

Abstract
Background:
The characteristic progression of Lewy pathology in Parkinson’s disease likely involves intercellular exchange and accumulation of misfolded α-synuclein amplified by a prion-like self-templating mechanism. Silencing of the α-synuclein gene could provide long-lasting disease modifying benefits by reducing the requisite substrate for the spreading aggregation.
Objectives:
Due to poor penetration of viral vectors across the blood-brain barrier, gene therapy for central nervous system disorders requires direct injections into affected brain regions, and invasiveness is further increased by the need for bilateral delivery to multiple brain regions. Here we test a non-invasive approach by combining low intensity MR-guided focused ultrasound and intravenous microbubbles that can transiently increase the access of brain impermeant therapeutic macromolecules to targeted brain regions.
Methods:
Transgenic mice expressing human α-synuclein were subjected to MR-guided focused ultrasound targeted to four brain regions (hippocampus, substantia nigra, olfactory bulb, and dorsal motor nucleus) in tandem with intravenous microbubbles and an adeno-associated virus serotype 9 vector bearing a short hairpin RNA sequence targeting the α-synuclein gene.
Results:
One month following treatment, α-synuclein immunoreactivity was decreased in targeted brain regions, whereas other neuronal markers such as synaptophysin or tyrosine hydroxylase were unchanged, and cell death and glial activation remained at basal levels.
Conclusions:
These results demonstrate that MR-guided focused ultrasound can effectively, non-invasively, and simultaneously deliver viral vectors targeting α-synuclein to multiple brain areas. Importantly, this approach may be useful to alter the progression of Lewy pathology along selected neuronal pathways, particularly as prodromal PD markers improve early diagnoses.
Keywords: Parkinson’s disease, Gene therapy, Adeno-associated virus, Inhibitory RNA, Blood-brain barrier
INTRODUCTION
Alpha-synuclein (α-syn) is an abundant presynaptic protein in mammalian brains1, 2 and a key pathogenic factor in Parkinson’s disease (PD) and related synucleinopathies. Aggregated and phosphorylated α-syn accumulates progressively in affected individuals causing selective neuronal dysfunction and degeneration3–6. Braak staging of PD Lewy pathology suggests that the initial appearance of α-syn aggregates in the central nervous system (CNS) occurs in the dorsal motor nucleus of the vagus (DMN) in the lower brainstem and in the olfactory bulb (OB), followed by a rostral progression of pathology into the midbrain and cerebral cortex7–9. These early manifestations of synucleinopathy correspond to prodromal non-motor features of PD, including constipation, due to impaired vagal innervation of the gastrointestinal tract, and a loss of olfaction10.
The spreading Lewy pathology is linked to α-syn’s ability to self-template in a manner similar to prions11–14. α-Syn can adopt stable conformations with high beta-sheet content and misfolded variants can confer their structure onto additional α-syn molecules. In multiple experimental cell and animal models, α-syn pathology spreads to nearby cells or along neuronal tracts following inoculation with either fibrillar α-syn or synucleinopathy-sourced brain homogenates15–22. The underlying process, initially postulated to explain Lewy pathology within fetal grafts implanted in PD patients23, 24, implicates a general spreading cascade whereby exchange of misfolded α-syn is coupled to its interaction with native α-syn in recipient neurons25.
The inherent requirement for α-syn expression in recipient neurons for permissive templating is consistent with PD genetics showing that elevated α-syn expression by gene multiplication raises the risk for PD26–28. Conversely, therapeutic down-regulation of α-syn expression may be a viable approach to delay or block spreading pathology in PD29. Several studies have reported α-syn knockdown in rodents or primates30–38 and as protection against dopaminergic degeneration induced by either rotenone or α-syn fibrils39–41. However, a key challenge to adapting putative therapeutics for clinical use is the low penetrability of the blood-brain barrier (BBB) to molecules larger than ~400 Da and of poor lipid solubility, thereby limiting the effectiveness of systemically delivered immuno- or gene therapies for brain disorders42. CNS entry of therapeutic molecules such as immunoglobulin, growth factors, or non-viral gene sequences are enhanced by conjugation to brain-penetrant ligands (molecular Trojan horse) recognized by an endogenous transporter43. For example, encapsulation of α-syn siRNA into exosomes labeled with a brain-targeting viral peptide34 or direct conjugation to a monoamine reuptake inhibitor37 reduced α-syn expression in brain neurons. One limitation of non-viral gene expression is its transient effect, such that treatments must be repeated to maintain gene regulation.
Stable gene expression lasting more than a decade can be safely achieved with viral vectors44, 45, although brain delivery requires intracerebral injections, which are invasive and carry risks of infection, hemorrhage, and neuronal damage around the injection site46. Moreover, the practicality of intracerebral injections is diminished by the need for multiple injections to cover an entire brain region or bilateral coverage in multiple brain regions. Alternatively, transcranial magnetic resonance (MR)-guided focused ultrasound (FUS) combined with microbubbles injected into the bloodstream can locally and transiently increase the permeability of the BBB for several hours after sonication47. This approach permits brain targeted delivery of systemically administered therapeutics, such as immunoglobulin or viral vectors, efficiently across the BBB without invasive surgery48–58. Here, we use FUS to deliver a virally-expressed short hairpin RNA (shRNA) to silence the α-syn gene in multiple discrete brain regions that are known to develop Lewy pathology in PD. Our study demonstrates the feasibility of simultaneous modulation of α-syn expression in distal brain regions without multiple invasive procedures.
METHODS
Virus
Recombinant adeno-associated virus serotype 9 (AAV9) was utilized to express shRNA targeting human α-syn to knockdown human α-syn gene expression in our transgenic mouse model. Plasmids containing a silencing sequence targeting nucleotides 299–309 of the human α-syn gene (AAV9-hSNCA-shRNA)33 were generously provided by Dr. Martha C. Bohn (Northwestern University). Control plasmids contain a scrambled sequence of the same nucleotides (AAV9-Scr-shRNA). Both vectors used in this study, and as previously reported59, contain short mir-30 microRNA insertions flanking the 5’ and 3’ ends of the silencing sequence, which can reduce cell death associated with hSNCA-shRNA. Upstream to the silencing cassette included the cytomegalovirus (CMV) promoter, a reporter turbo green fluorescent protein (turboGFP) sequence, and an internal ribosome entry site (IRES) for bicistronic expression. Single-stranded rAAV9 viruses expressing either hSNCA- or the Scr-shRNA were produced by Virovek. For each animal, virus was intravenously delivered via the tail vein as a single dose of 1.25 × 1010 VG/g, or approximately 3.75 × 1011 vector genomes per 30 g animal.
Animals
Transgenic (Tg) mice (3–4 months) lacking endogenous murine α-syn but expressing wild-type human α-syn were used in this study60, 61. Pan-neuronal expression of the human Tg α-syn is driven by the hamster prion promoter62–69. Brain α-syn protein levels are 1–1.5-fold of nonTg wild-type animals and the Tg animals do not manifest behavioural changes or brain abnormalities at the ages studied. 8 Tg mice received FUS targeting the hippocampus (HC) and substantia nigra (SN), and another 8 Tg mice received FUS targeting the OB and DMN. These FUS-targeted groups were subdivided into 2 virus treatment groups (n = 4 per group): AAV9-Scr-shRNA or AAV9-hSNCA-shRNA. Animal procedures were carried out in compliance with the Canadian Council on Animal Care and the Animals for Research Act of Ontario.
