

Abstract
Perinatal hypoxia-ischemia resulting in death or lifelong disabilities remains a major clinical disorder. Neonatal models of hypoxia-ischemia in rodents have enhanced our understanding of cellular mechanisms of neural injury in developing brain, but have limitations in simulating the range, accuracy, and physiology of clinical hypoxia-ischemia and the relevant systems neuropathology that contribute to the human brain injury pattern. Large animal models of perinatal hypoxia-ischemia, such as partial or complete asphyxia at the time of delivery of fetal monkeys, umbilical cord occlusion and cerebral hypoperfusion at different stages of gestation in fetal sheep, and severe hypoxia and hypoperfusion in newborn piglets, have largely overcome these limitations. In monkey, complete asphyxia produces preferential injury to cerebellum and primary sensory nuclei in brainstem and thalamus, whereas partial asphyxia produces preferential injury to somatosensory and motor cortex, basal ganglia, and thalamus. Mid-gestational fetal sheep provide a valuable model for studying vulnerability of progenitor oligodendrocytes. Hypoxia followed by asphyxia in newborn piglets replicates the systems injury seen in term newborns. Efficacy of post-insult hypothermia in animal models led to the success of clinical trials in term human neonates. Large animal models are now being used to explore adjunct therapy to augment hypothermic neuroprotection.
Keywords: Animal model, development, hypoxia-ischemia, neonate, selective neuronal vulnerability
Introduction
In this issue dedicated to the legacy of Dr. Richard J. Traystman, we devote this review to some of the work that he inspired on regulation of the cerebral circulation during development and the physiological and neurological impacts of global cerebral ischemia on the developing brain, such as that which occurs in perinatal hypoxia-ischemia (HI) and pediatric cardiac arrest. Early in his research, he championed the use of large animal models as clinical translation models to better understand the physiology and pathophysiology of acute insults to the brain. Here, we emphasize how the use of large animal models of HI has advanced our understanding of neonatal HI encephalopathy (HIE) and led to clinical therapeutic hypothermia.
HIE: Epidemiology and current treatments
Neonatal encephalopathy affects approximately 3 in 1000 births1 and is caused by infection and inflammation, global HI, and focal cerebral stroke. Some estimates suggest that nearly half of all neonatal encephalopathies can be attributed to HI.1 Despite advances in reducing childhood mortality from infections, the mortality rate from birth asphyxia remains high, and survivors often suffer persistent morbidity, including motor, cognitive, visual, hearing, attention, and executive function deficits.2,3 Current treatment for HI in term newborns is hypothermia. Clinical trials of hypothermia have demonstrated reduced mortality and/or improved neurologic outcome at 18–24 months.4-6 However, the number needed to treat to benefit one newborn ranges from 7 to 9 infants.7–10 About one-third to one-half had persistent neurologic abnormalities or low IQ at six to seven years of age.2,3 The incomplete neurologic recovery is attributed to the severity of the HI insult and the 2–6-h delay in initiating hypothermia after birth in many of the patients. Thus, interest persists in better understanding the mechanisms of HI-induced damage in the developing brain so that the number needed-to-treat can be reduced and additional therapeutics can be devised.
HIE animal modeling
Experimentally, global HI models have been the most studied in neonatal and fetal animals. However, it is noteworthy that many vascular, hematological, and hemodynamic differences between human and commonly used animals exist.11 The most commonly used models of HI in postnatal mice and rats involve permanent unilateral occlusion of a common carotid artery and transient systemic hypoxia.12 These models have provided valuable information on some molecular mechanisms of neuronal injury during development, as reviewed elsewhere,13–17 but its pertinence to human HIE-related neurodegeneration remains uncertain.18,19 Postnatal rodent models of HIE have important limitations. While the hypoxia component of the insult is systemic, the ischemic component is unilateral and incomplete resulting in focal stroke-like brain lesions. Thus, variable amounts of infarction often develop and impede the ability to study the selective neuronal cell death typically present in human HIE at term. There is also a lack of strict and reproducible concordance between the physiological insult and manifestation of brain injury. In neonatal mice particularly, animals might not develop overt brain damage despite the insult, and hypothermic treatment is generally not individualized.
There are major neuroanatomical drawbacks to using rodent models of HIE that can be remedied by large animals. The cerebral cortex of rodents is lissencephalic compared to large animals, and it lacks clear divisibility of cortical parcellation similar to human Brodmann areas (Figure 1). The gyrencephalic brains of large animals, such as monkey, sheep, and pig (Figure 1), are, at birth, 20–50 times the size of rodent brains (Figure 1) and permit the study of selective cortical area and neuronal vulnerability, regional white matter injury, and connectivity. The amount of white matter in postnatal rodents is small, and the ability to perform biochemical assays on white matter and cortical gray matter independently is not feasible. In contrast, infant monkeys20 and newborn pigs have white matter tractography similar to infant human (Figure 2 gross cross-sectional slabs of infant human and pig) that differ only in quantity. Experimental modeling of brain injury and development with large animals has many challenges. Experiments are more expensive to conduct. The animals are more costly on an individual basis, and the fiscal needs to support equipment and personnel teams for animal intensive care are great. However, this permits better whole-animal patient-like care, and physiological monitoring standardizes the model for generating highly reproducible and predictable outcomes for rigorous and transparent therapeutic preclinical testing in accordance with the STAIR guidelines.21
Figure 1.

