

Abstract
Comorbidities are a hallmark of stroke that both increase the incidence of stroke and worsen outcome. Hypertension is prevalent in the stroke population and the most important modifiable risk factor for stroke. Hypertensive disorders promote stroke through increased shear stress, endothelial dysfunction, and large artery stiffness that transmits pulsatile flow to the cerebral microcirculation. Hypertension also promotes cerebral small vessel disease through several mechanisms, including hypoperfusion, diminished autoregulatory capacity and localized increase in blood–brain barrier permeability. Preeclampsia, a hypertensive disorder of pregnancy, also increases the risk of stroke 4–5-fold compared to normal pregnancy that predisposes women to early-onset cognitive impairment. In this review, we highlight how comorbidities and concomitant disorders are not only risk factors for ischemic stroke, but alter the response to acute ischemia. We focus on hypertension as a comorbidity and its effects on the cerebral circulation that alters the pathophysiology of ischemic stroke and should be considered in guiding future therapeutic strategies.
Keywords: Hypertension, ischemic stroke, cerebral small vessel disease, preeclampsia, collateral circulation
Introduction
Stroke is a highly heterogeneous condition that occurs in all demographics. Unlike pediatric stroke, ischemic stroke in the adult is predominantly a disorder of comorbidities (e.g. hypertension, diabetes mellitus, hyperlipidemia) and/or altered coagulation states (e.g. pregnancy and preeclampsia). Stroke is the leading cause of long-term disability in the United States and is currently the 5th leading cause of death.1–4 The decline in stroke mortality over the last decade can be partly attributed to the aggressive treatment of hypertension and dyslipidemia that is common in the stroke population.3,5 In fact, hypertension is the most important modifiable risk factor for all types of stroke. The incidence of stroke increases proportionally with both systolic and diastolic pressure, increasing the relative risk 3.1-fold for men and 2.9-fold for women.6,7 Blood pressure also increases in the acute phase of stroke, through unexplained mechanisms, fueling clinical controversy over whether or not and to what extent it should be treated.
Hypertension also worsens stroke outcome. Patients with pre-existing hypertension have small amounts of salvageable tissue (penumbra) and larger infarctions compared to normotensive patients, although lower blood pressure is also detrimental in stroke.8–11 The risk of stroke in the setting of hypertension is also far-reaching. Hypertension is a primary driver of cerebral small vessel disease (CSVD) that leads to cognitive decline and lacunar stroke. In addition, recent studies have shown that in women with prior preeclampsia, a common hypertensive disorder of pregnancy, the long-term risk of stroke is increased 4–5-fold.12–14 The impact of hypertension on the cerebral circulation is therefore of profound importance to the stroke field.
Studies in animal models have demonstrated striking effects of hypertension on the cerebral circulation and have been invaluable in helping to understand the disease process that leads to increased small and large vessel disease risk and worsened outcome from stroke. Inward remodeling of small and large arteries decreases lumen diameters and vasodilatory reserve that if prolonged can cause hypoperfusion and hemodynamic compromise.15,16 Repeated mechanical stress during hypertension and degradation of elastin fibers in the vascular wall stiffens large arteries and transmits the pulsatile load downstream into the brain parenchyma.17,18 Endothelial dysfunction and diminished nitric oxide (NO) are also associated with hypertension that has negative consequences for the brain, including increasing cerebrovascular resistance (CVR) and diminished autoregulatory capacity.19 In addition, hypertension increases shear stress on the vascular endothelium – the force per unit area created when blood flows over the endothelium. Shear stress is directly related to the velocity of blood flow and viscosity, and inversely proportional to the cube of arterial radius.20 Under normal conditions, increased shear stress causes an adaptive vasodilatory response through increased NO production that serves to normalize shear stress.20–22 However, under hypertensive conditions that impair NO, the adaptive response to high shear stress is impaired, leading to endothelial damage and upregulation of atherogenic genes.20–22 Thus, increased shear stress on the cerebral endothelium during hypertension causes atheroma formation and subsequent atherosclerosis, an important underlying risk factor for large and small vessel occlusion.23,24 This review highlights basic, clinical and translational studies on the impact of hypertension on the cerebral circulation and how structural and functional alterations of cerebral vessels during hypertension promote stroke and stroke injury. Using hypertension as an example, we underscore how comorbidities or concomitant disorders are not solely risk factors for ischemic stroke, but affect the cerebral circulation that alters the response to acute ischemia. Consideration of these myriad effects on the pathophysiology of ischemic stroke may be instrumental in guiding future therapeutic strategies.
Cerebral blood flow autoregulation
The brain is an organ that regulates cerebral blood flow very effectively in the face of perfusion pressure fluctuations, a response that importantly enables the brain to match its metabolic demand to the blood supply.25 Autoregulation of cerebral blood flow was first described by Lassen26 in 1959 after analyzing a number of studies in the human in which cerebral blood flow was measured in response to altering blood pressure using hypotensive medication, tilting head-up, inhalation of carbon dioxide, sympathectomy or adrenalectomy mainly in hypertensive subjects. Lassen found that relatively constant cerebral blood flow could be maintained at systemic blood pressure between approximately 60 to 160 mm Hg.2 Similar cerebral blood flow autoregulation values have been reported in many other species, including dogs,27 baboons,28 monkeys,29 cats,30 lambs,31 pigs,32 rabbits,33 rats34 and mice.35 Above and below these limits, autoregulation is impaired and cerebral blood flow becomes dependent on arterial pressure in a linear fashion. When blood pressure is not within the limits of autoregulation, or autoregulation is impaired, there is considerable risk of brain injury. For example, when cerebral perfusion pressure falls below the lower limit of autoregulation, cerebral ischemia ensues that can cause permanent injury.36–38
The impact of chronic hypertension on the cerebral blood flow autoregulation was first reported in 1973 by Strandgaard et al.39 in a group of patients with essential hypertension. This study showed that both the upper and the lower limits of the cerebral blood flow autoregulation curve were shifted to the higher pressures when compared to normotensive humans.38 In rat models of renal and spontaneous hypertension, similar shift of both limits of the cerebral blood flow autoregulation curves was found.39–42 Harper and Bohlen41 used hemorrhagic hypotension and phenylephrine infusion to decrease and increase blood pressure, respectively, in spontaneously hypertensive rats (SHRs) and normotensive Wistar-Kyoto rats (WKY) to show the range of cerebral blood flow autoregulation increased from ∼60 to 150 mm Hg in WKY to ∼110 to 200 mm Hg in SHR. Figure 1 shows how the CBF autoregulatory curve is shifted to higher pressures in hypertension. Importantly, the shift in the autoregulatory curve to higher pressures is thought to be protective of the brain during chronic hypertension by limiting the damaging effects of high blood pressure to the downstream microcirculation. However, a shift in the lower threshold to higher pressures can be detrimental during acute hypotension in which the ability to maintain constant cerebral blood flow is diminished and therefore increases the susceptibility of ischemic injury. Interestingly, attempts have been made to normalize cerebral blood flow autoregulation during chronic hypertension, mostly in SHR. Long-term antihypertensive treatment with a vasodilator (budralazine) for nine weeks normalized the upper but not the lower limit of cerebral blood flow autoregulation in SHR.43 The calcium channel blocker benidipine treatment for eight days normalized both upper and lower limits of cerebral blood flow autoregulation in SHR.44 Acute angiotensin-converting enzyme (ACE) inhibitor captopril45 and both acute and longer-term (3–14 days) angiotensin type 1 receptor (AT1R) blocker candesartan45,46 also provided normalization of both the upper and lower thresholds of cerebral blood flow autoregulation in SHR.
Figure 1.
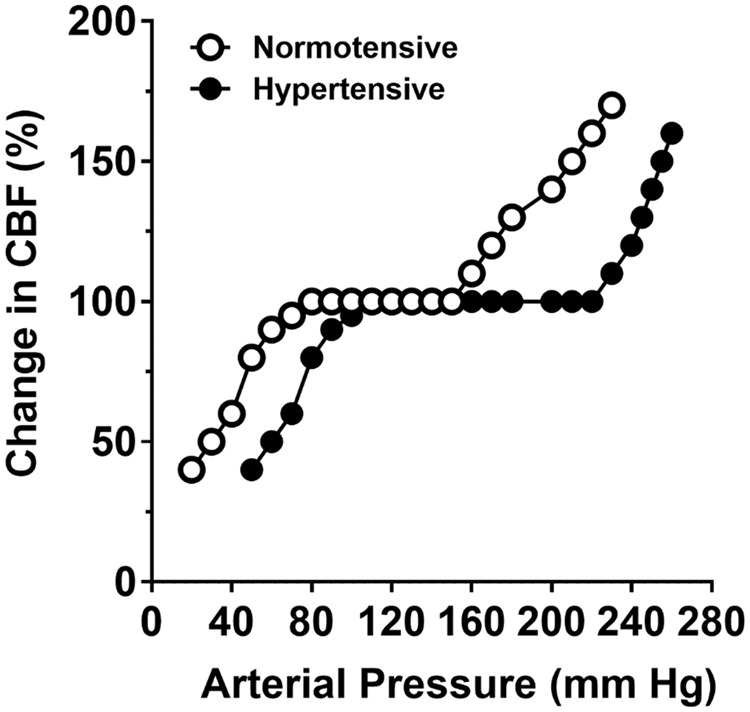
Graph showing curve of cerebral blood flow autoregulation in hypertensive SHR (closed circle) compared to normotensive WKY (open circle).
Ischemic stroke can impair cerebral blood flow autoregulation that is associated with worsened outcome. Early studies using radioisotope injection into the carotid artery found that autoregulation was impaired in the ipsilateral hemisphere; however, distinction between core infarction and penumbra was not made.47–49 More recent studies using transcranial Doppler to assess small fluctuations in blood pressure found that in patients with large ischemic strokes, autoregulation was significantly altered.50,51 In addition, patients with impaired autoregulation had worse clinical outcome than those with intact autoregulation.50,51 Other studies have shown little to no effect of stroke on cerebral blood flow autoregulation. Using single-photon emission computed tomography and repeated cerebral blood flow measurements after decreasing blood pressure with nimodipine, there was no change in cerebral blood flow, suggesting intact autoregulation.52 However, it is worth noting that substantially changing blood pressure in humans is difficult, and potentially dangerous especially in stroke patients. Therefore, defining the upper and lower limits of autoregulation and/or how effective it is in patients with existing brain injury is complex and risky. In addition, despite a plethora of animal studies defining autoregulation of CBF, there are only limited data in ischemic stroke patients for this reason.
Cerebral blood flow autoregulation is facilitated by changes in diameter of resistance blood vessels, leading to changes in vascular resistance and hence cerebral blood flow. Large artery resistance in the brain is high and therefore both large and small vessels participate in the autoregulation of cerebral blood flow.53 There may be several mechanisms by which ischemic stroke affects autoregulation of cerebral blood flow, including derangements in neuronal activation due to ischemic injury, biproducts of altered metabolism that are vasoactive and/or cause vascular dysfunction in response to ischemia and reperfusion. All brain blood vessels seem to have some degree of myogenic tone, which is tonic basal constriction to intraluminal pressure. Our own studies have shown that myogenic tone of middle cerebral arteries was decreased at post-ischemic reperfusion with the longer duration of ischemia, the greater loss of tone.54 As the myogenic response is a primary mechanism of autoregulation,55 the loss of tone in the MCA could contribute to impaired autoregulation during stroke. On the contrary, post-ischemic reperfusion caused an increase in tone in intraparenchymal arterioles,56 demonstrating the significant heterogeneity in vascular segments in the brain and the response to ischemia and reperfusion.
Structural remodeling during hypertension
One of the most important consequences of chronic hypertension is its effect on cerebral arteries and arterioles that increases CVR and causes hypoperfusion of the brain.15,16 Increased tone and structurally smaller lumens of pial vessels during hypertension are well-documented.57–61 These vascular changes associated with chronic hypertension increase the severity and extent of hypoperfusion and decrease the amount of penumbral tissue during ischemic stroke.62–66 Thus, understanding mechanisms by which chronic hypertension affects vascular function is important for stroke outcome as well as chronic hypoperfusion that can lead to stroke and cognitive decline.
