB, is typically 25 to 45 bp and contains not only the region at which strand-exchange occurs, but also the sequences corresponding to the 3′ end of the tRNA gene; tRNA gene integrity is thus maintained at the attachment junction (attL or attR) following integration (Fig. 5). In phage BPs, the common core is 35 bp long and positioned 33 codons from the 3′ end of the viral repressor gene. Following integration, the first base of the bacterial sequence joined at attL changes a cysteine codon (5′-TGT) to a translation terminal codon (5′-TGA), so the prophage-encoded form of the repressor lacks the C-terminal 33 amino acid residues of the virally encoded form (68).
Canonical tyrosine integration systems such as in phage lambda and mycobacteriophage L5 (69, 70) use complex attP sites containing secondary integrase-binding sites flanking the common core (i.e., arm-type Int-binding sites) and thus span up to 250 bp. Not only is there no evidence of similar arm-type binding sites in BPs or other integration-dependent systems, but the integrase proteins lack an N-terminal arm-type binding domain (68). As such, these are simple site-specific recombination systems reminiscent of FLP or Cre. This in turn raises the question of how directionality (i.e., integration versus excision) is regulated, because without the ability to use arm-type binding to form higher-order protein-DNA architectures, there is no obvious role for a recombination directionality factor such as Xis (71). It is likely that directionality is regulated at least in part by the availability of active integrase, because the integrases in these systems also contain an ssrA-like C-terminal proteolysis tag. Removing the tag leads to higher rates of excisive recombination even in the absence of additional phage-encoded proteins (68).
The relationship between the attP site and the repressor gene also results in an unusual expression pattern (Fig. 6). This is illustrated by transcriptome sequencing (RNAseq) analysis of an M. smegmatis lysogen carrying a prophage, the cluster N phage Panchino, which integrates at the attB-4 site overlapping a tRNA-lys gene (22). The truncated prophage form of the repressor (gene 33) is expressed as expected, but the host tRNA-lys gene is also expressed, generating RNA on both strands within the repressor gene (Fig. 6). These rightward transcripts terminate before they proceed through to gene 34, which is a candidate for a cro gene, whose expression could result in interference with lysogeny and induction of lytic growth. It is somewhat surprising that the transcripts originating from the tRNA do not act as antisense inhibitors of repressor expression. This unusual expression pattern is observed in all the integration-dependent systems that have been examined by RNAseq (63).
FIGURE 6.

Gene expression in a Panchino lysogen of M. smegmatis. RNA was isolated from a lysogen of mycobacteriophage Panchino and strand-specific RNAseq reads were mapped to the Panchino prophage. Only the genome segments at the ends of the prophage near attL and attR are shown. Sequence reads for forward and reverse DNA direction are shown, and the scales are 0 to 500 reads. Genes below the genome are transcribed leftward, and those above are transcribed rightward. Panchino uses an integration-dependent immunity system similar to that in phage BPs (see Fig. 5). Genes 31 to 28 are also expressed; the lytic genes are not expressed (not shown). Reproduced with permission from reference 63.
Finally, these immunity systems can be used to construct integration vectors for inserting genes into mycobacterial chromosomes at attB loci, as has been described for other integration vectors (72–74). However, they transform at very low frequency because of the ssrA-like tags on the integrase. Removal of these tags gives higher transformation frequencies, but this may be accompanied by instability of the integrated vector if the integrase gene is still present in the recombinant strains (68).
INSIGHTS INTO PHAGE EVOLUTION
Mosaicism: The Hallmark of Phage Genomes
Comparative genomic analysis of phages clearly shows them to have modular structures, with segments assembled to form distinct genomes (35, 75). This is evident at the nucleotide sequence level when comparing phages grouped within particular clusters or subclusters, where otherwise closely related segments are interspersed with regions that are unrelated or more distantly related. An example illustrated by some genomes grouped in subcluster A2 is shown in Fig. 7. It is strikingly common for the boundaries between shared and nonshared sequences to coincide with gene boundaries. A vast number of discontinuities are identifiable, and rarely are there signs of sequence homology that could have promoted recombination through a homology-dependent mechanism, although such events have been described (54). This has led to the suggestion that the recombination events are likely to predominantly occur without extensive sequence dependence but with selection for function (76). It is plausible that very small regions of homology (3 to 5 bp) are favored, and these could be mediated by phage-encoded recombinases, which often have relaxed sequence requirements (77–79).
FIGURE 7.

Genomic mosaicism in subcluster A2 phages. Mycobacteriophages are characteristically mosaic, with shared genome segments interspersed with nonhomologous regions. The genomes of 12 subcluster A2 phages are shown (Bactobuster, Che12, D29, Echild, Jaan, L5, Pukovnik, RedRock, Serenity, StarStuff, Turbido, and Updawg), with pairwise nucleotide sequence similarities shown as shadings between the genomes; shading is spectrum colored, with violet being the most closely related and red being just above the threshold E value of 10−5. Genes are shown as colored boxes, and genes within the same phamily are similarly colored. Note the segments with close sequence similarity (violet-shaded regions) interspersed with dissimilar regions (white-shaded segments).
Genomic mosaicism is also evident when comparing genes at the amino acid sequence level, where distant homologues can be identified (Fig. 8). There are numerous examples in which gene members of a phamily are present in two otherwise unrelated genomes, such that the flanking genes to its left and its right are quite different (Fig. 8). Although sometimes two or more genes may be coevolving and are shared between genomes, it is very common to observe single genes as units within the mosaic architecture. This mosaicism is particularly pervasive among the large number of small nonstructural genes, most of which are of unknown function. However, it is also evident in some of the virion structure and assembly genes, especially those located between the capsid and major tail subunit genes (Fig. 8).
FIGURE 8.

Single-gene mosaics in mycobacteriophage genomes. Segments of five phage genomes (Phrann, Corndog, Brujita, Squirty, and Gaia) are shown, which are unrelated at the nucleotide sequence level and are grouped in different clusters, as shown (in clusters, N, O, I1, F3, and X, respectively). Genes are shown as colored boxes, with genes in the same phamilies colored similarly and pham numbers shown above with the number of phamily members in parentheses. Pairwise-shared homologues are indicated with dumbbells. Note that the homologues are situated in different genomic contexts and the flanking genes are not homologous.
Mobile Elements
Although it is anticipated that mobile genetic elements contribute to mosaicism, transposons, introns, and inteins are relatively rare in mycobacteriophage genomes. There is an IS110-like element in phage Omega and some other cluster J phages (52), IS605-like elements in phage Llij and several other subcluster F1 phages (although inserted at several distinct locations), an IS1096-like element in the cluster G phage LouisV14, and an IS1380-like element in Guillsminger (subcluster K5). There are instances of introns (e.g., in Omega and other cluster J phages) (52) and inteins, especially in terminase large subunit genes (65). However, there are more genes appearing to code for stand-alone homing endonucleases or HNH proteins (65). These are expected to be mobile, and it is not unusual to find HNH genes sharing high levels of amino acid sequence and sometimes nucleotide sequence similarity located in different genomic contexts, indicative of recent mobility.
The most common mobile elements are small mycobacteriophage mobile elements (MPMEs) that form two main classes, MPME1 and MPME2, which share about 85% nucleotide identity with each other (80). These were first identified by comparison of genomes in cluster G, where some pairs of genomes are very closely related and differ primarily by insertion of the MPME (80). These comparisons reveal the sequence of the preintegration site and thus the consequences of insertion. The MPMEs are small (∼440 bp) and have short (11 bp) imperfectly conserved inverted repeats (IRs) at each end (IR right [IR-R] and IR left [IR-L]), and IR-R is joined to the target DNA. However, the junction at IR-L contains six additional base pairs that are not evidently part of the transposon and are not derived from the target sequence. Neither the origin of these nucleotides nor the mechanism giving rise to their inclusion is known. The MPMEs contain a small opening reading frame coding for a putative 125-residue protein, although it has no motifs commonly found in other transposases or recombinases (80). Many instances of these MPMEs have been identified (over 120 MPME1 and over 35 MPME2) in a variety of genomes (including those in clusters F, G, I, O, T, and W); within a group they are either completely identical or differ by one nucleotide difference. Thus, even though there has been no experimental demonstration of mobility, there is strong evidence that they are indeed mobile, and their small size and unusual features suggest that they warrant further investigation.
Multiple Modes of Evolution
The pervasive mosaicism of the genomes likely arises because of a relatively high level of horizontal genetic exchange relative to the mutational clock. However, the rates have not been simple to quantify, in part because of the challenges in determining the number of horizontal genetic exchange events and estimating mutation rates that likely differ among different phages, especially those that may deploy their own DNA replication machinery. Pairwise comparisons of NCBI phage reference sequences (whose annotations should be reasonably reliable), determining both nucleotide distances and shared gene content, shed some light on these parameters. Phages of all hosts were included (not just mycobacteriophages) (81). Traditional comparison methods are too computationally intensive to measure nucleotide distances, so a kmer-based Mash method was used (82). Although there is not universal agreement that Mash distances reflect nucleotide divergence, a variety of validation strategies supported its application in this analysis (81). Because phage genome diversity is high, most genomes are not related, and pairwise comparisons have Mash scores above 0.5 (Fig. 9) and are insufficiently similar to be informative (81). The second parameter is an estimate of gene content similarity (Fig. 9), which was determined by grouping the genes into phams and then determining what proportion of phams are shared pairwise between genome pairs (81).
FIGURE 9.

Modes of phage evolution. (A) Nucleotide distance (using Mash) and gene content dissimilarity (using phams from Phamerator) are plotted for ∼2.4 × 106 dsDNA phage comparisons to reveal two evolutionary modes, HGCF and LGCF (inset). The line at y = 2x is plotted for reference. Marginal frequency histograms emphasize densely plotted regions, with truncated y axes for viewability. (B–D) Cluster-specific intracluster (orange) and intercluster (black) comparisons are plotted as in Fig. 1A for actinobacteriophage clusters B (panel B), F (panel C), and K (panel D). Phages in clusters F and K are temperate, and phages in cluster B are lytic, as indicated. n, number of phages present in the specific cluster. Reproduced with permission from reference 81.