MR-guided FUS
Mice were anesthetized with isoflurane and a 26-gauge catheter was inserted into the tail vein. Mice were secured on an MRI-compatible sled and imaged using a 7.0 T MRI (BioSpin 7030, Bruker). Pre-sonication T2-weighted (T2w) scans were used to target ultrasound foci. In one set of animals (n = 8), FUS was targeted to the HC and the ipsilateral SN. In a second set of animals (n = 8), FUS was targeted bilaterally to the OB and DMN. FUS was conducted using the RK100 system (FUS Instruments Inc.). Ultrasound waveforms were generated using a spherically focused transducer (1.68 MHz, 75 mm diameter, 60 mm radius of curvature). A polyvinylidene difluoride hydrophone was positioned in the centre of the transducer, as illustrated in Fig. 1A.
Fig. 1: MRI-guided FUS-induced BBB opening and transgene expression following FUS-mediated delivery of AAV9-hSNCA-shRNA and AAV9-Scr-shRNA to targeted brain regions.

(A) Schematic of the FUS experimental setup. (B,C) Selected focal spots in the HC, SN, OB and DMN (indicated by purple, green, blue and red dots, respectively) for FUS-mediated BBB disruption. (D,E) Following FUS, T1-weighted MR images were taken to confirm increased BBB permeability at selected focal spots in the HC and SN in one cohort of animals, and (F,G) in the OB and DMN in a second cohort. There was no significant difference in (H,J) enhancement level and (I,K) mean peak pressure required to induce BBB opening between AAV9-hSNCA-shRNA (black bar) and AAV9-Scr-shRNA treated animals (white bar) for both targeting schemes. (L-W) Representative confocal images (60X magnification) of brain sections prepared from AAV9-hSNCA-shRNA and AAV9-Scr-shRNA injected animals to detect turboGFP immunofluorescence. For both experimental groups, viral-mediated turboGFP was restricted to the FUS-treated side in the (L,M) HC and (O,P) SN, and bilateral (R,S) OB and (U,V) DMN. In contrast, turboGFP was not detectable on the contralateral hemisphere in the (N) HC and (Q) SN, nor in the untreated (T) OB and (W) DMN. Statistics: Paired, two-tailed, student’s t-test. Data represent the mean ± SEM. n = 4 per group. TurboGFP, green; DAPI, blue. Scale bars: (D-G) 5 mm; and (L-W) 20 μm.
At the start of sonication, Definity microbubbles (0.02 mL/kg, Lantheus Medical Imaging) were injected via the tail vein. Standard BBB disruption parameters were used for sonication (10 ms bursts, 1 Hz pulse repetition frequency, 120 s duration). The applied acoustic pressure was incrementally increased with each pulse. Once subharmonic emissions were detected, the acoustic pressure was dropped by 50% and maintained for the rest of sonication, as described70. The peak pressure required for BBB opening was averaged across the focal spots per animal to calculate the mean peak pressure for each animal.
Immediately following FUS, a gadolinium-based MRI contrast agent (0.2 mL/kg Gadovist, Schering AG) was injected via the tail vein and T1w scans were obtained to assess BBB permeability. Mice subsequently received the viral solution in saline via the tail vein catheter and 200 μL of saline to facilitate transfer of the virus into the bloodstream.
The relative enhancement for each animal was calculated as pixel intensity in a 2 mm by 2 mm region on the post-sonication T1w MR images normalized to the intensity in a reference region of the brain. The enhancement values were averaged over all ultrasound foci per animal using a custom program (Matlab).
Immunohistochemistry
One month post-sonication, mice were deeply anesthetized with a mixture of ketamine (150 mg/kg) and xylazine (10 mg/kg). Animals were transcardially perfused with saline and fixed with 4% paraformaldehyde in 0.1 M phosphate buffer. Whole brains were sectioned axially at 40 μm using a sliding microtome.
For detection of transgene expression, free floating, brain sections were incubated in 10 mM sodium citrate buffer (pH 6.0) for 30 min at 80°C and then in blocking solution (5% goat serum, 0.25% Triton X-100 in PBS) for 2 h at room temperature (RT). Sections were incubated with anti-turboGFP antibody (PA5–22688, 1:500; Thermofisher Scientific) for 72 h at 4°C and then a secondary antibody for 2 h at RT, followed by 4’6’-diamidino-2-phenylindole (DAPI, D9542, 1:10 000; Sigma-Aldrich) in PBS for 10 min at RT.
To evaluate the extent of α-syn knockdown in the HC and SN, triple labeling for α-syn, synaptophysin and tyrosine hydroxylase (TH) was performed, while α-syn, synaptophysin and choline acetyltransferase (ChAT) was performed for the OB and DMN. Sections were incubated in blocking solution at RT for 2 h, followed by anti-α-syn (32–8100, 1:1000; Invitrogen), anti-synaptophysin (ab52636, 1:250; Abcam) and anti-TH (ab76442, 1:1000; Abcam) primary antibodies for 72 h at 4°C. Sections were incubated in secondary antibodies for 2 h, followed by DAPI for 10 min at RT.
To detect neuronal cell death associated with shRNA delivery, terminal deoxynucleotidyl transferase-mediated dUTP-fluorescein nick-end labeling (TUNEL), cleaved caspase-3 and neuronal nuclei (NeuN) immunofluorescent colabeling was evaluated. Briefly, TUNEL-positive cells were detected with an in situ cell death detection kit (DeadEnd Flurometric TUNEL system, Promega), according to the supplier’s instructions. Sections were incubated in blocking solution (10% goat serum, 0.3% Triton X-100) for 2 h at RT, and then for 24 h at 4°C with primary antibodies against cleaved caspase-3 (9661S, 1:300; Cell Signaling Technology) and NeuN (ABN90, 1:500; Millipore), followed by secondary antibodies for 24 h at 4°C.
To characterize the immune response to viral gene therapy, including the activation of glia, we performed double immunolabeling with ionized calcium-binding adapter molecule 1 (Iba-1) and glial fibrillary acidic protein (GFAP), for microglia and astrocytes, respectively. Sections were incubated in 10 mM sodium citrate buffer (pH 6.0) for 30 min at 80°C and then in blocking solution (5% donkey serum, 0.25% Triton X-100, PBS) for 2 h at RT. Sections were incubated with anti-Iba-1 antibody (019–19741, 1:1000; Wako Chemicals) and anti-GFAP (NB100–53809, 1:1000; Novus Biologicals) for 24 h at 4°C followed by secondary antibodies for 2 h and DAPI for 10 min at RT.
Imaging and quantification
Serial sections were processed in parallel and the regions of interest were applied to four serial sections throughout the HC, SN, OB and DMN, as defined using mouse stereotaxic coordinates71. All images shown were acquired using a Nikon A1 laser scanning confocal microscope (20X and 60X magnification objectives) coupled to NIS-Elements software (Nikon Instruments). Z-series of 0.7 µm optical section thickness were merged. All quantification of α-syn, synaptophysin, and TH immunoreactivity was done using images acquired at 20X magnification on a Zeiss spinning disk microscope coupled to the PV camera (Zeiss Axio Observer.Z1) and Zen software (Carl Zeiss). Tiled Z-stack images were obtained with 0.7 µm optical section thickness and projected to generate maximum intensity images. Signal intensity was examined in regions of interest at sonicated and corresponding non-sonicated structures in the brain. Pixel densitometry was analyzed with ImageJ software (NIH).