Comparative CNS gross neuroanatomy in neonatal mouse, pig, and human infant at 12 months’ age. The P7 mouse CNS is about 34 mm in length, while that of a P5 piglet is almost 32 cm. Similar to the human infant brain, the pig brain is gyrencephalic but the mouse brain is lissencephalic. The pig and human brain have clear cortical parcellation of the cerebral cortex into lobes that have some equivalence to Brodmann cortical areas.
Figure 2.
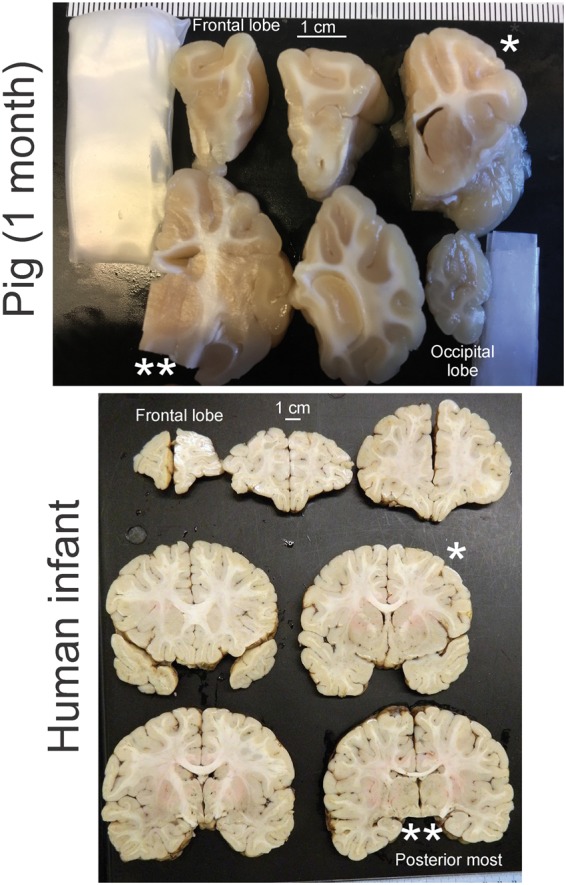
Comparative gross neuroanatomy of the pig (1 month of age) and human infant (12 months of age). The brains have been cut into coronal slabs from the anterior most part (the frontal lobes) to the posterior most part (the occipital lobes for the pig and the diencephalon-midbrain transition for human). The single asterisks (*) identify comparable anatomic levels in pig and human brains at the mid-striatum. The double asterisks (**) identify comparable levels at thalamus and midbrain. Note the encephalization of the pig as seen by the gyrencephalic cerebral cortex and corresponding major elaboration of white matter pathways that are grossly similar to the human infant brain.
Fetal monkey models of HIE
In 1959, Ranck and Windle22 studied birth asphyxia in non-human primate. They delivered five near-term (157–164 days’ gestation, term = 166 days) Macaca mulatta fetuses (rhesus monkeys) by hysterectomy and waited 11–16 min before opening the amniotic sac and resuscitating with pulmonary insufflation with oxygen. Subsequently, more than 50 other monkeys were studied.23 The clinical presentation of infant monkey HIE is variable. Some developed acute bilateral rigidity and then alternating flaccidity and rigidity, indicative of pathology in descending motor pathways, including corticospinal, rubrospinal, and reticulospinal tracts,24–28 and spinal pathology. They also exhibit attenuated vestibulo-ocular and pupillary light reflexes and oral motor dysfunction, consistent with brainstem pathology and the human infant dorsal brainstem syndromes.29,30 An intermittent paretic hand grasp reflex is present occasionally. Other infant monkeys show acute inability to right with diminished appendicular tone, inactivity, and hypo-responsiveness, but by six days muscle tone reappears and ambulation is present, though paretic and ataxic. Other monkeys display athetoid movements, inability to right, and status epilepticus.
