

Abstract
Global ischemia in childhood often leads to poor neurologic outcomes, including learning and memory deficits. Using our novel model of childhood cardiac arrest/cardiopulmonary resuscitation (CA/CPR), we investigate the mechanism of ischemia-induced cognitive deficits and recovery. Memory is impaired seven days after juvenile CA/CPR and completely recovers by 30 days. Consistent with this remarkable recovery not observed in adults, hippocampal long-term potentiation (LTP) is impaired 7–14 days after CA/CPR, recovering by 30 days. This recovery is not due to the replacement of dead neurons (neurogenesis), but rather correlates with brain-derived neurotrophic factor (BDNF) expression, implicating BDNF as the molecular mechanism underlying impairment and recovery. Importantly, delayed activation of TrkB receptor signaling reverses CA/CPR-induced LTP deficits and memory impairments. These data provide two new insights (1) endogenous recovery of memory and LTP through development may contribute to improved neurological outcome in children compared to adults and (2) BDNF-enhancing drugs speed recovery from pediatric cardiac arrest during the critical school ages.
Keywords: Global ischemia, hippocampus, brain-derived neurotrophic factor, synaptic plasticity, memory and cognition
Introduction
Great improvements in clinical resuscitation in recent years have led to improved survival rates following cardiac arrest. However, the vast majority of survivors experience motor and cognitive deficits,1–3 making the development of new therapies to improve neurologic outcome even more pressing. The prevailing notion continues to be that post-ischemic behavioral impairments are primarily due to cell death of vulnerable neuronal populations, such as hippocampal CA1 neurons, cerebellar Purkinje cells and striatal neurons. Indeed, numerous studies have identified acute strategies to decrease neuronal cell death after cerebral ischemia, with only therapeutic hypothermia translating to human use4,5 (albeit not for all age groups, see below). Therefore, due to the lack of translation of neuroprotective agents into human use,6 we have transitioned our focus to improving functional outcomes after ischemia, rather than protecting neurons from ischemic injury. To this end, studies of the function of surviving neurons after ischemia have shown that deficits in hippocampal plasticity and learning and memory are observed for months after ischemic insults7–11 and emerging therapies have targeted reversing synaptic plasticity deficits (neuro-restoration) to enhancing functional recovery from brain ischemia, effectively broadening the therapeutic window. While there are studies in experimental models of adult focal cerebral ischemia,12–14 it remains unclear whether similar plasticity deficits and interventions are observed following ischemia in the developing brain.
The impact of age on the brain’s response to cerebral ischemia remains understudied, with a particular scarcity of studies modeling childhood cardiac arrest. It is estimated that 16,000 children suffer cardiac arrest annually in the United States, with important differences observed compared to adults. To date, mild therapeutic hypothermia is the only therapy available to increase survival and improve outcomes for adults after cardiac arrest (CA/CPR) and neonates less than 6 h old.4,5 Unfortunately, recent clinical trials for hypothermia in children failed to improve outcome.15,16 Considering this and the idea that children have improved recovery compared to adults,17,18 there is increased importance in considering juveniles independently from adults or neonates. Thus, the current study takes advantage of our newly developed mouse model of childhood (juvenile) cardiac arrest to assess synaptic plasticity and memory deficits at acute and chronic time points to assess recovery following childhood cardiac arrest and potential therapeutic interventions.
Intact memory and synaptic plasticity in the hippocampus require N-methyl-D-aspartate (NMDA) receptor activity19,20 and brain-derived neurotrophic factor (BDNF) signaling through tyrosine kinase (Trk) B receptors.21,22 We have reported that impairments in LTP after CA/CPR are not due to alterations in NMDA receptor expression or function in either adult7 or juvenile mice.23 Alternatively, BDNF has been found to play a neuroprotective role after neonatal hypoxic-ischemic injury24,25 and likely plays a role in stem cell-induced regeneration of hippocampus in neonatal ischemic brain injury.26 In adult models, BDNF infusion preserved LTP after transient focal ischemia27 but did not alter brain injury after CA/CPR.28 These data indicate that the effects of BDNF may vary between different models of brain injury and across ages. The data presented here will establish the temporal, phenotypic and mechanistic impact of BDNF signaling on impaired memory and synaptic function after juvenile cardiac arrest. Further, we present a novel therapeutic strategy following cardiac arrest centered on recovery of memory and synaptic function by stimulating BDNF–TrkB signal pathway at time points beyond those critical in neuronal death following cardiac arrest in the developing brain.
Methods
Experimental animals
All experimental protocols were approved by the University of Colorado-Denver Institutional Animal Care and Use Committee (IACUC) and conformed to the National Institutes of Health guidelines for care and use of animals. Male C57Bl/6 20-25 day old (PND 20-25, prepubertal) mice (Charles River Laboratory) were used for this study. These mice were weaned and not with dam at the time of experiment. The mice were housed in a standard 12 h light and 12 h dark cycle and had free access to food and water. All experiments in the study adhered to the ARRIVE guidelines for animal experiments. Mice were randomly assigned to experimental groups and the investigator was blinded as stated below. A total of 128 mice were used for this study. The average age of CA/CPR was 22.5 ± 0.2 days (n = 69). The average age for sham surgeries was 22.6 ± 0.3 days (n = 59). Five mice were removed from the study after CA/CPR and one was removed from the sham group due to premature death.
Cardiac arrest and cardiopulmonary resuscitation
Cardiac arrest in juvenile mice was performed as previously described.29 Briefly, mice were anesthetized using 3% isoflurane and maintained with 1.5–2% isoflurane in 25% fraction of inspired oxygen (FiO2) via face-mask. Body temperature was maintained at 37℃ using a heat lamp and heating pad while being monitored with temperature probes placed into the left ear canal and rectum. For drug administration, a PE-10 catheter was inserted into the right internal jugular vein and flushed with heparinized 0.9% normal saline solution. Animals were endotracheally intubated using a 24G intravenous catheter and connected to a mouse ventilator (Minivent, Hugo Sachs Elektronik, March-Hugstetten, Germany) set to a respiratory rate of 160 breaths per minute. Cardiac function was monitored throughout the experiment with electrocardiography (EKG). Cardiac arrest was induced by injection of 30 µL of 0.5 M KCl via the jugular catheter and confirmed by asystole on EKG and absence of spontaneous breathing. The endotracheal tube was disconnected from the ventilator during cardiac arrest and no spontaneous breathing was observed. During this time, anesthesia was not being delivered. Body warming was ceased 1 min prior to cardiac arrest. During cardiac arrest, the pericranial temperature was maintained at 37.5 ± 0.5℃ by using a water-filled coil. Body temperature was allowed to fall spontaneously to 35℃. Resuscitation was begun 8 min after the initiation of cardiac arrest by slow injection of 0.2–0.5 mL of epinephrine (16 µg epinephrine/mL 0.9% saline), chest compressions at a rate of approximately 300 min−1 and resumption of ventilation with 100% FiO2 at a rate of 210 breaths/min. Chest compressions were stopped upon return of spontaneous circulation (ROSC), defined as electrical evidence of cardiac contractions. If ROSC was not achieved within 2 min of CPR initiation, resuscitation was stopped and the animal was excluded from the study. Five minutes following ROSC, FiO2 was decreased to 50%. When the spontaneous respiratory rate was 30 breaths/min, the ventilator was adjusted to 150 breaths/min, and when the animals had at least 60 spontaneous breaths/min, the endotracheal tube was removed. Temperature probes and intravascular catheters were removed and the surgical wounds were closed.