Statistical analysis
Prism software (GraphPad Software Inc.) was used for statistical analysis and graph generation. All graphs are presented as mean ± SEM. Statistical significance was set at p < 0.05. Power analyses were performed using G*Power software72.
RESULTS
MRI enhancement confirms BBB permeability in sonicated brain regions
FUS was targeted using T2w MRI scans either unilaterally to the HC and SN in one cohort of animals or bilaterally to the OB and DMN in a second cohort (Fig. 1B,C). Post-sonication, gadolinium enhancement was measured from T1w images of targeted brain areas (Fig. 1D-G), reflecting the relative amount of FUS-induced BBB opening73. We found no significant difference in enhancement level between AAV9-hSNCA-shRNA and AAV9-Scr-shRNA treated animals, indicating comparable FUS-mediated increase in BBB permeability between the treatment groups (Fig. 1H,J). Normalized to the reference region, the mean increase in voxel intensity in sonicated regions was 43% ± 17% and 47% ± 13% in the AAV9-hSNCA-shRNA and AAV9-Scr-shRNA injected groups, respectively (p = 0.62).
Similarly, there was no difference in the mean acoustic energy delivered to the FUS-targeted regions between treatment groups (Fig. 1I,K). For the AAV9-hSNCA-shRNA and AAV9-Scr-shRNA groups, respectively, the mean peak pressure during sonication was 1.0 ± 0.2 and 1.1 ± 0.2 (p = 0.52). These data suggest that consistent BBB opening was achieved by using similar acoustic pressures in both treatment groups.
Reduction in α-syn expression after FUS-mediated delivery of silencing vector
The AAV9 vectors contained a reporter turboGFP sequence to verify transgene expression and location to FUS-targeted brain regions. All sonicated areas showed immunoreactivity to turboGFP (Fig. 1L-W). In contrast, there was no turboGFP expression in corresponding contralateral regions of brains which were not targeted by FUS. These results confirm the poor penetration of the virus across an intact BBB at an intravenous dose of 1.25 × 1010 VG/g, and that AAV9 delivery to the brain is selectively increased by FUS application. Varying levels of turboGFP expression were evident between brain areas, possibly reflecting regional neuronal versus non-neuronal populations and differences in afferent projections containing nerve terminals enriched in α-syn and presynaptic marker synaptophysin. In adult mice, AAV9 displays neuronal tropism, particularly when delivered to the brain and spinal cord using intraparenchymal injections or systemic administration combined with FUS48, 51, 74–77. Here, the amount and localization of turboGFP-positive cells found in the hippocampal formation are comparable to previously reported using scAAV9-GFP51. The targeting of the OB, SN and DMN also results in a subpopulation of neurons expressing turboGFP and suggest that α-syn is suppressed within that subpopulation of neurons transduced.
One month following FUS-mediated delivery of the AAV9 vectors, α-syn immunoreactivity was observed in the FUS-targeted regions receiving the control Scr-shRNA (Fig. 2,3; red). The α-syn expression was a diffuse, punctate pattern matching that of synaptophysin, consistent with its preferentially presynaptic localization as observed in previous studies78–85. In comparison, FUS-targeted regions of animals treated with hSNCA-shRNA had lower α-syn immunoreactivity. Synaptophysin expression was similar between the two treatments (Fig. 2E,3E), suggesting that synaptic integrity remains unaltered following FUS gene delivery. Therefore, to account for anatomical variations in the regions of interest, we normalized the intensity of α-syn immunofluorescence to that of synaptophysin (Fig. 2F-G,3F-G). Regions of interest in the SN and DMN were identified by the presence of dopaminergic (TH-positive) and cholinergic (ChAT-positive) soma (Supplemental Fig. 1). Relative to FUS-mediated delivery of AAV9-Scr-shRNA, we found a significant decrease in the α-syn/synaptophysin ratio following AAV9-hSNCA-shRNA treatment in the FUS-targeted HC (p = 0.021), SN (p = 0.012), and OB (p = 0.018). Similarly, we noted that α-syn levels in the DMN were 1.6 ± 0.3 and 2.6 ± 0.4, when treated with AAV9-hSNCA-shRNA and AAV9-Scr-shRNA, respectively (p = 0.081, t-test, n = 4). Based on these results, a power analysis reveals that sample sizes of 6 animals per treatment group have an 80% power to detect differences with a significance level of 0.05 (two-tailed).
Fig. 2: α-Syn knockdown following FUS delivery of AAV9-hSNCA-shRNA to the hippocampus and substantia nigra.

(A-D) Representative low (20X) and high (60X) magnification confocal images of brain sections immunolabeled for α-syn, synaptophysin and DAPI following FUS delivery of either AAV9-hSNCA-shRNA or AAV9-Scr-shRNA. Mean α-syn immunoreactivity was significantly decreased in animals that received AAV9-hSNCA-shRNA (black bar) compared to AAV9-Scr-shRNA (white bar) in the (A, B, F) HC and (C, D, G) SN. Dashed outline in C,D denotes area with TH-positive cells. Measurements of the α-syn (red) immunofluorescence signal are expressed as a percentage of the synaptophysin signal in the same region of interest. There was no significant difference in synaptophysin (green) immunoreactivity between AAV9-hSNCA-shRNA (black bar) and AAV9-Scr-shRNA injected animals (white bar) with FUS targeted to the (A, B, E) HC and (C, D, E) SN. Statistics: Paired, two-tailed, student’s t-test. Data represent the mean ± SEM. n = 4 per group. Synaptophysin, green; α-syn, red; DAPI, blue. Scale bars: (A-D) 20x magnification, 100 μm; and 60x magnification, 20 μm.
Fig. 3: α-Syn knockdown following FUS delivery of AAV9-hSNCA-shRNA to the olfactory bulbs and dorsal motor nucleus.

(A-D) Representative low (20X) and high (60X) magnification confocal images of brain sections immunolabeled for α-syn, synaptophysin and DAPI following FUS delivery of either AAV9-hSNCA-shRNA or AAV9-Scr-shRNA. Mean α-syn immunoreactivity was significantly decreased in animals that received AAV9-hSNCA-shRNA (black bar) compared to AAV9-Scr-shRNA (white bar) in the (A, B, F) OB and (C, D, G) DMX. Dashed outline in C,D denotes area with ChAT-positive cells. Measurements of the α-syn (red) immunofluorescence signal are expressed as a percentage of the synaptophysin signal in the same region of interest. There was no significant difference in synaptophysin (green) immunoreactivity between AAV9-hSNCA-shRNA (black bar) and AAV9-Scr-shRNA injected animals (white bar) with FUS targeted to the (A, B, E) OB and (C, D, E) DMX. Statistics: Paired, two-tailed, student’s t-test. Data represent the mean ± SEM. n = 4 per group. Synaptophysin, green; α-syn, red; DAPI, blue. Scale bars: (A-D) 20x magnification, 100 μm; and 60x magnification, 20 μm.
There are reports of a decline in nigral TH expression following suppression of α-syn in the SN32, 36, 38, raising the possibility of neurotoxicity associated with low α-syn levels. However, despite the ~60% reduction in α-syn, there was no change in TH immunoreactivity in the SN (Fig. 4), whether in comparison to the control shRNA treatment (p = 0.181), or to the contralateral hemisphere (p = 0.773).