Chronic hypertension causes profound changes in the cerebral circulation that impacts cerebral blood flow, one of them being structural remodeling. Both hypertrophy and inward remodeling of cerebral arteries and arterioles are commonly observed in the cerebral circulation of animal models of hypertension, mostly in SHR and stroke-prone SHR (SHRSP). Hypertrophy of the vessel wall is defined as increased wall thickness, increased wall cross-sectional areas, or increased wall-to-lumen ratio.67 Inward remodeling, on the other hand, was first defined as smaller external diameter in cerebral arterioles by Baumbach and Heistad in 1989.68 Later inward remodeling was re-defined and standardized as smaller lumen diameter, which is more important in the context of vascular resistance because lumen diameter is the most important determinant of resistance to flow.67 Hypertension-induced hypertrophy and inward remodeling are considered protective and also detrimental in cerebral arterioles. Law of LaPlace describes wall tension in a thinned-wall cylindrical vessel by the formula: wall tension = intraluminal pressure × radius and therefore wall stress is wall tension/wall thickness. In this relationship, increased intraluminal pressure, as seen in chronic hypertension, increases wall tension and wall stress. Blood vessels can therefore normalize wall tension and wall stress by decreasing the radius and increasing wall thickness, respectively. In the brain circulation, it has been shown that cerebral arterioles in SHRSP underwent reduced radius (inward remodeling) and increased wall thickness (wall hypertrophy).68,69 Normalized wall tension and wall stress are believed to protect against damaging effect of hypertension on the wall of cerebral vasculature. On the other hand, hypertrophy and inward remodeling of cerebral arteries and arterioles limits vasodilator reserve, i.e. the capacity of the vessel to dilate. In addition, vessel wall hypertrophy of cerebral arteries and arterioles increases the contractile smooth muscle mass and therefore enhances vasoconstrictor responsiveness.70,71 Inward remodeling also reduces maximum diameter of cerebral arterioles during chronic hypertension and therefore reduces the capacity to dilate during acute hypotension.72
The underlying mechanisms of hypertension-induced hypertrophy and inward remodeling in cerebral arterioles have been extensively studied. Strong evidence now suggests that increased angiotensin II (Ang II) in hypertension, and not hypertension itself, is the main determinant of inward remodeling in cerebral arteries and arterioles, based on a number of observations. First, low-dose use of the ACE-inhibitor perindopril inhibited inward remodeling in SHRSP equally effective when compared to high-dose perindopril although low-dose perindopril was only half as effective as high dose in reducing blood pressure.73 Second, the beta-adrenergic receptor blocker propranolol did not attenuate inward remodeling although it was as effective as low-dose perindopril in reducing blood pressure in SHRSP.73 Third, low- and high-dose Ang II infusion for 28 days in mice was equally effective in producing inward remodeling in cerebral arterioles although low-dose Ang II did not significantly increase blood pressure.74 Fourth, treatment with indapamide, a thiazide-like diuretic, reduced blood pressure in SHR but failed to attenuate inward remodeling in cerebral arterioles.75 Further mechanistic studies using a high-dose Ang II infusion model in transgenic knockout (KO) mice showed that caveolin-1-mediated activation of matrix metalloproteinase-9 was responsible for inward remodeling of cerebral arterioles.76
Vasoconstrictor stimuli during hypertension
In addition to structural remodeling that limits the vasodilatory capacity of cerebral arteries and arterioles, reactivity to a number of vasoactive agents is different in SHRSP compared to WKY. For example, cerebral arterioles dilate to serotonin in WKY but constrict in SHRSP.77 In addition, constriction of cerebral arterioles to norepinephrine is significantly greater in SHR compared to WKY.70 These results suggest that increased constriction in cerebral arterioles during chronic hypertension may be partly related to increased sympathetic nerve density and/or catecholamine outflow.78 On the other hand, dilation of cerebral arterioles to vasodilators is impaired in SHRSP versus WKY, including adenosine diphosphate,77 acetylcholine,79 bradykinin and calcium ionophore A23187.80 These results are indicative of endothelial dysfunction in cerebral arteries and arterioles in chronic hypertension that may be related to reduced NO,81 increased oxidative stress81,82 and/or reduced prostaglandins productions.83
Ang II
The renin-angiotensin system (RAS) has an important role in the pathophysiology of hypertension. Ang II is the primary hormone of the RAS that is elevated in the circulation and brain in most forms of hypertension and has been implicated as the major cause of oxidative stress, endothelial dysfunction and increased tone in cerebral arteries mainly through the actions of AT1R activation.84–89 AT1R activation increases oxidative stress through increased nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity, a major producer of superoxide (O2−) that causes vasoconstriction and endothelial dysfunction.88,89 The effect of Ang II on the cerebral circulation has been reviewed in detail by De Silva and Faraci.90 Importantly, O2− production in response to Ang II was shown to be decreased by AT1R blockers.91–93 Interestingly, there appear to be sex differences in the response to AngII. Ang II-treated male mice deficient in manganese superoxide dismutase (MnSOD) had significant endothelial dysfunction in cerebral arteries, whereas female mice were unaffected, suggesting females are protected from the detrimental effects of Ang II.94
AT1R blockade has also been shown to improve stroke outcome in hypertensive animals and reduce ischemic stroke severity in humans.95–100 The detrimental effect of AT1R activation in stroke was also demonstrated by studies showing reduced infarction in mice genetically deficient in the AT1R (AT1aR−/−) and increased in mice with Ang II infusion.97–99 The beneficial effects of AT1R blockade on ischemic stroke are not necessarily due to its antihypertensive effects as the same effect was not seen in which blood pressure was lowered through other means (hydralazine or nimodipine), or was not decreased by treatment.100–102 In fact, the mechanism by which AT1R blockade is beneficial to stroke outcome is likely multifactorial. AT1R inhibition was shown to decrease stroke-induced upregulation of brain Ang II and matrix metalloproteinase activation in normotensive rats, suggesting one means by which AT1R inhibition improves outcome may be by preventing edema formation and hemorrhagic transformation.100 In hypertensive animals, AT1R blockade administered 3 h and 24 h after ischemia reduced infarct size and was associated with decreased Rho-kinase activation that was shown to be increased in cerebral microvessels.102,103 In a head-to-head comparison of AT1R blockade with telmisartan vs. ACE inhibition with Ramipril or the combination of the two given to spontaneously hypertensive stroke-prone rats (SHRSP), both had beneficial effects on stroke incidence and survival, but only telmisartan or the combination of telmisartan and Ramipril improved neurological outcome after acute middle cerebral artery occlusion (MCAO).104 These results demonstrate that decreasing blood pressure can lower stroke incidence and enhance survival, but that AT1R blockade is most beneficial for stroke outcome, likely through its effect on preventing NADPH oxidase-induced O2− production.
Endothelin
Endothelin-1 (ET-1) is a potent vasoconstrictor peptide synthesized and released mostly by endothelial cells in the cerebral vasculature.105 ET-1 regulates vascular tone through the activation of two specific receptor subtypes, ETA and ETB receptors.106 Under physiological conditions, there is a balance between the vasoconstrictor effect induced by ETA receptors on vascular smooth muscle and the vasodilator mechanism mediated by activation of ETB receptors on endothelium to produce NO.107 ET-1 has been implicated in the pathophysiology of hypertension, including in the brain and cerebral circulation through the actions of Ang II. In SHRSP, ET-1 levels in plasma are increased as are expression of ET-1 and ETA receptors in the brain.108,109 In addition, chronic ET-1 receptor inhibition partially prevents Ang II-induced hypertension.110 Importantly, Ang II increases expression of ET-1 in several tissues including smooth muscle that is prevented by AT1R antagonism.110,111 Under normal physiological conditions, NO inhibits the release of ET-1.112 However, conditions that decrease NO, such as hypertension, may enhance ET-1 release and its vasoconstrictor actions in an autocrine feed-forward mechanism.113,114 The effect of Ang II and ET-1 is also altered after stroke. Ang II potentiates vasoconstriction to ET-1 in cerebral arteries after ischemia and reperfusion but not under non-ischemic control conditions.115
Activation of ET-1 receptors during ischemia and reperfusion has been shown to produce reactive oxygen (ROS) and nitrogen (RONS) species in an endothelium-dependent manner, that may be enhanced during hypertension in which ET-1 is elevated.116 A previous study found that brain parenchymal arterioles (PAs) constricted to sarafotoxin, a selective ETB receptor agonist, only after tMCAO that was prevented by FeTMPyP to scavenge ONOO−, suggesting that ET-1 activation of ETB receptors on endothelium produces ONOO− to cause vasoconstriction.117 The constriction to ETB activation was not due to de novo expression of ETB receptors on the smooth muscle, as has been shown in MCA at longer time periods of ischemia and reperfusion.118 In vivo, ETB receptor antagonism has been shown to decrease infarction 24 h after permanent MCAO if given as a pretreatment.119,120 When ETB receptors were blocked 2 h post-occlusion, there was significant protection at seven days that was associated with upregulation of ETB, but not ETA receptors.121
Ion channel activation in endothelium and smooth muscle
The cerebral endothelium also has a prominent role in regulation of vascular tone, mainly through the actions of NO and endothelium-derived hyperpolarization (EDH), or release of an EDH factor. Under resting conditions, basal NO production by the cerebral endothelium inhibits resting tone and thus affects cerebral blood flow.122,123 The mechanism of EDH-mediated vasodilation occurs via increased endothelial cell calcium that activates small- and intermediate-conductance calcium-activated potassium (SKCa and IKCa) channels, expressed in endothelium, causing K+ efflux and membrane hyperpolarization that is transferred to smooth muscle via myoendothelial gap junctions.124–127 While EDH does not appear to contribute to resting tone in cerebral arteries, we and others found that inhibition of SKCa/IKCa channels caused constriction of brain PAs, suggesting that unlike MCA, basal EDH is present in PAs that inhibits resting tone and contributes to CBF regulation.128,129 In addition, inhibition of SKCa/IKCa channels in vivo results in decreased CBF,129 consistent with the concept that SKCa/IKCa channels are active in PAs under basal conditions and have a prominent influence on CBF through EDH. Several studies have shown that brain arterioles are less sensitive to NS309, a potent activator of SKCa/IKCa channels in SHR and SHRSP.130,131 Because SKCa/IKCa activity appears to be basally active and inhibit tone in PAs, their impairment during hypertension could contribute to increased tone and CVR that leads to hypoperfusion chronically and also incomplete reperfusion in the acute phase of stroke.
Other ion channels may be affected by hypertension and ischemic stroke to promote vascular dysregulation as well. Kir2.x channels are heterogeneously expressed in smooth muscle and endothelium depending on the vessel type.132,133 In cerebrovascular smooth muscle, Kir2.1 is expressed and opens with modest increases in extracellular potassium [K+]o (≤ 20 mM), causing outward movement of K+ and hyperpolarization that closes L-type Ca+2 channels to cause potent vasodilation.132,134 Kir2.1 channels have a key role in the vascular response to neuronal activation and in response to ischemia, as [K+]o in the brain increases from resting levels of 3–5 mM to 10–12 mM or greater that causes potent vasodilation of PAs through the activation of Kir channels.131,132 Kir channels may also have an important role in limiting infarct expansion through their vasodilating effect. Several studies have investigated Kir channel function in MCAs after ischemia and reperfusion.135,136 Marrelli et al. found impaired dilation to [K+]o in MCA after 2 h ischemia with 24 h reperfusion.137 Subsequent patch clamp studies of isolated smooth muscle from MCAs found decreased Kir currents that positively correlated with infarction, suggesting a protective role of Kir channel activity during stroke.138 Kir channels appear to be affected by hypertension as well and may contribute to impaired neurovascular coupling.139 In addition, Bastide et al.140 found a progressive decrease in Kir current density in myocytes from SHRSP, suggesting a role for Kir channels in development of stroke as well.
Collateral perfusion and hypertension
It is well-established in models of chronic hypertension that the brain is more susceptible to ischemic injury than aged-matched normotensive counterparts. Numerous studies have shown that infarction is larger in genetic models of hypertension (SHR and SHRSP) and in renal hypertension.62,63,130,141–143 One of the primary mechanisms by which infarction is larger in hypertension is the relative lack of salvageable tissue in hypertensive states, i.e. poor collateral status. Although several collateral systems exists within the brain vasculature, the pial collaterals or leptomeningeal anastomoses (LMAs) appear to be the most important for stroke outcome.144–146 Pial collaterals are distal connections between major arterial territories that can promote flow from one region to the other during an occlusion. During MCAO, pial collaterals between the ACA and MCA territories can sustain flow to the penumbra, a region of constrained blood supply that is salvageable if reperfusion occurs promptly or neuroprotective agents are given to prevent neuronal cell death.144–148 Pial collaterals sustain flow to the penumbra, and are therefore a strong predictor of recanalization, reperfusion and favorable clinical outcome.149–151
In addition to having small amounts of salvageable tissue during MCAO, SHR and SHRSP have been shown to have rapid evolution of infarction that encompasses the penumbra. McCabe et al.63 used magnetic resonance imaging (MRI) to measure relative cerebral blood flow and quantitative apparent diffusion coefficient (ADC) maps to show that SHRSP had larger ADC lesion areas than normotensive WKY rats after just 1 h MCAO (Figure 2). In addition, SHRSP had a significantly larger perfusion deficit and perfusion-diffusion mismatch at 1 h after MCAO that was considerably less in WKY, demonstrating less salvageable tissue in SHRSP.63 The small amount of mismatch in SHRSP as early as 1 h after MCAO indicates the ischemic injury occurs rapidly in this strain. Other studies have also shown that the ischemic lesion is maximal at 1 h after distal MCAO in SHR that correlated with final infarct at 24 h.152,153
Figure 2.
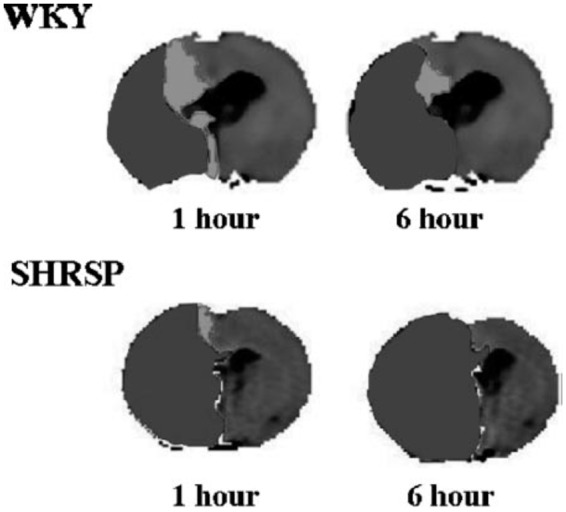
Representative thresholded CBF map with ADC abnormality (black) overlaid highlighting the DWI-PWI mismatch (shown in white) at 1 and 6 h after stroke in a WKY (top) and SHRSP (bottom). Used with permission from McCabe et al. Stroke 2009; 40: 3864–3868.