Acute hippocampal slice preparation
Hippocampal slices were prepared at 7, 14 or 30 days after recovery from CA/CPR or sham surgeries. Mice were anesthetized with 3% isoflurane in an O2-enriched chamber. Mice were transcardially perfused with ice-cold (2–5℃) oxygenated (95% O2/5% CO2) artificial cerebral spinal fluid (aCSF) for 2 min prior to decapitation. The brains were then extracted and placed in the same aCSF. The composition of aCSF was the following (in mmol/L): 126 NaCl, 2.5 KCl, 25 NaHCO3, 1.3 NaH2PO4, 2.5 CaCl2, 1.2 MgCl2, and 12 glucose.7 Horizontal hippocampal slices (300 µm thick) were cut with a Vibratome 1200 (Leica) and transferred to a holding chamber containing aCSF for at least 1 h before recording.
Electrophysiology
Synaptically evoked field potentials were recorded from hippocampal CA1 slices that were placed on a temperature-controlled (31 ± 0.5℃) interface chamber perfused with aCSF at a rate of 1.5 mL/min.Field excitatory post-synaptic potentials (fEPSPs) were produced by stimulating the Schaffer collaterals and recording in the stratum radiatum of the CA1 region. The fEPSPs were adjusted to 50% of the maximum slope and test pulses were evoked every 20 s. Paired pulse responses were recorded using a 50 ms interpulse interval (20 Hz) and expressed as a ratio of the slopes of the second pulse over the first pulse. A 20 min stable baseline was established before delivering a theta burst stimulation (TBS) train of four pulses delivered at 100 Hz in 30 ms bursts repeated 10 times with 200 ms interburst intervals.7 Following TBS, the fEPSP was recorded for 60 min. The averaged 10 min slope from 50 to 60 min after TBS was divided by the average of the 10 min baseline (set to 100%) prior to TBS to determine the amount of potentiation. Analog fEPSPs were amplified (1000×) and filtered through a pre-amplifier (Model LP511 AC, Grass Instruments) at 1.0 kHz, digitized at 10 kHz and stored on computer for later offline analysis (Clampfit 10.4, Axon Instruments). The derivative (dV/dT) of the initial fEPSP slope was measured. For time course graphs, normalized fEPSP slope values were averaged and plotted as the percent change from baseline. In experiments using 7,8 dihydroxyflavone (7,8 DHF, Tocris), the drug was made in 5 mM aliquots in DMSO and diluted in ACSF to final concentrations as indicated in the results. Electrophysiology studies using 7,8 DHF were performed in paired fashion. Specifically, acute slices were obtained from mice seven days after cardiac arrest or sham surgeries and were superfused with aCSF (control) or aCSF + 7,8 DHF. Experiments were only included in final analysis if data were obtained under control and 7,8 DHF exposed slices from the same animal. Two electrophysiolgists (RD and JO) independently verified all LTP results in this report.
Behavioral test
The contextual fear conditioning (CFC) paradigm was utilized as a hippocampal-dependent memory task.30 The apparatus consisted of two fear conditioning chambers with shock grid floors, consisting of 16 stainless steel rods connected to a shock generator (Colbourn Instruments, Model H13-15, Whitehall, PA, USA). Mice were transported in white buckets during the training and testing sessions. During training, mice were allowed to habituate to the conditioning chamber for two separate 2-min pre-exposure sessions followed by a foot shock (2-s/1.0 mA electric shock) immediately after the second exposure. Following shock, mice were returned to their home cages. Memory was tested 24 h later by transporting mice in white buckets and placing back into the fear conditioning chambers. Memory was determined by percentage of freezing behavior, measured in 10 s intervals across a 5-min test by a blinded observer and was defined as the absence of movement except for heart beat/respiration. Two studies were conducted independently. Mice were randomly assigned to groups. In study 1, mice underwent CA/CPR or sham surgery, and CFC was performed 7 or 30 days after the surgery. In study 2, mice underwent CA/CPR or sham surgeries and 7,8 DHF or vehicle was injected intraperitoneally on day 6 after surgery and CFC was performed 24 h after injection. Study was randomized and blinded; 7,8 DHF was made and coded before given to a second person who injected the agent into the mice without their knowledge of the surgery performed on the mice. The person performing behavior was not aware of the surgery performed or if the mouse received 7,8 DHF or vehicle, and blinding was intact through analysis of the experiment. The primary author then decoded the data.
Western blot analysis
Single whole hippocampi were homogenized in N-PER (Thermo Scientific, Rockford, IL, USA), and nuclei and debris were removed by centrifugation (1000g ×10 min). Total protein content was measured by using a microplate bicinchoninic acid assay (Thermo Scientific, Rockford, IL, USA), and protein was resolved via electrophoresis on 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis gels for 1 h at 150 V. Protein was transferred to a polyvinylidene difluoride membrane for 1 h at 100 V, and incubated at room temperature (22℃) with gentle rocking in 5% milk in Tris-buffered saline with Tween (140 mM NaCl, 20 mM Tris (pH 7.4) and 0.1% Tween 20). Blots were incubated with anti-BDNF (1:2000 dilution in 1% milk; Millipore, Billerica, MA, USA) or anti-β-actin (1:5000 in 1% milk; Sigma-Aldrich, St Louis, MO, USA) overnight at 4℃, and washed five times for 5 min each in Tris-buffered saline with Tween, followed by a 1 h incubation with a horseradish peroxidase-conjugated goat anti-mouse (1:4000; Thermo Scientific) for membranes treated with anti-BDNF or anti-β-actin at room temperature. Blots were then washed in Tris-buffered saline with Tween five times for 5 min each, and bands were detected using SuperSignal chemiluminescent substrate kits (Thermo Scientific) and a ChemiDocTM MP Imaging System (Bio-Rad, Hercules, CA, USA). Quantification of integrated volume of bands was performed using IMAGE LAB software version 4.0 (Bio-Rad). Mice were randomly assigned to surgery and treatment groups and experimenter was blinded throughout Western blot procedures.