The neuropathology at two to nine days of recovery was impressive.22 No gross pathology, including large hemorrhage, was seen; it was microscopic and distributed bilaterally throughout the neuraxis. A pattern of selective neuronal injury that primarily involved primary sensory nuclei, including inferior colliculus, superior olive, medial geniculate nucleus, ventral posterior thalamus, gracile and medial cuneate nuclei, and vestibular nuclei.30 Other brainstem regions with prominent injury included the principal sensory and motor nuclei of the trigeminal nerve. Similar brainstem and thalamic damage occurs in term human HIE.31–33 However, in human HIE infants, interestingly, some of the reduced motor activity, hypotonia, flaccidity, and depressed or absent deep tendon reflexes can be due to spinal cord injury.34 Basal ganglia damage was found in globus pallidus, the caudal half of the putamen, mirroring the damage seen in term human HIE.32,33,35 Minor neuronal loss was evident in the monkey dentate gyrus, but most of the hippocampus appeared normal, unlike the vulnerability of the neonatal human hippocampus, particularly after clinical seizures.36 Loss of cerebellar Purkinje neurons was largely restricted to the vermis. Damage to the cerebral cortex involved isolated degeneration of pyramidal neurons with necrotic-like cytology. Cerebral and cerebellar cortical pathology is often less severe than subcortical injury in human HIE,31 but seizure presence clearly correlates with the amount of cortical necrosis.37
The HIE monkeys also exhibited white matter damage. Myelin degeneration was found in a variety of white matter tracts, including the internal capsule. The myelin degeneration appeared to coincide with axonal degeneration. Individual oligodendrocytes in degenerating white matter showed cytopathology.22 White matter pathology is particularly relevant to human neonatal HIE,32,33,35 although early work could not attribute perinatal leukoencephalopathy to a single etiology.38
Faro and Windle23 studied another cohort (n = 12) of neonatal HIE monkeys survived for 10 months to about 9 years of age. Interestingly, these monkeys show minimal delayed damage to hippocampus. Progressive pathology was not evident in the putamen, globus pallidus or cerebellum. In contrast, the spinal cord showed atrophy of the dorsal horns and intermediolateral columns, and the brainstem pathology evolved, particularly in cochlear nuclei. The thalamus had protracted secondary pathology including the anterior ventral, ventral anterior and centromedial nuclei and the pulvinar; in human, lesions in the pulvinar cause attentional deficits, spatial neglect syndromes, and hemiagnosia.39 The primary motor and somatosensory cortices underwent apparent transneuronal degeneration and atrophy.23
These monkeys underwent behavioral testing during their lifetime. At six to seven months of age, HIE monkeys (five HIE and five controls) did not show differences in object discrimination, delayed response, and perseveration tasks; however, fine motor and visual-spatial skills were impaired.40 HIE monkey also had less emotionality and reactivity than age-matched controls.40,41 When 8–10-year-old HIE monkeys were tested on a memory delayed-response task, the monkeys were normal with no imposed delay, but with 5 s and longer delays, significant memory deficits appeared.42
Ronald Myers extended the work in monkey HIE on over 300 term fetuses during the 1960s and 1970s.43–46 His fundamental achievements were showing that: total asphyxia causes brainstem pathology; partial asphyxia causes severe brain edema and neocortical damage; partial asphyxia without any major increase in PCO2 or decrease in pH causes white matter injury; and partial followed by total asphyxia, as might occur during breech delivery or prolapse of the umbilical cord, causes basal ganglia damage. He reported that complete asphyxia produced by preventing ventilation after occluding the umbilical cord resulted in loss of arterial pressure by 10–12 min, indicating that this model was a model of incomplete followed by complete ischemia. Extending asphyxia to 25 min produced more extensive damage in brainstem and thalamus. He noted that this pattern of injury more closely resembled that of human infants who suffered complete ischemia from cardiac arrest than that typically observed in term neonates who suffered intrapartum asphyxia. He went on to test different models of partial asphyxia without loss of cardiac pulsations, such as partial occlusion of the maternal descending aorta, partial ablation of the placenta, and umbilical cord occlusion.44 His work suggested a threshold of approximately 3 ml O2/dL arterial O2 content and a pH <7.1 for depressing fetal cardiac function, arterial pressure, and cerebral O2 transport sufficiently to produce significant neuropathology. With arterial O2 content sustained for at least 30 min in the 0.5–3 ml O2/dL range, paracentral gyrus of neocortex and basal ganglia were found to be selectively vulnerable with little damage to brainstem. Thus, the distribution of brain injury from partial asphyxia was quite distinct from the distribution of injury from complete asphyxia (Table 1). Moreover, the pattern of injury from partial asphyxia more closely resembled the typical pattern of injury seen in term human autopsy tissue47,48 and in survivors.32,49–51 Other salient observations were (1) that injury sufficiently severe to produce neocortical panlaminar necrosis was associated with seizures and augmented injury in hippocampus, (2) regional selective vulnerability tended to correlate with regional metabolic rate rather than correlating with vascular watershed regions, (3) preterm fetal monkeys required longer durations of complete asphyxia to achieve similar brainstem and cerebellar injury as term fetuses, and (4) overnight fasting of the mother before inducing complete asphyxia attenuated fetal brain lactate accumulation and edema.
Table 1.
Severity grade of neuronal selective vulnerability in different brain regions among the major large animal perinatal models of hypoxia-ischemia.
| Model | Brainstem sensory nuclei and motor tracts | Cerebellum | Thalamus sensory nuclei | Striatum | Hippocampus | Cerebral cortex |
|---|---|---|---|---|---|---|
| Fetal monkey | ||||||
| Total asphyxia | +++ | ++ | +++ | + | – | + |
| Partial asphyxia | – | – | + | ++ | ++ | +++ |
| Fetal sheep | ||||||
| Periodic UCO (term) | – | ++ | ++ | ++ | – | ++ |
| Steady UCO (preterm) | ++ | ++ | ||||
| Cerebral ischemia (term) | – | + | + | ++ | +++ | +++ |
| Newborn piglet | ||||||
| Severe hypoxia | + | +++ | + | ++ | +++ | +++ |
| BCAO+hypoxia | – | – | ++ | +++ | ++ | ++ |
| Hypoxia+asphyxia | + | – | ++ | +++ | – | ++ |
Note: +, ++, +++, mild, moderate, and severe neurodegeneration, respectively. –, unremarkable.
UCO: umbilical cord occlusion; BCAO: bilateral carotid artery occlusion.