Other studies have shown that SHR and SHRSP have greater depth of ischemia during MCAO and poorer reperfusion that also likely contribute to larger infarction in these models.130,142 Our laboratory used hydrogen clearance to measure absolute levels of cerebral blood flow in the peri-infarct region of normotensive Wistar rats and SHRSP during transient MCAO and reperfusion. During filament occlusion, the perfusion deficit was significantly greater in SHRSP (Figure 3(a)). In addition, when cerebral blood flow was restored by filament withdrawal, reperfusion was incomplete in both strains for the 2 h that it was measured. The increase in perfusion deficit and impaired reperfusion was also shown by Kang et al.142 using MRI in SHR. Figure 3(b) shows infarction, measured as early as 2 h after reperfusion was significantly larger in SHRSP. While more extensive perfusion deficits and poor collateral status are important contributors to increased infarction volume in SHR and SHRSP, other factors may also have a role in increasing the sensitivity of the brain to ischemia during hypertension, independent of collateral status, including increased release of glutamate,154 production of vasoconstrictor agents such as 20-hydroxyeicosatetraenoic acid (20-HETE),155 and increased ROS.156
Figure 3.

Effect of hypertension on CBF and infarct volume. (a) Cerebral blood flow at baseline and during ischemia and reperfusion in Wistar and SHRSP using hydrogen clearance. Flow measurements were taken 30 and 60 min after MCAO and 15, 30, 60 and 90 min after reperfusion. SHRSP had greater perfusion deficit than Wistar during MCAO and both groups had incomplete reperfusion; (b) brain infarct volume measured using TTC after 2 h of ischemia and 2 h of reperfusion in the same animals as in (a). Results are mean ± SEM. *p < 0.05 vs. Wistar; ^p < 0.05 vs. baseline, using t-test. Used with permission from Cipolla et al. J Cereb Blood Flow Metab 2017; 37: 1276–1285.
Poor collateral status in models of hypertension has mostly been attributed to structurally smaller lumen diameters of LMAs or fewer pial collateral vessels that limits flow to the penumbra. However, we recently demonstrated the functional capabilities of pial collaterals using an isolated vessel approach. Pial collaterals that connected ACA and MCA vascular territories were dissected and mounted on glass microcannulae and their reactivity to increased intravascular pressure and other vasoactive agents were measured in young (18 week old) and aged (48 week old) male normotensive WKY rats and hypertensive SHR. Figure 4(a) shows a photomicrograph of an LMA – a distal branch of the ACA and MCA arteries – that was dissected and mounted on glass cannulas (Figure 4(b)). When intravascular pressure was increased, LMAs from WKY rats behaved relatively passively and increased diameter as the pressure was increased (Figure 5(a)). The relatively low level of tone and dilatory response of LMAs to increased pressure would be conducive to rapid bidirectional flow and important for facilitating collateral perfusion. Importantly, LMAs from hypertensive SHR were highly vasoactive in response to pressure (Figure 5(a)) and constricted in a myogenic manner. In both strains, aging did not appear to affect the response to increased pressure (Figure 5(b) and (c)). We also compared the response of LMAs to pial arterioles that did not anastomose (non-LMA). Pial arterioles from both WKY and SHR were highly reactive to pressure and were more vasoconstricted than LMAs from WKY rats (Figure 5(d)). These results importantly demonstrate the heterogeneity of vascular function in the cerebral circulation and that LMAs under normotensive conditions are less vasoconstricted than non-LMAs that would be conducive to retrograde flow during an occlusion. However, the highly vasoconstricted nature of LMAs from SHR likely contributes to the increased perfusion deficit and poor collateral status that has been shown in numerous studies. These results also suggest that salvaging penumbral tissue by opening vasoconstricted pial collaterals is indeed, possible. In fact, we recently demonstrated the ability of Sanguinate™, a carboxyhemoglobin gas transfer agent, to increase collateral flow in SHR and decrease early infarction.157
Figure 4.

(a) Diagram showing LMA (circle) reconnecting distal branches of MCA and ACA. (b) Photomicrograph of isolated and pressurized LMA on glass cannula. Used with permission from Chan et al. Stroke 2016; 47: 1618–1625.
Figure 5.

Effects of hypertension on myogenic response of LMAs. (a) Myogenic response of LMAs over a range of pressure in young WKY and SHR. LMAs from WKY behaved relatively passively, while LMAs from SHR constricted in response to increase in pressure. Myogenic reactivity of LMAs in young versus aged WKY (b) and SHR (c). Note that myogenic response of LMAs was not changed in aging in either strain. (d) Myogenic reactivity of LMAs comparing LMAs to non-LMAs in WKY and SHR. Non-LMAs were more myogenic than LMAs from WKY, demonstrating the uniqueness of LMAs in facilitating collateral perfusion during cerebral pial artery occlusion. *p < 0.05 vs. WKY18-LMA; ^p < 0.05 vs. WKY18-non-LMA using t-test or one-way ANOVA with post hoc Bonferroni test for multiple comparisons. Used with permission from Chan et al. Stroke 2016; 47: 1618–1625.
Collateral arteriogenesis and hypertension
The primary collaterals, which include the pial arteries of the Circle of Willis, immediately redirect flow in an event of one or more occlusions within the four extracranial arteries (two carotid and two vertebral arteries) that perfuse the brain. Long-term increases in flow cause structural enlargement in cerebral collaterals, termed arteriogenesis. In a rat model of three-artery occlusion (one carotid and two vertebral), the ipsilateral posterior cerebral artery enlarged by 39% by one week and 72% by three weeks due to a substantial increase in flow from the sole carotid artery.158 In another model of bilateral common artery occlusion and arteriovenous fistula in rats, arteriogenesis of the posterior cerebral artery occurred due to increased flow from the vertebral arteries.159 The same study showed that arteriogenesis was due to shear stress-mediated activation of transient receptor potential vanilloid-4 (TRPV4) channels in endothelium.159
LMAs also undergo arteriogenesis in response to increased flow during chronic occlusions. In normotensive WKY rats, LMAs underwent arteriogenesis (diameter increased by 50%) one month after MCAO.160 However, LMAs from SHRSP were narrower with less blood flow and larger infarct compared to WKY one month after MCAO.160 Other studies showed impairment of arteriogenesis in LMAs from SHR subjected to unilateral common artery occlusion that was normalized by antihypertensive treatment.161,162 Impaired arteriogenesis of pial collaterals may be due to decreased TRPV4 expression that has been shown to be decreased in SHRSP compared to WKY.163
Hypertensive CSVD
CSVD significantly increases the chance of a future stroke,164–166 and is a major cause of lacunar infarction,167–169 vascular dementia and cognitive and motor impairment.169–177 CSVD is caused by a number of factors, including genetics, comorbidities, and diet, yet chronic hypertension is the most common risk factor for CSVD.178 In fact, many clinical studies reported that the majority, between 70 to 80%, of CSVD patients are hypertensive.167,171,174,177 Due to the cerebral small vessels being too small to visualize in clinical imaging,179 CSVD is commonly inferred and diagnosed by white matter lesions (WMLs), lacunes, microbleeds, and increased perivascular space seen on MRI.180,181 Studies on CSVD have largely relied on autopsy studies in deceased human brains, which are limited to pathologies of end-stage CSVD and commonly combined with pathologies of other brain diseases. Hypertensive CSVD affects all small vessels in the brain, including arterioles, capillaries, and venules but most data available are in the small perforating arterioles. Classical studies by Fisher showed mostly occlusive lipohyalinosis lesions of small perforating arteries associated with lacunar infarct in 18 autopsy human brains.182,183 Lipohyalinosis is defined by Fisher as occlusion of small perforating arteries by lipid and hyaline materials. Later studies showed that most lipohyalinosis in hypertensive CSVD is characterized by thickening of the vessel wall and narrowing of lumen due to loss of smooth muscle that was replaced by fibrohyaline (fibrin and hyaline) materials.184,185 Other less common features of hypertensive CSVD are atherosclerosis and microaneurysms.184,185 It is believed that occlusive lipohyalinosis lesions of small perforating arteries cause a chronic state of hypoperfusion as evidenced by increased hypoxia-inducing factor-1 in autopsy human brains with WML.186 More recent studies in hypertensive CSVD patients demonstrated that these patients have impaired vascular reactivity leading to the loss of autoregulation,187–189 impaired neurovascular coupling190,191 and increased blood–brain barrier (BBB) permeability.192–195 Although these studies are important, the pathological changes at earlier stages of hypertensive CSVD and the processes by which hypertension affects small vessels to promote CSVD, deserve further investigation.
The presence of hypertensive CSVD and WML is often asymptomatic and even neglected.196–198 However, the presence of WML is a predictor of CSVD progression and new formation of lacunes.199 More importantly, severity of WML is not just a predictor of future stroke but also a predictor of stroke outcome including infarct progression and poor cognitive performance.164–166 Increased infarct volume after acute ischemic stroke has also been shown in models of hypertension (SHR and SHRSP).200,201 Lacunar stroke comprises about 25% all first-time ischemic stroke and therefore finding treatment for hypertensive CSVD to reduce stroke burden is of great importance.202
Peroxisome proliferator-activated receptor gamma and hypertensive CSVD
In addition to antihypertensive treatments, new treatments that specifically normalize the adverse effects of hypertension on target cerebral small vessels at early stages of the disease are important. For example, the transcription factor peroxisome proliferator-activated receptor gamma (PPARγ) may be a therapeutic target for hypertensive CSVD. Dominant negative mutations of PPARγ in humans at P467L or V290M cause insulin resistance and hypertension.203 Pharmacological activation of PPARγ by thiazolidinediones lowers blood pressure in various animal models of hypertension, including SHR and AngII infusion in rats.204,205 In the cerebral circulation, genetic knock-in mutation of PPARγ at P467L caused endothelial dysfunction, inward remodeling and increased wall thickness in cerebral arterioles.206 In studies using mice with endothelium-specific interference of PPARγ function, a high-fat diet caused endothelial dysfunction in basilar arteries but not aorta, suggesting PPARγ may be more involved in the protection of the cerebral circulation.207 In cerebral small vessels, PPARγ activation by rosiglitazone normalized L-NAME-induced increased blood pressure, myogenic tone and hypertrophic inward remodeling in third-order posterior cerebral arteries.208 Rosiglitazone also normalized L-NAME-induced capillary rarefaction in the brain.209 Moreover, relaxin, a hormone related to pregnancy, selectively caused outward remodeling of PAs and not the upstream middle cerebral arteries in normotensive female rats through activation of PPARγ.209 Another study showed that treatment of relaxin increased distensibility of PAs in young female SHR.210 Collectively, the protective effect of PPARγ activation selectively in cerebral small vessels during hypertension may be a therapeutic target for hypertensive CSVD.
Preeclampsia
Preeclampsia is a hypertensive disorder of pregnancy characterized by de novo hypertension after the 20th week of gestation with evidence of organ damage. Preeclampsia complicates up to 10% of pregnancies worldwide and is one of the greatest causes of maternal and fetal morbidity and mortality.211–213 Compared to normal pregnancy, preeclampsia is characterized by increased peripheral vascular resistance and hypertension, endothelial dysfunction, hypercoagulation, and increased vascular stiffness.214–218 Although preeclampsia is defined as a disorder of pregnancy, its consequences are far reaching and can extend decades into the postpartum years. Preeclampsia is a major risk factor for future cardiovascular and cerebrovascular diseases including hypertension, heart failure and stroke.12–14,214–219 Preeclampsia is also associated with brain atrophy and cognitive decline later in life.217,218 The cognitive defects and decline positively correlate with lesions in deep brain structures (i.e. WMLs).220–224 Importantly, formerly preeclamptic women can develop cognitive impairment and WMLs as early as in their 30s, suggesting preeclampsia predisposes the brain to early-onset cognitive impairment and white matter damage.220,221,225–228
Dyslipidemia and vessel stiffness in preeclampsia
There are several mechanisms by which preeclampsia could promote cerebrovascular disease. Preeclampsia is a state of dyslipidemia in which the normally elevated low-density lipoprotein (LDL) cholesterol during pregnancy is exaggerated.229 Importantly, very low-density LDL (vLDL) is increased in preeclamptic women that is highly susceptible to oxidation to form oxidized LDL (oxLDL).229 Levels of oxLDL are over 200% increase in serum of women with early-onset preeclampsia (diagnosis prior to 34 weeks’ gestation).230 oxLDL binds the LOX-1 receptor with high affinity to activate NADPH oxidase to generate superoxide (O2−), a primary mechanism of atherogenesis.230 Studies in humans have shown that arteries from women with preeclampsia have increased oxLDL and LOX-1 expression, suggesting this is a mechanism of endothelial dysfunction and oxidative stress.231
Preeclamptic women also have increased large artery stiffness that may precede clinical symptoms. In a prospective study, carotid-femoral and carotid-radial pulse wave velocity were increased at 22 weeks’ gestation in women who subsequently went on to develop preeclampsia.232 The importance of finding women with preeclampsia have increased large vessel stiffness is that it is an independent predictor of cerebrovascular disease and stroke.233 Peripheral arterial stiffness occurs with aging, hypertension and dyslipidemia that also promote microvascular damage by transmitting pulsatile pressure to distal microvessels.17,18 The brain is highly susceptible to microvascular damage associated with peripheral arterial stiffening because it is an organ with high flow and low impedance, thus allowing the pulsatile load to be transmitted more deeply into the brain parenchyma.17,18 The propagation of high pulsatile flow to the cerebral microvasculature is thought to promote remodeling and cause microvascular damage that underlies white matter hyperintensities, cerebral microbleeds and lacunar infarcts.17,58,234 In addition, clinical studies have shown strong associations between arterial stiffness and vascular dementia, suggesting cerebral microvascular remodeling secondary to enhanced arterial stiffness may be a causative factor in neurocognitive decline.58,235,236 Importantly, preeclamptic women have increased peripheral arterial stiffness, including enhanced carotid pulse wave velocity and augmentation index as well as increased carotid intima-media thickness237,238 that persists postpartum.214,239,240 Although the mechanism by which arterial stiffness occurs in preeclampsia is not known, oxLDL appears to have a central role in promoting arterial stiffness outside of pregnancy. Elevated oxLDL levels are associated with higher arterial stiffness in aging, independent of cardiovascular disease risk factors,241 and in patients with peripheral arterial disease.242 While the presence of high oxLDL is, in and of itself, a marker of high oxidative stress, several studies have demonstrated that arterial stiffness is associated with LOX-1 binding and increase oxidative stress in patients with hypertension, metabolic syndrome and aging.243–247
Our own studies found mild hippocampus-dependent cognitive impairment in a rat model of preeclampsia.248 Hippocampal arterioles, isolated with the internal transverse hippocampal artery (Figure 6(a) and (b)), were smaller and stiffer than those from normal pregnant or nonpregnant rats (Figure 6(c) and (d)).249 In addition, while hippocampal blood flow was not different between groups basally, the experimental preeclamptic rats lacked the normal hyperemic response to seizure. The importance of these findings is that women with preeclampsia are more susceptible to seizure (termed eclampsia) and the lack of a hyperemic response during seizure may increase seizure-induced damage to hippocampal neurons through hypoxic ischemic injury. In addition, whether hippocampal arteriolar remodeling and increased stiffness was due to high oxLDL and/or increased large vessel stiffness is not clear, but if this effect is persistent years after pregnancy, it could contribute to early onset cognitive impairment that has been shown to occur in these women.