This analysis provides an interesting insight, in that there appear to be two major modes of phage genome evolution, one in which gene content varies broadly in proportion to nucleotide distances (low gene content flux [LGCF]) and a second in which gene content appears to vary substantially more over shorter nucleotide distance changes (high gene content flux [HGCF]) (81) (Fig. 9A). In general, lytic phages follow the LGCF evolutionary mode, whereas temperate phages distribute between the LGCF and HGCF modes. Moreover, phage genomes fall into one of the two modes based on their genomic type, such that clusters of phages tend to follow one or the other but not both modes (81). This is well illustrated by three clusters of mycobacteriophages (Fig. 9B,C,D).Cluster B phages are lytic, do not form stable lysogens, and do not contain integrase or repressor genes. These generally distribute throughout the LGCF profile (Fig. 9B). In contrast, clusters F and K are both temperate (Fig. 9C,D), stable lysogens can be recovered, and repressors and integrases have been characterized (73, 83). Cluster F genomes clearly distribute in the HGCF mode (Fig. 9C), whereas cluster K phages lie in the LGCF mode (Fig. 9D). The drivers of these evolutionary modes are unclear, but it is plausible that the HGCF temperate phages strongly participate in the acquisition of genes that benefit their host when expressed in lysogeny and may contribute directly to pathogenesis (81).
Evolution and Host Range
The great genetic diversity of the mycobacteriophages, with many types sharing little or no DNA sequence information, is unexpected, given that they all infect the same host bacterial strain and therefore should be in genetic communication with each other. A simple model to explain this is that phage host preferences are quite mutable, and there is a low barrier to switching from one host to another (84). Moreover, there must be strong pressure to do so, because phages must mutate to survive viral attack, and phages must coevolve to access hosts for replication. We thus envisage pathways by which phages migrate across bacterial host landscapes, such that phages pursuing different paths have different access to the large common gene pool (Fig. 10).
FIGURE 10.

A model for mycobacteriophage diversity. The diversity of mycobacteriophages isolated on M. smegmatis mc2155 can be explained by a model in which (i) phages can readily infect a new bacterial host, by either a switch or an expansion of host range, and (ii) there is a highly diverse bacterial population, including many closely related strains, in the environments from which the phages are isolated. As such, phages with distinctly different genome sequences and GC contents infecting distantly related bacterial hosts, such as those to the left (red) or right (blue) extremes of a spectrum of hosts, can migrate across a microbial landscape through multiple steps. Each host switch occurs at a relatively high frequency (∼1 in 105 particles, or an average of about one every 103 bursts of lytic growth) and much faster than either amelioration of phage GC content to its new host or genetic recombination. Two phages (such as those shown in red and blue) can thus “arrive” at a common host (M. smegmatis mc2155) but be of distinctly different types (clusters, subclusters, and singletons). The variety of hosts is shown two-dimensionally for simplicity; the actual relationships among bacteria in environments such as soil and compost is likely to be considerably more complicated. Because host range variation is a common feature of bacteriophages, the model predicts that a high degree of phage diversity will be seen for any particular host if the microbial population from which the phages are isolated is also highly diverse and rich in closely related strains. Reproduced with permission from reference 84.
Several lines of evidence support this model. First, several examples demonstrate that mycobacteriophages can expand their host ranges at reasonably high frequencies (e.g., 10−5), including acquiring the ability to infect M. tuberculosis or a different strain of M. smegmatis (84). In each case, this requires just a single base pair change resulting in single amino acid substitutions in phage tail proteins. Second, characterization of the cluster U phage Patience identified features consistent with the hypothesis that it acquired the ability to infect mycobacteriophage hosts relatively recently (85). Patience has an unusually low G+C content (50.3%), substantially different than most other mycobacteriophages (Table 1) and its M. smegmatis host (67.3%). Perhaps not surprisingly, it also has a radically different codon usage profile than M. smegmatis, including preferred use of some codons that are only rarely used in the host. A seemingly obvious route to adapting to this situation would be for Patience to acquire tRNA genes that would recognize the rare codons, but this has not happened, and Patience has just a single tRNA gene (tRNA-gln). Instead, the genome appears to be in the process of adapting to its new host, such that the most highly expressed genes are more quickly adapting to match the coding profiles of host genes (85).
DRIVERS OF MYCOBACTERIAL AND MYCOBACTERIOPHAGE DYNAMICS
Prophage-Mediated Defense against Viral Attack
Because bacteria are under constant attack from viral infection, there is a strong selection for survival and acquisition of systems for viral defense. These include the well-studied restriction-modification and CRISPR-Cas systems, but there are numerous examples of defense by abortive infection, and a plethora of new defense systems have been identified (86, 87). Some mycobacterial strains—including M. tuberculosis—have CRISPR-Cas-like arrays, although their functionality in defense and spacer acquisition is unclear (88); mycobacterial restriction-modifications have also been described (89–92). However, M. smegmatis mc2155, which has been used extensively for phage isolation, appears to be free of both restriction and CRISPR-Cas systems (93).
Mycobacteriophages have been useful for demonstrating that phage defense systems can also be expressed from the prophages of temperate phages (63). This phenomenon has also been described for Pseudomonas phages and is likely to be widespread (94). Although it has been known for some time that phages and plasmids can carry restriction and abortive infection systems (95, 96), the number and variety of prophage-encoded systems is quite impressive. These have been best characterized in the mycobacteriophages within cluster N but are predicted to be present in many other temperate phages and other phages of actinobacterial hosts (63). It is simple to envisage how such defense systems would play key roles in microbial dynamics, because lysogeny is common, and prophage acquisition and recruitment of critical defense systems can occur at relatively high frequency in bacterial populations (97).
The presence of prophage-mediated defense systems is suggested from the expression patterns of lysogenic strains, as indicated by RNAseq analysis. The expression pattern of part of the Panchino genome is shown in Fig. 6, which clearly illustrates that the repressor is not the only phage gene being expressed during lysogeny. Up to four additional genes are expressed (28 to 31), and the expression of gene 28 is several times greater than the repressor (Fig. 6). Bioinformatic analysis strongly predicts the gp28 is a multisubunit restriction system, containing hsdR-, hsdM-, and hsdS-like domains (63). Genes 30 and 31 are expressed at more modest levels and are of unknown function, although gp31 is a predicted membrane protein. Although prophage-expressed genes could mediate a multitude of physiological responses in the general category of lysogenic conversion, the restriction system is strongly implicated in viral defense.
The availability of the large collection of diverse phages that infect M. smegmatis mc2155 facilitates the characterization of such defense systems. For example, determination of the plating efficiencies of about 100 phages shows that about one-third of them have a reduction of 10−5-fold or greater plating efficiency on the Panchino lysogen relative to the parent strain. When Panchino 28 alone is expressed from a plasmid, the plating efficiencies of the Panchino lysogen are largely reproduced (63). The cluster N phages are homoimmune and show strong superinfection immunity to each other (Fig. 11A), and we note that Panchino gp28 does not alter infection of Panchino itself or other cluster N phages; thus, this defense system is heterotypic with regard to its specificity for the attacking phages. Presumably, genes 29, 30, and 31 could be involved in viral defense too, but if a phage(s) targeted by these is not present in the set that was tested, it could be readily overlooked.
FIGURE 11.

Prophage-mediated defense against phage attack. (A) Phage defense plaque assays. Ten-fold serial dilutions of phages Giles (control), MichelleMyBell (MMB), Phrann, and Tweety were plated onto lawns of a nonlysogen (mc2155) and M. smegmatis lysogens of Phrann [mc2155(Phrann)] and MichelleMyBell [mc2155(MMB)]. The Phrann and MMB lysogens confer superinfection immunity to Phrann and MMB, because these are part of cluster N, and all of the cluster N phages are homoimmune, coding for closely related repressors. Tweety (cluster F) is unrelated to Phrann and MMB, and the Phrann and MMB lysogens defend against Tweety, and the efficiency of plating is greatly reduced relative to the wild-type strain. A defense escape mutant (DEM205) overcomes the Phrann defense and plates efficiently on the Phrann lysogen but remains subject to MMB-mediated defense. DEM205 contains a mutational change in gene 54, but it is a gain of function mutant, because a Tweety mutant in which gene 54 has been deleted (Δ54) is still targeted by the Phrann defense. (B) Relationships among phage-encoded (p)ppGpp synthetase-like proteins. Phrann gp29 is a homologue of RelA/SpoT proteins with similarity to the (p)ppGpp synthetase domain of Streptococcus equisimilis RelA, including five conserved motifs (1 to 5). The cluster F phage Squirty encodes a related protein (gp29) sharing the N-terminal 124 residues with Phrann gp29 but with divergent C-termini. MMB gp29 is not predicted to be related to RelSeq but shares its C-terminus with Squirty gp29. MMB gp30 and Squirty gp30 are closely related and are both predicted to be membrane localized. (C) Models for prophage-mediated viral defense. An integrated prophage (red line) confers defense against viral attack through numerous mechanisms, either homotypically (i.e., against the same or closely related viruses) or heterotypically (i.e., against unrelated phages). Homotypic defense includes repressor-mediated immunity (repressor, red circle) and superinfection exclusion (blue circle) against itself (red phage). Heterotypic defense includes an exclusion-like system illustrated by Charlie gp32 defense against Che9c (blue phage) and restriction against many viruses (illustrated by the green phage) by Panchino gp28. Defense is also mediated by a predicted (p)ppGpp synthetase (e.g., Phrann gp29; gold circle), which we propose is kept in an inactive form (gold circle with crossed lines) by an inhibitor (purple circle), which for Squirty gp30 is membrane localized. Lytic growth by specific phages activates the defense through early lytic protein, which is proposed to dissociate the (p)ppGpp synthetase from its inhibitor, enabling rapid accumulation of (p)ppGpp and growth arrest. Tweety encodes a counterdefense system (gp54) that may prevent activation of (p)ppGpp synthesis. Reproduced with permission from reference 63.
The Panchino 28 to 31 genes are centrally located in the genome between the immunity cassette and the lysis genes (63). Even though the genomes in cluster N are closely related to each other, this central region is highly variable, and many of these genes are expressed from the prophages (63). Approximately 3 to 8 genes are expressed from each of 11 cluster N genomes, corresponding to over 25 phamilies, and the determination of plating efficiencies shows that all of the 11 prophages defend against at least one other phage (63). In the case of phage Charlie, the specificity is quite impressive, and the Charlie prophage defends against only one of the tested phages, Che9c. A Charlie derivative in which gene 32 has been deleted using Bacteriophage Recombineering of Electroporated DNA (BRED) engineering (98) loses its defense against Che9c, and a recombinant plasmid expressing gp32 recapitulates the defense (63). Charlie gp32 is a small (121-residue) protein that is predicted to be membrane localized and could plausibly act by exclusion, preventing Che9c DNA injection. Consistent with this is the observation that Charlie gp32 not only inhibits Che9c plaque formation, but also interferes with Che9c lysogenization. It is plausible that Charlie gp32 would defend against other phages that use similar host proteins for DNA entry, but these are rare in our collection.