Fetal sheep models of HIE
Fetal sheep can undergo survival surgery in utero for implantation of catheters, electroencephalographic (EEG) electrodes, and other instrumentation, thus providing insights into developmental physiology. Brain development in sheep is more advanced at birth compared to humans.52 To approximate human brain development at term, fetal sheep typically are studied at ∼0.85 gestation, whereas they can be studied at 0.5–0.75 gestation to model preterm human brain. Cerebral metabolic rate of oxygen (CMRO2), cerebral blood flow (CBF), and the relative change in CBF during intrauterine hypoxia increase between 0.6 and 0.9 gestation.53–56 CMRO2 is not limited by the low intrauterine oxygenation state because increasing fetal arterial PO2 to near postnatal levels does not increase CMRO2,57 although pial arterioles do constrict to limit brain hyperoxia.58 Importantly, reductions in fetal or postnatal arterial O2 content down to ∼2 mmol/L increase CBF sufficiently to sustain CMRO2 as long as arterial hypotension does not occur.56,59,60 Mean arterial blood pressure (MABP) increases from ∼35 mmHg at 0.6 gestation to ∼45 mmHg at 0.9 gestation,53,55,61 and autoregulation of CBF to increases in MABP has been described in fetal sheep at 0.8 gestation.62 However, the autoregulatory range for decreases in cerebral perfusion pressure is limited, especially when fetal arterial O2 saturation is <50%.63–65 Thus, a combination of severe hypoxia and arterial hypotension is needed for brain damage.66,67
Umbilical cord occlusion in fetal sheep
Intermittent umbilical cord occlusion has been used in fetal sheep to simulate what may occur during periodic uterine contractions and provides reproducible histopathology. In a study, Clapp et al.68 used fetal sheep at 0.8–0.9 gestation with umbilical cord partial occlusion for 1 min every 3 min (33% duty cycle) for 2 h. Fetal acidemia did not occur, yet white matter lesions were evident.68 Thus, white matter vulnerability can extend into late gestation when the insult consists of brief repetitive bouts of cerebral hypoxia.
Peter Gluckman, Alistair Gunn and coworkers in the 1990s studied more severe umbilical cord occlusion. De Haan et al.69 used umbilical cord occlusions lasting 1 min every 2.5 min or lasting 2 min every 5 min (40% duty cycles) until persistent arterial hypotension occurred (average of 145 min and 120 min, respectively). This insult produced severe acidemia (pH of 6.83) and gray matter injury. Indeed, selective neuronal cell loss was more consistent than in a model of uterine artery occlusion.70 With 2-min occlusions every 5 min, the loss of EEG occurred more quickly and neuronal injury was worse than with the 1-min occlusions every 2.5 min. With the prolonged cycling of intermittent occlusion lasting ∼2 h, fetal arterial hypotension became sustained,71 and the incidence of seizure activity became more prominent and was associated with infarcts in parasagittal cortex, thalamus, and cerebellum. In a different paradigm using 5 min occlusions every 30 min for 2 h, neuronal loss became more prominent in striatum.72,73 In contrast, a single umbilical cord occlusion lasting 10 min produced hippocampal injury with little striatal injury in near-term fetal sheep74; neither region displayed pathology when the occlusion was produced at 0.6 gestation.75 At 0.7 gestation, sustained occlusion of the umbilical cord for 25 min produces injury to striatal and CA1/2 hippocampal neurons and subcortical and periventricular oligodendrocytes; the striatal and oligodendrocyte injury was found to be ameliorated by administration of a peptide to block connexin hemichannels protected,76 whereas the hippocampal injury could be ameliorated with an NMDA receptor antagonist.77 Glutamate receptors are highly enriched in fetal sheep white matter oligodendrocytes and undergo developmental regulation.78 Thus, the pattern of injury after sustained or intermittent umbilical cord occlusion depends on the developmental stage, the timing of the occlusion and reperfusion phases, and on whether arterial hypotension becomes severe and prolonged (Table 1).
Cerebral ischemia in fetal sheep
Another commonly used HI model in fetal sheep involves ligation of vertebral-carotid arterial anastomoses and placement of occluders on both common carotid arteries. At 0.85 gestation, bilateral carotid occlusion for 10 or 20 min produced selective neuronal necrosis, whereas occlusion for 30 or 40 min produced epileptiform EEG activity starting at 7–9 h associated with laminar necrosis in parasagittal cortex and substantial hippocampal damage.79,80 Moderate damage was also seen in striatum, and mild damage was often present in thalamus and cerebellum (Table 1). The 30-min occlusion duration is typically used to produce robust neuronal injury. As in the umbilical cord occlusion model, intermittent bouts of ischemia preferentially augmented injury in the striatum.81 The cerebral ischemia model has the advantage of being able to investigate cerebral therapeutics without the variability encountered from impaired cardiac function in the umbilical cord occlusion model. For example, benefits have been demonstrated with delayed administration of an NMDA receptor antagonist,82 insulin-like growth factor-1,83 and a connexin hemichannel peptide blocker.84
White matter injury in preterm fetal sheep
Human preterms born between 23 and 32 weeks show greater vulnerability to injury in white matter than gray matter. In human autopsy tissue, most oligodendrocyte lineage cells in this age span are beyond the early progenitor mitotic and migration stage and are NG2 proteoglycan- and O4-positive.85 After 30 weeks’ gestation, these late progenitor cells differentiate into immature oligodendrocytes that are O1- and O4-positive and then into mature myelin basic protein-positive oligodendrocytes. Thus, the period of increased periventricular white matter vulnerability in human development corresponds to the period of increased late progenitor oligodendrocytes prior to differentiation into immature oligodendrocytes. These late stage progenitor oligodendrocytes are thought to be particularly vulnerable to oxidative stress, as evident by increased F2-isoprostanes.86