Figure 6.

Effect of experimental preeclampsia (ePE) on passive structural and biomechanical properties of HAs. (a) Diagram showing the arterial blood supply to the hippocampus. (b) Photomicrograph of isolated and pressurized HA branching off the internal transverse hippocampal artery. (c) Passive inner diameters over a range of pressures. (d) Stress–strain curves as a measure of vessel stiffness; a shift to the left demonstrates increased stiffness. *p < 0.05 vs. Nonpreg; ** p < 0.01 vs. Nonpreg; ^^p < 0.01 vs. Nonpreg and Preg by one-way ANOVA with post hoc Bonferroni test for multiple comparisons. Used with permission from Coyle. Exp Neurol 1976; 52: 447–458 and Johnson and Cipolla. J Cerebr Blood Flow Metab 2017; 37: 2857–2869.
Stroke in preeclampsia
Hypertensive disorders of pregnancy, most notably preeclampsia, is a major cause of stroke in young women, a trend that is increasing.250,251 Different subtypes of stroke occur in preeclamptic women, including ischemic and hemorrhagic, demonstrating the heterogeneity of both stroke and preeclampsia.250,251 Normotensive pregnancy alone is a state associated with increases in coagulation factor levels and decreases in endogenous anti-coagulation, promoting a hypercoagulable state.252–254 The hypercoagulability during pregnancy is thought to ensure control of bleeding during birth and placental separation. In preeclamptic women, this normal prothrombotic state further exaggerated.255–257 Endothelial dysfunction, combined with hemostatic disturbances likely contribute to some of the stroke risk in these women, especially cerebral venous thrombosis and intracerebral hemorrhage. Although pregnancy alone increases stroke risk, other factors associated with preeclampsia further increase this risk, including hypertension, obesity and metabolic syndrome that are common features and comorbidities of preeclamptic women.258–262 In addition, arterial and venous vascular remodeling during pregnancy and the postpartum state, combined with dramatic fluctuations in plasma volume and cardiac output, likely contributes to risk of subarachnoid hemorrhage and aneurysm rupture.
The risk of ischemic stroke in preeclamptic women is not limited to the index pregnancy. Several case-controlled studies have demonstrated that women with a history of preeclampsia were associated with increased risk of stroke remote from pregnancy.263,264 The mechanism by which prior preeclampsia increases stroke risk years after pregnancy is not clear. While it is possible that endothelial dysfunction and hemostatic dysregulation associated with preeclampsia persist later in life, another possibility is that women who develop preeclampsia have a predisposition to cardiovascular disease risk that the physiological stress of pregnancy unmasks, i.e. preeclampsia is a manifestation of overall cardiovascular and cerebrovascular disease risk. Understanding the underlying causes of increased stroke risk in preeclampsia during pregnancy and years later is a challenge. However, it is clear that women with prior preeclampsia, especially early-onset preeclampsia, are at increased risk of cerebrovascular disease and stroke. In fact, the American Heart Association recommends that a history of preeclampsia be considered a major risk factor for heart disease and stroke.265
Summary and conclusions
Chronic hypertension has a profound impact on the cerebral circulation, summarized in Figure 7. In general, hypertension is associated with high levels of Ang II and AT1R activation in the blood vessels and brain that increases superoxide production and diminishes the normal vasodilatory influence of NO. There is also an increased vasoconstrictor influence (e.g. ET-1) that together with decreased endothelial NO increases CVR and shifts both the low and high limits of the autoregulatory curve to higher pressures. During acute ischemic stroke, the smaller lumen diameters and increased vasoconstriction of cerebral arteries and arterioles can increase perfusion deficit and impair collateral perfusion, leading to increased infarction. In addition, if prolonged, the increased CVR can cause hypoperfusion and contribute to CSVD, a leading contributor to stroke. Hypertension also increases pulse pressure and shear stress on the endothelium that can cause BBB disruption and vascular wall damage, both contributors to CSVD. Preeclampsia, a common hypertensive disorder of pregnancy, is associated with high levels of oxLDL and LOX-1 activation that increases BBB permeability that can promote seizure (eclampsia). In addition, women with prior preeclampsia have an increased risk of stroke and early-onset cognitive impairment, that is not seen in women with gestational hypertension, suggesting preeclampsia is a unique and more devastating form of hypertension. Figure 7 summarizes the major effects of hypertension on the cerebral circulation and some of the molecular and biochemical mechanisms of dysfunction, many of which can be considered targets for therapeutic intervention.
Figure 7.

Summary figure showing potential impacts of hypertension on the cerebral circulation that may increase the chance of future stroke and/or worsen stroke outcomes.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Roger VL, Go AS, Lloyd-Jones DM, et al. American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics – 2011 update: a report from the American Heart Association. Circulation 2011; 123: e18–e209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mozzafarian D, Benjamin EJ, Go AS, et al. On behalf of the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics – 2016 update: a report from the American Heart Association. Circulation 2016; 133: e38–e360. [DOI] [PubMed] [Google Scholar]
- 3.Chobanian AV, Bakris GL, Black HR, et al. National Heart, Lung, Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee. The seventh report of the joint national committee on prevention, detection, evaluation, and treatment of high blood pressure: the JNC 7 report. JAMA 2003; 289: 2560–2572. [DOI] [PubMed] [Google Scholar]
- 4.Lewington S, Clarke R, Qizilbash N, et al. Prospective Studies Collaboration. Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet 2002; 360: 1903–1913. [DOI] [PubMed] [Google Scholar]
- 5.Lackland DT, Carey RM, Conforto AB, et al. Implications of recent clinical trials and hypertension guidelines on stroke and future cerebrovascular research. Stroke 2018; 49: 772–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kannel WB, Wolf PA, Verter J, et al. Epidemiologic assessment of the role of blood pressure in stroke. The Framingham study. JAMA 1970; 214: 301–310. [PubMed] [Google Scholar]
- 7.Kannel WB, Wolf PA, McGee DL, et al. Systolic blood pressure, arterial rigidity, and risk of stroke. The Framingham study. JAMA 1981; 245: 1225–1229. [PubMed] [Google Scholar]
- 8.Lima FO, Furie KL, Silva GS, et al. The pattern of leptomeningeal collaterals on CT angiography is a strong predictor of long-term functional outcome in stroke patients with large vessel intracranial occlusion. Stroke 2010; 41: 2316–2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Menon BK, Smith EE, Coutts SB, et al. Leptomeningeal collaterals are associated with modifiable metabolic risk factors. Ann Neurol 2013; 74: 241–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ahmed N, Wahlgren N, Brainin M, et al. SITS Investigators. Relationship of blood pressure, antihypertensive therapy, and outcome in ischemic stroke treated with intravenous thrombolysis: retrospective analysis from Safe Implementation of Thrombolysis in Stroke-International Stroke Thrombolysis Register (SITS-ISTR). Stroke 2009; 40: 2442–2449. [DOI] [PubMed] [Google Scholar]
- 11.Leonardi-Bee J, Bath PM, Phillips SJ, et al. IST Collaborative Group. Blood pressure and clinical outcomes in the International Stroke Trial. Stroke 2002; 33: 1315–1320. [DOI] [PubMed] [Google Scholar]
- 12.Brown DW, Dueker N, Jamieson DJ, et al. Preeclampsia and the risk of ischemic stroke among young women: results from the Stroke Prevention in Young Women Study. Stroke 2006; 37: 1055–1059. [DOI] [PubMed] [Google Scholar]
- 13.Bellamy L, Casas JP, Hingorani AD, et al. Pre-eclampsia and risk of cardiovascular disease and cancer in later life: systematic review and meta-analysis. BMJ 2007; 335: 974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McDonald SD, Malinowski A, Zhou Q, et al. Cardiovascular sequelae of preeclampsia/eclampsia: a systematic review and meta-analyses. Am Heart J 2008; 156: 918–930. [DOI] [PubMed] [Google Scholar]
- 15.Strandgaard S. Autoregulation of cerebral circulation in hypertension. Acta Neurol Scand 1978; 57: 1–82. [PubMed] [Google Scholar]
- 16.Muller M, van der Graaf Y, Visseren FL, et al. SMART Study Group. Hypertension and longitudinal changes in cerebral blood flow: the SMART-MR study. Ann Neurol 2012; 71: 825–833. [DOI] [PubMed] [Google Scholar]
- 17.Cooper LL, Mitchell GF. Aortic stiffness, cerebrovascular dysfunction, and memory. Pulse 2016; 4: 69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mitchell GF. Effects of central arterial aging on the structure and function of the peripheral vasculature: implications for end-organ damage. J Appl Physiol 2008; 105: 1652–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baumbach GL, Faraci FM, Heistad DD. Effects of local reduction in pressure on endothelium-dependent responses of cerebral arterioles. Stroke 1994; 25: 1456–1461. [DOI] [PubMed] [Google Scholar]
- 20.Humphrey JD. Mechanisms of arterial remodeling in hypertension: coupled roles of wall shear and intraluminal stress. Hypertension 2008; 52: 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davies PF, Polacek DC, Handen JS, et al. A spatial approach to transcriptional profiling: mechanotransduction and the focal origin of atherosclerosis. Trends Biotechnol 1999; 17: 347–351. [DOI] [PubMed] [Google Scholar]
- 22.Davies PF. Hemodynamic shear stress and endothelium in cardiovascular pathophysiology. Nat Clin Pract Cardiovasc Med 2009; 6: 16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kher N, Marsh JD. Pathobiology of atherosclerosis – a brief review. Semin Thromb Hemost 2004; 30: 665–672. [DOI] [PubMed] [Google Scholar]
- 24.Gradinaru D, Borsa C, Ionescu C, et al. Oxidized LDL and NO synthesis – biomarkers of endothelial dysfunction and ageing. Mech Ageing Dev 2015; 151: 101–113. [DOI] [PubMed] [Google Scholar]
- 25.Raichle ME, Gusnard DA. Appraising the brain's energy budget. Proc Natl Acad Sci U S A 2002; 99: 10237–10239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lassen NA. Cerebral blood flow and oxygen consumption in man. Physiol Rev 1959; 39: 183–238. [DOI] [PubMed] [Google Scholar]
- 27.Harper AM. Autoregulation of cerebral blood flow: influence of the arterial blood pressure on the blood flow through the cerebral cortex. J Neurol Neurosurg Psychiatry 1966; 29: 398–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Strandgaard S, MacKenzie ET, Sengupta D, et al. Upper limit of autoregulation of cerebral blood flow in the baboon. Circ Res 1974; 34: 435–440. [DOI] [PubMed] [Google Scholar]
- 29.Grubb RL, Raichle ME, Phelps ME, et al. Effects of increased intracranial pressure on cerebral blood volume, blood flow, and oxygen utilization in monkeys. J Neurosurg 1975; 43: 385–398. [DOI] [PubMed] [Google Scholar]
- 30.MacKenzie ET, Strandgaard S, Graham DI, et al. Effects of acutely induced hypertension in cats on pial arteriolar caliber, local cerebral blood flow, and the blood-brain barrier. Circ Res 1976; 39: 33–41. [DOI] [PubMed] [Google Scholar]
- 31.Borel CO, Backofen JE, Koehler RC, et al. Cerebral blood flow autoregulation during intracranial hypertension in hypoxic lambs. Am J Physiol 1987; 253: H1342–H1348. [DOI] [PubMed] [Google Scholar]
- 32.Stange K, Lagerkranser M, Sollevi A. Effect of adenosine-induced hypotension on the cerebral autoregulation in the anesthetized pig. Acta Anaesthesiol Scand 1989; 33: 450–457. [DOI] [PubMed] [Google Scholar]