Fig. 4: Tyrosine hydroxylase expression is not reduced in the substantia nigra following FUS delivery of AAV9-hSNCA-shRNA.
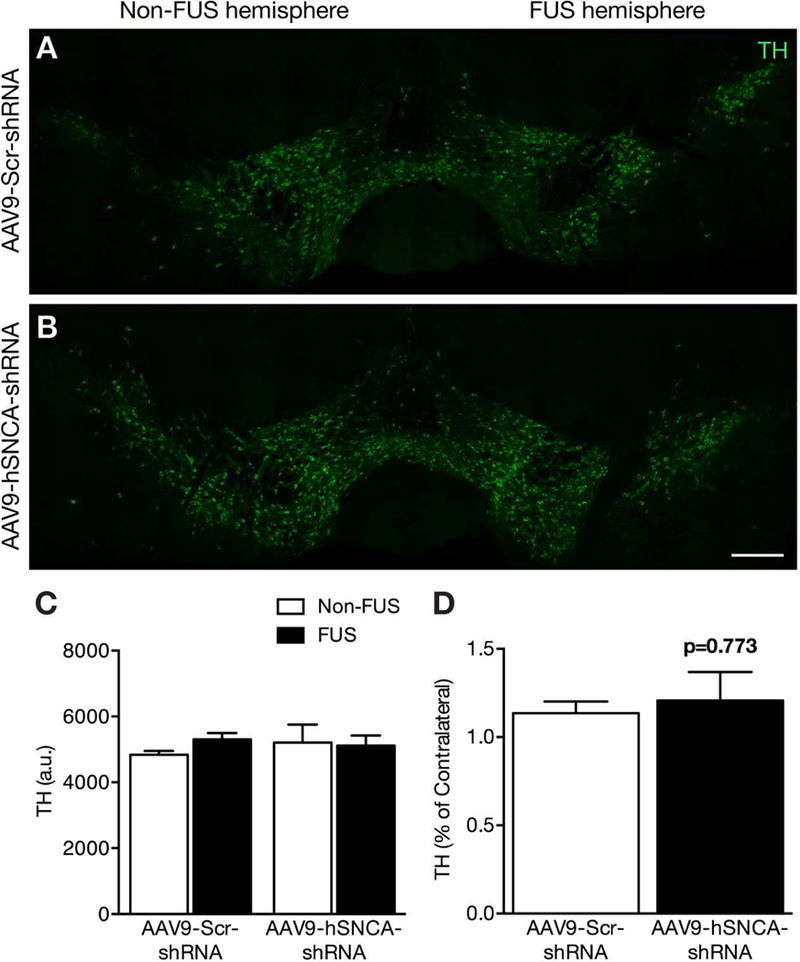
(A,B) TH expression was measured in nigral dopaminergic neurons following sonication to the SN. (C,D) There was no significant difference between AAV9-hSNCA-shRNA (black bar) and AAV9-Scr-shRNA injected animals (white bar), and comparing the FUS-treated side to the contralateral non-FUS hemisphere. Statistics: Paired, two-tailed, student’s t-test. Data represent the mean ± SEM. n = 4 per group. Tyrosine Hydroxylase, green. Scale bars: (A-B): 250 μm.
Absence of cell death or glial activation one month following FUS treatment
As measures of potential neuronal damage, we evaluated markers of cell death and glial activation in brain areas targeted with FUS and the AAV9 vectors. There was no discernible increase in TUNEL or cleaved caspase-3 staining due to FUS or α-syn knockdown in any brain region evaluated (Fig. 5A-L). The OB displayed some neuronal apoptosis in all groups (Fig. 5G-I), which is related to normal granule cell turnover86–88. Moreover, the distribution and intensity of glial markers including astrocytes (GFAP) and microglia/macrophages (Iba-1)89–91 were comparable in FUS-targeted areas between mice treated with Scr-shRNA or hSNCA-shRNA, and hSNCA-shRNA without FUS (Fig. 5M-X; Supplemental Fig. 2–5). These results are consistent with previous studies that reported a transient increase in glial activation and inflammatory markers after FUS treatment, using parameters that minimized cell death, and a subsequent return to baseline levels50, 55, 92–95. In accord with the unchanged TH staining in Fig. 4, the absence of cell death markers after one month of transgene expression further indicates a lack of neurotoxicity associated with α-syn gene silencing.
Fig. 5: MRI-guided FUS gene delivery does not induce neuronal apoptosis or inflammation.

(A-L) Representative confocal images of brain sections immunolabeled for TUNEL (green), cleaved caspase-3 (Cas3,red) and NeuN (blue). TUNEL- and cleaved caspase-3 positive cells were not detected in sonicated AAV9-Scr-shRNA or AAV9-hSNCA-shRNA-transduced areas of the (A, B) HC, (D, E) SN, (G, H) OB (relative to basal expression levels), and (J, K) DMN. (C, F, I, L) Immunostaining in non-FUS areas showed a similar pattern of apoptotic labeling. (M-X) Confocal images of brain sections depicting Iba-1(green), GFAP (red), and nuclei (DAPI, blue) immunolabeled cells. There was no difference in expression between the sonicated AAV9-hSNCA-shRNA and AAV9-Scr-shRNA treatment in the (M, N) HC, (P, Q) SN, (S, T) OB, and (V, W) DMN, as compared to immunolabeling in corresponding non-FUS regions (O, R, U, X). Scale bar: 100μm.
DISCUSSION
There are multiple difficulties associated with pharmacological treatment of CNS disorders. Primary among these is the inability of many drugs to cross the BBB at therapeutic levels. This limitation can be circumvented with transcranial MRI-guided FUS, which increases the permeability of the BBB locally and transiently for up to 6 hours when combined with intravenous microbubbles96. We and others have shown that FUS is a safe and practical technology for the therapeutic delivery of macromolecules, genes and cells into the CNS50, 52–55, 57. It is effective in a broad spectrum of species, from rodents to humans, to target selectively brain regions as small as 1–2 mm to an entire hemisphere.