To better elucidate the mechanisms of this developmental vulnerability, Back et al.87 have studied fetal sheep at 0.65 gestation, when they have a preponderance of late progenitor oligodendrocytes that are vulnerable to HI.88 Using the cerebral ischemia model at this age in which periventricular CBF is reduced by 90% for durations of 30–45 min, the severity of white matter injury can be varied.89 Indeed, as in humans, the injury severity in preterm fetal sheep can vary from dispersed late progenitor cell death, microregions of necrosis, or widespread necrosis leading to cyst formation. Necrotic lesions were associated with damage to axons, as assessed by decreased neurofilament staining, increased β-amyloid precursor staining, axonal swelling, and the appearance of spheroids, whereas dispersed progenitor cell death exhibited a paucity of axonal damage and no change in the distribution of axon diameters.90 Moreover, periventricular white matter vulnerability in the preterm fetal sheep is not related to vascular border zones, but rather to the oligodendrocyte lineage expressed at the period of heightened vulnerability.91 As in humans, white matter injury in fetal sheep at this stage was not accompanied by profound gray matter injury,92 which could have confounded interpretation of white matter injury. Likewise, the transition from early to late progenitors in rabbit brain occurs rapidly between E22 and E25, at which time white matter is selectively vulnerability to hypoxia, further supporting enhanced vulnerability of the late progenitors.93
With insults that do not generate massive white matter necrosis in mid-gestation fetal sheep, late stage progenitors increased over a two-week period in microregions of periventricular white matter, whereas pre-myelinating immature oligodendrocytes decreased.94 Thus, it appears that the late progenitors proliferate but fail to mature into myelin producing oligodendrocytes after cerebral ischemia. Proliferation of late progenitor cells has also been observed in autopsy tissue from preterm infants who survived two to three weeks after birth.95 Therefore, preterm fetal sheep exhibit strong parallel histopathology and biochemistry with that seen in human preterm autopsy tissue, thereby supporting use of this model for preclinical evaluation of therapies targeting preterm white matter injury.
Neonatal piglet models of HIE
Newborn piglets provide a model of HIE relevant to term human newborns for many reasons. The peak brain growth spurt in piglets and humans both occur near the time of birth, while rodents have a major postnatal-skewed growth spurt.52 Similar to humans, brain weight at birth in piglets is approximately 35–40% of that of adult pigs.96,97 Pigs and humans share many gross neuroanatomical complexities (Figures 1 and 2) and myelination patterns.98 Furthermore, the genomes, including synteny, gene order, and DNA methylation, of human and pig have important similarities.99,100
Severe hypoxia model with suppressed EEG in piglets
Marianne Thoresen et al.101 developed a severe hypoxia model in which anesthetized newborn piglets are orally intubated and mechanically ventilated with ∼6% O2 for as long as 45 min. The inspired O2 is titrated to keep the EEG amplitude <7 µV while avoiding cardiac arrest. By three days of recovery, neuronal injury is prominent in parts of cerebral cortex, subcortical white matter and hippocampus and is also evident in basal ganglia, cerebellum, and thalamus. Seizure activity often occurs within several hours and is associated with worse neurodegeneration. This model has been used to study the benefit of hypothermia102 and xenon inhalation.103
Hypoxia + bilateral carotid occlusion model in piglets
In this model, both carotid arteries are occluded and the inspired O2 is decreased to produce systemic hypoxia.104 The model can be refined by using phosphorus magnetic resonance spectroscopy to monitor high-energy phosphates and titrate the inspired O2 to decrease cerebral ATP to low levels.105,106 The model produces damage in parasagittal cortex, striatum, thalamus, and hippocampus. This model has been used to demonstrate neuroprotection with the use of 2-iminobiotin,107,108 allopurinol,109 deferroximine,109 amilioride,106 melatonin,110 xenon,111 argon,112 cannabidiol.113–115 and remote ischemic postconditioning.116,117
Hypoxia + complete asphyxia model in piglets
This model was developed in Dr. Traystman’s lab by Drs. Hans Hennes and Ansgar Brambrink118,119 and has been used by the authors for many years. This piglet injury model is most relevant to asphyxia in the full-term neonate.120,121 It combines systemic hypoxia followed by complete asphyxia as a model of worsening oxygenation of the entire body. In developing this model, it was found that airway occlusion of 7–8 min alone was not sufficient to produce significant brain damage, whereas longer durations of asphyxia produced prolonged asystole and difficulties in resuscitating the heart. However, ventilation with 10% O2 for 30–45 min before 7–8 min of airway occlusion led to reproducible regionally selective brain damage.
Our piglet model has been pursued because of its relevance to HIE in human newborns and children.121 HI in one-week-old piglets preferentially damages primary sensory and forebrain motor systems.122 This neuropathology is progressive and is not static. The cerebral cortex and basal ganglia are highly vulnerable (Table 1). Based on electrophysiological mapping,123,124 the neocortical area that is most vulnerable to HI corresponds to motor cortex and primary somatosensory cortex. The most vulnerable region of piglet striatum (i.e. central putamen) appears to be the sensorimotor-recipient region, based on known corticostriatal connectivity in other mammals.125 In diencephalon, thalamic relay nuclei for somatosensory (ventral posterior nucleus), visual (lateral geniculate nucleus), auditory (medial geniculate nucleus), and motor (ventral anterior/lateral) systems are consistently damaged. In brainstem, visual (superior colliculus) and auditory (inferior colliculus) relay nuclei are predisposed to injury.