- 33.Florence G, Seylaz J. Rapid autoregulation of cerebral blood flow: a laser-Doppler flowmetry study. J Cereb Blood Flow Metab 1992; 12: 674–680. [DOI] [PubMed] [Google Scholar]
- 34.Hernandez MJ, Brennan RW, Bowman GS. Cerebral blood flow autoregulation in the rat. Stroke 1978; 9: 150–154. [DOI] [PubMed] [Google Scholar]
- 35.Dalkara T, Irikura K, Huang Z, et al. Cerebrovascular responses under controlled and monitored physiological conditions in the anesthetized mouse. J Cereb Blood Flow Metab 1995; 15: 631–638. [DOI] [PubMed] [Google Scholar]
- 36.Hossmann KA. Pathophysiology and therapy of experimental stroke. Cell Mol Neurobiol 2006; 26: 1057–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hossmann KA. Viability thresholds and the penumbra of focal ischemia. Ann Neurol 1994; 36: 557–565. [DOI] [PubMed] [Google Scholar]
- 38.Roggendorf W, Cervos-Navarro J, Lazaro-Lacalle MD. Ultrastructure of venules in the cat brain. Cell Tissue Res 1978; 192: 461–474. [DOI] [PubMed] [Google Scholar]
- 39.Strandgaard S, Olesen J, Skinhoj E, et al. Autoregulation of brain circulation in severe arterial hypertension. BMJ 1973; 1: 507–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barry DI, Strandgaard S, Graham DI, et al. Cerebral blood flow in rats with renal and spontaneous hypertension: resetting of the lower limit of autoregulation. J Cereb Blood Flow Metab 1982; 2: 347–353. [DOI] [PubMed] [Google Scholar]
- 41.Harper SL, Bohlen HG. Microvascular adaptation in the cerebral cortex of adult spontaneously hypertensive rats. Hypertension 1984; 6: 408–419. [DOI] [PubMed] [Google Scholar]
- 42.Hoffman WE, Albrecht RF, Miletich DJ. The influence of aging and hypertension on cerebral autoregulation. Brain Res 1981; 214: 196–199. [DOI] [PubMed] [Google Scholar]
- 43.Tanaka S, Tanaka M, Akashi A. Influence of antihypertensive treatment with budralazine on autoregulation of cerebral blood flow in spontaneously hypertensive rats. Stroke 1989; 20: 1724–1729. [DOI] [PubMed] [Google Scholar]
- 44.Ikeda J, Yao K, Matsubara M. Effects of benidipine, a long-lasting dihydropyridine-ca2+ channel blocker, on cerebral blood flow autoregulation in spontaneously hypertensive rats. Biol Pharm Bull 2006; 29: 2222–2225. [DOI] [PubMed] [Google Scholar]
- 45.Barry DI, Paulson OB, Jarden JO, et al. Effects of captopril on cerebral blood flow in normotensive and hypertensive rats. Am J Med 1984; 76: 79–85. [DOI] [PubMed] [Google Scholar]
- 46.Nishimura Y, Ito T, Saavedra JM. Angiotensin II at(1) blockade normalizes cerebrovascular autoregulation and reduces cerebral ischemia in spontaneously hypertensive rats. Stroke 2000; 31: 2478–2486. [DOI] [PubMed] [Google Scholar]
- 47.Hoedt-Rasmussen K, Skinhoj E, Paulson O, et al. Regional cerebral blood flow in acute apoplexy. The “luxury perfusion syndrome” of brain tissue. Arch Neurol 1967; 17: 271–281. [DOI] [PubMed] [Google Scholar]
- 48.Agnoli A, Fieschi C, Bozzao L, et al. Autoregulation of cerebral blood flow. Studies during drug-induced hypertension in normal subjects and in patients with cerebral vascular diseases. Circulation 1968; 38: 800–812. [DOI] [PubMed] [Google Scholar]
- 49.Paulson OB. Regional cerebral blood flow in apoplexy due to occlusion of the middle cerebral artery. Neurology 1970; 20: 63–77. [DOI] [PubMed] [Google Scholar]
- 50.Reinhard M, Wihler C, Roth M, et al. Cerebral autoregulation dynamics in acute ischemic stroke after rtPA thrombolysis. Cerebrovasc Dis 2008; 26: 147–155. [DOI] [PubMed] [Google Scholar]
- 51.Reinhard M, Rutsch S, Lambeck J, et al. Dynamic cerebral autoregulation associates with infarct size and outcome after ischemic stroke. Acta Neurol Scand 2011; 125: 156–162. [DOI] [PubMed] [Google Scholar]
- 52.Pozzilli C, Di Piero V, Pantano P, et al. Influence of nimodipine on cerebral blood flow in human cerebral ischaemia. J Neurol 1989; 236: 199–202. [DOI] [PubMed] [Google Scholar]
- 53.Faraci FM, Heistad DD. Regulation of large cerebral arteries and cerebral microvascular pressure. Circ Res 1990; 66: 8–17. [DOI] [PubMed] [Google Scholar]
- 54.Cipolla MJ, McCall A, Lessov N, et al. Reperfusion decreases myogenic reactivity and alters middle cerebral artery function after focal cerebral ischemia in rats. Stroke 1997; 28: 176–180. [DOI] [PubMed] [Google Scholar]
- 55.Osol G, Halpern W. Myogenic properties of cerebral blood vessels in normotensive and hypertensive rats. Am J Physiol 1985; 249: H914–H921. [DOI] [PubMed] [Google Scholar]
- 56.Cipolla MJ, Chan SL, Sweet JG, et al. Postischemic reperfusion causes smooth muscle calcium sensitization and vasoconstriction of parenchymal arterioles. Stroke 2014; 45: 2425–2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Halabi CM, Beyer AM, de Lange WJ, et al. Interference with PPAR gamma function in smooth muscle causes vascular dysfunction and hypertension. Cell Metab 2008; 7: 215–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fujishima M, Ibayashi S, Fujii K, et al. Cerebral blood flow and brain function in hypertension. Hypertens Res 1995; 18: 111–117. [DOI] [PubMed] [Google Scholar]
- 59.Dunn WR, Wallis SJ, Gardiner SM. Remodeling and enhanced myogenic tone in cerebral resistance arteries isolated from genetically hypertensive Brattleboro rats. J Vasc Res 1998; 35: 18–26. [DOI] [PubMed] [Google Scholar]
- 60.Cipolla MJ, DeLance N, Vitullo L. Pregnancy prevents hypertensive remodeling of cerebral arteries: a potential role in the development of eclampsia. Hypertension 2006; 47: 619–626. [DOI] [PubMed] [Google Scholar]
- 61.Baumbach GL, Dobrin PB, Hart MN, et al. Mechanics of cerebral arterioles in hypertensive rats. Circ Res 1988; 62: 56–64. [DOI] [PubMed] [Google Scholar]
- 62.Letourneur A, Roussel S, Toutain J, et al. Impact of genetic and renovascular chronic arterial hypertension on the acute spatiotemporal evolution of the ischemic penumbra: a sequential study with MRI in the rat. J Cereb Blood Flow Metab 2011; 31: 504–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McCabe C, Gallagher L, Gsell W, et al. Differences in the evolution of the ischemic penumbra in stroke-prone spontaneously hypertensive and Wistar-Kyoto rats. Stroke 2009; 40: 3864–3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dunn KM, Nelson MT. Neurovascular signaling in the brain and the pathological consequences of hypertension. Am J Physiol 2014; 306: H1–H14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reid E, Graham D, Lopez-Gonzalez MR, et al. Penumbra detection using PWI/DWI mismatch MRI in a rat stroke model with and without comorbidity: comparison of methods. J Cereb Blood Flow Metab 2012; 32: 1765–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Heistad DD, Mayhan WG, Coyle P, et al. Impaired dilatation of cerebral arterioles in chronic hypertension. Blood Vessels 1990; 27: 258–262. [DOI] [PubMed] [Google Scholar]
- 67.Mulvany MJ, Baumbach GL, Aalkjaer C, et al. Vascular remodeling. Hypertension 1996; 28: 505–506. [PubMed] [Google Scholar]
- 68.Baumbach GL, Heistad DD. Remodeling of cerebral arterioles in chronic hypertension. Hypertension 1989; 13: 968–972. [DOI] [PubMed] [Google Scholar]
- 69.Hart MN, Heistad DD, Brody MJ. Effect of chronic hypertension and sympathetic denervation on wall/lumen ratio of cerebral vessels. Hypertension 1980; 2: 419–423. [DOI] [PubMed] [Google Scholar]
- 70.Folkow B, Hallback M, Lundgren Y, et al. Background of increased flow resistance and vascular reactivity in spontaneously hypertensive rats. Acta Physiol Scand 1970; 80: 93–106. [DOI] [PubMed] [Google Scholar]
- 71.Baumbach GL, Heistad DD. Adaptive changes in cerebral blood vessels during chronic hypertension. J Hypertens 1991; 9: 987–991. [DOI] [PubMed] [Google Scholar]
- 72.Heistad DD, Baumbach GL. Cerebral vascular changes during chronic hypertension: good guys and bad guys. J Hypertens 1992; 10: S71–S75. [PubMed] [Google Scholar]
- 73.Chillon JM, Baumbach GL. Effects of an angiotensin-converting enzyme inhibitor and a beta-blocker on cerebral arterioles in rats. Hypertension 1999; 33: 856–861. [DOI] [PubMed] [Google Scholar]
- 74.Chan SL, Baumbach GL. Deficiency of nox2 prevents angiotensin ii-induced inward remodeling in cerebral arterioles. Front Physiol 2013; 4: 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chillon JM, Baumbach GL. Effects of indapamide, a thiazide-like diuretic, on structure of cerebral arterioles in hypertensive rats. Hypertension 2004; 43: 1092–1097. [DOI] [PubMed] [Google Scholar]
- 76.Chan SL, Umesalma S, Baumbach GL. Epidermal growth factor receptor is critical for angiotensin ii-mediated hypertrophy in cerebral arterioles. Hypertension 2015; 65: 806–812. [DOI] [PubMed] [Google Scholar]
- 77.Mayhan WG, Faraci FM, Heistad DD. Responses of cerebral arterioles to adenosine 5'-diphosphate, serotonin, and the thromboxane analogue u-46619 during chronic hypertension. Hypertension 1988; 12: 556–561. [DOI] [PubMed] [Google Scholar]
- 78.Lee TJ, Saito A. Altered cerebral vessel innervation in the spontaneously hypertensive rat. Circ Res 1984; 55: 392–403. [DOI] [PubMed] [Google Scholar]
- 79.Mayhan WG, Faraci FM, Heistad DD. Impairment of endothelium-dependent responses of cerebral arterioles in chronic hypertension. Am J Physiol 1987; 253: H1435–H1440. [DOI] [PubMed] [Google Scholar]
- 80.Baumbach GL, Faraci FM, Heistad DD. Effects of local reduction in pressure on endothelium-dependent responses of cerebral arterioles. Stroke 1994; 25: 1456–1461. [DOI] [PubMed] [Google Scholar]
- 81.Chan SL, Baumbach GL. Nox2 deficiency prevents hypertension-induced vascular dysfunction and hypertrophy in cerebral arterioles. Int J Hypertens 2013; 2013: 793630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yang ST, Mayhan WG, Faraci FM, et al. Mechanisms of impaired endothelium-dependent cerebral vasodilatation in response to bradykinin in hypertensive rats. Stroke 1991; 22: 1177–1182. [DOI] [PubMed] [Google Scholar]
- 83.Yang ST, Mayhan WG, Faraci FM, et al. Endothelium-dependent responses of cerebral blood vessels during chronic hypertension. Hypertension 1991; 17: 612–618. [DOI] [PubMed] [Google Scholar]
- 84.Saavedra JM, Nishimura Y. Angiotensin and cerebral blood flow. Cell Mol Neurobiol 1999; 19: 553–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res 2000; 86: 494–501. [DOI] [PubMed] [Google Scholar]
- 86.Dzau VJ. Tissue angiotensin and pathophysiology of vascular disease: a unifying hypothesis. Hypertension 2001; 37: 1047–1052. [DOI] [PubMed] [Google Scholar]
- 87.Nickenig G, Harrison DG. The AT1-type angiotensin receptor in oxidative stress and atherosclerosis, part I: oxidative stress and atherogenesis. Circulation 2002; 105: 393–396. [DOI] [PubMed] [Google Scholar]
- 88.Rajagopalan S, Kurz S, Munzel T, et al. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. J Clin Invest 1996; 97: 1916–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Didion SP, Faraci FM. Angiotensin II produces superoxide-mediated impairment of endothelial function in cerebral arterioles. Stroke 2003; 34: 2038–2042. [DOI] [PubMed] [Google Scholar]
- 90.De Silva TM, Faraci FM. Microvascular dysfunction and cognitive impairment. Cell Mol Neurobiol 2016; 36: 241–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Griendling KK, Ushio-Fukai M. NADH/NADPH oxidase and vascular function. Trends Cardiovasc Med 1997; 7: 301–307. [DOI] [PubMed] [Google Scholar]
- 92.Pueyo ME, Arnal JF, Rami J, et al. Angiotensin stimulates the production of NO and peroxynitrite in endothelial cells. Am J Physiol 1998; 274: C214–C220. [DOI] [PubMed] [Google Scholar]
- 93.Wattanapitayakul SK, Weinstein DM, Holycross BJ, et al. Endothelial dysfunction and peroxynitrite formation are early events in angiotensin-induced cardiovascular disorders. FASEB J 2000; 14: 271–278. [DOI] [PubMed] [Google Scholar]
- 94.Chrissobolis S, Faraci FM. Sex differences in protection against angiotensin II-induced endothelial dysfunction by manganese superoxide dismutase in the cerebral circulation. Hypertension 2010; 55: 905–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Miyamoto N, Tanaka Y, Ueno Y, et al. Benefits of prestroke use of angiotensin type 1 receptor blockers on ischemic stroke severity. J Stroke Cerebrovasc Dis 2012; 21: 363–368. [DOI] [PubMed] [Google Scholar]