Several of the cluster N prophages defend against phage Tweety (cluster F), including Phrann, and three closely related phages: Xerxes, Carcharodon, and MichelleMyBell (Fig. 11) (63). For this second group, a responsible gene was suggested from comparative genomic analysis, because Pipsqueaks is a closely related phage, but has a small deletion in the region corresponding to MichelleMyBell gene 29 (and its relatives), and Pipsqueaks does not defend against Tweety. Deletion of genes 29 and 30 from MichelleMyBell abrogates Tweety defense, and a plasmid carrying genes 29 and 30 reproduces the defense. When using extrachromosomal vectors, it was not possible to clone MichelleMyBell gene 29 without gene 30, suggesting that high levels of gp29 are toxic and that gp30 alleviates the toxicity, akin to the behaviors of toxin-antitoxin systems. MichelleMyBell gp30 is predicted to be membrane localized, but the biochemical function of gp30 is unclear.
Genetic analysis of phage Phrann showed that Phrann 29 and 30 are required for defense against Tweety (note that the similarity of gene names is coincidental and does not indicate homology) (Fig. 11). These share some of the behaviors of the MichelleMyBell defense genes in that Phrann gp29 is toxic in the absence of Phrann gp30, but they are not related at the sequence level. Phrann gp29 is strongly predicted to be a (p)ppGpp synthetase with similarity to RelA family proteins (63). This poses a conundrum in that (p)ppGpp is used by bacteria as an alarmone to promote arrest of cell growth, and yet it is expressed well in a Phrann lysogen (63). However, the lysogen grows normally, and it is probable that gp30 interacts with gp29 and maintains it in an inactive state, consistent with its anti-toxic behavior. This model is attractive because the Phrann gp29/30 proteins would then be poised to be activated upon infection by an attacking phage (Fig. 11). Because the defense is specific to only a subset of the phages tested (including TM4 and Gaia, in addition to Tweety), these phages presumably code for a function that dissociates Phrann gp29 and gp30, resulting in activation of gp29, synthesis of (p)ppGpp, growth arrest, and inhibition of phage replication (63). Interestingly, Phrann gp29/30 defends against Tweety lytic growth but does not interfere with Tweety lysogenization, consistent with a post-DNA injection sensing mechanism. It is unclear what genes or sequences identify Tweety, TM4, and Gaia for targeting by this defense system, but early lytic genes are implicated (see below) (63).
MichelleMyBell gp29/30 likely acts similarly to Phrann, in spite of the lack of sequence similarity between the products (Fig. 11B). In an unusual set of relationships, the C-terminal half of MichelleMyBell gp29 is closely related (92% amino acid identity) to gp29 encoded by phage Squirty (cluster F). However, the N-terminal half of Squirty gp29 is 100% identical to Phrann gp29 (Fig. 11B). Thus, although Phrann gp29 and MichelleMyBell gp29 are not related to each other, it is plausible that MichelleMyBell is also involved in alarmone synthesis (63).
Mycobacteriophage Counterdefense
Phages often carry counterdefense systems such as antirestriction and anti-CRISPR genes (99, 100), so it should not be surprising that mycobacteriophages have systems to counter prophage-mediated defenses. A particularly intriguing system is encoded by Tweety, which was revealed by isolating defense escape mutants (DEMs) that overcome either Phrann-mediated or MichelleMyBell-mediated defense (63). Many of these DEMs have genomic changes in gene 54, a peculiar gene coding for a protein with multiple copies of a tetrapeptide repeat (Fig. 12). The DEMs typically have changes in the numbers of repeats, usually fewer but sometimes more (63). DEMs isolated for their escape of Phrann defense remain subject to MichelleMyBell defense, and vice versa (C. Gauthier and G.F. Hatfull, unpublished observations), suggesting that gp54 is being “tuned” to specifically counter the different defenses. The hypothesis that these mutants are gain-of-function derivatives rather than loss-of-activation mutants is illustrated by the observation that gene 54 can be deleted and Tweety is still subject to both Phrann and MichelleMyBell defenses (63).
FIGURE 12.

Variations in tetrapeptide repeats in Tweety gp54. Gene 54 of phage Tweety codes for a 374-residue protein in which a unique N-terminal (U-L) and C-terminal (U-R) flank a long series of tetrapeptide repeats. The number of repeats in the original sequenced genome is 48, although the derivative used for defense studies (wt-N) shown here only has 40. Each of the repeats has a sequence AAXX, where XX is either WS, GY, QS, GS, or WY, and the sequence at each repeat is shown. The organization of nine defense escape mutants (DEMs) are shown below, each of which efficiently overcomes defense mediated by the Phrann prophage. They all remain subject to defense mediated by the MichelleMyBell prophage. Six of the DEM mutants (DEM10, DEM200, DEM201, DEM202, DEM204, DEM205) have lost some of the repeats but also have altered repeat sequences, as indicated. DEM202 also has an insertion of five additional repeat copies. DEM203 has two insertions, one with two repeat units and the other with four repeat units.
Tweety gp54 plays no known role other than in its counterdefense capacity, but homologues are present in about 40 other cluster F phages, all with different numbers of tetrapeptide repeats. The repeats are composed of the sequence alanine-alanine-XX, where XX is either WS, GY, QS, GS, or WY (Fig. 12). The three-dimensional structure of this type of repeat is not known, but because the DEMs have variations in the numbers of repeats, it is plausible that it forms an extended structure, positioning the unique N- and C-terminal domains in configurations that can bind to gp29 and gp30 and prevent their dissociation. The mechanism by which expansion and contraction of the repeat number happens is also not known, but it is noteworthy that in addition to changes in repeat number, many of the mutants also have changes in the sequence of the repeat itself (Fig. 12). However, the changes in the repeat are always in the XX motif and always substituted by one of the tetrapeptide repeats found elsewhere. For example, DEM201, a mutant that overcomes Phrann defense, has lost 11 copies of the repeat, but 6 of the individual repeats are altered (Fig. 12). Not only are the 6 repeats not contiguous, but only 1 pair of the substituted sequences is present elsewhere, suggesting that 5 distinct mutational events created that derivative, in addition to the deletion of 11 copies. Because the repeated nature is also reflected as a 12-bp nucleotide sequence repeat, these variations could arise from replication errors and slippage, although it would appear to occur at a relatively high frequency. Whether the repeat substitutions play any functional role in counterdefense is unclear.
Finally, we note that although Tweety 54 homologues are predominantly found in cluster F phages, there is a related gene in the subcluster A1 phage, Nerujay (Nerujay gene 74) (Fig. 13). The unique N- and C-terminal regions are closely related to Tweety gp54 (90% and 80% amino acid identity, respectively), but Nerujay contains only 10 copies of the repeat, and only 2 tetrapeptide sequences are present, AAWS and AAGS. Nerujay is the only cluster A phage to carry such a gene, and it is notably absent from phage DD5, which is closely related in this region at the DNA sequence level (Fig. 13). A simple evolutionary explanation is that Nerujay acquired gene 74 relatively recently as insertion of a 796-bp fragment, perhaps derived from a cluster F phage similar to Ibhubesi, whose homologue (54) is identical in length to Nerujay 74 and shares 90% nucleotide sequence identity. Nerujay gp74 may also be active in counterdefense, but identifying which defense system it counters is a substantial challenge.
FIGURE 13.

Genomes with potential counterdefense systems. Tweety gene 54 is active in counterdefense against Phrann (see Figs. 11, 12), and there are homologues in many other cluster F phages. The only phage outside of cluster F with a closely related homologue is the subcluster A1 phage, Nerujay. All other cluster A phages, including DD5, lack a Tweety 54 homologue. Segments of the genome maps are aligned with pairwise nucleotide sequence similarity shown as colored shading, as described for Fig. 7. In this region, all of the DD5 and Nerujay genes are transcribed leftward, and the Tweety genes are transcribed rightward.
Exclusion during Lytic Growth
Many viruses have been reported to exclude superinfection of other viruses during either lytic or lysogenic growth. Some well-studied examples include the T4 imm gene and P22 sieA, both of which are small membrane proteins that confer homotypic exclusion (101, 102). Coliphage HK97 also codes for a small membrane protein that is prophage expressed and prevents HK97 superinfection (103). It is not unusual for some of the small genes of unknown function in mycobacteriophages to be predicted membrane proteins that could perform such functions (e.g., there are eight predicted membrane proteins in the right arm of phage Nerujay), but a specific role for these in exclusion has not been shown. However, a WhiB-like regulator encoded in phage TM4 is implicated in homotypic exclusion, plausibly through regulation of host genes (104). In a recent example, superinfection exclusion was shown to be mediated by interference of a host protein used for efficient infection by some other phages (Fig. 14) (62).
FIGURE 14.
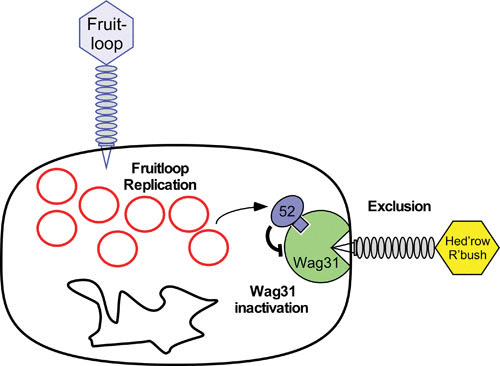
A model for Fruitloop gp52-mediated superinfection exclusion of Wag31-dependent phages. This model proposes a role for gp52 expressed by lytically growing phage Fruitloop in binding to and inactivating the host Wag31 (DivIVA) protein, which is essential and localizes to the growing pole of the cell. Some phages, such as subcluster B2 Hedgerow and Rosebush depend on Wag31 for efficient infection, such that gp52-mediated inactivation of Wag31 excludes them from superinfecting. Replicating Fruitloop genomes are shown as red circles alongside the bacterial chromosome (black circle), and Fruitloop gp52 is shown in blue, binding to Wag31, shown in green. Not drawn to scale. Reproduced with permission from reference 62.
This superinfection exclusion system was discovered through use of a screen for mycobacteriophage genes that are toxic to M. smegmatis when overexpressed (62). Approximately 5 to 10% of genes are expected to have this toxic property (105–108), and we identified gene 52 of phage Fruitloop (cluster F) as being highly toxic when overexpressed. Fruitloop gp52 interacts directly with the essential host protein Wag31 (DivIVA) and inactivates it, resulting in cell death (Fig. 14). Fruitloop 52 is expressed early in lytic growth and is not essential for plaque formation, and its toxicity is unlikely to be relevant for phage growth. Rather, it was reasoned that some mycobacteriophages depend on Wag31 for efficient infection, because it is polar localized and present both cytoplasmically and extracytoplasmically (109, 110). A search for phages inhibited by Fruitloop gp52 expression identified two subcluster B2 phages, Hedgerow and Rosebush, that are affected and presumably use Wag31 for efficient infection (62). Hedgerow and Rosebush are not related to Fruitloop at the nucleotide sequence level, and it is not evident that they have followed similar evolutionary paths as Fruitloop. It is plausible that mycobacteriophages code for many similar systems, targeting different aspects of mycobacteria that other phages exploit to gain entry.