This regional distribution of neocortical and subcortical injury is important conceptually because it indicates that the formation of HIE in newborns is not a random and static process but, rather, is highly organized and topographic, targeting preferentially regions that function in sensory-motor integration and control of movement. This distribution of neonatal brain damage is dictated possibly by regional connectivity, function, and mitochondrial activity.122 This theory is called the connectivity-metabolism hypothesis for brain damage in newborns.122,126 The pattern of brain damage in HI piglets bears a close resemblance to that found in perinatal asphyxia in humans31,120,127,128 and nonhuman primates.22,46
The selective vulnerability of the basal ganglia in this model is profound.122,126 The putamen is the most vulnerable. The death of striatal neurons after HI in piglets is categorically necrosis,129 contrasting with findings in neonatal rat striatum after HI.130,131 Nevertheless, despite the necrosis, this neurodegeneration in piglet striatum evolves with a specific temporal pattern of subcellular organelle damage and biochemical defects.129,132,133 Damage to the Golgi apparatus and rough endoplasmic reticulum (ER) occurs at 3–12 h, while most mitochondria appear intact until 12 h. Mitochondria undergo an early suppression of metabolic activity, then a transient burst of activity at 6 h after the insult, followed by mitochondrial failure.129 Cytochrome c is depleted at 6 h after HI, failing to accumulate in the cytosol compartment, and is not restored thereafter. Lysosomal destabilization occurs within 3–6 h after HI consistent with the lack of evidence for autophagy in striatum. By 3-h recovery, glutathione levels are reduced in striatum.129,132 Peroxynitrite-mediated oxidative damage to membrane proteins occurs at 3–12 h after HI, and the Golgi apparatus and cytoskeleton are early targets for extensive tyrosine nitration. Striatal neurons sustain hydroxyl radical damage to DNA and RNA within 6 h after HI. The early emergence of this injury coincides with elevated NMDA receptor phosphorylation, recruitment of neuronal NOS to the synaptic/plasma membrane, and prominent oxidative damage by 5 min after reoxygenation.133 This work demonstrates that neuronal necrosis in the striatum after HI in piglets evolves rapidly and suggests that this injury would be difficult to protect against in piglet.129,133 Early implemented interventions will thus be required to protect the basal ganglia region from HI.
In parasagittal sensorimotor cortex, neuronal cell death progresses more slowly than in basal ganglia in this model. Whereas some neuronal cell death can be detected between 6 and 24 h, the majority of the neurons die between 24 and 48 h.134 Morphologically, the cells appear to undergo a form of necrosis rather than classical apoptosis. When the delayed cell death in cortex is severe, the piglets develop clinical seizures between 24 and 48 h, a time of onset that is similar to that seen in human newborns with severe HIE.118,122 The occurrence of clinical seizures is associated with panlaminar necrosis in somatosensory cortex and augmented neuronal loss in hippocampus.122
HIE mechanisms of neuronal vulnerability in piglet putamen
Because loss of neurons is rapid and most severe in putamen in the piglet model of hypoxia + asphyxia, we conducted a series of studies to further investigate the mechanisms and potential therapeutic targets. The effects of various interventions described below on the number of surviving viable neurons in putamen are summarized in Table 2.
Table 2.
Viable neurons in piglet putamen after hypoxia+asphyxia (HI) with various interventions (% of sham; mean ±SD).
| Treatment | Start of treatment | Viable neurons |
|---|---|---|
| No drug | 20 ± 9 | |
| AMPA antagonist | Pre- and post-HI | 20 ± 18 |
| D1 antagonist | 5 min post-HI | 39 ± 18 |
| Endaravone | 30 min post-HI | 39 ± 19 |
| EUK-134 | 30 min post-HI | 41 ± 17 |
| A2A antagonist | 5 min post-HI | 43 ± 11 |
| NMDA antagonist | Pre-HI | 45 ± 18 |
| 16-h hypothermia + 4-h rewarming | 4 h post-HI | 46 ± 21 |
| EUK + 16-h hypothermia + 4-h rewarming | EUK at 30 min Hypothermia at 4 h | 47 ± 25 |
| ASIC1a antagonist | Pre-HI into ventricle | 47 ± 10 |
| 20-HETE synthesis inhibitor | 5 min post-HI | 52 ± 20 |
| 17-h hypothermia + 4-h rewarming | 3 h post-HI | 61 ± 7 |
| Sigma receptor ligand | 5 min post-HI | 62 ± 19 |
| 20-HETE synthesis inhibitor + 17-h hypothermia + 4-h rewarming | Inhibitor at 5 min post-HI Hypothermia at 4 h | 70 ± 6 |
| ASIC1a + NMDA antagonists | Pre-HI into ventricle | 79 ± 19 |
| 20-h hypothermia + 4-h rewarming | 5 min post-HI | 99 ± 8 |
20-HETE: 20-hydroxyeicosatetraenoic; HI: hypoxia-ischemia.