Immunohistochemistry
To label proliferative cells, the thymidine analog 5-bromo-2-deoxyuridine (BrdU, 50 mg/kg, Sigma-Aldrich, St. Louis, MO, USA) was administered intraperitoneally on days 2 to 4 after CA/CPR or sham, a time point shown to report maximum proliferation after ischemia.31–33 Thirty days after surgery, mice were transfused transcardially with 4% paraformaldehyde under deep anesthesia to disclose the survival and migration of pattern of BrdU + cells. Cell death was assessed at seven days after CA/CPR by a terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL, Cell Death Detection Kit, Roche) assay and co-labeled with Hoechst (DNA/nuclear counterstain). For BDNF expression, staining of 50 µm sections consisted of phosphate-buffered saline washes (1 × PBS, 3 × 5 min), 2 h incubation in blocking serum (10% normal donkey serum in 0.3% Triton X-100), 48 h incubation at 4℃ in primary antibody, PBS washes (3 × 5 min), 1 h incubation in secondary antibody, PBS washes (3 × 5 min), mounting and coverslip with anti-fade mounting medium (Vectashield, H-1000). Primary antibodies used were rabbit anti-brain-derived neurotrophic factor (BDNF, 1:500, Millipore, AB1779) and mouse anti-neuronal nuclei (NeuN, 1:500, Millipore, MAB377), with Alexa Fluor 488 or 594-conjugated IgG (1:600; Jackson Immuno) secondary antibody. Confocal microscopy was used to confirm co-localization of BDNF and NeuN using an Olympus FV1000 laser scanning confocal microscope and Olympus Fluoview imaging software (Center Valley, PA, USA). The Cell Counter plug-in on Fiji software34 was used for cell count analyses of medial hippocampus, averaged across the left and right hemisphere. Mice were randomly assigned to surgery and treatment groups, and experimenter was blinded throughout IHC experiments.
Statistical analysis
All data are presented as mean ± SEM. Sample size and power analyses were performed using previous data generated in our laboratory. To determine group size for LTP recordings and to observe a 40% change in LTP between two groups with a standard deviation of 20 and an alpha error of 5% and a beta error of 80%, a group of six slices per group are required, with no more than two slices per animal used in analysis per condition. For behavior studies, to observe a 25% change in object exploration between two groups with a standard deviation of 15%, with an alpha error of 5% and a beta error of 80%, eight animals per group are required for behavior experiments. Statistical analysis was performed using Student’s t-test for two-group comparisons and one-way ANOVA with Tukey post-hoc test for comparison of multiple groups. The effect of 7,8 DHF on LTP after CA/CPR was analyzed by paired t-test. Differences were considered statistically significant at two-tailed p < 0.05.
Results
Recovery of learning impairment after CA/CPR
To determine the effect of global ischemia on hippocampal function, we exposed juvenile mice (postnatal days 20–25) to 8 min of CA/CPR29 and performed contextual fear conditioning to analyze hippocampal-dependent cognition and memory.35,36 We found that seven days after sham surgery, mice exhibited freezing behavior in 51 ± 5.2% of time epochs (n = 8, Figure 1(a)) 24 h after training, indicating intact memory. However, mice seven days after CA/CPR demonstrated decreased freezing (24 ± 6.4%, n = 9, p < 0.05), indicating impairment in memory. Mice tested 30 days after recovery from CA/CPR had recovery of freezing behavior (52 ± 5.3%, n = 7), which was similar to both seven-day sham (above) and 30-day sham mice (52 ± 7.2%, n = 7). These data indicate that juvenile mice have memory dysfunction through development that shows pronounced recovery in adulthood.
Figure 1.
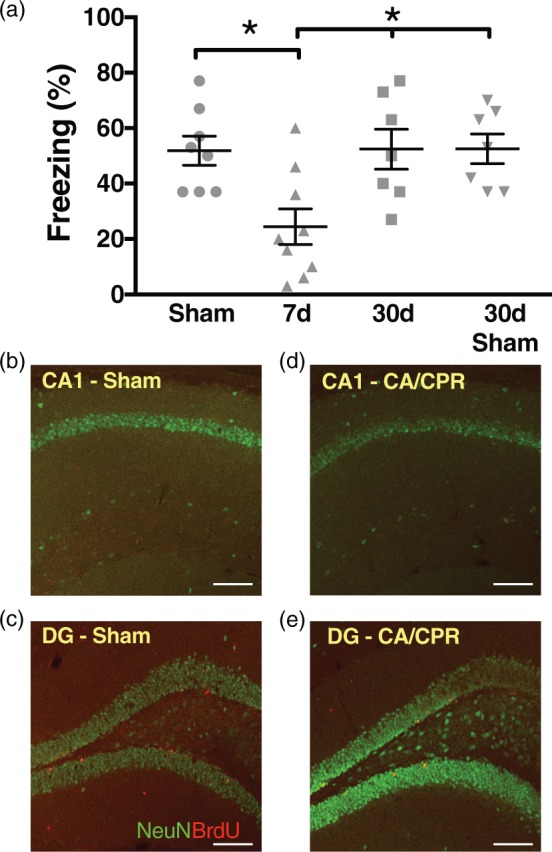
Learning and memory is transiently impaired after CA/CPR. (a) Quantification of freezing behavior 24 h after contextual fear conditioning in a novel environment after cardiac arrest. Each point plotted represents each animal tested. * p< 0.05 compared to sham or 30-day data. (b–e) High magnification images of hippocampal CA1 and dentate gyrus (DG) 30 days after CA/CPR or sham surgeries labeled with NeuN (green) to identify neurons and anti-BrdU (red) to identify newborn neurons. No BrdU signal was identified in CA1 indicating lack of newborn neurons. (b) CA1 after sham surgery; (c) dentate gyrus after sham surgery; (d) CA1 after CA/CPR; (e) dentate gyrus after CA/CPR. Scale bar = 100 µm.
CA/CPR: cardiac arrest/cardiopulmonary resuscitation; NeuN: neuronal nuclei; BrdU: 5-bromo-2-deoxyuridine.
We have previously shown that this model of CA/CPR in juvenile mice produces equivalent delayed neuronal death to that observed in adults,29 with the peak occurring three days after ischemic injury. Consistent with our previous findings, Supplemental Figure 1 shows extensive neuronal cell death in mice seven days after juvenile CA/CPR. One possible explanation for the recovery in memory ability in juvenile mice not observed in adults is increased neurogenesis and replacement of CA1 neurons after global ischemia in the weeks after CA/CPR. Therefore, we tested whether neurogenesis occurs which may account for the endogenous recovery of memory acquisition in the juvenile mouse. 5′-bromo-2′-deoxyuridine (BrdU), a marker for proliferating cells, was injected intraperitoneally 2–4 days after CA/CPR or sham surgeries when it has been reported that there is maximum proliferation of new neurons after ischemia,31–33 and the brains were collected on postoperative day 30 for analysis. We did not observe any post-ischemic newborn neurons (BrdU + cells) repopulating the CA1 region 30 days after recovery from juvenile CA/CPR, although BrdU + cells were observed in dentate gyrus (Figure 1(b to e)). Therefore, neurogenesis does not appear to account for the endogenous recovery of memory ability.