Compatibility and Incompatibility
Most of the temperate mycobacteriophages can be grouped into homoimmune classes and have closely related repressors. The large majority of these carry integration systems, targeting a variety of attB sites. In general, if two phages are heteroimmune and use different attB sites for integration, they are fully compatible and there is no obvious impediment to the formation of double lysogens. This is not unexpected, and the sequences of large numbers of bacterial genomes show that they commonly carry two or more prophages. If phages are heteroimmune but use the same attB site, then they may compete with each other for stable lysogeny.
A subset of temperate mycobacteriophages do not have integration systems but instead encode parABS systems similar to those used by plasmids to promote segregation into daughter cells at division (57, 58). These phages, all of which group in cluster A but span 10 subclusters, replicate extrachromosomally at low copy number and, as such, are subject to incompatibility mediated potentially by both the prophage replication system and the partitioning systems themselves. None of the replication systems have yet to be described, but there is good evidence of incompatibility among phages with similar parABS systems (58). For example, a double lysogen of phages RedRock and Alma, both of which have parABS systems, can be constructed but not stably maintained without loss of one of the prophages (58). In contrast, a double lysogen of RedRock and Bxb1, which has a canonical integration system, stably maintains both prophages.
Repressor Theft
The immunity repressors have been well studied in the cluster A phages (107), although there is substantial sequence variation reflecting a variety of heteroimmune groups. Unexpectedly, a variety of other phages in other clusters carry closely related copies of some of these repressors (65). These related gene copies are found in lytic phages that do not form lysogens, including some cluster C phages, and also in temperate phages (e.g., cluster F and cluster J), where they are present in addition to their canonical repressors. For example, Fruitloop (cluster F1) has a canonical repressor (gene 44), which confers immunity to itself and some other cluster F1 phages, and the stolen repressor (37), which shares 97% amino acid identity with the Bxb1 repressor. Fruitloop gene 37 is expressed in a lysogen, although at a somewhat lower level than the canonical repressor (62), but confers immunity to at least some cluster A1 phages such as SkiPole and Pinto (Deborah Jacobs-Sera and GFH, unpublished observations). Because the cluster C phages do not form lysogens, the repressor may block superinfection of cluster A1-like phages during lytic growth.
EXPLOITATION OF MYCOBACTERIOPHAGES
Using Recombinant Phages for Efficient Delivery
Bacteriophages have the inherent ability to efficiently inject their DNA into host bacterial cells. Mycobacteria do not undergo natural transformation at high frequency, and electroporation is effective, but only a subset of cells take up DNA. In contrast, infection with mycobacteriophages at a multiplicity of infection greater than three generally results in DNA injection into every cell in the culture. This property was exploited early on in the development of mycobacterial genetic systems (12) and has been further developed for the delivery of transposons, reporter genes, and allelic exchange substrates (111–115). Phages are useful tools for the delivery of reporter genes such as luciferase or florescent proteins in rapid diagnostic assays for tuberculosis drug susceptibility in the clinic (116–119) but are also useful for understanding mycobacterial physiology. For example, dual reporter phages have been exploited to identify and analyze persister cells in M. tuberculosis (120).
Mycobacteriophage Tools for Mycobacterial Genetics
Mycobacteriophages also offer a rich reservoir of genes and cassettes for use in mycobacterial genetics. One application that has found widespread use is the development of integration-proficient vectors that transform mycobacteria by stable chromosomal integration using phage integrases. Integration vectors have been constructed from a variety of phages, including Adephagia, Brujita, L5, Bxb1, Tweety, Giles, Omega, and Ms6 (52, 68, 72–74, 83, 121, 122). These typically transform efficiently, and the transformants are quite stable, except if newly introduced genes are deleterious to growth and the integrase gene is retained in the recombinants. Most of these vectors work in both fast- and slow-growing strains, because the attB sites—especially those used by the tyrosine integrases—tend to be well conserved.
A second tool with broad applicability is a mycobacteria-specific recombineering system utilizing mycobacteriophage-encoded recombination systems (123). The most widely used system is that derived from mycobacteriophage Che9c, which has been used for both dsDNA and ssDNA recombineering (78, 79, 124). For dsDNA recombineering, both the exonuclease and DNA pairing genes (Che9c 60 and 61, respectively) are required, but for ssDNA recombineering, only 61 is needed. The recombineering systems work in both M. smegmatis and M. tuberculosis, and recombination is sufficiently efficient that it can be used to introduce point mutations into the genome of M. tuberculosis (125). Many newly identified recombination systems are present in the multitude of mycobacteriophages sequenced since the Che9c system was developed, and some of these may be worth investigating for improved ease of use or efficiency.
The recombineering system has been especially useful for engineering the phages themselves (98, 123, 126). When phage DNA and a mutagenic DNA substrate are coelectroporated into recombineering-proficient cells, recombination is efficient, and following recovery, individual plaques can be screened by PCR for the presence of the mutant allele. Typically ∼10% of progeny are mixed and contain both wild-type and mutant alleles, and a homogenous mutant derivative can be identified by PCR of plaques after replating. Point mutations can be introduced relatively easily, provided a PCR scheme is used that can distinguish between the mutant and wild-type alleles (68). The main limitation of the BRED approach is the question of whether plaques can be recovered following transfection in M. smegmatis (98). Many phages do this quite efficiently, and sufficient plaques can be readily recovered for screening. However, some phages transfect at very low efficiency or not at all, and these tend to be phages with larger genomes. Whether genome size per se is the principle factor or whether some of these phages use encapsidated phage proteins for infection is unclear.
Mycobacteriophages have also provided non-antibiotic-based selectable markers for mycobacteria, taking advantage of immunity repressor genes that confer immunity to superinfection, and lytic derivatives of the temperate phages that efficiently kill mycobacteria. The most well-developed system is based on the cluster K phage Adephagia, which infects both M. smegmatis and M. tuberculosis (127). Plasmids carrying the Adephagia repressor (gene 43) can be introduced by electroporation and selected in the presence of an Adephagia derivative from which gene 43 has been deleted. The transformants carry an integrated copy of the Adephagia genome, with the additional advantage that the plasmid cannot be lost without lytic induction and death, thus ensuring plasmid stability. If the presence of the prophage is undesirable, a second phage derivative can be used in which both the repressor (43) and the integrase gene (41) have been deleted (127).
Mycobacterial plasmids based of the oriM origin of replication derived from plasmid pAL5000 (128–131) are somewhat unstable in the absence of selection (72), especially if recombinant genes they are carrying are not well tolerated. Addition of the phage-encoded parABS systems to these plasmids adds considerable stability to their maintenance (58).
Therapeutic Potential
The possibility of using mycobacteriophages for the treatment of mycobacterial infections has been postulated for some time, although little progress has been made (10). Although tuberculosis remains a major public health concern in many parts of the world, effective treatment could be achieved with current antibiotics, a robust health care infrastructure, and monitoring. However, the emergence of multiply and extensively drug-resistant tuberculosis infections demands new approaches, and phage applications may warrant attention. It would seem unlikely that mycobacteriophages could achieve sterilization from an M. tuberculosis infection and a long-term cure. But some gains might be achieved from combinations with antibiotics that could extend their effectiveness, lower required dosages, reduce the prevalence of resistance, provide relief in late stages of infection, or help prevent transmission. An alternative approach would be to use phages prophylactically, to protect coworkers or family members from infection when in close contact with an infected patient (10).
Therapeutic utility is likely to require a cocktail of phages to minimize the incidence of phage resistance, just as antibiotics must be used in combination for tuberculosis treatment. However, the key property of phages in such a cocktail is that resistance to one phage does not confer resistance to the other phages. Unfortunately, very little is known about either the specific receptors used for mycobacteriophage infection or the mechanisms of resistance (132), and this is an area that needs further investigation. Furthermore, relatively few of the phages isolated on M. smegmatis infect M. tuberculosis, and they generally group in a small number of clusters or subclusters (84). As such, they may use a rather limited set of receptors, and cross-resistance may limit the numbers that could be combined together into a suitable cocktail. Moreover, early phage typing studies suggested differences in phage sensitivities among clinical isolates of M. tuberculosis, and this needs to be revisited now that we have a deeper understanding of the genetic diversity of M. tuberculosis strains (133).
Infections of nontuberculosis mycobacteria may also represent possible targets for phage therapy. Attractive targets are Mycobacterium abscessus infections associated with cystic fibrosis, which are often highly refractory to antibiotic therapies. Relatively little is known about the phage susceptibility profiles of M. abscessus clinical isolates, but a substantial proportion of sequenced M. abscessus strains carry one or more prophages (134, 135), which could code for viral defense systems. Suitable mycobacteriophage cocktails could conceivably be used to reduce the bacterial load in cystic fibrosis patients or to prevent infection following lung transplantation.
PERSPECTIVES
Mycobacteriophages have provided a wealth of insights into viral diversity and evolution and played indispensable roles in the development of mycobacterial genetics. We anticipate that the advances in mycobacteriophage genomics will slow somewhat, in large part because we are nearing saturation in the isolation of phages that infect M. smegmatis. However, as the emphasis shifts to phages that infect other mycobacterial strains and other Actinobacteria, new and different types of genomes are likely to be discovered. We anticipate that the coming years will see some progress toward defining the functions of more mycobacteriophage genes. The number of uncharacterized genes is vast, but the methods are available for their genetic, biochemical, and structural characterization, and the limiting factor may be the development of suitable assays. Surrogate approaches such as screening for toxicity (62, 106) and finding interacting partners (136, 137) should be useful strategies. Because many of the phage genes may play roles in microbial dynamics, the availability of such a large collection offers a very substantial resource.
Finally, the mycobacteriophages occupy a unique niche in how they have been isolated and characterized, mostly by early-career undergraduate students and faculty at institutions spread across the United States and the world (34). These students have not only made fantastic contributions to this field, but the fun, curious, and sometimes entertaining names of the phages they discovered will continue to amuse. The number of phages that infect actinobacterial hosts likely far exceeds the numbers of students who will isolate and characterize them over the coming years, and I can’t wait to see the novelties, surprises, and potential utilities that emerge.
ACKNOWLEDGMENTS
I thank Bekah Dedrick, Krista Freeman, Becky Garlena, Christian Gauthier, Carlos Guerrero, Debbie Jacobs-Sera, Ching-Chung Ko, Travis Mavrich, Welkin Pope, Dan Russell, and Katie Wetzel for comments on the manuscript and for other help and insights. The Mycobacteriophage_899 database is available at http://phamerator.webfactional.com/databases_Hatfull. I thank the National Institutes of Health and the Howard Hughes Medical Institute for support and all the student phage hunters and SEA-PHAGES faculty and staff for their amazing contributions to mycobacteriophageology.