In this study, we show that a single intravenous injection of AAV9 encoding α-syn inhibitory RNA, delivered to the brain using FUS in combination with microbubbles, is sufficient to decrease α-syn expression in FUS-targeted brain regions. Based on the Braak histopathological staging of PD, we sonicated the OB, DMN, SN, and HC, which represent a spectrum of brain regions affected from early to late PD8, 97, 98. The FUS-mediated increase in BBB permeability was confirmed by the diffusion of MRI contrast agent gadolinium around target areas and by the robust expression of the reporter turboGFP at one-month post-treatment, neither of which was detectable in non-sonicated hemispheres. Each FUS focal spot demarcated by gadolinium enhancement (Fig. 1D,F) is approximately 1 mm in diameter laterally and 3–4 mm along the beam, which is challenging to dissect without contamination from surrounding tissue expressing normal levels of α-syn. Therefore, to validate the FUS-mediated knockdown of α-syn in multiple discrete brain areas, we relied on immunofluorescence which offers superior spatial resolution compared to western blotting to detect changes in protein levels. α-Syn immunoreactivity was decreased in FUS-targeted areas of animals treated with hSNCA-shRNA relative to control Scr-shRNA. The overall extent of the α-syn knockdown varied depending on the brain region, as transgene expression is influenced by local differences in transduction efficiency, cell-specific tropism, and the ratio of cell soma to axons99. Transduction efficiency was consistent with our previous reports48, 51 and the graded expression of turboGFP in cells within each brain region implies a corresponding range of α-syn knockdown in those cells. In the current study, FUS targeting was intentionally limited to small brain volumes to establish proof of concept. FUS exposure to broader brain areas51, coupled with improvements in AAV vectors, including capsid, promoter and transgene, could further enhance therapeutic efficacy. Nevertheless, the decrease was clearly selective for α-syn expression because synaptophysin, which is also highly expressed in nerve terminals, was unchanged by the shRNA treatment and was therefore used to normalize α-syn within each region of interest. The α-syn shRNA reduced α-syn expression by at least 50% (p < 0.05) in the HC, SN, and OB, and by 40% (p = 0.08) in the DMN. A power analysis indicates that a total of 6 animals per treatment group would be required to reach the recommended power level of 0.80 with alpha set to 0.05 (one-tailed). Previous reports using stereotaxic injections to deliver α-syn interfering RNA or antisense oligonucleotides also effectively reduced transgenic and endogenous brain α-syn in rodents and primates30, 31, 33, 34, 39–41. Although the 35–60% knockdown in α-syn expression in those studies had minimal adverse effects, higher levels of α-syn gene silencing appeared to induce some nigral toxicity causing a loss of TH expression32, 36, 38. In contrast, we did not detect any loss of TH immunoreactivity in the SN following sonication and α-syn shRNA, either in comparison to the sonicated SN with control shRNA or to the contralateral non-sonicated SN in hSNCA-shRNA injected animals. Moreover, markers of apoptotic cell death or glial activation were not notably changed one month after the virus or FUS treatments, in accord with our previous studies50, 94, 95.
Idiopathic PD is an ideal neurodegenerative disorder for gene therapy: α-syn pathology, neurodegeneration, and dopamine loss are confined to discrete brain regions, and clinical motor symptoms are quantifiable. As the identification of prodromal motor and non-motor symptoms increasingly refine early diagnoses of PD10, 100–104, drug delivery to appropriate brain regions will be relevant for initiating disease-modifying treatments as early as possible. The ability to target specific brain regions for gene therapy, as shown here, may prevent spreading Lewy pathology following α-syn knockdown, and also rescue failing neurons when combined with regeneration strategies using growth factors. Previous Phase I-II clinical trials used viral vectors to express growth factors (neurturin, GDNF) or increase dopamine biosynthesis (TH, aromatic L-amino-acid decarboxylase, and GTP cyclohydrolase-1)105–107. These trials involved small numbers of patients with fairly advanced disease and revealed only transient or modest benefits despite long-lasting and well-tolerated transgene expression. Several current Phase I-II trials are targeting α-syn (NPT200–11, Neuropore Therapeutics; PRX002, Biotech Prothena; AFFITOPE-PD01A, AFFiRiS AG; BIIB-054, Biogen; anti-aggregant EGCG, Ludwig Maximillians University) (ClinicalTrials.gov). Whether these treatments, in single or combination therapies, will provide sufficient neuroprotection and regeneration for clinical improvement at early disease stages remains to be determined. Nevertheless, it is likely that enhanced delivery across the BBB will be beneficial therapeutically. For example, recent FUS-mediated GDNF expression in rat brain was shown to protect against 6-hydroxydopamine toxicity56, 58, and a Phase I clinical trial to evaluate low intensity FUS in Alzheimer patients has recently been completed (NCT02986932, ClinicalTrials.gov).
Our results represent an opportunity to alter the infiltration of α-syn pathology into the CNS prior to its effects on the SN. To our knowledge, not only is this the first report targeting the OB and the DMN regions for α-syn gene silencing, but also that multiple distal and discrete brain regions were targeted simultaneously. One limitation of our study is that we did not assess therapeutic benefits of α-syn knockdown. Studies are underway to determine the therapeutic window for neuroprotection and the rescue of motor symptoms after initiation of Lewy pathology in α-syn fibril seeding models. We anticipate that the choice of regimen and brain region will be aided by ongoing advances in prodromal biomarkers and imaging diagnostics. The use of FUS to deliver therapeutics in a targeted manner could open a range of opportunities for the treatment of synucleinopathies, particularly with complementary approaches including gene silencing, immunotherapy, anti-aggregants, and growth factors.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Martha C. Bohn (Northwestern University, Evanston, IL, USA) for providing the anti-α-syn and scrambled shRNA. This work was supported by the Weston Brain Institute and the Canadian Institutes of Health Research (CIHR) operating grants to AT (MOP130321 & PJT148736), IA (FRN137064), and KH (FRN 119312). KH was also funded by Canada Research Chair Program and the National Institute of Biomedical Imaging and Bioengineering of the National Institutes of Health (R01 EB003268). KX and FN received graduate scholarships from CIHR and the Queen Elizabeth II Graduate Scholarship in Science and Technology (QEII-GSST) Program, respectively. We thank Kelly Coultes for tissue extraction and processing; Paul Nagy and Alison Burgess for technical assistance with FUS experiments; Tammy Langman and Shawna Rideout-Gros for help with animal care.
Financial disclosure: This work was supported by the Weston Brain Institute and the Canadian Institutes of Health Research (CIHR) operating grants to AT (MOP130321 & PJT148736), IA (FRN137064), and KH (FRN 119312). KH was also funded by Canada Research Chair Program and the National Institute of Biomedical Imaging and Bioengineering of the National Institutes of Health (R01 EB003268). KX and FN received graduate scholarships from CIHR and the Queen Elizabeth II Graduate Scholarship in Science and Technology (QEII-GSST) Program, respectively.