Impairment of synaptic plasticity following juvenile CA/CPR endogenously recovers
It has been demonstrated that learning behavior correlates with LTP in the hippocampus.37–41 Given the lack of neurogenesis in hippocampal CA1 30 days after CA/CPR to explain the endogenous recovery of memory, we next turned to LTP to determine the cellular mechanisms that may account for the memory recovery observed following juvenile CA/CPR. We previously reported that CA/CPR in juvenile mice impairs synaptically evoked LTP when examining extracellular field recordings in acute hippocampal slices prepared seven days after ischemia.23 Here we confirm LTP impairment after CA/CPR in juvenile mice by comparing the percent increase in synaptic response (in relation to baseline, set at 100%) in ischemic animals to sham controls. In slices from juvenile mice seven days after sham surgery, a brief theta burst stimulus (40 pulses, 100 Hz) resulted in LTP that increased the slope of the fEPSP to 156 ± 5.3% of baseline (baseline set to 100%, n = 8, Figure 2) after 60 min. In contrast, recordings obtained in the sub-acute recovery phase (7 and 14 days) exhibited impairment of LTP compared to sham; 7 day: 110 ± 5.7% (n = 8); 14 day: 119 ± 9.0% (n = 6), both p < 0.05 compared to sham control (Figure 2). When slices were prepared 30 days after CA/CPR, there was complete recovery of LTP (155 ± 5.3%, n = 8, Figure 2), mirroring the recovery of memory behavior at 30 days. Further, to assure that there was no change in synaptic plasticity through development from juvenile to young adult, sham 30-day mice were also examined and exhibited similar LTP to control shams above (153 ± 7.3%, n = 8). This recovery of LTP is in contrast to our recent studies in adults in which we observed that impairment persists for at least 30 days after CA/CPR.7
Figure 2.

Global cerebral ischemia transiently impairs synaptic plasticity. (a) Time course of fEPSP slope (mean ± SEM) from control male mice (n = 8, black), mice at seven days (n = 8, blue), 14 days (n = 7, grey) or 30 days (n = 8, red) after CA/CPR or 30 days after sham surgery (n = 8, green). Arrow indicates timing of theta burst stimulation (40 pulses, 100 Hz). (b) Quantification of change in fEPSP slope after 60 min following TBS normalized to baseline, set at 100% (mean ± SEM). (c) Quantification of the paired pulse ratio of the sham, 7, 14 and 30 days after CA/CPR and 30-day shams. (d) Input–output curve showing normalized fEPSP slope plotted against stimulus intensity (fEPSP slope is normalized to the maximum of each recording). Each point represents a hippocampal slice that was recorded with no more than two slices per animal used. *p < 0.05 compared with control mice.
CA/CPR: cardiac arrest/cardiopulmonary resuscitation; TBS: theta burst stimulation.
To assess the effect of CA/CPR on the intrinsic properties of excitability and presynaptic release probability, paired-pulse stimulation and input output responses were analyzed. Similar paired-pulse facilitation was seen between groups: pulse 2/pulse 1 ratios of 1.52 ± 0.06 for 7 d sham, 1.53 ± 0.05 for 7 d CA/CPR, 1.44 ± 0.08 for 14 d CA/CPR, 1.53 ± 0.05 for 30 d CA/CPR, and 1.46 ± 0.07 for 30 d sham (p > 0.05 by ANOVA, Figure 2(c)). Input–output responses revealed slopes of 1.86 ± 0.14 for 7 d sham, 1.95 ± 0.04 for 7 d CA/CPR, 2.11 ± 0.23 for 14 d CA/CPR, 1.97 ± 0.09 for 30 d CA/CPR and 2.03 ±0.07 for 30 d sham (p = 0.76 by linear regression analysis, Figure 2(d)). Therefore, the CA/CPR-induced alterations in LTP do not appear to be caused by changes in pre-synaptic excitability or probability of release, implicating post-synaptic mechanisms.
BDNF expression transiently decreases after CA/CPR
It is well accepted that BDNF signaling is required for synaptic plasticity.22,42 We therefore tested if BDNF expression changes during the sub-acute (7 days) and chronic (30 days) time periods after juvenile CA/CPR. Figure 3 reveals a 40 ± 0.11% decrease in mature BDNF (∼15 kDa) in hippocampus seven days after CA/CPR (n = 5), measured by normalized BDNF/β-actin ratio, compared to seven-day sham (n = 4, p < 0.05 by one-way ANOVA). However, BDNF expression recovered in mice 30 days after CA/CPR (n = 4) or sham surgery (n = 4) similar to seven days after sham and was significantly higher than 7 d CA/CPR (p < 0.05 by one-way ANOVA). We went on to confirm that there is decreased BDNF in hippocampal CA1 neurons seven days after CA/CPR using immunohistochemistry. CA1 neurons that were co-labeled with NeuN (neuronal marker) and BDNF were quantified seven days after CA/CPR and showed that there were less BDNF-positive neurons seven days after CA/CPR compared to sham mice (Figure 3), consistent with the Western blot analysis and impaired synaptic function at seven days after CA/CPR. The number of BDNF-positive neurons in CA1 was not different at 30 days compared to seven days after CA/CPR, possibly indicating a non-neuronal source of BDNF measured in the Western blots. Together, these data suggest that the memory impairment and diminished synaptic function correlate with decreased neuronal BDNF in juvenile mice sub-acutely after CA/CPR.
Figure 3.

Brain-derived neurotrophic factor (BDNF) expression is decreased after cardiac arrest. (a) Representative Western blot analysis of mature BDNF (∼15 kDa) and β-actin in sham and cardiac arrest mice. (b) Quantification of the normalized ratio of BDNF to β-actin in whole hippocampus after cardiac arrest. Each point represents the numbers of animals used. *p < 0.05 compared with control and 30-day mice. (c) Representative images of CA1 neurons co-labeled with NeuN (green) and BDNF (red) from seven-day sham, 30-day sham, seven-day CA/CPR, and 30-day CA/CPR. (d) Quantification of NeuN+/BDNF + cells in CA1 region of hippocampus (Sham 7 d n = 8; sham 30 d n = 8; CA/CPR 7 d n = 9; CA/CPR 30 d n = 10; *p < 0.05 by two-way ANOVA). Scale bar = 100 µm.
CA/CPR: cardiac arrest/cardiopulmonary resuscitation; NeuN: neuronal nuclei.
Administration of TrkB receptor agonist reverses synaptic impairment
To test whether the decline in BDNF observed following CA/CPR is a causative contributor to impaired LTP, we sought to stimulate the BDNF receptor tyrosine kinase (TrkB) receptors22,42 at time points well beyond that of neuronal death29 that would be relevant to the impairments described above. Therefore, we tested whether the selective TrkB agonist 7,8 DHF can reverse synaptic impairment at seven days after CA/CPR. 7,8 DHF is a potent agonist of TrkB that can cross the blood–brain barrier after oral or intraperitoneal administration.43 In paired experiments (slices from same animal used for control and 7,8 DHF recordings) using acute hippocampal slices from mice seven days after sham surgeries, 7,8 DHF showed no effect on LTP (Figure 4(c)). However, in paired experiments using hippocampal slices from mice seven days after CA/CPR, 7,8 DHF (250 nM) rescued synaptic impairment (105 ± 5% in vehicle vs. 143 ± 10% with 7,8 DHF, n = 6 each, p < 0.05, paired t-test). There was no difference in paired-pulse stimulation after 30-min wash of 7,8 DHF (data not shown). We found no further difference when using 1 µM 7,8 DHF (102 ± 2% without 7,8 DHF vs. 132 ± 8% with 7,8 DHF, n = 5 each, p < 0.05, paired t-test). Therefore, stimulating the TrkB receptor at delayed time points rescued LTP after CA/CPR.