REFERENCES
- 1.Gardner GM, Weiser RS. 1947. A bacteriophage for Mycobacterium smegmatis. Proc Soc Exp Biol Med 66:205–206 10.3181/00379727-66-16037. [PubMed] [DOI] [PubMed] [Google Scholar]
- 2.Whittaker E. 1950. Two bacteriophages for Mycobacterium smegmatis. Can J Public Health 41:431–436. [PubMed] [PubMed] [Google Scholar]
- 3.Froman S, Will DW, Bogen E. 1954. Bacteriophage active against virulent Mycobacterium tuberculosis. I. Isolation and activity. Am J Public Health Nations Health 44:1326–1333 10.2105/AJPH.44.10.1326. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grange JM. 1975. Proceedings: bacteriophage typing of strains of Mycobacterium tuberculosis isolated in south-east England. J Med Microbiol 8:ix. [PubMed] [Google Scholar]
- 5.Jones WD Jr. 1975. Phage typing report of 125 strains of “Mycobacterium tuberculosis”. Ann Sclavo 17:599–604. [PubMed] [PubMed] [Google Scholar]
- 6.Snider DE Jr, Jones WD, Good RC. 1984. The usefulness of phage typing Mycobacterium tuberculosis isolates. Am Rev Respir Dis 130:1095–1099. [PubMed] [DOI] [PubMed] [Google Scholar]
- 7.Goode D. 1983. Bacteriophage typing of strains of Mycobacterium tuberculosis from Nepal. Tubercle 64:15–21 10.1016/0041-3879(83)90045-4. [PubMed] [DOI] [PubMed] [Google Scholar]
- 8.Hatfull GF. 2006. Mycobacteriophages, p 602–620. In Calendar R (ed), The Bacteriophages. Oxford University Press, New York, NY. [Google Scholar]
- 9.Hatfull GF. 2010. Mycobacteriophages: genes and genomes. Annu Rev Microbiol 64:331–356 10.1146/annurev.micro.112408.134233. [PubMed] [DOI] [PubMed] [Google Scholar]
- 10.Hatfull GF. 2014. Mycobacteriophages: windows into tuberculosis. PLoS Pathog 10:e1003953 10.1371/journal.ppat.1003953. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jacobs WR Jr. 2014. Gene transfer in Mycobacterium tuberculosis: shuttle phasmids to enlightenment. Microbiol Spectr 2:2 10.1128/microbiolspec.MGM2-0037-2013. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jacobs WR Jr, Tuckman M, Bloom BR. 1987. Introduction of foreign DNA into mycobacteria using a shuttle phasmid. Nature 327:532–535 10.1038/327532a0. [PubMed] [PubMed] [DOI] [PubMed] [Google Scholar]
- 13.Snapper SB, Lugosi L, Jekkel A, Melton RE, Kieser T, Bloom BR, Jacobs WR Jr. 1988. Lysogeny and transformation in mycobacteria: stable expression of foreign genes. Proc Natl Acad Sci U S A 85:6987–6991 10.1073/pnas.85.18.6987. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hatfull GF, Sarkis GJ. 1993. DNA sequence, structure and gene expression of mycobacteriophage L5: a phage system for mycobacterial genetics. Mol Microbiol 7:395–405 10.1111/j.1365-2958.1993.tb01131.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 15.Hatfull GF. 1994. Mycobacteriophage L5: A toolbox for tuberculosis. ASM News 60:255–260. [Google Scholar]
- 16.Russell DA, Hatfull GF. 2017. PhagesDB: the actinobacteriophage database. Bioinformatics 33:784–786 10.1093/bioinformatics/btw711. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mizuguchi Y. 1984. Mycobacteriophages, p 641–662. In Kubica GP, Wayne LG (ed), The Mycobacteria: A Sourcebook, Part A. Marcel Dekker, Inc., New York, NY. [Google Scholar]
- 18.Hatfull GF. 2008. Bacteriophage genomics. Curr Opin Microbiol 11:447–453 10.1016/j.mib.2008.09.004. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hatfull GF. 1999. Mycobacteriophages, p 38–58. In Ratledge C, Dale J (ed), Mycobacteria: Molecular Biology and Virulence. Chapman and Hall, London, UK. 10.1002/9781444311433.ch3 [DOI] [Google Scholar]
- 20.Hatfull GF. 2000. Molecular genetics of mycobacteriophages, p 37–54. In Hatfull GF, Jacobs WR Jr (ed), Molecular Genetics of the Mycobacteria. ASM Press, Washington, DC. [Google Scholar]
- 21.Hatfull GF. 2004. Mycobacteriophages and tuberculosis, p 203–218. In Eisenach K, Cole ST, Jacobs WR Jr, McMurray D (ed), Tuberculosis. ASM Press, Washington, DC. [Google Scholar]
- 22.Hatfull GF. 2012. The secret lives of mycobacteriophages. Adv Virus Res 82:179–288 10.1016/B978-0-12-394621-8.00015-7. [PubMed] [DOI] [PubMed] [Google Scholar]
- 23.Hatfull GF, Jacobs WR Jr. 1994. Mycobacteriophages: cornerstones of mycobacterial research, p 165–183. In Bloom BR (ed), Tuberculosis: Pathogenesis, Protection and Control. ASM, Washington, DC. 10.1128/9781555818357.ch12 [DOI] [Google Scholar]
- 24.McNerney R. 1999. TB: the return of the phage. a review of fifty years of mycobacteriophage research. Int J Tuberc Lung Dis 3:179–184. [PubMed] [PubMed] [Google Scholar]
- 25.Jacobs WR Jr, Snapper SB, Tuckman M, Bloom BR. 1989. Mycobacteriophage vector systems. Rev Infect Dis 11(Suppl 2):S404–S410 10.1093/clinids/11.Supplement_2.S404. [PubMed] [DOI] [PubMed] [Google Scholar]
- 26.Sarkis GJ, Hatfull GF. 1998. Mycobacteriophages. Methods Mol Biol 101:145–173. [PubMed] [DOI] [PubMed] [Google Scholar]
- 27.Hatfull GF. 2014. Molecular genetics of mycobacteriophages. Microbiol Spectr 2:1–36 10.1128/microbiolspec.MGM2-0032-2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rohwer F, Segall AM. 2015. In retrospect: a century of phage lessons. Nature 528:46–48 10.1038/528046a. [PubMed] [DOI] [PubMed] [Google Scholar]
- 29.Hatfull GF. 2015. Innovations in undergraduate science education: going viral. J Virol 89:8111–8113 10.1128/JVI.03003-14. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hanauer DI, Jacobs-Sera D, Pedulla ML, Cresawn SG, Hendrix RW, Hatfull GF. 2006. Inquiry learning. Teaching scientific inquiry. Science 314:1880–1881 10.1126/science.1136796. [DOI] [PubMed] [Google Scholar]
- 31.Hatfull GF, Racaniello V. 2014. PHIRE and TWiV: experiences in bringing virology to new audiences. Annu Rev Virol 1:37–53 10.1146/annurev-virology-031413-085449. [PubMed] [DOI] [PubMed] [Google Scholar]
- 32.Hanauer D, Hatfull GF, Jacobs-Sera D. 2009. Active Assessment: Assessing Scientific Inquiry. Springer, New York, NY. 10.1007/978-0-387-89649-6_1 [DOI] [Google Scholar]
- 33.Jordan TC, Burnett SH, Carson S, Caruso SM, Clase K, DeJong RJ, Dennehy JJ, Denver DR, Dunbar D, Elgin SC, Findley AM, Gissendanner CR, Golebiewska UP, Guild N, Hartzog GA, Grillo WH, Hollowell GP, Hughes LE, Johnson A, King RA, Lewis LO, Li W, Rosenzweig F, Rubin MR, Saha MS, Sandoz J, Shaffer CD, Taylor B, Temple L, Vazquez E, Ware VC, Barker LP, Bradley KW, Jacobs-Sera D, Pope WH, Russell DA, Cresawn SG, Lopatto D, Bailey CP, Hatfull GF. 2014. A broadly implementable research course in phage discovery and genomics for first-year undergraduate students. MBio 5:e01051-13 10.1128/mBio.01051-13. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hanauer DI, Graham MJ, Betancur L, Bobrownicki A, Cresawn SG, Garlena RA, Jacobs-Sera D, Kaufmann N, Pope WH, Russell DA, Jacobs WR Jr, Sivanathan V, Asai DJ, Hatfull GF, Hatfull GF, SEA-PHAGES. 2017. An inclusive Research Education Community (iREC): impact of the SEA-PHAGES program on research outcomes and student learning. Proc Natl Acad Sci U S A 114:13531–13536 10.1073/pnas.1718188115. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pedulla ML, Ford ME, Houtz JM, Karthikeyan T, Wadsworth C, Lewis JA, Jacobs-Sera D, Falbo J, Gross J, Pannunzio NR, Brucker W, Kumar V, Kandasamy J, Keenan L, Bardarov S, Kriakov J, Lawrence JG, Jacobs WR Jr, Hendrix RW, Hatfull GF. 2003. Origins of highly mosaic mycobacteriophage genomes. Cell 113:171–182 10.1016/S0092-8674(03)00233-2. [PubMed] [DOI] [PubMed] [Google Scholar]
- 36.Cresawn SG, Bogel M, Day N, Jacobs-Sera D, Hendrix RW, Hatfull GF. 2011. Phamerator: a bioinformatic tool for comparative bacteriophage genomics. BMC Bioinformatics 12:395 10.1186/1471-2105-12-395. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klyczek KK, et al. 2017. Tales of diversity: genomic and morphological characteristics of forty-six Arthrobacter phages. PLoS One 12:e0180517 10.1371/journal.pone.0180517. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klyczek KK, Jacobs-Sera D, Adair TL, Adams SD, Ball SL, Benjamin RC, Bonilla JA, Breitenberger CA, Daniels CJ, Gaffney BL, Harrison M, Hughes LE, King RA, Krukonis GP, Lopez AJ, Monsen-Collar K, Pizzorno MC, Rinehart CA, Staples AK, Stowe EL, Garlena RA, Russell DA, Cresawn SG, Pope WH, Hatfull GF, SEA-PHAGES Program, PHIRE Program. 2018. Complete genome sequences of 44 Arthrobacter phages. Genome Announc 6:e01474-17 10.1128/genomeA.01474-17. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ford ME, Sarkis GJ, Belanger AE, Hendrix RW, Hatfull GF. 1998. Genome structure of mycobacteriophage D29: implications for phage evolution. J Mol Biol 279:143–164 10.1006/jmbi.1997.1610. [PubMed] [DOI] [PubMed] [Google Scholar]
- 40.Ford ME, Stenstrom C, Hendrix RW, Hatfull GF. 1998. Mycobacteriophage TM4: genome structure and gene expression. Tuber Lung Dis 79:63–73 10.1054/tuld.1998.0007. [PubMed] [DOI] [PubMed] [Google Scholar]