- 96.Engelhorn T, Goerike S, Doerfler A, et al. The angiotensin II type 1-receptor blocker candesartan increases cerebral blood flow, reduces infarct size, and improves neurologic outcome after transient cerebral ischemia in rats. J Cereb Blood Flow Metab 2004; 24: 467–474. [DOI] [PubMed] [Google Scholar]
- 97.Nagai M, Terao S, Vital SA, et al. Role of blood cell-associated angiotensin II type 1 receptors in the cerebral microvascular response to ischemic stroke during angiotensin-induced hypertension. Exp Transl Stroke Med 2011; 3: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Walther T, Olah L, Harms C, et al. Ischemic injury in experimental stroke depends on angiotensin II. FASEB J 2002; 16: 169–176. [DOI] [PubMed] [Google Scholar]
- 99.Iwai M, Liu H-W, Chen R, et al. Possible inhibition of focal cerebral ischemia by angiotensin II type 2 receptor stimulation. Circulation 2004; 110: 843–848. [DOI] [PubMed] [Google Scholar]
- 100.Hosomi N, Nishiyama A, Ban CR, et al. Angiotensin type 1 receptor blockage improves ischemic injury following transient focal cerebral ischemia. Neuroscience 2005; 134: 225–231. [DOI] [PubMed] [Google Scholar]
- 101.Kozak W1, Kozak A, Johnson MH, et al. Vascular protection with candesartan after experimental acute stroke in hypertensive rats: a dose-response study. J Pharmacol Exp Ther 2008; 326: 773–782. [DOI] [PubMed] [Google Scholar]
- 102.Omura-Matsuoka E1, Yagita Y, Sasaki T, et al. Postischemic administration of angiotensin II type 1 receptor blocker reduces cerebral infarction size in hypertensive rats. Hypertens Res 2009; 32: 548–553. [DOI] [PubMed] [Google Scholar]
- 103.Ito T, Yamakawa H, Bregonzio C, et al. Protection against ischemia and improvement of cerebral blood flow in genetically hypertensive rats by chronic pretreatment with an angiotensin II AT1 antagonist. Stroke 2002; 33: 2297–2303. [DOI] [PubMed] [Google Scholar]
- 104.Thoene-Reineke C1, Rumschüssel K, Schmerbach K, et al. Prevention and intervention studies with telmisartan, ramipril and their combination in different rat stroke models. PLoS One 2011; 6: e23646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ergul A. Endothelin-1 and endothelin receptor antagonists as potential cardiovascular therapeutic agents. Pharmacotherapy 2002; 22: 54–65. [DOI] [PubMed] [Google Scholar]
- 106.Marasciulo FL, Montagnani M, Potenza MA. Endothelin-1: the yin and yang on vascular function. Curr Med Chem 2006; 13: 1655–1665. [DOI] [PubMed] [Google Scholar]
- 107.Thorin E, Clozel M. The cardiovascular physiology and pharmacology of endothelin-1. Adv Pharmacol 2010; 60: 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jesmin S, Zaedi S, Maeda S, et al. Endothelin antagonism suppresses plasma and cardiac endothelin-1 levels in SHRSPs at the typical hypertensive stage. Exp Biol Med 2006; 231: 919–924. [PubMed] [Google Scholar]
- 109.Jesmin S, Maeda S, Mowa CN, et al. Antagonism of endothelin action normalizes altered levels of VEGF and its signaling in the brain of stroke-prone spontaneously hypertensive rat. Eur J Pharmacol 2007; 574: 158–171. [DOI] [PubMed] [Google Scholar]
- 110.d'Uscio LV, Shaw S, Barton M, et al. Losartan but not verapamil inhibits angiotensin II-induced tissue endothelin-1 increase: role of blood pressure and endothelial function. Hypertension 1998; 31: 1305–1310. [DOI] [PubMed] [Google Scholar]
- 111.Barton M, Shaw S, d'Uscio LV, et al. Angiotensin II increases vascular and renal endothelin-1 and functional endothelin converting enzyme activity in vivo: role of ETA receptors for endothelin regulation. Biochem Biophys Res Commun 1997; 238: 861–865. [DOI] [PubMed] [Google Scholar]
- 112.Luscher TF, Yang Z, Tschudi M, et al. Interaction between endothelin-1 and endothelium-derived relaxing factor in human arteries and veins. Circ Res 1990; 66: 1088–1094. [DOI] [PubMed] [Google Scholar]
- 113.Alonso D, Radomski MW. The nitric oxide-endothelin-1 connection. Heart Fail Rev 2003; 8: 107–115. [DOI] [PubMed] [Google Scholar]
- 114.Nguyen TD, Vequaud P, Thorin E. Effects of endothelin receptor antagonists and nitric oxide on myogenic tone and alpha-adrenergic-dependent contractions of rabbit resistance arteries. Cardiovasc Res 1999; 43: 755–761. [DOI] [PubMed] [Google Scholar]
- 115.Salcedo A, Fernández N, García Villalón AL, et al. Role of angiotensin II in the response to endothelin-1 of goat cerebral arteries after ischemia-reperfusion. Vascul Pharmacol 2009; 50: 160–165. [DOI] [PubMed] [Google Scholar]
- 116.Lankhorst S, Kappers MH, van Esch JH, et al. Hypertension during vascular endothelial growth factor inhibition: focus on nitric oxide, endothelin-1, and oxidative stress. Antioxid Redox Signal 2014; 20: 135–145. [DOI] [PubMed] [Google Scholar]
- 117.Cipolla MJ, Sweet JG, Gokina NI, et al. Mechanisms of enhanced basal tone of brain parenchymal arterioles during early postischemic reperfusion: role of ET-1-induced peroxynitrite generation. J Cereb Blood Flow Metab 2013; 33: 1486–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Stenman E, Malmsjӧ M, Uddman E, et al. Cerebral ischemia upregulates vascular endothelin ET(B) receptors in rat. Stroke 2002; 33: 2311–2316. [DOI] [PubMed] [Google Scholar]
- 119.Chuquet J, Benchenane K, Toutain J, et al. Selective blockade of endothelin-B receptors exacerbates ischemic brain damage in the rat. Stroke 2002; 33: 3019–3025. [DOI] [PubMed] [Google Scholar]
- 120.Leonard MG, Briyal S, Gulati A. Endothelin B receptor agonist, IRL-1620, reduces neurological damage following permanent middle cerebral artery occlusion in rats. Brain Res 2011; 1420: 48–58. [DOI] [PubMed] [Google Scholar]
- 121.Leonard MG, Briyal S, Gulati A. Endothelin B receptor agonist, IRL-1620, provides long-term neuroprotection in cerebral ischemia in rats. Brain Res 2012; 1464: 14–23. [DOI] [PubMed] [Google Scholar]
- 122.Andresen J, Shafi NI, Bryan RM., Jr Endothelial influences on cerebrovascular tone. J Appl Physiol 2006; 100: 318–327. [DOI] [PubMed] [Google Scholar]
- 123.Faraci FM, Brian JE. Nitric oxide and the cerebral circulation. Stroke 1994; 25: 692–703. [DOI] [PubMed] [Google Scholar]
- 124.Luksha L, Nisell H, Luksha N, et al. Endothelium-derived hyperpolarizing factor in preeclampsia: heterogeneous contribution, mechanisms, and morphological prerequisites. Am J Physiol 2008; 294: R510–R519. [DOI] [PubMed] [Google Scholar]
- 125.Ledoux J, Werner ME, Brayden JE, et al. Calcium-activated potassium channels and the regulation of vascular tone. Physiology 2006; 21: 69–79. [DOI] [PubMed] [Google Scholar]
- 126.McNeish AJ, Dora KA, Garland CJ. Possible role for K+ in endothelium-derived hyperpolarizing factor-linked dilatation in rat middle cerebral artery. Stroke 2005; 36: 1526–1532. [DOI] [PubMed] [Google Scholar]
- 127.Sonkusare SK, Bonev AD, Ledoux J, et al. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science 2012; 336: 597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cipolla MJ, Smith J, Kohlmeyer MM, et al. SKCa and IKCa channels, myogenic tone, and vasodilator responses in middle cerebral arteries and parenchymal arterioles: effect of ischemia and reperfusion. Stroke 2009; 40: 1451–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hannah RM, Dunn KM, Bonev AD, et al. Endothelial SK(Ca) and IK(Ca) channels regulate brain parenchymal arteriolar diameter and cortical cerebral blood flow. J Cereb Blood Flow Metab 2011; 31: 1175–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Cipolla MJ, Sweet JG, Chan SL. Effect of hypertension and peroxynitrite decomposition with FeTMPyP on CBF and stroke outcome. J Cereb Blood Flow Metab 2017; 37: 1276–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chan SL, Sweet JG, Bishop N, et al. Pial collateral reactivity during hypertension and aging: understanding the function of collaterals for stroke therapy. Stroke 2016; 47: 1618–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Nilius B, Viana F, Droogmans G. Ion channels in vascular endothelium. Annu Rev Physiol 1997; 59: 145–170. [DOI] [PubMed] [Google Scholar]
- 133.Quayle JM, Nelson MT, Standen NB. ATP-sensitive and inwardly rectifying potassium channels in smooth muscle. Physiol Rev 1997; 77: 1165–1232. [DOI] [PubMed] [Google Scholar]
- 134.Smith PD, Brett SE, Luykenaar KD, et al. KIR channels function as electrical amplifiers in rat vascular smooth muscle. J Physiol 2008; 586: 1147–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Filosa JA, Bonev AD, Straub SV, et al. Local potassium signaling couples neuronal activity to vasodilation in the brain. Nat Neurosci 2006; 9: 1397–1403. [DOI] [PubMed] [Google Scholar]
- 136.Gido G, Kristian T, Siesjo BK. Extracellular potassium in a neocortical core area after transient focal ischemia. Stroke 1997; 28: 206–210. [DOI] [PubMed] [Google Scholar]
- 137.Marrelli SP, Johnson TD, Khorovets A, et al. Altered function of inward rectifier potassium channels in cerebrovascular smooth muscle after ischemia/reperfusion. Stroke 1998; 29: 1469–1474. [DOI] [PubMed] [Google Scholar]
- 138.Bastide M, Bordet R, Pu Q, et al. Relationship between inward rectifier potassium current impairment and brain injury after cerebral ischemia/reperfusion. J Cereb Blood Flow Metab 1999; 19: 1309–1315. [DOI] [PubMed] [Google Scholar]
- 139.Chan SL, Cipolla MJ. Conducted vasodilation in brain parenchymal arterioles is impaired during chronic hypertension. FASEB J 2015; 29: 949.7. [Google Scholar]
- 140.Bastide M, Ouk T, Pétrault O, et al. Time-induced progressive alteration of Kir current in cerebral smooth muscle cells of stroke-prone spontaneously hypertensive rats. Int J Hypertens 2013; 2013: 849750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Barone FC, Clark RK, Feuerstein G, et al. Quantitative comparison of magnetic resonance imaging (MRI) and histologic analyses of focal ischemic damage in the rat. Brain Res Bull 1991; 26: 285–291. [DOI] [PubMed] [Google Scholar]
- 142.Kang BT, Leoni RF, Silva AC. Impaired CBF regulation and high CBF threshold contribute to the increased sensitivity of spontaneously hypertensive rats to cerebral ischemia. Neuroscience 2014; 269: 223–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Dogan A, Baskaya MK, Rao VL, et al. Intraluminal suture occlusion of the middle cerebral artery in spontaneously hypertensive rats. Neurol Res 1998; 20: 265–270. [DOI] [PubMed] [Google Scholar]
- 144.Coyle P, Heistad DD. Development of collaterals in the cerebral circulation. Blood Vessels 1991; 28: 183–189. [DOI] [PubMed] [Google Scholar]
- 145.Shuaib A, Butcher K, Mohammad AA, et al. Collateral blood vessels in acute ischemic stroke: a potential therapeutic target. Lancet Neurol 2011; 10: 909–921. [DOI] [PubMed] [Google Scholar]
- 146.Zhang H, Prabhakar P, Sealock R, et al. Wide genetic variation in the native pial collateral circulation is a major determinant of variation in severity of stroke. J Cereb Blood Flow Metab 2010; 30: 923–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Astrup J, Siesjo BK, Symon L. Thresholds in cerebral ischemia – the ischemic penumbra. Stroke 1981; 12: 723–725. [DOI] [PubMed] [Google Scholar]
- 148.Jung S, Gilgen M, Slotboom J, et al. Factors that determine penumbral tissue loss in acute ischaemic stroke. Brain 2013; 136: 3554–3560. [DOI] [PubMed] [Google Scholar]
- 149.Liebeskind DS, Jahan R, Nogueira RG, et al. Impact of collaterals on successful revascularization in Solitaire FR with the intention for thrombectomy. Stroke 2014; 45: 2036–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lima FO, Furie KL, Silva GS, et al. The pattern of leptomeningeal collaterals on CT angiography is a strong predictor of long-term functional outcome in stroke patients with large vessel intracranial occlusion. Stroke 2010; 41: 2316–2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bang OY, Saver JL, Kim SJ, et al. Collateral flow predicts response to endovascular therapy for acute ischemic stroke. Stroke 2011; 42: 693–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Legos JJ, Lenhard SC, Haimbach RE, et al. SB 234551 selective ET(A) receptor antagonism: perfusion/diffusion MRI used to define treatable stroke model, time to treatment and mechanism of protection. Exp Neurol 2008; 212: 53–62. [DOI] [PubMed] [Google Scholar]