Abbreviations:
- AAV9
adeno-associated virus serotype 9
- BBB
blood-brain barrier
- CMV
cytomegalovirus
- DMN
dorsal motor nucleus of the vagus
- FUS
focused ultrasound
- GFAP
glial fibrillary acidic protein
- HC
hippocampus
- hSNCA
human α-syn gene
- Iba-1
ionized calcium-binding adapter molecule 1
- IRES
internal ribosome entry site
- OB
olfactory bulb
- SN
substantia nigra
- shRNA
short hairpin RNA
- Scr
scrambled
- TH
Tyrosine hydroxylase
- turboGFP
turbo green fluorescent protein
- T1w
T1 weighted
- T2w
T2 weighted
- VG
vector genomes
Footnotes
Conflicts of interest:
REFERENCES
- 1.Iwai A, Masliah E, Yoshimoto M, et al. The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 1995;14(2):467–475. [DOI] [PubMed] [Google Scholar]
- 2.Clayton DF, George JM. The synucleins: a family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci 1998;21(6):249–254. [DOI] [PubMed] [Google Scholar]
- 3.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature 1997;388(6645):839–840. [DOI] [PubMed] [Google Scholar]
- 4.Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. ProcNatlAcadSciUSA 1998;95(11):6469–6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Braak H, Braak E. Pathoanatomy of Parkinson’s disease. JNeurol 2000;247 Suppl 2:II3–10. [DOI] [PubMed] [Google Scholar]
- 6.Fujiwara H, Hasegawa M, Dohmae N, et al. alpha-Synuclein is phosphorylated in synucleinopathy lesions. NatCell Biol 2002;4(2):160–164. [DOI] [PubMed] [Google Scholar]
- 7.Del Tredici K, Rub U, de Vos RA, Bohl JR, Braak H. Where does parkinson disease pathology begin in the brain? JNeuropatholExpNeurol 2002;61(5):413–426. [DOI] [PubMed] [Google Scholar]
- 8.Braak H, Tredici KD, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. NeurobiolAging 2003;24(2):197–211. [DOI] [PubMed] [Google Scholar]
- 9.Braak H, Del Tredici K. Nervous system pathology in sporadic Parkinson disease. Neurology 2008;70(20):1916–1925. [DOI] [PubMed] [Google Scholar]
- 10.Postuma RB, Berg D. Advances in markers of prodromal Parkinson disease. Nat Rev Neurol 2016;12(11):622–634. [DOI] [PubMed] [Google Scholar]
- 11.Conway KA, Harper JD, Lansbury PT Jr. Fibrils formed in vitro from alpha-synuclein and two mutant forms linked to Parkinson’s disease are typical amyloid. Biochemistry 2000;39(10):2552–2563. [DOI] [PubMed] [Google Scholar]
- 12.Hardy J Expression of normal sequence pathogenic proteins for neurodegenerative disease contributes to disease risk: ‘permissive templating’ as a general mechanism underlying neurodegeneration. Biochemical Society Transactions 2005;33:578–581. [DOI] [PubMed] [Google Scholar]
- 13.Olanow CW, Prusiner SB. Is Parkinson’s disease a prion disorder?1. ProcNatlAcadSciUSA 2009;106(31):12571–12572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Angot E, Steiner JA, Hansen C, Li JY, Brundin P. Are synucleinopathies prion-like disorders? Lancet Neurol 2010;9(11):1128–1138. [DOI] [PubMed] [Google Scholar]
- 15.Luk KC, Song C, O’Brien P, et al. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells2. ProcNatlAcadSciUSA 2009;106(47):20051–20056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Desplats P, Lee HJ, Bae EJ, et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein3. ProcNatlAcadSciUSA 2009;106(31):13010–13015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hansen C, Angot E, Bergstrom AL, et al. alpha-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells1. JClinInvest 2011;121(2):715–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Volpicelli-Daley LA, Luk KC, Patel TP, et al. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death1. Neuron 2011;72(1):57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luk KC, Kehm V, Carroll J, et al. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012;338(6109):949–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nonaka T, Watanabe ST, Iwatsubo T, Hasegawa M. Seeded aggregation and toxicity of {alpha}-synuclein and tau: cellular models of neurodegenerative diseases1. JBiolChem 2010;285(45):34885–34898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Watts JC, Giles K, Oehler A, et al. Transmission of multiple system atrophy prions to transgenic mice. Proceedings of the National Academy of Sciences of the United States of America 2013;110(48):19555–19560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Woerman AL, Stohr J, Aoyagi A, et al. Propagation of prions causing synucleinopathies in cultured cells. Proceedings of the National Academy of Sciences of the United States of America 2015;112(35):E4949–4958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li JY, Englund E, Holton JL, et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation2. NatMed 2008;14(5):501–503. [DOI] [PubMed] [Google Scholar]
- 24.Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease3. NatMed 2008;14(5):504–506. [DOI] [PubMed] [Google Scholar]
- 25.Olanow CW, Brundin P. Parkinson’s disease and alpha synuclein: is Parkinson’s disease a prion-like disorder? Mov Disord 2013;28(1):31–40. [DOI] [PubMed] [Google Scholar]
- 26.Singleton AB, Farrer M, Johnson J, et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science 2003;302(5646):841. [DOI] [PubMed] [Google Scholar]
- 27.Chartier-Harlin MC, Kachergus J, Roumier C, et al. alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004;364(9440):1167–1169. [DOI] [PubMed] [Google Scholar]
- 28.Ibanez P, Bonnet AM, Debarges B, et al. Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet 2004;364(9440):1169–1171. [DOI] [PubMed] [Google Scholar]
- 29.Brundin P, Dave KD, Kordower JH. Therapeutic approaches to target alpha-synuclein pathology. Exp Neurol 2017. [DOI] [PMC free article] [PubMed]
- 30.Lewis J, Melrose H, Bumcrot D, et al. In vivo silencing of alpha-synuclein using naked siRNA. Mol Neurodegener 2008;3:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McCormack AL, Mak SK, Henderson JM, Bumcrot D, Farrer MJ, Di Monte DA. Alpha-synuclein suppression by targeted small interfering RNA in the primate substantia nigra. PloS one 2010;5(8):e12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gorbatyuk OS, Li S, Nash K, et al. In vivo RNAi-mediated alpha-synuclein silencing induces nigrostriatal degeneration2. MolTher 2010;18(8):1450–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khodr CE, Sapru MK, Pedapati J, et al. An alpha-synuclein AAV gene silencing vector ameliorates a behavioral deficit in a rat model of Parkinson’s disease, but displays toxicity in dopamine neurons. Brain Res 2011;1395:94–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cooper JM, Wiklander PB, Nordin JZ, et al. Systemic exosomal siRNA delivery reduced alpha-synuclein aggregates in brains of transgenic mice. Mov Disord 2014;29(12):1476–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khodr CE, Becerra A, Han Y, Bohn MC. Targeting alpha-synuclein with a microRNA-embedded silencing vector in the rat substantia nigra: positive and negative effects. Brain Res 2014;1550:47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Collier TJ, Redmond DE Jr., Steece-Collier K, Lipton JW, Manfredsson FP. Is Alpha-Synuclein Loss-of-Function a Contributor to Parkinsonian Pathology? Evidence from Non-human Primates. Front Neurosci 2016;10:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alarcon-Aris D, Recasens A, Galofre M, et al. Selective alpha-Synuclein Knockdown in Monoamine Neurons by Intranasal Oligonucleotide Delivery: Potential Therapy for Parkinson’s Disease. Mol Ther 2018;26(2):550–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Benskey MJ, Sellnow RC, Sandoval IM, Sortwell CE, Lipton JW, Manfredsson FP. Silencing Alpha Synuclein in Mature Nigral Neurons Results in Rapid Neuroinflammation and Subsequent Toxicity. Front Mol Neurosci 2018;11:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zharikov AD, Cannon JR, Tapias V, et al. shRNA targeting alpha-synuclein prevents neurodegeneration in a Parkinson’s disease model. J Clin Invest 2015;125(7):2721–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cole T, Paumier K, Collier T, et al. Snca targeted antisense oligonucleotides modulate progression of pathological Ddeposition in alpha synuclein rodent transmission models of Parkinson’s Disease. 142nd Annual Meeting of The American Neurological Association; 2017; San Diego. [Google Scholar]