Glutamate receptors
In piglet striatum, the GluN1 subunit undergoes phosphorylation at the PKA-dependent site Ser897 and at the PKC-dependent sites Ser 890 and Ser 896.133,135,136 Phosphorylation has been detected as early as 5 min after reoxygenation and persists for 3–24 h. These observations suggest changes in function and trafficking of NMDA receptors that might contribute to excitotoxicity. It should be noted that loss of astrocytes and glutamate transporter expression occur by 24 h when most of the neurons are already dead.137 Pretreatment with an NMDA receptor antagonist provides only partial protection of putamen neurons from HI,138 whereas pretreatment with an AMPA receptor antagonist failed to provide neuroprotection (Table 2).118 Thus, NMDA receptors play a significant role in neurodegeneration in putamen after HI.
Dopamine 1 receptor and adenosine 2 A receptor
Medium spiny neurons comprise over 90% of the neurons in striatum.20 Those that project to substantia nigra are enriched in dopamine D1 receptors, and those that project to globus pallidus are enriched in adenosine A2A receptors. These receptors amplify glutamatergic signaling139 and thus have the potential to amplify excitotoxicity. Microdialysis experiments in neonatal piglets demonstrated that severe hypoxia without ischemia can increase extracellular dopamine140 and adenosine.141 Large increases in extracellular dopamine also occur during asphyxia and persist into the early reoxygenation period.136
When a D1 receptor antagonist was infused in piglets at 5 min of reoxygenation, the number of viable neurons at four days was increased to 39% (Table 2), indicating that a considerable amount of the dopamine-induced damage is initiated after reoxygenation.136 Likewise, post-treatment with an A2A receptor antagonist preserved 43% of putamen neurons.142 The subpopulation of neurons protected by the A2A receptor antagonist was primarily striatopallidal neurons, whereas the D1 antagonist preferentially protected striatonigral neurons.
The D1 and A2A receptors share some common downstream signaling in their respective medium spiny neuron populations (Figure 3). They both stimulate adenylate cyclase, cAMP formation, PKA activity, and phosphorylation of PKA-sensitive sites on specific target proteins. In addition, Thr34 on the dopamine- and adenylate cyclase-regulated phosphoprotein of 32 kDa (DARPP-32) is phosphorylated that, in turn, decreases the activity of protein phosphatase-1 (PP1).143,144 By inhibiting PP1 activity, the phosphorylation of the PKA-dependent sites that would normally be dephosphorylated by PP1 instead is sustained. For example, Ser897 on the GluN1R subunit of the NMDA receptor is phosphorylated by PKA,145 and its phosphorylation state is increased within 5 min of reoxygenation in piglet putamen.133 This phosphorylation at GluN1R Ser897 is associated with Thr34 phosphorylation on DARPP-32, and both are inhibited by treatment with a D1 or A2A receptor antagonist.136,142 Another known PKA target is Ser943 on Na,K-ATPase, the phosphorylation of which reduces enzymatic activity.146 Phosphorylation at this site is also increased 3 h after reoxygenation in piglet putamen, and this increase is inhibited by infusion of a D1 or A2A receptor antagonist.136,142 Furthermore, Na,K-ATPase activity decreases at 3 h, and this decrease precedes the loss of the α− and β−subunits of the enzyme.132 This early loss of enzyme activity was attenuated by infusion of a D1 or A2A receptor antagonist.136,142 Because decreased Na,K-ATPase activity can lead to cell depolarization and increased intracellular Na accumulation, these effects of D1 and A2A receptors likely contribute to an augmentation of neuronal excitotoxicity during the reoxygenation period after HI.
Figure 3.

Schematic diagram of some of the signaling pathways involved in death of putamen medium spiny neurons in the piglet hypoxia + asphyxia model. Dashed line = inhibitory effect; D1R: dopamine receptor-1; A2AR: adenosine receptor 2A; NKA: Na,K-ATPase; DARPP-32: dopamine and adenylate cyclase regulated phosphoprotein; PP1: protein phosphatase 1; ASIC1a: acid-sensitive ion channel 1a; σ1R: σ-1 receptor; GluNR1: NMDA receptor subunit 1; GluNR2: NMDA receptor subunit 2; NO: nitric oxide; nNOS: neuronal NO synthase; O2–: superoxide anion; ONOO–: peroxynitrite; PSD95: postsynaptic density-95; AA: arachidonic acid.
20-hydroxyeicosatetraenoic acid
Arachidonic acid is mobilized during ischemia, and O2-dependent hydroxylation of the ω-carbon by cytochrome P450 4A enzymes during reoxygenation produces the lipid bioactive molecule, 20-hydroxyeicosatetraenoic (20-HETE).147–149 Direct toxic actions of 20-HETE in neurons are supported by protection from oxygen–glucose deprivation in primary cultured neurons150 and in hippocampal slices151 with 20-HETE synthesis inhibitors and antagonists and by expression of cytochrome P450 4A synthetic enzymes in mouse, rat, and piglet neurons.152 To test the possible role of 20-HETE in neonatal HI, piglets subjected to hypoxia + asphyxia were treated with the 20-HETE synthesis inhibitor HET0016 after reoxygenation. Survival of putamen neurons increased to 52% (Table 2) without an effect on recovery of CBF.153 In piglet putamen, α-subunit of Na,K-ATPase pSer23 increases 3 h after HI, and the increase is blocked HET0016. HET0016 also partially restores Na,K-ATPase activity.153 HET0016 also blocked the increased phosphorylation of GluN1R at its PKC-sensitive site, Ser896. Moreover, infusion of 20-HETE into non-ischemic piglet putamen via microdialysis recapitulated the increased phosphorylation of Ser896 on GluN1 and Ser23 on Na,K-ATPase. Thus, 20-HETE signaling through PKC and dopamine/adenosine signaling through PKA appear to act in concert on similar targets in striatal medium spiny neurons during reoxygenation after HI (Figure 3).