Figure 4.

TrkB receptor agonist rescues synaptic impairment when applied seven days after CA/CPR. (a) Experimental design for LTP experiments. (b) Time course of fEPSP slope from mice seven days after CA/CPR (black, n = 6) and paired slices bath applied with 7,8 dihydroxyflavone (7,8 DHF, 250 nM, n = 6, red). Arrow indicates timing of theta burst stimulation (TBS, 40 pulses, 100 Hz). (c) Quantification of change in fEPSP slope after 60 min following TBS normalized to baseline. Sham and seven-day CA/CPR slices were paired with and without 7,8 DHF and paired experiments are indicated by the line between the pairs. (d) Experimental design for contextual fear conditioning in relation to 7,8 DHF injection. (e) Quantification of freezing behavior 24 h after contextual fear conditioning in a novel environment in sham or cardiac arrest mice administered vehicle or 7,8 DHF (5 mg/kg). Each dot represents one animal used for behavior. *p < 0.05 by paired t-test for electrophysiology and ANOVA for behavior.
LTP: long-term potentiation; CA/CPR: cardiac arrest/cardiopulmonary resuscitation.
Finally, to test whether this finding could be extended to memory impairments in vivo, contextual fear conditioning studies were conducted in mice that were administered a single dose of 7,8 DHF (5 mg/kg, ip)44 or vehicle at six days after CA/CPR and underwent contextual fear conditioning testing 24 h later. 7,8 DHF had no effect on freezing in sham mice compared to vehicle (vehicle 57 ± 10% (n = 6), 7,8 DHF 56 ± 5% (n = 6), Figure 4). In contrast, CA/CPR mice that received vehicle froze in 33 ± 6% (n = 8) of time epochs, whereas mice that received 7,8 DHF froze in 55 ± 3% of time epochs (n = 8, p < 0.05, Figure 4), indicating reversal of memory impairments by stimulation of TrKB receptors at delayed time points.
Discussion
We present here the first evidence of endogenous recovery of memory and synaptic function after global ischemia in the developing brain (Figure 5). We further show that endogenous functional recovery is not due to replacement of new neurons in hippocampal CA1 (neurogenesis, consistent with previous data in cardiac arrest45 although controversial in some other models of transient global ischemia46–48) or changes in pre-synaptic excitability or probability of release, but rather correlates with a transient decrease and recovery of BDNF expression (Figure 5). Together, these important observations are consistent with data showing that among survivors of cardiac arrest in children, global ischemic brain injury often leads to significant neurologic dysfunction,1–3 including impaired memory and executive cognitive function3,49–53 that can persist through school age.54 The lack of neurogenesis we report here is consistent with previous global cerebral models, although some have found small numbers of BrdU-labeled cells after 2 - or 4 vessel occlusion studies 35–90 days after ischemia. In conjunction with the observation that children may be capable of some degree of repair after ischemic brain injury,55 the novel observation of endogenous recovery is important because: (1) our data indicate functional impairment during important developmental time points that is reversible and (2) spontaneous recovery following brain injury in the young represents a unique developmental plasticity not observed following global ischemic brain injury in the adult.7,8 Our data suggest that there are important differences that exist between adults who suffer ischemic injury and those who have ischemic injuries when young and then are left to recover and mature to adulthood. It is well accepted that several days in a young mouse likely represents several months to years of neurodevelopment in a child.56 While many children may not achieve full recovery to pre-injury levels as suggested by the data in our mouse model, our data support the remarkable plasticity potential of the young brain and reinforces the role that BDNF appears to play.
Figure 5.

Model of time courses of neurodevelopment between mice and humans in relation to timing of global ischemia. Impairment of memory and synaptic plasticity is transient, but likely represents several years in a child during which intervention can occur. This model shows the corresponding decrease and recovery in BDNF which contributes to the impairment and endogenous recovery of memory and synaptic function.
BDNF: brain-derived neurotrophic factor; CA/CPR: cardiac arrest/cardiopulmonary resuscitation.
BDNF is a well-studied neurotrophic factor important in the development of neuronal networks and learning and memory, exerting effects on neuronal plasticity, synapse development and dendritic sprouting22 through the regulation by multiple endocrine and neurotransmitter pathways.57 Indeed, there is abundant evidence for the role of BDNF–TrkB signaling in normal synaptic plasticity via various downstream mediators.22,57 The requirement of BDNF for synaptic plasticity makes it impossible to block the endogenous recovery reported here by antagonizing the TrkB receptor, as this would result in impaired LTP and memory in sham animals.22 Therefore, it is impossible to assess the effect of inhibiting TrkB receptors on spontaneous functional recovery following CA/CPR in juveniles. In contrast, the role for BDNF–TrkB signaling following ischemia is less clear. Prior studies have found that BDNF and TrkB mRNA levels58 are increased in the first hours after ischemia in adults and acute BDNF infusions immediately after experimental stroke decreased histological injury, improved LTP and functional recovery (memory and motor behavior).27,59,60 Further, hypothermia after adult CA/CPR can potentiate BDNF levels 24 h after reperfusion, potentially playing a role in the beneficial effects of this therapy.61 However, BDNF infusion does not improve survival or provide neuroprotection when given soon after CA/CPR in adult rats28,62 possibly due to poor bioavailability and lipophilic nature. Juvenile rats exposed to hypoxia-ischemia demonstrate unchanged BDNF mRNA levels in the hours after ischemia, after which BDNF decreased in CA1 neurons of hippocampus ipsilateral to carotid ligation at three and five days after ischemia.63 These data are consistent with the decreased BDNF expression we describe here in juvenile mice days after CA/CPR and highlights the importance of studying the developing brain apart from adult brain. While it remains an open question whether decreased BDNF expression is involved in impairment of synaptic plasticity in adults after CA/CPR,7 it is likely that this signaling is unique to the juvenile brain given that spontaneous recovery does not occur in the mature brain. To our knowledge, no other studies have investigated the changes in BDNF expression at chronic time points (30 days) after global ischemia as we have in the present study.