- 153.Chandra S, White RF, Everding D, et al. Use of diffusion-weighted MRI and neurological deficit scores to demonstrate beneficial effects of isradipine in a rat model of focal ischemia. Pharmacology 1999; 58: 292–299. [DOI] [PubMed] [Google Scholar]
- 154.Gemba T, Matsunaga K, Ueda M. Changes in extracellular concentration of amino acids in the hippocampus during cerebral ischemia in stroke-prone SHR, stroke-resistant SHR and normotensive rats. Neurosci Lett 1992; 135: 184–188. [DOI] [PubMed] [Google Scholar]
- 155.Dunn KM, Renic M, Flasch AK, et al. Elevated production of 20-HETE in the cerebral vasculature contributes to severity of ischemic stroke and oxidative stress in spontaneously hypertensive rats. Am J Physiol 2008; 295: H2455–H2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Riedel MW, Anneser F, Haberl RL. Different mechanisms of L-arginine induced dilation of brain arterioles in normotensive and hypertensive rats. Brain Res 1995; 671: 21–26. [DOI] [PubMed] [Google Scholar]
- 157.Cipolla MJ, Linfante I, Abuchowski A, et al. Pharmacologically increasing collateral perfusion during acute stroke using a carboxyhemoglobin gas transfer agent (Sanguinate™) in spontaneously hypertensive rats. J Cereb Blood Flow Metab 2018; 38: 755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Busch HJ, Buschmann IR, Mies G, et al. Arteriogenesis in hypoperfused rat brain. J Cereb Blood Flow Metab 2003; 23: 621–628. [DOI] [PubMed] [Google Scholar]
- 159.Schierling W, Troidl K, Apfelbeck H, et al. Cerebral arteriogenesis is enhanced by pharmacological as well as fluid-shear-stress activation of the trpv4 calcium channel. Eur J Vasc Endovasc Surg 2011; 41: 589–596. [DOI] [PubMed] [Google Scholar]
- 160.Coyle P, Heistad DD. Development of collaterals in the cerebral circulation. Blood Vessels 1991; 28: 183–189. [DOI] [PubMed] [Google Scholar]
- 161.Sugiyama Y, Yagita Y, Yukami T, et al. Granulocyte colony-stimulating factor fails to enhance leptomeningeal collateral growth in spontaneously hypertensive rats. Neurosci Lett 2014; 564: 16–20. [DOI] [PubMed] [Google Scholar]
- 162.Omura-Matsuoka E, Yagita Y, Sasaki T, et al. Hypertension impairs leptomeningeal collateral growth after common carotid artery occlusion: restoration by antihypertensive treatment. J Neurosci Res 2011; 89: 108–116. [DOI] [PubMed] [Google Scholar]
- 163.Seki T, Goto K, Kiyohara K, et al. Downregulation of endothelial transient receptor potential vanilloid type 4 channel and small-conductance of ca2+-activated k+ channels underpins impaired endothelium-dependent hyperpolarization in hypertension. Hypertension 2017; 69: 143–153. [DOI] [PubMed] [Google Scholar]
- 164.Vermeer SE, Hollander M, van Dijk EJ, et al. Silent brain infarcts and white matter lesions increase stroke risk in the general population: the Rotterdam scan study. Stroke 2003; 34: 1126–1129. [DOI] [PubMed] [Google Scholar]
- 165.Lau KK, Li L, Schulz U, et al. Total small vessel disease score and risk of recurrent stroke: validation in 2 large cohorts. Neurology 2017; 88: 2260–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Uiterwijk R, Staals J, Huijts M, et al. Framingham stroke risk profile is related to cerebral small vessel disease progression and lower cognitive performance in patients with hypertension. J Clin Hypertens 2018; 20: 240–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Benisty S, Gouw AA, Porcher R, et al. Location of lacunar infarcts correlates with cognition in a sample of non-disabled subjects with age-related white-matter changes: the LADIS study. J Neurol Neurosurg Psychiatry 2009; 80: 478–483. [DOI] [PubMed] [Google Scholar]
- 168.Gouw AA, van der Flier WM, Fazekas F, et al. Progression of white matter hyperintensities and incidence of new lacunes over a 3-year period: the leukoaraiosis and disability study. Stroke 2008; 39: 1414–1420. [DOI] [PubMed] [Google Scholar]
- 169.Gouw AA, van der Flier WM, Pantoni L, et al. On the etiology of incident brain lacunes: longitudinal observations from the ladis study. Stroke 2008; 39: 3083–3085. [DOI] [PubMed] [Google Scholar]
- 170.Inzitari D, Pracucci G, Poggesi A, et al. Changes in white matter as determinant of global functional decline in older independent outpatients: three year follow-up of ladis (leukoaraiosis and disability) study cohort. BMJ 2009; 339: 279–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Jokinen H, Kalska H, Ylikoski R, et al. Mri-defined subcortical ischemic vascular disease: baseline clinical and neuropsychological findings. The LADIS study. Cereb Dis 2009; 27: 336–344. [DOI] [PubMed] [Google Scholar]
- 172.Jokinen H, Kalska H, Ylikoski R, et al. Longitudinal cognitive decline in subcortical ischemic vascular disease – the LADIS study. Cereb Dis 2009; 27: 384–391. [DOI] [PubMed] [Google Scholar]
- 173.Mungas D, Harvey D, Reed BR, Jagust WJ, et al. Longitudinal volumetric MRI change and rate of cognitive decline. Neurology 2005; 65: 565–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Price CC, Jefferson AL, Merino JG, et al. Subcortical vascular dementia: integrating neuropsychological and neuroradiologic data. Neurology 2005; 65: 376–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.van der Flier WM, van Straaten EC, Barkhof F, et al. Small vessel disease and general cognitive function in nondisabled elderly: the LADIS study. Stroke 2005; 36: 2116–2120. [DOI] [PubMed] [Google Scholar]
- 176.van Swieten JC, Staal S, Kappelle LJ, et al. Are white matter lesions directly associated with cognitive impairment in patients with lacunar infarcts? J Neurol 1996; 243: 196–200. [DOI] [PubMed] [Google Scholar]
- 177.Inzitari D, Simoni M, Pracucci G, et al. Risk of rapid global functional decline in elderly patients with severe cerebral age-related white matter changes: the LADIS study. Arch Intern Med 2007; 167: 81–88. [DOI] [PubMed] [Google Scholar]
- 178.Khan U, Porteous L, Hassan A, et al. Risk factor profile of cerebral small vessel disease and its subtypes. J Neurol Neurosurg Psychiatry 2007; 78: 702–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Smith EE, Beaudin AE. New insights into cerebral small vessel disease and vascular cognitive impairment from MRI. Curr Opin Neurol 2018; 31: 36–43. [DOI] [PubMed] [Google Scholar]
- 180.Wardlaw JM, Smith C, Dichgans M. Mechanisms of sporadic cerebral small vessel disease: insights from neuroimaging. Lancet Neurol 2013; 12: 483–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Wardlaw JM, Smith EE, Biessels GJ, et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol 2013; 12: 822–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Fisher CM. Lacunes: small, deep cerebral infarcts. Neurology 1965; 15: 774–784. [DOI] [PubMed] [Google Scholar]
- 183.Fisher CM. The vascular lesion in lacunae. Trans Am Neurol Assoc 1965; 90: 243–245. [PubMed] [Google Scholar]
- 184.Pantoni L. Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol 2010; 9: 689–701. [DOI] [PubMed] [Google Scholar]
- 185.Lammie GA. Hypertensive cerebral small vessel disease and stroke. Brain Pathol 2002; 12: 358–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Fernando MS, Simpson JE, Matthews F, et al. White matter lesions in an unselected cohort of the elderly: molecular pathology suggests origin from chronic hypoperfusion injury. Stroke 2006; 37: 1391–1398. [DOI] [PubMed] [Google Scholar]
- 187.Birns J, Jarosz J, Markus HS, et al. Cerebrovascular reactivity and dynamic autoregulation in ischaemic subcortical white matter disease. J Neurol Neurosurg Psychiatry 2009; 80: 1093–1098. [DOI] [PubMed] [Google Scholar]
- 188.Purkayastha S, Fadar O, Mehregan A, et al. Impaired cerebrovascular hemodynamics are associated with cerebral white matter damage. J Cereb Blood Flow Metab 2014; 34: 228–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Guo ZN, Xing Y, Wang S, et al. Characteristics of dynamic cerebral autoregulation in cerebral small vessel disease: diffuse and sustained. Sci Rep 2015; 5: 15269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Lin WH, Hao Q, Rosengarten B, et al. Impaired neurovascular coupling in ischemic stroke patients with large or small vessel disease. Eur J Neurol 2011; 18: 731–736. [DOI] [PubMed] [Google Scholar]
- 191.Schroeter ML, Cutini S, Wahl MM, et al. Neurovascular coupling is impaired in cerebral microangiopathy – an event-related stroop study. Neuroimage 2007; 34: 26–34. [DOI] [PubMed] [Google Scholar]
- 192.Hainsworth AH, Fisher MJ. A dysfunctional blood-brain barrier and cerebral small vessel disease. Neurology 2017; 88: 420–421. [DOI] [PubMed] [Google Scholar]
- 193.Wardlaw JM. Blood-brain barrier and cerebral small vessel disease. J Neurol Sci 2010; 299: 66–71. [DOI] [PubMed] [Google Scholar]
- 194.Wardlaw JM, Doubal FN, Valdes-Hernandez M, et al. Blood-brain barrier permeability and long-term clinical and imaging outcomes in cerebral small vessel disease. Stroke 2013; 44: 525–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Zhang CE, Wong SM, van de Haar HJ, et al. Blood-brain barrier leakage is more widespread in patients with cerebral small vessel disease. Neurology 2017; 88: 426–432. [DOI] [PubMed] [Google Scholar]
- 196.Hatazawa J, Shimosegawa E, Satoh T, et al. Subcortical hypoperfusion associated with asymptomatic white matter lesions on magnetic resonance imaging. Stroke 1997; 28: 1944–1947. [DOI] [PubMed] [Google Scholar]
- 197.Kirkpatrick JB, Hayman LA. White-matter lesions in MR imaging of clinically healthy brains of elderly subjects: possible pathologic basis. Radiology 1987; 162: 509–511. [DOI] [PubMed] [Google Scholar]
- 198.Gunning-Dixon FM, Raz N. The cognitive correlates of white matter abnormalities in normal aging: a quantitative review. Neuropsychology 2000; 14: 224–232. [DOI] [PubMed] [Google Scholar]
- 199.Ay H, Arsava EM, Rosand J, et al. Severity of leukoaraiosis and susceptibility to infarct growth in acute stroke. Stroke 2008; 39: 1409–1413. [DOI] [PubMed] [Google Scholar]
- 200.Rewell SS, Fernandez JA, Cox SF, et al. Inducing stroke in aged, hypertensive, diabetic rats. J Cereb Blood Flow Metab 2010; 30: 729–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Dawson DA, Ruetzler CA, Hallenbeck JM. Temporal impairment of microcirculatory perfusion following focal cerebral ischemia in the spontaneously hypertensive rat. Brain Res 1997; 749: 200–208. [DOI] [PubMed] [Google Scholar]
- 202.Bamford J, Sandercock P, Jones L, et al. The natural history of lacunar infarction: the Oxfordshire Community Stroke Project. Stroke 1987; 18: 545–551. [DOI] [PubMed] [Google Scholar]
- 203.Barroso I, Gurnell M, Crowley VE, et al. Dominant negative mutations in human ppargamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature 1999; 402: 880–883. [DOI] [PubMed] [Google Scholar]
- 204.Wakino S, Hayashi K, Kanda T, et al. Peroxisome proliferator-activated receptor gamma ligands inhibit rho/rho kinase pathway by inducing protein tyrosine phosphatase SHP-2. Circ Res 2004; 95: e45–55. [DOI] [PubMed] [Google Scholar]
- 205.Diep QN, El Mabrouk M, Cohn JS, et al. Structure, endothelial function, cell growth, and inflammation in blood vessels of angiotensin II-infused rats: role of peroxisome proliferator-activated receptor-gamma. Circulation 2002; 105: 2296–2302. [DOI] [PubMed] [Google Scholar]