- 41.Mediavilla J, Jain S, Kriakov J, Ford ME, Duda RL, Jacobs WR Jr, Hendrix RW, Hatfull GF. 2000. Genome organization and characterization of mycobacteriophage Bxb1. Mol Microbiol 38:955–970 10.1046/j.1365-2958.2000.02183.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 42.Hatfull GF, Pedulla ML, Jacobs-Sera D, Cichon PM, Foley A, Ford ME, Gonda RM, Houtz JM, Hryckowian AJ, Kelchner VA, Namburi S, Pajcini KV, Popovich MG, Schleicher DT, Simanek BZ, Smith AL, Zdanowicz GM, Kumar V, Peebles CL, Jacobs WR Jr, Lawrence JG, Hendrix RW. 2006. Exploring the mycobacteriophage metaproteome: phage genomics as an educational platform. PLoS Genet 2:e92 10.1371/journal.pgen.0020092. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hatfull GF, Jacobs-Sera D, Lawrence JG, Pope WH, Russell DA, Ko CC, Weber RJ, Patel MC, Germane KL, Edgar RH, Hoyte NN, Bowman CA, Tantoco AT, Paladin EC, Myers MS, Smith AL, Grace MS, Pham TT, O’Brien MB, Vogelsberger AM, Hryckowian AJ, Wynalek JL, Donis-Keller H, Bogel MW, Peebles CL, Cresawn SG, Hendrix RW. 2010. Comparative genomic analysis of 60 mycobacteriophage genomes: genome clustering, gene acquisition, and gene size. J Mol Biol 397:119–143 10.1016/j.jmb.2010.01.011. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grose JH, Casjens SR. 2014. Understanding the enormous diversity of bacteriophages: the tailed phages that infect the bacterial family Enterobacteriaceae. Virology 468-470:421–443 10.1016/j.virol.2014.08.024. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pope WH, Bowman CA, Russell DA, Jacobs-Sera D, Asai DJ, Cresawn SG, Jacobs WR Jr, Hendrix RW, Lawrence JG, Hatfull GF, Science Education Alliance Phage Hunters Advancing Genomics and Evolutionary Science, Phage Hunters Integrating Research and Education, Mycobacterial Genetics Course. 2015. Whole genome comparison of a large collection of mycobacteriophages reveals a continuum of phage genetic diversity. eLife 4:e06416 10.7554/eLife.06416. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pope WH, Mavrich TN, Garlena RA, Guerrero-Bustamante CA, Jacobs-Sera D, Montgomery MT, Russell DA, Warner MH, Hatfull GF, Science Education Alliance-Phage Hunters Advancing Genomics and Evolutionary Science (SEA-PHAGES). 2017. Bacteriophages of Gordonia spp. display a spectrum of diversity and genetic relationships. MBio 8:e01069-17 10.1128/mBio.01069-17. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pope WH, Montgomery MT, Bonilla JA, Dejong R, Garlena RA, Guerrero Bustamante C, Klyczek KK, Russell DA, Wertz JT, Jacobs-Sera D, Hatfull GF. 2017. Complete genome sequences of 38 Gordonia sp. bacteriophages. Genome Announc 5:e01143-16 10.1128/genomeA.01143-16. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marinelli LJ, Fitz-Gibbon S, Hayes C, Bowman C, Inkeles M, Loncaric A, Russell DA, Jacobs-Sera D, Cokus S, Pellegrini M, Kim J, Miller JF, Hatfull GF, Modlin RL. 2012. Propionibacterium acnes bacteriophages display limited genetic diversity and broad killing activity against bacterial skin isolates. MBio 3:e00279-12 10.1128/mBio.00279-12. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dyson ZA, Tucci J, Seviour RJ, Petrovski S. 2015. Lysis to kill: evaluation of the lytic abilities, and genomics of nine bacteriophages infective for Gordonia spp. and their potential use in activated sludge foam biocontrol. PLoS One 10:e0134512 10.1371/journal.pone.0134512. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Delcher AL, Bratke KA, Powers EC, Salzberg SL. 2007. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23:673–679 10.1093/bioinformatics/btm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Borodovsky M, Lomsadze A. 2011. Gene identification in prokaryotic genomes, phages, metagenomes, and EST sequences with GeneMarkS suite. Curr Protoc Bioinformatics Chapter 4:Unit 4 5 1-17. [PubMed] [DOI] [PubMed] [Google Scholar]
- 52.Pope WH, Jacobs-Sera D, Best AA, Broussard GW, Connerly PL, Dedrick RM, Kremer TA, Offner S, Ogiefo AH, Pizzorno MC, Rockenbach K, Russell DA, Stowe EL, Stukey J, Thibault SA, Conway JF, Hendrix RW, Hatfull GF. 2013. Cluster J mycobacteriophages: intron splicing in capsid and tail genes. PLoS One 8:e69273 10.1371/journal.pone.0069273. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mageeney C, Pope WH, Harrison M, Moran D, Cross T, Jacobs-Sera D, Hendrix RW, Dunbar D, Hatfull GF. 2012. Mycobacteriophage Marvin: a new singleton phage with an unusual genome organization. J Virol 86:4762–4775 10.1128/JVI.00075-12. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dedrick RM, Mavrich TN, Ng WL, Hatfull GF. 2017. Expression and evolutionary patterns of mycobacteriophage D29 and its temperate close relatives. BMC Microbiol 17:225 10.1186/s12866-017-1131-2. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Payne KM, Hatfull GF. 2012. Mycobacteriophage endolysins: diverse and modular enzymes with multiple catalytic activities. PLoS One 7:e34052 10.1371/journal.pone.0034052. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Payne K, Sun Q, Sacchettini J, Hatfull GF. 2009. Mycobacteriophage Lysin B is a novel mycolylarabinogalactan esterase. Mol Microbiol 73:367–381 10.1111/j.1365-2958.2009.06775.x. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stella EJ, de la Iglesia AI, Morbidoni HR. 2009. Mycobacteriophages as versatile tools for genetic manipulation of mycobacteria and development of simple methods for diagnosis of mycobacterial diseases. Rev Argent Microbiol 41:45–55. [PubMed] [PubMed] [Google Scholar]
- 58.Dedrick RM, Mavrich TN, Ng WL, Cervantes Reyes JC, Olm MR, Rush RE, Jacobs-Sera D, Russell DA, Hatfull GF. 2016. Function, expression, specificity, diversity and incompatibility of actinobacteriophage parABS systems. Mol Microbiol 101:625–644 10.1111/mmi.13414. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dedrick RM, Marinelli LJ, Newton GL, Pogliano K, Pogliano J, Hatfull GF. 2013. Functional requirements for bacteriophage growth: gene essentiality and expression in mycobacteriophage Giles. Mol Microbiol 88:577–589 10.1111/mmi.12210. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Halleran A, Clamons S, Saha M. 2015. Transcriptomic characterization of an infection of Mycobacterium smegmatis by the cluster A4 mycobacteriophage Kampy. PLoS One 10:e0141100 10.1371/journal.pone.0141100. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fan X, Duan X, Tong Y, Huang Q, Zhou M, Wang H, Zeng L, Young RF III, Xie J. 2016. The global reciprocal reprogramming between mycobacteriophage SWU1 and Mycobacterium reveals the molecular strategy of subversion and promotion of phage infection. Front Microbiol 7:41 10.3389/fmicb.2016.00041. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ko CC, Hatfull GF. 2018. Mycobacteriophage Fruitloop gp52 inactivates Wag31 (DivIVA) to prevent heterotypic superinfection. Mol Microbiol 108:443–460 10.1111/mmi.13946. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dedrick RM, Jacobs-Sera D, Bustamante CA, Garlena RA, Mavrich TN, Pope WH, Reyes JC, Russell DA, Adair T, Alvey R, Bonilla JA, Bricker JS, Brown BR, Byrnes D, Cresawn SG, Davis WB, Dickson LA, Edgington NP, Findley AM, Golebiewska U, Grose JH, Hayes CF, Hughes LE, Hutchison KW, Isern S, Johnson AA, Kenna MA, Klyczek KK, Mageeney CM, Michael SF, Molloy SD, Montgomery MT, Neitzel J, Page ST, Pizzorno MC, Poxleitner MK, Rinehart CA, Robinson CJ, Rubin MR, Teyim JN, Vazquez E, Ware VC, Washington J, Hatfull GF. 2017. Prophage-mediated defence against viral attack and viral counter-defence. Nat Microbiol 2:16251 10.1038/nmicrobiol.2016.251. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brown KL, Sarkis GJ, Wadsworth C, Hatfull GF. 1997. Transcriptional silencing by the mycobacteriophage L5 repressor. EMBO J 16:5914–5921 10.1093/emboj/16.19.5914. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pope WH, et al. 2011. Expanding the diversity of mycobacteriophages: insights into genome architecture and evolution. PLoS One 6:e16329 10.1371/journal.pone.0016329. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bandhu A, Ganguly T, Jana B, Mondal R, Sau S. 2010. Regions and residues of an asymmetric operator DNA interacting with the monomeric repressor of temperate mycobacteriophage L1. Biochemistry 49:4235–4243 10.1021/bi9020956. [PubMed] [DOI] [PubMed] [Google Scholar]
- 67.Nesbit CE, Levin ME, Donnelly-Wu MK, Hatfull GF. 1995. Transcriptional regulation of repressor synthesis in mycobacteriophage L5. Mol Microbiol 17:1045–1056 10.1111/j.1365-2958.1995.mmi_17061045.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 68.Broussard GW, Oldfield LM, Villanueva VM, Lunt BL, Shine EE, Hatfull GF. 2013. Integration-dependent bacteriophage immunity provides insights into the evolution of genetic switches. Mol Cell 49:237–248 10.1016/j.molcel.2012.11.012. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Landy A. 2015. The lambda integrase site-specific recombination pathway. Microbiol Spectr 3:MDNA3-0051-2014. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Peña CE, Lee MH, Pedulla ML, Hatfull GF. 1997. Characterization of the mycobacteriophage L5 attachment site, attP. J Mol Biol 266:76–92 10.1006/jmbi.1996.0774. [PubMed] [DOI] [PubMed] [Google Scholar]