Our data in this study and previous studies7,23,64 indicate relative recovery of basal synaptic transmission following CA/CPR, as indicated by field recordings of input–output curves and paired-pulse ratios. This indicates robust compensatory mechanisms, which are capable of restoring synaptic transmission, albeit not to pre-injury levels. Indeed, previous work has shown increased dendritic arborization following global ischemia,65,66 consistent with our electrophysiology data of compensation. Thus, we believe that following neuronal cell death, the network compensates. However, the newly compensated network exhibits impaired synaptic plasticity (and learning and memory behavior), which we are able to target in the current study by enhancing BDNF–TrkB signaling with 7,8 DHF. While recent in vitro binding assays have suggested that the ability of 7,8 DHF to activate TrkB receptors is limited,67,68 our results are consistent with a large body of literature showing 7,8 DHF activates TrkB receptors in vivo. Further, we do not think that the effects of 7,8 DHF are due to neurogenesis or angiogenesis due to the short exposure of the drug in our experimental design. Therefore, our data using delayed administration of 7,8 DHF is consistent with the observation that juvenile CA/CPR causes reduced BDNF expression, that can be targeted to enhance recovery following global cerebral ischemia in the young.
One reason that many pre-clinical studies of ischemia have failed to translate in clinical studies may be due to the focus on preventing neuronal death. Here, we show that a neurorestorative strategy can rescue functional impairments at time points significantly after neuronal death has occurred, dramatically increasing the therapeutic window after global ischemia. The strategy to administer 7,8 DHF at time points after CA/CPR-induced cell death to restore memory and synaptic function is novel following cardiac arrest. Our finding is an important advance in the development of new therapies for memory deficits, consistent with recent studies showing efficacy of delayed interventions to improve functional outcomes following brain ischemia. Clarkson et al. have previously targeted recovery of cortical neuronal networks after focal stroke. Their initial work targeted tonic GABA signaling three days after stroke, resulting in reversal of motor impairments, possibly by normalization of excitability in peri-infarct tissue.69 Interestingly, Clarkson et al. also administered positive allosteric modulator to stimulate AMPA receptor activity as a positive allosteric modulator (termed AMPAkine) five days after stroke to improve limb motor control.70 Importantly, there was no effect on infarct size, indicating the rescue effect is on the recovering tissue. Further, AMPAkines were found to enhance BDNF signaling in peri-infarct cortex.70 Recently, histone deacetylase 2 has also been shown to be a novel target that provides a prolonged therapeutic window after focal ischemia.13 These findings, combined with those presented here, suggest that BDNF signaling is a compelling target to reverse impairments after global ischemia, leading to potential new applications of agents known to enhance BDNF levels.71,72
In conclusion, we provide the first direct evidence that the developing brain has the endogenous ability to recover learning and memory function, highlighting the difference between the developing brain and previously described impairments of the mature brain. The mechanism for the initial impairment and subsequent recovery is the decrease and recovery of BDNF–TrkB signaling. Despite this endogenous recovery, memory and plasticity deficits for any amount of time likely reflect impaired learning potential during critical school age years. Therefore, our strategy of stimulating the TrkB receptor several days after cardiac arrest in juvenile animals leading to recovered learning and memory impairments provide the possibility of dramatically altering the learning potential of the young brain after ischemia. Further, we identify an intervention window far longer than what is currently used as a clinical standard of care that can improve functional outcomes in the developing brain at sub-acute time points after global cerebral ischemia. Thus, this work establishes a novel therapeutic strategy to improve functional consequences of global ischemia that can be delivered well after cardiac arrest-induced neuronal death to recover function in surviving neurons of the developing brain.
Supplemental Material
Supplemental material for Juvenile cerebral ischemia reveals age-dependent BDNF–TrkB signaling changes: Novel mechanism of recovery and therapeutic intervention by Robert M Dietz, James E Orfila, Krista M Rodgers, Olivia P Patsos, Guiying Deng, Nicholas Chalmers, Nidia Quillinan, Richard J Traystman and Paco S Herson in Journal of Cerebral Blood Flow & Metabolism
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: R01NS092645, R01NS046072, 1K08NS097586.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
RMD and JEO contributed equally to this article. RMD: Study conception and design, acquisition of data, analysis and interpretation of data, drafting of manuscript. JEO: Acquisition of data, analysis and interpretation of data, critical revision. KMR: Acquisition of data, analysis and interpretation of data, drafting of manuscript. OPP: Acquisition of data, analysis and interpretation of data, drafting of manuscript. GD: Acquisition of data, analysis and interpretation of data, drafting of manuscript. NC: Acquisition of data, analysis and interpretation of data, drafting of manuscript. NQ: Study conception and design, critical revision. RJT: Study conception and design, critical revision. PSH: Study conception and design, analysis and interpretation of data, critical revision.
Supplementary material
Supplementary material for this paper can be found at the journal website: http://journals.sagepub.com/home/jcb.
References
- 1.Michiels EA, Dumas F, Quan L, et al. Long-term outcomes following pediatric out-of-hospital cardiac arrest*. Pediatr Crit Care Med 2013; 14: 755–760. [DOI] [PubMed] [Google Scholar]
- 2.Moler FW, Donaldson AE, Meert K, et al. Multicenter cohort study of out-of-hospital pediatric cardiac arrest. Crit Care Med 2011; 39: 141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schindler MB, Bohn D, Cox PN, et al. Outcome of out-of-hospital cardiac or respiratory arrest in children. N Engl J Med 1996; 335: 1473–1479. [DOI] [PubMed] [Google Scholar]
- 4.Arrich J, Holzer M, Havel C, et al. Hypothermia for neuroprotection in adults after cardiopulmonary resuscitation. Cochrane Database Syst Rev 2012; 9: CD004128. [DOI] [PubMed] [Google Scholar]