- 41.Luna E, Decker SC, Riddle DM, et al. Differential alpha-synuclein expression contributes to selective vulnerability of hippocampal neuron subpopulations to fibril-induced toxicity. Acta Neuropathol 2018. [DOI] [PMC free article] [PubMed]
- 42.Pardridge WM. CSF, blood-brain barrier, and brain drug delivery. Expert Opin Drug Deliv 2016;13(7):963–975. [DOI] [PubMed] [Google Scholar]
- 43.Pardridge WM. Targeted delivery of protein and gene medicines through the blood-brain barrier. Clin Pharmacol Ther 2015;97(4):347–361. [DOI] [PubMed] [Google Scholar]
- 44.Tuszynski MH, Yang JH, Barba D, et al. Nerve Growth Factor Gene Therapy: Activation of Neuronal Responses in Alzheimer Disease. JAMA Neurol 2015. [DOI] [PMC free article] [PubMed]
- 45.Sehara Y, Fujimoto KI, Ikeguchi K, et al. Persistent Expression of Dopamine-Synthesizing Enzymes 15 Years After Gene Transfer in a Primate Model of Parkinson’s Disease. Hum Gene Ther Clin Dev 2017;28(2):74–79. [DOI] [PubMed] [Google Scholar]
- 46.Coune PG, Schneider BL, Aebischer P. Parkinson’s disease: gene therapies. Cold Spring Harb Perspect Med 2012;2(4):a009431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Burgess A, Hynynen K. Microbubble-Assisted Ultrasound for Drug Delivery in the Brain and Central Nervous System. Adv Exp Med Biol 2016;880:293–308. [DOI] [PubMed] [Google Scholar]
- 48.Weber-Adrian D, Thevenot E, O’Reilly MA, et al. Gene delivery to the spinal cord using MRI-guided focused ultrasound. Gene Ther 2015. [DOI] [PMC free article] [PubMed]
- 49.Burgess A, Dubey S, Yeung S, et al. Alzheimer disease in a mouse model: MR imaging-guided focused ultrasound targeted to the hippocampus opens the blood-brain barrier and improves pathologic abnormalities and behavior. Radiology 2014;273(3):736–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jordao JF, Thevenot E, Markham-Coultes K, et al. Amyloid-beta plaque reduction, endogenous antibody delivery and glial activation by brain-targeted, transcranial focused ultrasound. Exp Neurol 2013;248:16–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thevenot E, Jordao JF, O’Reilly MA, et al. Targeted delivery of scAAV9 to the brain using MRI-guided focused ultrasound. Hum Gene Ther 2012. [DOI] [PMC free article] [PubMed]
- 52.Burgess A, Ayala-Grosso CA, Ganguly M, Jordao JF, Aubert I, Hynynen K. Targeted delivery of neural stem cells to the brain using MRI-guided focused ultrasound to disrupt the blood-brain barrier. PloS one 2011;6(11):e27877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jordao JF, Ayala-Grosso CA, Markham K, et al. Antibodies targeted to the brain with image-guided focused ultrasound reduces amyloid-beta plaque load in the TgCRND8 mouse model of Alzheimer’s disease. PloS one 2010;5(5):e10549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nisbet RM, Van der Jeugd A, Leinenga G, Evans HT, Janowicz PW, Gotz J. Combined effects of scanning ultrasound and a tau-specific single chain antibody in a tau transgenic mouse model. Brain 2017;140(5):1220–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leinenga G, Gotz J. Scanning ultrasound removes amyloid-beta and restores memory in an Alzheimer’s disease mouse model. Sci Transl Med 2015;7(278):278ra233. [DOI] [PubMed] [Google Scholar]
- 56.Fan CH, Ting CY, Lin CY, et al. Noninvasive, Targeted, and Non-Viral Ultrasound-Mediated GDNF-Plasmid Delivery for Treatment of Parkinson’s Disease. Sci Rep 2016;6:19579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Samiotaki G, Acosta C, Wang S, Konofagou EE. Enhanced delivery and bioactivity of the neurturin neurotrophic factor through focused ultrasound-mediated blood--brain barrier opening in vivo. J Cereb Blood Flow Metab 2015;35(4):611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mead BP, Kim N, Miller GW, et al. Novel Focused Ultrasound Gene Therapy Approach Noninvasively Restores Dopaminergic Neuron Function in a Rat Parkinson’s Disease Model. Nano Lett 2017;17(6):3533–3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Han Y, Khodr CE, Sapru MK, Pedapati J, Bohn MC. A microRNA embedded AAV alpha-synuclein gene silencing vector for dopaminergic neurons. Brain Res 2011;1386:15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wislet-Gendebien S, D’Souza C, Kawarai T, et al. Cytosolic proteins regulate alpha-synuclein dissociation from presynaptic membranes. JBiolChem 2006;281(43):32148–32155. [DOI] [PubMed] [Google Scholar]
- 61.Wislet-Gendebien S, Visanji NP, Whitehead SN, et al. Differential regulation of wild-type and mutant alpha-synuclein binding to synaptic membranes by cytosolic factors1. BMCNeurosci 2008;9:92. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 62.Chishti MA, Yang DS, Janus C, et al. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. JBiolChem 2001;276(24):21562–21570. [DOI] [PubMed] [Google Scholar]
- 63.Citron M, Westaway D, Xia W, et al. Mutant presenilins of Alzheimer’s disease increase production of 42- residue amyloid beta-protein in both transfected cells and transgenic mice. NatMed 1997;3(1):67–72. [DOI] [PubMed] [Google Scholar]
- 64.DeArmond SJ, Sanchez H, Yehiely F, et al. Selective neuronal targeting in prion disease. Neuron 1997;19(6):1337–1348. [DOI] [PubMed] [Google Scholar]
- 65.Oesch B, Westaway D, Walchli M, et al. A cellular gene encodes scrapie PrP 27–30 protein. Cell 1985;40(4):735–746. [DOI] [PubMed] [Google Scholar]
- 66.Scott M, Foster D, Mirenda C, et al. Transgenic mice expressing hamster prion protein produce species-specific scrapie infectivity and amyloid plaques. Cell 1989;59(5):847–857. [DOI] [PubMed] [Google Scholar]
- 67.Westaway D, DeArmond SJ, Cayetano-Canlas J, et al. Degeneration of skeletal muscle, peripheral nerves, and the central nervous system in transgenic mice overexpressing wild-type prion proteins. Cell 1994;76(1):117–129. [DOI] [PubMed] [Google Scholar]
- 68.Telling GC, Haga T, Torchia M, Tremblay P, DeArmond SJ, Prusiner SB. Interactions between wild-type and mutant prion proteins modulate neurodegeneration in transgenic mice. Genes Dev 1996;10(14):1736–1750. [DOI] [PubMed] [Google Scholar]
- 69.Telling GC, Scott M, Mastrianni J, et al. Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell 1995;83(1):79–90. [DOI] [PubMed] [Google Scholar]
- 70.O’Reilly MA, Hynynen K. Blood-brain barrier: real-time feedback-controlled focused ultrasound disruption by using an acoustic emissions-based controller. Radiology 2012;263(1):96–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Franklin KBJ, Paxinos G. Paxinos and Franklin’s The mouse brain in stereotaxic coordinates. Fourth edition. ed. Amsterdam: Academic Press, an imprint of Elsevier, 2013. [Google Scholar]