- 71.Lewis JA, Hatfull GF. 2001. Control of directionality in integrase-mediated recombination: examination of recombination directionality factors (RDFs) including Xis and Cox proteins. Nucleic Acids Res 29:2205–2216 10.1093/nar/29.11.2205. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee MH, Pascopella L, Jacobs WR Jr, Hatfull GF. 1991. Site-specific integration of mycobacteriophage L5: integration-proficient vectors for Mycobacterium smegmatis, Mycobacterium tuberculosis, and bacille Calmette-Guérin. Proc Natl Acad Sci U S A 88:3111–3115 10.1073/pnas.88.8.3111. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pham TT, Jacobs-Sera D, Pedulla ML, Hendrix RW, Hatfull GF. 2007. Comparative genomic analysis of mycobacteriophage Tweety: evolutionary insights and construction of compatible site-specific integration vectors for mycobacteria. Microbiology 153:2711–2723 10.1099/mic.0.2007/008904-0. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Morris P, Marinelli LJ, Jacobs-Sera D, Hendrix RW, Hatfull GF. 2008. Genomic characterization of mycobacteriophage Giles: evidence for phage acquisition of host DNA by illegitimate recombination. J Bacteriol 190:2172–2182 10.1128/JB.01657-07. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hatfull GF. 2015. Dark matter of the biosphere: the amazing world of bacteriophage diversity. J Virol 89:8107–8110 10.1128/JVI.01340-15. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hendrix RW, Smith MC, Burns RN, Ford ME, Hatfull GF. 1999. Evolutionary relationships among diverse bacteriophages and prophages: all the world’s a phage. Proc Natl Acad Sci U S A 96:2192–2197 10.1073/pnas.96.5.2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Martinsohn JT, Radman M, Petit MA. 2008. The lambda red proteins promote efficient recombination between diverged sequences: implications for bacteriophage genome mosaicism. PLoS Genet 4:e1000065 10.1371/journal.pgen.1000065. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.van Kessel JC, Hatfull GF. 2007. Recombineering in Mycobacterium tuberculosis. Nat Methods 4:147–152 10.1038/nmeth996. [PubMed] [DOI] [PubMed] [Google Scholar]
- 79.van Kessel JC, Hatfull GF. 2008. Efficient point mutagenesis in mycobacteria using single-stranded DNA recombineering: characterization of antimycobacterial drug targets. Mol Microbiol 67:1094–1107 10.1111/j.1365-2958.2008.06109.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 80.Sampson T, Broussard GW, Marinelli LJ, Jacobs-Sera D, Ray M, Ko CC, Russell D, Hendrix RW, Hatfull GF. 2009. Mycobacteriophages BPs, Angel and Halo: comparative genomics reveals a novel class of ultra-small mobile genetic elements. Microbiology 155:2962–2977 10.1099/mic.0.030486-0. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mavrich TN, Hatfull GF. 2017. Bacteriophage evolution differs by host, lifestyle and genome. Nat Microbiol 2:17112 10.1038/nmicrobiol.2017.112. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ondov BD, Treangen TJ, Melsted P, Mallonee AB, Bergman NH, Koren S, Phillippy AM. 2016. Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol 17:132 10.1186/s13059-016-0997-x. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pope WH, Ferreira CM, Jacobs-Sera D, Benjamin RC, Davis AJ, DeJong RJ, Elgin SCR, Guilfoile FR, Forsyth MH, Harris AD, Harvey SE, Hughes LE, Hynes PM, Jackson AS, Jalal MD, MacMurray EA, Manley CM, McDonough MJ, Mosier JL, Osterbann LJ, Rabinowitz HS, Rhyan CN, Russell DA, Saha MS, Shaffer CD, Simon SE, Sims EF, Tovar IG, Weisser EG, Wertz JT, Weston-Hafer KA, Williamson KE, Zhang B, Cresawn SG, Jain P, Piuri M, Jacobs WR Jr, Hendrix RW, Hatfull GF. 2011. Cluster K mycobacteriophages: insights into the evolutionary origins of mycobacteriophage TM4. PLoS One 6:e26750 10.1371/journal.pone.0026750. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jacobs-Sera D, Marinelli LJ, Bowman C, Broussard GW, Guerrero Bustamante C, Boyle MM, Petrova ZO, Dedrick RM, Pope WH, Modlin RL, Hendrix RW, Hatfull GF, Science Education Alliance Phage Hunters Advancing Genomics And Evolutionary Science Sea-Phages Program. 2012. On the nature of mycobacteriophage diversity and host preference. Virology 434:187–201 10.1016/j.virol.2012.09.026. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pope WH, Jacobs-Sera D, Russell DA, Rubin DH, Kajee A, Msibi ZN, Larsen MH, Jacobs WR Jr, Lawrence JG, Hendrix RW, Hatfull GF. 2014. Genomics and proteomics of mycobacteriophage patience, an accidental tourist in the Mycobacterium neighborhood. MBio 5:e02145 10.1128/mBio.02145-14. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Doron S, Melamed S, Ofir G, Leavitt A, Lopatina A, Keren M, Amitai G, Sorek R. 2018. Systematic discovery of antiphage defense systems in the microbial pangenome. Science 359:359 10.1126/science.aar4120. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ofir G, Melamed S, Sberro H, Mukamel Z, Silverman S, Yaakov G, Doron S, Sorek R. 2018. DISARM is a widespread bacterial defence system with broad anti-phage activities. Nat Microbiol 3:90–98 10.1038/s41564-017-0051-0. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.He L, Fan X, Xie J. 2012. Comparative genomic structures of Mycobacterium CRISPR-Cas. J Cell Biochem 113:2464–2473 10.1002/jcb.24121. [PubMed] [DOI] [PubMed] [Google Scholar]
- 89.Jones WD Jr, Greenberg J. 1977. Host modification and restriction with a mycobacteriophage isolated from a pseudolysogenic Mycobacterium chelonei. J Gen Microbiol 99:389–395 10.1099/00221287-99-2-389. [PubMed] [DOI] [PubMed] [Google Scholar]
- 90.Shankar S, Tyagi AK. 1992. MhaAI, a novel isoschizomer of PstI from Mycobacterium habana recognizing 5′-CTGCA/G-3′. Nucleic Acids Res 20:2891 10.1093/nar/20.11.2891. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shankar S, Tyagi AK. 1992. MfoAI, a novel isoschizomer of HaeIII from Mycobacterium fortuitum recognizing 5′-GG/CC-3′. Nucleic Acids Res 20:2890 10.1093/nar/20.11.2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shankar S, Tyagi AK. 1993. MchAI and MchAII, two class-II restriction endonucleases from Mycobacterium chelonei. Gene 132:119–123 10.1016/0378-1119(93)90523-6. [PubMed] [DOI] [PubMed] [Google Scholar]
- 93.Snapper SB, Melton RE, Mustafa S, Kieser T, Jacobs WR Jr. 1990. Isolation and characterization of efficient plasmid transformation mutants of Mycobacterium smegmatis. Mol Microbiol 4:1911–1919 10.1111/j.1365-2958.1990.tb02040.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 94.Bondy-Denomy J, Qian J, Westra ER, Buckling A, Guttman DS, Davidson AR, Maxwell KL. 2016. Prophages mediate defense against phage infection through diverse mechanisms. ISME J 10:2854–2866 10.1038/ismej.2016.79. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fineran PC, Blower TR, Foulds IJ, Humphreys DP, Lilley KS, Salmond GP. 2009. The phage abortive infection system, ToxIN, functions as a protein-RNA toxin-antitoxin pair. Proc Natl Acad Sci U S A 106:894–899 10.1073/pnas.0808832106. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chopin MC, Chopin A, Bidnenko E. 2005. Phage abortive infection in lactococci: variations on a theme. Curr Opin Microbiol 8:473–479 10.1016/j.mib.2005.06.006. [PubMed] [DOI] [PubMed] [Google Scholar]
- 97.Knowles B, Silveira CB, Bailey BA, Barott K, Cantu VA, Cobián-Güemes AG, Coutinho FH, Dinsdale EA, Felts B, Furby KA, George EE, Green KT, Gregoracci GB, Haas AF, Haggerty JM, Hester ER, Hisakawa N, Kelly LW, Lim YW, Little M, Luque A, McDole-Somera T, McNair K, de Oliveira LS, Quistad SD, Robinett NL, Sala E, Salamon P, Sanchez SE, Sandin S, Silva GG, Smith J, Sullivan C, Thompson C, Vermeij MJ, Youle M, Young C, Zgliczynski B, Brainard R, Edwards RA, Nulton J, Thompson F, Rohwer F. 2016. Lytic to temperate switching of viral communities. Nature 531:466–470 10.1038/nature17193. [PubMed] [DOI] [PubMed] [Google Scholar]
- 98.Marinelli LJ, Piuri M, Swigonová Z, Balachandran A, Oldfield LM, van Kessel JC, Hatfull GF. 2008. BRED: a simple and powerful tool for constructing mutant and recombinant bacteriophage genomes. PLoS One 3:e3957 10.1371/journal.pone.0003957. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tock MR, Dryden DT. 2005. The biology of restriction and anti-restriction. Curr Opin Microbiol 8:466–472 10.1016/j.mib.2005.06.003. [PubMed] [DOI] [PubMed] [Google Scholar]
- 100.Bondy-Denomy J, Pawluk A, Maxwell KL, Davidson AR. 2013. Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature 493:429–432 10.1038/nature11723. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lu MJ, Henning U. 1989. The immunity (imm) gene of Escherichia coli bacteriophage T4. J Virol 63:3472–3478. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Susskind MM, Botstein D, Wright A. 1974. Superinfection exclusion by P22 prophage in lysogens of Salmonella typhimurium. III. Failure of superinfecting phage DNA to enter sieA+ lysogens. Virology 62:350–366 10.1016/0042-6822(74)90398-5. [PubMed] [DOI] [PubMed] [Google Scholar]
- 103.Cumby N, Edwards AM, Davidson AR, Maxwell KL. 2012. The bacteriophage HK97 gp15 moron element encodes a novel superinfection exclusion protein. J Bacteriol 194:5012–5019 10.1128/JB.00843-12. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rybniker J, Nowag A, van Gumpel E, Nissen N, Robinson N, Plum G, Hartmann P. 2010. Insights into the function of the WhiB-like protein of mycobacteriophage TM4: a transcriptional inhibitor of WhiB2. Mol Microbiol 77:642–657 10.1111/j.1365-2958.2010.07235.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 105.Rybniker J, Plum G, Robinson N, Small PL, Hartmann P. 2008. Identification of three cytotoxic early proteins of mycobacteriophage L5 leading to growth inhibition in Mycobacterium smegmatis. Microbiology 154:2304–2314 10.1099/mic.0.2008/017004-0. [DOI] [PubMed] [Google Scholar]