- 5.Jacobs SE, Berg M, Hunt R, et al. Cooling for newborns with hypoxic ischaemic encephalopathy. Cochrane Database Syst Rev 2013; 1: CD003311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herson PS, Traystman RJ. Animal models of stroke: translational potential at present and in 2050. Future Neurol 2014; 9: 541–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Orfila JE, Shimizu K, Garske AK, et al. Increasing small conductance Ca(2+) -activated potassium channel activity reverses ischemia-induced impairment of long-term potentiation. Eur J Neurosci 2014; 40: 3179–3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang L, Hsu JC, Takagi N, et al. Transient global ischemia alters NMDA receptor expression in rat hippocampus: correlation with decreased immunoreactive protein levels of the NR2A/2B subunits, and an altered NMDA receptor functionality. J Neurochem 1997; 69: 1983–1994. [DOI] [PubMed] [Google Scholar]
- 9.Liu Z, Zhao W, Xu T, et al. Alterations of NMDA receptor subunits NR1, NR2A and NR2B mRNA expression and their relationship to apoptosis following transient forebrain ischemia. Brain Res 2010; 1361: 133–139. [DOI] [PubMed] [Google Scholar]
- 10.Kiryk A, Pluta R, Figiel I, et al. Transient brain ischemia due to cardiac arrest causes irreversible long-lasting cognitive injury. Behav Brain Res 2011; 219: 1–7. [DOI] [PubMed] [Google Scholar]
- 11.Cohan CH, Neumann JT, Dave KR, et al. Effect of cardiac arrest on cognitive impairment and hippocampal plasticity in middle-aged rats. PloS One 2015; 10: e0124918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carmichael ST. Brain excitability in stroke: the yin and yang of stroke progression. Arch Neurol 2012; 69: 161–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin YH, Dong J, Tang Y, et al. Opening a new time window for treatment of stroke by targeting HDAC2. J Neurosci 2017; 37: 6712–6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Orfila JE, Grewal H, Dietz RM, et al. Delayed inhibition of tonic inhibition enhances functional recovery following experimental ischemic stroke. J Cereb Blood Flow Metab Epub ahead of print 2017. 271678X17750761. 2017/12/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moler FW, Silverstein FS, Holubkov R, et al. Therapeutic hypothermia after out-of-hospital cardiac arrest in children. N Engl J Med 2015; 372: 1898–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moler FW, Silverstein FS, Holubkov R, et al. Therapeutic hypothermia after in-hospital cardiac arrest in children. N Engl J Med 2017; 376: 318–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hickey RW, Painter MJ. Brain injury from cardiac arrest in children. Neurol Clin 2006; 24: 147–158. [DOI] [PubMed] [Google Scholar]
- 18.Rice D, Barone S., Jr Critical periods of vulnerability for the developing nervous system: evidence from humans and animal models. Environ Health Perspect 2000; 108(Suppl 3): 511–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nicoll RA, Malenka RC. Contrasting properties of two forms of long-term potentiation in the hippocampus. Nature 1995; 377: 115–118. [DOI] [PubMed] [Google Scholar]
- 20.Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron 2004; 44: 5–21. [DOI] [PubMed] [Google Scholar]
- 21.Figurov A, Pozzo-Miller LD, Olafsson P, et al. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature 1996; 381: 706–709. [DOI] [PubMed] [Google Scholar]
- 22.Minichiello L. TrkB signalling pathways in LTP and learning. Nat Rev Neurosci 2009; 10: 850–860. [DOI] [PubMed] [Google Scholar]
- 23.Dietz RM, Deng G, Orfila JE, et al. Therapeutic hypothermia protects against ischemia-induced impairment of synaptic plasticity following juvenile cardiac arrest in sex-dependent manner. Neuroscience 2016; 325: 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Almli CR, Levy TJ, Han BH, et al. BDNF protects against spatial memory deficits following neonatal hypoxia-ischemia. Exp Neurol 2000; 166: 99–114. [DOI] [PubMed] [Google Scholar]
- 25.Han BH, Holtzman DM. BDNF protects the neonatal brain from hypoxic-ischemic injury in vivo via the ERK pathway. J Neurosci 2000; 20: 5775–5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Donega V, Nijboer CH, van Tilborg G, et al. Intranasally administered mesenchymal stem cells promote a regenerative niche for repair of neonatal ischemic brain injury. Exp Neurol 2014; 261: 53–64. [DOI] [PubMed] [Google Scholar]
- 27.Kiprianova I, Sandkuhler J, Schwab S, et al. Brain-derived neurotrophic factor improves long-term potentiation and cognitive functions after transient forebrain ischemia in the rat. Exp Neurol 1999; 159: 511–519. [DOI] [PubMed] [Google Scholar]
- 28.Callaway CW, Ramos R, Logue ES, et al. Brain-derived neurotrophic factor does not improve recovery after cardiac arrest in rats. Neurosci Lett 2008; 445: 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deng G, Yonchek JC, Quillinan N, et al. A novel mouse model of pediatric cardiac arrest and cardiopulmonary resuscitation reveals age-dependent neuronal sensitivities to ischemic injury. J Neurosci Meth 2014; 222: 34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rudy JW, O'Reilly RC. Conjunctive representations, the hippocampus, and contextual fear conditioning. Cogn Affect Behav Neurosci 2001; 1: 66–82. [DOI] [PubMed] [Google Scholar]
- 31.Zhang RL, Zhang ZG, Zhang L, et al. Proliferation and differentiation of progenitor cells in the cortex and the subventricular zone in the adult rat after focal cerebral ischemia. Neuroscience 2001; 105: 33–41. [DOI] [PubMed] [Google Scholar]
- 32.Takasawa K, Kitagawa K, Yagita Y, et al. Increased proliferation of neural progenitor cells but reduced survival of newborn cells in the contralateral hippocampus after focal cerebral ischemia in rats. J Cereb Blood Flow Metab 2002; 22: 299–307. [DOI] [PubMed] [Google Scholar]
- 33.Kawai T, Takagi N, Miyake-Takagi K, et al. Characterization of BrdU-positive neurons induced by transient global ischemia in adult hippocampus. J Cereb Blood Flow Metab 2004; 24: 548–555. [DOI] [PubMed] [Google Scholar]
- 34.Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 2012; 9: 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu W, Wang L, Zhang L, et al. Isoflurane preconditioning neuroprotection in experimental focal stroke is androgen-dependent in male mice. Neuroscience 2010; 169: 758–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Uchida M, Palmateer JM, Herson PS, et al. Dose-dependent effects of androgens on outcome after focal cerebral ischemia in adult male mice. J Cereb Blood Flow Metab 2009; 29: 1454–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stuchlik A. Dynamic learning and memory, synaptic plasticity and neurogenesis: an update. Front Behav Neurosci 2014; 8: 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bruel-Jungerman E, Davis S, Laroche S. Brain plasticity mechanisms and memory: a party of four. Neuroscientist 2007; 13: 492–505. [DOI] [PubMed] [Google Scholar]
- 39.Whitlock JR, Heynen AJ, Shuler MG, et al. Learning induces long-term potentiation in the hippocampus. Science 2006; 313: 1093–1097. [DOI] [PubMed] [Google Scholar]
- 40.Pastalkova E, Serrano P, Pinkhasova D, et al. Storage of spatial information by the maintenance mechanism of LTP. Science 2006; 313: 1141–1144. [DOI] [PubMed] [Google Scholar]