- 206.Beyer AM, Baumbach GL, Halabi CM, et al. Interference with ppargamma signaling causes cerebral vascular dysfunction, hypertrophy, and remodeling. Hypertension 2008; 51: 867–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Beyer AM, de Lange WJ, Halabi CM, et al. Endothelium-specific interference with peroxisome proliferator activated receptor gamma causes cerebral vascular dysfunction in response to a high-fat diet. Circ Res 2008; 103: 654–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Cipolla MJ, Bishop N, Vinke RS, et al. Ppar{gamma} activation prevents hypertensive remodeling of cerebral arteries and improves vascular function in female rats. Stroke 2010; 41: 1266–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Chan SL, Cipolla MJ. Relaxin causes selective outward remodeling of brain parenchymal arterioles via activation of peroxisome proliferator-activated receptor-gamma. FASEB J 2011; 25: 3229–3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Chan SL, Sweet JG, Cipolla MJ. Treatment for cerebral small vessel disease: effect of relaxin on the function and structure of cerebral parenchymal arterioles during hypertension. FASEB J 2013; 27: 3917–3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Wallis AB, Saftlas AF, Hsia J, et al. Secular trends in the rates of preeclampsia, eclampsia, and gestational hypertension. Am J Hypertens 2008; 21: 521–526. [DOI] [PubMed] [Google Scholar]
- 212.Roberts JM, Cooper DW. Pathogenesis and genetics of pre-eclampsia. Lancet 2001; 357: 53–56. [DOI] [PubMed] [Google Scholar]
- 213.Duley L. Maternal mortality associated with hypertensive disorders of pregnancy in Africa, Asia, Latin America and the Caribbean. BJOG 1992; 99: 547–553. [DOI] [PubMed] [Google Scholar]
- 214.Christensen M, Kronborg CS, Eldrup N, et al. Preeclampsia and cardiovascular disease risk assessment – do arterial stiffness and atherosclerosis uncover increased risk ten years after delivery? Pregnancy Hypertens 2016; 6: 110–104. [DOI] [PubMed] [Google Scholar]
- 215.Lykke JA, Langhoff-Roos J, Sibai BM, et al. Hypertensive pregnancy disorders and subsequent cardiovascular morbidity and type 2 diabetes mellitus in the mother. Hypertension 2009; 53: 944–951. [DOI] [PubMed] [Google Scholar]
- 216.Ray JG, Vermeulen MJ, Schull MJ, et al. Cardiovascular health after maternal placental syndromes (CHAMPS): population-based retrospective cohort study. Lancet 2005; 366: 1797–1803. [DOI] [PubMed] [Google Scholar]
- 217.Bellamy L, Casas JP, Hingorani AD, et al. Pre-eclampsia and risk of cardiovascular disease and cancer in later life: systematic review and meta-analysis. BMJ 2007; 335: 974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.McDonald SD, Malinowski A, Zhou Q, et al. Cardiovascular sequelae of preeclampsia/eclampsia: a systematic review and meta-analyses. Am Heart J 2008; 156: 918–930. [DOI] [PubMed] [Google Scholar]
- 219.Ghossein-Doha C, van Neer J, Wissink B, et al. Pre-eclampsia: an important risk factor for asymptomatic heart failure. Ultrasound Obstet Gynecol 2017; 49: 143–149. [DOI] [PubMed] [Google Scholar]
- 220.Aukes AM, De Groot JC, Wiegman MJ, et al. Long-term cerebral imaging after pre-eclampsia. BJOG 2012; 119: 1117–1122. [DOI] [PubMed] [Google Scholar]
- 221.Aukes AM, Wessel I, Dubois AM, et al. Self-reported cognitive functioning in formerly eclamptic women. Am J Obstet Gynecol 2007; 197: 365.e1–365.e6. [DOI] [PubMed] [Google Scholar]
- 222.Aukes AM, de Groot JC, Aarnoudse JG, et al. Brain lesions several years after eclampsia. Am J Obstet Gynecol 2009; 200: 504.e1–5. [DOI] [PubMed] [Google Scholar]
- 223.Wiegman MJ, Zeeman GG, Aukes AM, et al. Regional distribution of cerebral white matter lesions years after preeclampsia and eclampsia. Obstet Gynecol 2014; 123: 790–795. [DOI] [PubMed] [Google Scholar]
- 224.Mielke MM, Milic NM, Weissgerber TL, et al. Impaired cognition and brain atrophy decades after hypertensive pregnancy disorders. Circ Cardiovasc Qual Outcomes 2016; 9: S70–S76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Postma IR, Bouma A, de Groot JC, et al. Cerebral white matter lesions, subjective cognitive failures, and objective neurocognitive functioning: a follow-up study in women after hypertensive disorders of pregnancy. J Clin Exp Neuropsychol 2016; 38: 585–98. [DOI] [PubMed] [Google Scholar]
- 226.Siepmann T, Boardman H, Bilderbeck A, et al. Long-term cerebral white and gray matter changes after preeclampsia. Neurology 2017; 88: 1256–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Thorsrud A, Kerty E. Combined retinal and cerebral changes in a pre-eclamptic woman. Acta Ophthalmol 2009; 87: 925–926. [DOI] [PubMed] [Google Scholar]
- 228.Schimp VL, Hallak M, Puder KS, et al. Multiple brain infarcts associated with severe preeclampsia. J Stroke Cerebrovasc Dis 2001; 10: 244–246. [DOI] [PubMed] [Google Scholar]
- 229.Hubel CA, Lyall F, Weissfeld L, et al. Small low-density lipoproteins and vascular cell adhesion molecule-1 are increased in association with hyperlipidemia in preeclampsia. Metabolism 1998; 47: 1281–1288. [DOI] [PubMed] [Google Scholar]
- 230.Schreurs MP, Hubel CA, Bernstein IM, et al. Increased oxidized low-density lipoprotein causes blood-brain barrier disruption in early-onset preeclampsia through LOX-1. FASEB J 2013; 27: 1254–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Sankaralingam S, Xu Y, Sawamura T, et al. Increased lectin-like oxidized low-density lipoprotein receptor-1 expression in the maternal vasculature of women with preeclampsia: role for peroxynitrite. Hypertension 2009; 53: 270–277. [DOI] [PubMed] [Google Scholar]
- 232.Savvidou MD, Kaihura C, Anderson JM, et al. Maternal arterial stiffness in women who subsequently develop pre-eclampsia. PLoS One 2011; 6: e18703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.van Sloten TT, Protogerou AD, Henry RM, et al. Association between arterial stiffness, cerebral small vessel disease and cognitive impairment: a systematic review and meta-analysis. Neurosci Biobehav Rev 2015; 53: 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Saji N, Toba K, Sakurai T. Cerebral small vessel disease and arterial stiffness: tsunami effect in the brain? Pulse 2016; 3: 182–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Tsao CW, Himali JJ, Beiser AS, et al. Association of arterial stiffness with progression of subclinical brain and cognitive disease. Neurology 2016; 86: 619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Zhong W, Cruickshanks KJ, Schubert CR, et al. Pulse wave velocity and cognitive function in older adults. Alzheimer Dis Assoc Disord 2014; 28: 44–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Yuan LJ, Xue D, Duan YY, et al. Carotid arterial intima–media thickness and arterial stiffness in pre-eclampsia: analysis with a radiofrequency ultrasound technique. Ultrasound Obstet Gynecol 2013; 42: 644–652. [DOI] [PubMed] [Google Scholar]
- 238.Stergiotou I, Crispi F, Valenzuela-Alcaraz B, et al. Patterns of maternal vascular remodeling and responsiveness in early- versus late-onset preeclampsia. Am J Obstet Gynecol 2013; 209: 558.e1–558.e14. [DOI] [PubMed] [Google Scholar]
- 239.Orabona R, Sciatti E, Vizzardi E, et al. Endothelial dysfunction and vascular stiffness in women with previous pregnancy complicated by early or late pre-eclampsia. Ultrasound Obstet Gynecol 2017; 49: 116–123. [DOI] [PubMed] [Google Scholar]
- 240.Orabona R, Sciatti E, Vizzardi E, et al. Elastic properties of ascending aorta in women with previous pregnancy complicated by early- or late-onset pre-eclampsia. Ultrasound Obstet Gynecol 2016; 47: 316–323. [DOI] [PubMed] [Google Scholar]
- 241.Brinkley TE, Nicklas BJ, Kanaya AM, et al. Plasma oxidized low-density lipoprotein levels and arterial stiffness in older adults: the health, aging, and body composition study. Hypertension 2009; 53: 846–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Zagura M, Kals J, Kilk K, et al. Metabolomic signature of arterial stiffness in male patients with peripheral arterial disease. Hypertens Res 2015; 38: 840–846. [DOI] [PubMed] [Google Scholar]
- 243.Ogura S, Kakino A, Sato Y, et al. Lox-1: the multifunctional receptor underlying cardiovascular dysfunction. Circulation 2009; 73: 1993–1999. [DOI] [PubMed] [Google Scholar]
- 244.Mitra S, Goyal T, Mehta JL. Oxidized LDL, LOX-1 and atherosclerosis. Cardiovasc Drugs Ther 2011; 25: 419–429. [DOI] [PubMed] [Google Scholar]
- 245.Kim M, Yoo HJ, Kim M, et al. Associations among oxidative stress, Lp-PLA2 activity and arterial stiffness according to blood pressure status at a 3.5-year follow-up in subjects with prehypertension. Atherosclerosis 2017; 257: 179–185. [DOI] [PubMed] [Google Scholar]
- 246.Otsuki T, Maeda S, Mukai J, et al. Association between plasma sLOX-1 concentration and arterial stiffness in middle-aged and older individuals. J Clin Biochem Nutr 2015; 57: 151–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Kim M, Kim M, Yoo HJ, et al. Age-specific determinants of pulse wave velocity among metabolic syndrome components, inflammatory markers, and oxidative stress. J Atheroscler Thromb 2018; 25: 178–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Johnson AC, Whitbeck A, Miller J, et al. Impaired cognitive function in experimental preeclampsia. Repro Sci 2018; 25(1 Supplement): 242A. [Google Scholar]
- 249.Johnson AC, Cipolla MJ. Altered hippocampal arteriole structure and function in a rat model of preeclampsia: potential role in impaired seizure-induced hyperemia. J Cereb Blood Flow Metab 2017; 37: 2857–2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Kuklina EV, Tong X, Bansil P, et al. Trends in pregnancy hospitalizations that included a stroke in the United States from 1994 to 2007: reasons for concern? Stroke 2011; 42: 2564–2570. [DOI] [PubMed] [Google Scholar]
- 251.McDermott M, Miller EC, Rundek T, et al. Preeclampsia: association with posterior reversible encephalopathy syndrome and stroke. Stroke 2018; 49: 524–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Uchikova EH, Ledjev II. Changes in haemostasis during normal pregnancy. Eur J Obstet Gynecol Reprod Biol 2005; 119: 185–188. [DOI] [PubMed] [Google Scholar]
- 253.Holmes VA, Wallace JM. Haemostasis in normal pregnancy: a balancing act? Biochem Soc Trans 2005; 33: 428–432. [DOI] [PubMed] [Google Scholar]
- 254.Brenner B. Haemostatic changes in pregnancy. Thromb Res 2004; 114: 409–414. [DOI] [PubMed] [Google Scholar]
- 255.McKay DG. Hematologic evidence of disseminated intravascular coagulation in eclampsia. Obstet Gynecol Surv 1972; 27: 399–417. [DOI] [PubMed] [Google Scholar]
- 256.Brown MA. The physiology of pre-eclampsia. Clin Exp Pharmacol Physiol 1995; 22: 781–791. [DOI] [PubMed] [Google Scholar]
- 257.Arias F, Mancilla-Jimenez R. Hepatic fibrinogen deposits in pre-eclampsia. Immunofluorescent evidence. N Engl J Med 1976; 295: 578–582. [DOI] [PubMed] [Google Scholar]
- 258.Kittner SJ, Stern BJ, Feeser BR, et al. Pregnancy and the risk of stroke. N Engl J Med 1996; 335: 768–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Lanska DJ, Kryscio RJ. Risk factors for peripartum and postpartum stroke and intracranial venous thrombosis. Stroke 2000; 31: 1274–1282. [DOI] [PubMed] [Google Scholar]
- 260.Sharshar T, Lamy C, Mas JL. Incidence and causes of strokes associated with pregnancy and puerperium. A study in public hospitals of Ile de France. Stroke in Pregnancy Study Group. Stroke 1995; 26: 930–936. [DOI] [PubMed] [Google Scholar]
- 261.Wiebers DO. Ischemic cerebrovascular complications of pregnancy. Arch Neurol 1985; 42: 1106–1113. [DOI] [PubMed] [Google Scholar]
- 262.Hooijschuur MC, Ghossein-Doha C, Al-Nasiry S, et al. Maternal metabolic syndrome, preeclampsia, and small for gestational age infancy. Am J Obstet Gynecol 2015; 213: 370. [DOI] [PubMed] [Google Scholar]
- 263.Wilson BJ, Watson MS, Prescott GJ, et al. Hypertensive diseases of pregnancy and risk of hypertension and stroke in later life: results from cohort study. BMJ 2003; 326: 845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Brown MC, Best KE, Pearce MS, et al. Cardiovascular disease risk in women with pre-eclampsia: systematic review and meta-analysis. Eur J Epidemiol 2013; 28: 1–19. [DOI] [PubMed] [Google Scholar]
- 265.Mosca L, Benjamin EJ, Berra K, et al. Effectiveness-based guidelines for the prevention of cardiovascular disease in women – 2011 update: a guideline from the American Heart Association. Circulation 2011; 123: 1243–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]