- 106.Rybniker J, Krumbach K, van Gumpel E, Plum G, Eggeling L, Hartmann P. 2011. The cytotoxic early protein 77 of mycobacteriophage L5 interacts with MSMEG_3532, an l-serine dehydratase of Mycobacterium smegmatis. J Basic Microbiol 51:515–522 10.1002/jobm.201000446. [PubMed] [DOI] [PubMed] [Google Scholar]
- 107.Donnelly-Wu MK, Jacobs WR Jr, Hatfull GF. 1993. Superinfection immunity of mycobacteriophage L5: applications for genetic transformation of mycobacteria. Mol Microbiol 7:407–417 10.1111/j.1365-2958.1993.tb01132.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 108.Liu J, Dehbi M, Moeck G, Arhin F, Bauda P, Bergeron D, Callejo M, Ferretti V, Ha N, Kwan T, McCarty J, Srikumar R, Williams D, Wu JJ, Gros P, Pelletier J, DuBow M. 2004. Antimicrobial drug discovery through bacteriophage genomics. Nat Biotechnol 22:185–191 10.1038/nbt932. [PubMed] [DOI] [PubMed] [Google Scholar]
- 109.Meniche X, Otten R, Siegrist MS, Baer CE, Murphy KC, Bertozzi CR, Sassetti CM. 2014. Subpolar addition of new cell wall is directed by DivIVA in mycobacteria. Proc Natl Acad Sci U S A 111:E3243–E3251 10.1073/pnas.1402158111. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mukherjee P, Sureka K, Datta P, Hossain T, Barik S, Das KP, Kundu M, Basu J. 2009. Novel role of Wag31 in protection of mycobacteria under oxidative stress. Mol Microbiol 73:103–119 10.1111/j.1365-2958.2009.06750.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 111.Bardarov S, Bardarov S Jr, Pavelka MS Jr, Sambandamurthy V, Larsen M, Tufariello J, Chan J, Hatfull G, Jacobs WR Jr. 2002. Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology 148:3007–3017 10.1099/00221287-148-10-3007. [PubMed] [DOI] [PubMed] [Google Scholar]
- 112.Bardarov S Jr, Dou H, Eisenach K, Banaiee N, Ya S, Chan J, Jacobs WR Jr, Riska PF. 2003. Detection and drug-susceptibility testing of M. tuberculosis from sputum samples using luciferase reporter phage: comparison with the Mycobacteria Growth Indicator Tube (MGIT) system. Diagn Microbiol Infect Dis 45:53–61 10.1016/S0732-8893(02)00478-9. [DOI] [PubMed] [Google Scholar]
- 113.Bardarov S, Kriakov J, Carriere C, Yu S, Vaamonde C, McAdam RA, Bloom BR, Hatfull GF, Jacobs WR Jr. 1997. Conditionally replicating mycobacteriophages: a system for transposon delivery to Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 94:10961–10966 10.1073/pnas.94.20.10961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jacobs WR Jr, Barletta RG, Udani R, Chan J, Kalkut G, Sosne G, Kieser T, Sarkis GJ, Hatfull GF, Bloom BR. 1993. Rapid assessment of drug susceptibilities of Mycobacterium tuberculosis by means of luciferase reporter phages. Science 260:819–822 10.1126/science.8484123. [PubMed] [DOI] [PubMed] [Google Scholar]
- 115.Piuri M, Jacobs WR Jr, Hatfull GF. 2009. Fluoromycobacteriophages for rapid, specific, and sensitive antibiotic susceptibility testing of Mycobacterium tuberculosis. PLoS One 4:e4870 10.1371/journal.pone.0004870. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.O’Donnell MR, Pym A, Jain P, Munsamy V, Wolf A, Karim F, Jacobs WR Jr, Larsen MH. 2015. A novel reporter phage to detect tuberculosis and rifampin resistance in a high-HIV-burden population. J Clin Microbiol 53:2188–2194 10.1128/JCM.03530-14. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jain P, Hartman TE, Eisenberg N, O’Donnell MR, Kriakov J, Govender K, Makume M, Thaler DS, Hatfull GF, Sturm AW, Larsen MH, Moodley P, Jacobs WR Jr. 2012. ɸ(2)GFP10, a high-intensity fluorophage, enables detection and rapid drug susceptibility testing of Mycobacterium tuberculosis directly from sputum samples. J Clin Microbiol 50:1362–1369 10.1128/JCM.06192-11. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yu X, Gu Y, Jiang G, Ma Y, Zhao L, Sun Z, Jain P, O’Donnell M, Larsen M, Jacobs WR Jr, Huang H. 2016. Evaluation of a high-intensity green fluorescent protein fluorophage method for drug- resistance diagnosis in tuberculosis for isoniazid, rifampin, and streptomycin. Front Microbiol 7:922 10.3389/fmicb.2016.00922. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Rondón L, Piuri M, Jacobs WR Jr, de Waard J, Hatfull GF, Takiff HE. 2011. Evaluation of fluoromycobacteriophages for detecting drug resistance in Mycobacterium tuberculosis. J Clin Microbiol 49:1838–1842 10.1128/JCM.02476-10. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jain P, Weinrick BC, Kalivoda EJ, Yang H, Munsamy V, Vilcheze C, Weisbrod TR, Larsen MH, O’Donnell MR, Pym A, Jacobs WR Jr. 2016. Dual-reporter mycobacteriophages (Φ2DRMs) reveal preexisting Mycobacterium tuberculosis persistent cells in human sputum. MBio 7:e01023-16 10.1128/mBio.01023-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kim AI, Ghosh P, Aaron MA, Bibb LA, Jain S, Hatfull GF. 2003. Mycobacteriophage Bxb1 integrates into the Mycobacterium smegmatis groEL1 gene. Mol Microbiol 50:463–473 10.1046/j.1365-2958.2003.03723.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 122.Freitas-Vieira A, Anes E, Moniz-Pereira J. 1998. The site-specific recombination locus of mycobacteriophage Ms6 determines DNA integration at the tRNA(Ala) gene of Mycobacterium spp. Microbiology 144:3397–3406 10.1099/00221287-144-12-3397. [PubMed] [DOI] [PubMed] [Google Scholar]
- 123.van Kessel JC, Marinelli LJ, Hatfull GF. 2008. Recombineering mycobacteria and their phages. Nat Rev Microbiol 6:851–857 10.1038/nrmicro2014. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.van Kessel JC, Hatfull GF. 2008. Mycobacterial recombineering. Methods Mol Biol 435:203–215 10.1007/978-1-59745-232-8_15. [PubMed] [DOI] [PubMed] [Google Scholar]
- 125.Singh V, Dhar N, Pató J, Kolly GS, Korduláková J, Forbak M, Evans JC, Székely R, Rybniker J, Palčeková Z, Zemanová J, Santi I, Signorino-Gelo F, Rodrigues L, Vocat A, Covarrubias AS, Rengifo MG, Johnsson K, Mowbray S, Buechler J, Delorme V, Brodin P, Knott GW, Aínsa JA, Warner DF, Kéri G, Mikušová K, McKinney JD, Cole ST, Mizrahi V, Hartkoorn RC. 2017. Identification of aminopyrimidine-sulfonamides as potent modulators of Wag31-mediated cell elongation in mycobacteria. Mol Microbiol 103:13–25 10.1111/mmi.13535. [PubMed] [DOI] [PubMed] [Google Scholar]
- 126.Marinelli LJ, Hatfull GF, Piuri M. 2012. Recombineering: a powerful tool for modification of bacteriophage genomes. Bacteriophage 2:5–14 10.4161/bact.18778. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Petrova ZO, Broussard GW, Hatfull GF. 2015. Mycobacteriophage-repressor-mediated immunity as a selectable genetic marker: adephagia and BPs repressor selection. Microbiology 161:1539–1551 10.1099/mic.0.000120. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gicquel-Sanzey B, Moniz-Pereira J, Gheorghiu M, Rauzier J. 1989. Structure of pAL5000, a plasmid from M. fortuitum and its utilization in transformation of mycobacteria. Acta Leprol 7(Suppl 1):208–211. [PubMed] [PubMed] [Google Scholar]
- 129.Ranes MG, Rauzier J, Lagranderie M, Gheorghiu M, Gicquel B. 1990. Functional analysis of pAL5000, a plasmid from Mycobacterium fortuitum: construction of a “mini” mycobacterium-Escherichia coli shuttle vector. J Bacteriol 172:2793–2797 10.1128/jb.172.5.2793-2797.1990. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Rauzier J, Moniz-Pereira J, Gicquel-Sanzey B. 1988. Complete nucleotide sequence of pAL5000, a plasmid from Mycobacterium fortuitum. Gene 71:315–321 10.1016/0378-1119(88)90048-0. [PubMed] [DOI] [PubMed] [Google Scholar]
- 131.Villar CA, Benitez J. 1992. Functional analysis of pAL5000 plasmid in Mycobacterium fortuitum. Plasmid 28:166–169 10.1016/0147-619X(92)90047-E. [PubMed] [DOI] [PubMed] [Google Scholar]
- 132.Chen J, Kriakov J, Singh A, Jacobs WR Jr, Besra GS, Bhatt A. 2009. Defects in glycopeptidolipid biosynthesis confer phage I3 resistance in Mycobacterium smegmatis. Microbiology 155:4050–4057 10.1099/mic.0.033209-0. [PubMed] [DOI] [PubMed] [Google Scholar]
- 133.Boritsch EC, Brosch R. 2016. Evolution of Mycobacterium tuberculosis: new insights into pathogenicity and drug resistance. Microbiol Spectr 4:TBTB2-0020-2016. [PubMed] [DOI] [PubMed] [Google Scholar]
- 134.Sassi M, Bebeacua C, Drancourt M, Cambillau C. 2013. The first structure of a mycobacteriophage, the Mycobacterium abscessus subsp. bolletii phage Araucaria. J Virol 87:8099–8109 10.1128/JVI.01209-13. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sassi M, Gouret P, Chabrol O, Pontarotti P, Drancourt M. 2014. Mycobacteriophage-drived diversification of Mycobacterium abscessus. Biol Direct 9:19 10.1186/1745-6150-9-19. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Mehla J, Dedrick RM, Caufield JH, Siefring R, Mair M, Johnson A, Hatfull GF, Uetz P. 2015. The protein interactome of mycobacteriophage Giles predicts functions for unknown proteins. J Bacteriol 197:2508–2516 10.1128/JB.00164-15. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Mehla J, Dedrick RM, Caufield JH, Wagemans J, Sakhawalkar N, Johnson A, Hatfull GF, Uetz P. 2017. Virus-host protein-protein interactions of mycobacteriophage Giles. Sci Rep 7:16514 10.1038/s41598-017-16303-7. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]