- 41.Bliss TV, Collingridge GL, Laroche S. ZAP and ZIP, a story to forget. Science 2006; 313: 1058–1059. [DOI] [PubMed] [Google Scholar]
- 42.Patterson SL, Abel T, Deuel TA, et al. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron 1996; 16: 1137–1145. [DOI] [PubMed] [Google Scholar]
- 43.Wang B, Wu N, Liang F, et al. 7,8-dihydroxyflavone, a small-molecule tropomyosin-related kinase B (TrkB) agonist, attenuates cerebral ischemia and reperfusion injury in rats. J Mol Histol 2014; 45: 129–140. [DOI] [PubMed] [Google Scholar]
- 44.Sanz-Garcia A, Knafo S, Pereda-Perez I, et al. Administration of the TrkB receptor agonist 7,8-dihydroxyflavone prevents traumatic stress-induced spatial memory deficits and changes in synaptic plasticity. Hippocampus 2016; 26: 1179–1188. [DOI] [PubMed] [Google Scholar]
- 45.Keilhoff G, John R, Langnaese K, et al. Triggered by asphyxia neurogenesis seems not to be an endogenous repair mechanism, gliogenesis more like it. Neuroscience 2010; 171: 869–884. [DOI] [PubMed] [Google Scholar]
- 46.Salazar-Colocho P, Lanciego JL, Del Rio J, et al. Ischemia induces cell proliferation and neurogenesis in the gerbil hippocampus in response to neuronal death. Neurosci Res 2008; 61: 27–37. [DOI] [PubMed] [Google Scholar]
- 47.Bendel O, Bueters T, von Euler M, et al. Reappearance of hippocampal CA1 neurons after ischemia is associated with recovery of learning and memory. J Cereb Blood Flow Metab 2005; 25: 1586–1595. [DOI] [PubMed] [Google Scholar]
- 48.Silasi G, Colbourne F. Therapeutic hypothermia influences cell genesis and survival in the rat hippocampus following global ischemia. J Cereb Blood Flow Metab 2011; 31: 1725–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bunch TJ, White RD, Smith GE, et al. Long-term subjective memory function in ventricular fibrillation out-of-hospital cardiac arrest survivors resuscitated by early defibrillation. Resuscitation 2004; 60: 189–195. [DOI] [PubMed] [Google Scholar]
- 50.Mateen FJ, Josephs KA, Trenerry MR, et al. Long-term cognitive outcomes following out-of-hospital cardiac arrest: a population-based study. Neurology 2011; 77: 1438–1445. [DOI] [PubMed] [Google Scholar]
- 51.O'Reilly SM, Grubb NR, O'Carroll RE. In-hospital cardiac arrest leads to chronic memory impairment. Resuscitation 2003; 58: 73–79. [DOI] [PubMed] [Google Scholar]
- 52.Meaney PA, Nadkarni VM, Cook EF, et al. Higher survival rates among younger patients after pediatric intensive care unit cardiac arrests. Pediatrics 2006; 118: 2424–2433. [DOI] [PubMed] [Google Scholar]
- 53.Nitta M, Iwami T, Kitamura T, et al. Age-specific differences in outcomes after out-of-hospital cardiac arrests. Pediatrics 2011; 128: e812–e820. [DOI] [PubMed] [Google Scholar]
- 54.Del Castillo J, Lopez-Herce J, Matamoros M, et al. Long-term evolution after in-hospital cardiac arrest in children: Prospective multicenter multinational study. Resuscitation 2015; 96: 126–134. [DOI] [PubMed] [Google Scholar]
- 55.Anderson V, Spencer-Smith M, Wood A. Do children really recover better? Neurobehavioural plasticity after early brain insult. Brain 2011; 134: 2197–2221. [DOI] [PubMed] [Google Scholar]
- 56.Semple BD, Blomgren K, Gimlin K, et al. Brain development in rodents and humans: identifying benchmarks of maturation and vulnerability to injury across species. Prog Neurobiol 2013; 106–107: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tapia-Arancibia L, Rage F, Givalois L, et al. Physiology of BDNF: focus on hypothalamic function. Front Neuroendocrinol 2004; 25: 77–107. [DOI] [PubMed] [Google Scholar]
- 58.Merlio JP, Ernfors P, Kokaia Z, et al. Increased production of the TrkB protein tyrosine kinase receptor after brain insults. Neuron 1993; 10: 151–164. [DOI] [PubMed] [Google Scholar]
- 59.Beck T, Lindholm D, Castren E, et al. Brain-derived neurotrophic factor protects against ischemic cell damage in rat hippocampus. J Cereb Blood Flow Metab 1994; 14: 689–692. [DOI] [PubMed] [Google Scholar]
- 60.Schabitz WR, Berger C, Kollmar R, et al. Effect of brain-derived neurotrophic factor treatment and forced arm use on functional motor recovery after small cortical ischemia. Stroke 2004; 35: 992–997. [DOI] [PubMed] [Google Scholar]
- 61.D'Cruz BJ, Fertig KC, Filiano AJ, et al. Hypothermic reperfusion after cardiac arrest augments brain-derived neurotrophic factor activation. J Cereb Blood Flow Metab 2002; 22: 843–851. [DOI] [PubMed] [Google Scholar]
- 62.Popp E, Padosch SA, Vogel P, et al. Effects of intracerebroventricular application of brain-derived neurotrophic factor on cerebral recovery after cardiac arrest in rats. Crit Care Med 2004; 32: S359–S365. [DOI] [PubMed] [Google Scholar]
- 63.Dragunow M, Beilharz E, Mason B, et al. Brain-derived neurotrophic factor expression after long-term potentiation. Neurosci Lett 1993; 160: 232–236. [DOI] [PubMed] [Google Scholar]
- 64.Deng G, Orfila JE, Dietz RM, et al. Autonomous CaMKII activity as a drug target for histological and functional neuroprotection after resuscitation from cardiac arrest. Cell Rep 2017; 18: 1109–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ruan YW, Han XJ, Shi ZS, et al. Remodeling of synapses in the CA1 area of the hippocampus after transient global ischemia. Neuroscience 2012; 218: 268–277. [DOI] [PubMed] [Google Scholar]
- 66.Ruan YW, Zou B, Fan Y, et al. Dendritic plasticity of CA1 pyramidal neurons after transient global ischemia. Neuroscience 2006; 140: 191–201. 2006/03/15. [DOI] [PubMed] [Google Scholar]
- 67.Todd D, Gowers I, Dowler SJ, et al. A monoclonal antibody TrkB receptor agonist as a potential therapeutic for Huntington's disease. PloS One 2014; 9: e87923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Boltaev U, Meyer Y, Tolibzoda F, et al. Multiplex quantitative assays indicate a need for reevaluating reported small-molecule TrkB agonists. Sci Signal 2017; 10: 1–10. [DOI] [PubMed] [Google Scholar]
- 69.Clarkson AN, Huang BS, Macisaac SE, et al. Reducing excessive GABA-mediated tonic inhibition promotes functional recovery after stroke. Nature 2010; 468: 305–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Clarkson AN, Overman JJ, Zhong S, et al. AMPA receptor-induced local brain-derived neurotrophic factor signaling mediates motor recovery after stroke. J Neurosci 2011; 31: 3766–3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nibuya M, Morinobu S, Duman RS. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosc 1995; 15: 7539–7547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Castren E, Rantamaki T. The role of BDNF and its receptors in depression and antidepressant drug action: Reactivation of developmental plasticity. Dev Neurobiol 2010; 70: 289–297. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material for Juvenile cerebral ischemia reveals age-dependent BDNF–TrkB signaling changes: Novel mechanism of recovery and therapeutic intervention by Robert M Dietz, James E Orfila, Krista M Rodgers, Olivia P Patsos, Guiying Deng, Nicholas Chalmers, Nidia Quillinan, Richard J Traystman and Paco S Herson in Journal of Cerebral Blood Flow & Metabolism