- 72.Faul F, Erdfelder E, Lang AG, Buchner A. G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods 2007;39(2):175–191. [DOI] [PubMed] [Google Scholar]
- 73.Yang FY, Horng SC, Lin YS, Kao YH. Association between contrast-enhanced MR images and blood-brain barrier disruption following transcranial focused ultrasound. J Magn Reson Imaging 2010;32(3):593–599. [DOI] [PubMed] [Google Scholar]
- 74.Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol 2009;27(1):59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Duque S, Joussemet B, Riviere C, et al. Intravenous administration of self-complementary AAV9 enables transgene delivery to adult motor neurons. Mol Ther 2009;17(7):1187–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Watakabe A, Ohtsuka M, Kinoshita M, et al. Comparative analyses of adeno-associated viral vector serotypes 1, 2, 5, 8 and 9 in marmoset, mouse and macaque cerebral cortex. Neurosci Res 2015;93:144–157. [DOI] [PubMed] [Google Scholar]
- 77.Timbie KF, Mead BP, Price RJ. Drug and gene delivery across the blood-brain barrier with focused ultrasound. J Control Release 2015;219:61–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kachroo A, Schwarzschild MA. Adenosine A2A receptor gene disruption protects in an alpha-synuclein model of Parkinson’s disease. Ann Neurol 2012;71(2):278–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Amschl D, Neddens J, Havas D, et al. Time course and progression of wild type alpha-synuclein accumulation in a transgenic mouse model. BMC Neurosci 2013;14:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Recasens A, Dehay B, Bove J, et al. Lewy body extracts from Parkinson disease brains trigger alpha-synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol 2014;75(3):351–362. [DOI] [PubMed] [Google Scholar]
- 81.Flores-Cuadrado A, Ubeda-Banon I, Saiz-Sanchez D, de la Rosa-Prieto C, Martinez-Marcos A. Hippocampal alpha-synuclein and interneurons in Parkinson’s disease: Data from human and mouse models. Mov Disord 2016;31(7):979–988. [DOI] [PubMed] [Google Scholar]
- 82.Bassil F, Guerin PA, Dutheil N, et al. Viral-mediated oligodendroglial alpha-synuclein expression models multiple system atrophy. Mov Disord 2017;32(8):1230–1239. [DOI] [PubMed] [Google Scholar]
- 83.Kim YC, Miller A, Lins LC, et al. RNA Interference of Human alpha-Synuclein in Mouse. Front Neurol 2017;8:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Frahm S, Melis V, Horsley D, et al. Alpha-Synuclein transgenic mice, h-alpha-SynL62, display alpha-Syn aggregation and a dopaminergic phenotype reminiscent of Parkinson’s disease. Behav Brain Res 2018;339:153–168. [DOI] [PubMed] [Google Scholar]
- 85.Chu Y, Buchman AS, Olanow CW, Kordower JH. Do subjects with minimal motor features have prodromal Parkinson disease? Ann Neurol 2018;83(3):562–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Najbauer J, Leon M. Olfactory experience modulated apoptosis in the developing olfactory bulb. Brain Res 1995;674(2):245–251. [DOI] [PubMed] [Google Scholar]
- 87.Imayoshi I, Sakamoto M, Ohtsuka T, et al. Roles of continuous neurogenesis in the structural and functional integrity of the adult forebrain. Nat Neurosci 2008;11(10):1153–1161. [DOI] [PubMed] [Google Scholar]
- 88.Liu H, Guthrie KM. Neuronal replacement in the injured olfactory bulb. Exp Neurol 2011;228(2):270–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Eng LF. Glial fibrillary acidic protein (GFAP): the major protein of glial intermediate filaments in differentiated astrocytes. J Neuroimmunol 1985;8(4–6):203–214. [DOI] [PubMed] [Google Scholar]
- 90.Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S. Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Res Mol Brain Res 1998;57(1):1–9. [DOI] [PubMed] [Google Scholar]
- 91.Imai Y, Kohsaka S. Intracellular signaling in M-CSF-induced microglia activation: role of Iba1. Glia 2002;40(2):164–174. [DOI] [PubMed] [Google Scholar]
- 92.Kovacs ZI, Kim S, Jikaria N, et al. Disrupting the blood-brain barrier by focused ultrasound induces sterile inflammation. Proceedings of the National Academy of Sciences of the United States of America 2017;114(1):E75–E84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kinoshita M, McDannold N, Jolesz FA, Hynynen K. Noninvasive localized delivery of Herceptin to the mouse brain by MRI-guided focused ultrasound-induced blood-brain barrier disruption. Proceedings of the National Academy of Sciences of the United States of America 2006;103(31):11719–11723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.McMahon D, Bendayan R, Hynynen K. Acute effects of focused ultrasound-induced increases in blood-brain barrier permeability on rat microvascular transcriptome. Sci Rep 2017;7:45657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.McMahon D, Hynynen K. Acute Inflammatory Response Following Increased Blood-Brain Barrier Permeability Induced by Focused Ultrasound is Dependent on Microbubble Dose. Theranostics 2017;7(16):3989–4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hynynen K, McDannold N, Vykhodtseva N, Jolesz FA. Noninvasive MR imaging-guided focal opening of the blood-brain barrier in rabbits. Radiology 2001;220(3):640–646. [DOI] [PubMed] [Google Scholar]
- 97.Braak H, Rub U, Steur ENHJ, Del Tredici K, De Vos RAI. Cognitive status correlates with neuropathologic stage in Parkinson disease. Neurology 2005;64(8):1404–1410. [DOI] [PubMed] [Google Scholar]
- 98.Hall H, Reyes S, Landeck N, et al. Hippocampal Lewy pathology and cholinergic dysfunction are associated with dementia in Parkinson’s disease. Brain 2014;137(Pt 9): 2493–2508. [DOI] [PubMed] [Google Scholar]
- 99.Aschauer DF, Kreuz S, Rumpel S. Analysis of transduction efficiency, tropism and axonal transport of AAV serotypes 1, 2, 5, 6, 8 and 9 in the mouse brain. PloS one 2013;8(9):e76310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ross GW, Abbott RD, Petrovitch H, Tanner CM, White LR. Pre-motor features of Parkinson’s disease: the Honolulu-Asia Aging Study experience. Parkinsonism Relat Disord 2012;18 Suppl 1:S199–202. [DOI] [PubMed] [Google Scholar]
- 101.Schrag A, Horsfall L, Walters K, Noyce A, Petersen I. Prediagnostic presentations of Parkinson’s disease in primary care: a case-control study. Lancet Neurol 2015;14(1):57–64. [DOI] [PubMed] [Google Scholar]
- 102.Berg D, Postuma RB, Adler CH, et al. MDS research criteria for prodromal Parkinson’s disease. Mov Disord 2015;30(12):1600–1611. [DOI] [PubMed] [Google Scholar]
- 103.Darweesh SK, Verlinden VJ, Stricker BH, Hofman A, Koudstaal PJ, Ikram MA. Trajectories of prediagnostic functioning in Parkinson’s disease. Brain 2017;140(2):429–441. [DOI] [PubMed] [Google Scholar]
- 104.Breen DP, Lang AE. Tracking the course of prodromal Parkinson’s disease. Brain 2017;140(2):259–262. [DOI] [PubMed] [Google Scholar]
- 105.O’Connor DM, Boulis NM. Gene therapy for neurodegenerative diseases. Trends Mol Med 2015;21(8):504–512. [DOI] [PubMed] [Google Scholar]
- 106.Piguet F, Alves S, Cartier N. Clinical Gene Therapy for Neurodegenerative Diseases: Past, Present, and Future. Hum Gene Ther 2017;28(11):988–1003. [DOI] [PubMed] [Google Scholar]
- 107.Bartus RT, Johnson EM Jr. Clinical tests of neurotrophic factors for human neurodegenerative diseases, part 1: Where have we been and what have we learned? Neurobiol Dis 2017;97(Pt B):156–168. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.