Acid-sensitive ion channel 1a
The acid-sensitive ion channel 1a (ASIC1a) permits Ca2+ and Na+ entry when tissue pH falls below 7.0.154 The role of ASIC1a in neonatal HI injury has been investigated in piglets by injecting the peptide inhibitor psalmotoxin into the lateral ventricle.138 Pretreatment was performed to allow time for drug diffusion deep into putamen. The inhibitor was found to decrease markers of oxidative and nitrative stress at 3 h of recovery and to salvage medium spiny neurons to an extent similar to that of the NMDA receptor antagonist MK-801.138 Interestingly, intraventricular injection of psalmotoxin and MK-801 produced additive neuroprotection with 79% of putamen neurons preserved compared to only 20% in the vehicle group (Table 2). A tissue pH electrode on the cortical surface registered a pH as low as 6.7 in early reoxygenation period followed by a recovery over the next 0.5 h of reoxygenation. Intraventricular injection of psalmotoxin at 15 min of reoxygenation did not provide significant protection, presumably because tissue pH in putamen recovered to the same extent as cortex by this time. Thus, the contribution of ASIC1a channels to medium spiny neuronal injury is comparable to that of NMDA receptors, but the injury process attributable to ASIC1a channels is set in motion during HI and early reoxygenation period.
Sigma-1 receptor
Sigma-1 receptors are located on the ER and are thought to provide inter-organelle signaling. Work from Dr. Traystman’s laboratory showed that a ligand of sigma receptors, 4-phenyl-1-(4-phenylbutyl) piperidine (PBPP), attenuates activation of NOS by NMDA155 and focal ischemia156 and is neuroprotective against ischemic stroke in adult cat157 and rat.158,159 It also protects neonatal mice from excitotoxicity.160 To investigate whether PBPP would be neuroprotective in neonatal HI, piglets were treated with PBPP at 5 min after HI, a time when nNOS is already enriched in the membrane fraction.133 PBPP was found to block the HI-induced Ser847 phosphorylation of neuronal NOS and the consequent association with GluN2 and PSD-95 in the plasma membrane.161 PBPP did not affect the association of GluN2 with PSD-95, suggesting that the drug mitigates neuronal NOS trafficking to the postsynaptic membrane rather than trafficking of GluN2 (Figure 3). The treatment preserved 62% of the medium spiny neurons, which was moderately more effective than the 44% preservation seen with MK-801 pretreatment (Table 2).
Antioxidants
Oxidative and nitrative stress is a hallmark of reperfusion injury. After reoxygenation from hypoxia + asphyxia, the putamen consistently displays markers of oxidative and nitrative stress, such as protein carbonyl formation, protein tyrosine nitration, and nucleic acid staining for 8-hydroxy-2-deoxyguanosine and 8-nitrogunosine.133,136,138,142,153,161 To directly test the role of oxidative stress, two different antioxidants were administered: EUK-134, a scavenger of superoxide, hydrogen peroxide, NO, and peroxynitrite; and edaravone, a scavenger of hydroxyl radical and NO.162 These agents attenuated the HI-induced increases in carbonyl formation, tyrosine nitration, 8-hydroxy-2-deoxyguanosine, and 8-nitrogunosine. Although each antioxidant improved survival of putamen neurons, the survival was not superior to any of the individual ion channel or receptor antagonist (Table 2). The incomplete neuroprotection may have been due to the 30-min delay in antioxidant administration after initiating reoxygenation, incomplete scavenging by these antioxidants, or to cell death mechanisms acting independently of oxidative and nitrative stress. Collectively, our interventions implicate multiple cytotoxic pathways that engage rapid neurodegeneration in newborn putamen after HI and, accordingly, a multi-target approach may be necessary for successful therapeutics.
Therapeutic hypothermia
Moderate hypothermia (33–34℃) is a multi-target therapy and is now the standard of care for term newborns diagnosed with HIE. This practice arose from the long history of body cooling for therapeutic application163 and many perinatal studies in rodent and large animal species that led to three successful large clinical trials.4–6 Most studies in P7–P10 rodents are limited to 3–6 h of hypothermia,164–166 although gavage feeding at 6-h intervals has been used to extend hypothermia through 26 h.167 Fetal sheep, newborn piglets, and newborn monkeys provide the opportunity to study more prolonged periods of hypothermia without disrupting the supply of metabolic substrates while monitoring blood glucose levels. In fetal sheep, novel methodology was utilized to selectively cool the skull via a coiled water jacket with only a small decrease in fetal core temperature.168 Results in fetal sheep at 0.85 gestation (approximately equivalent to term human newborn) showed that 72 h of head cooling initiated 90 min after cerebral ischemia reduced the incidence of delayed seizure activity, improved recovery of EEG, and increased neuronal survival in parasagittal cortex, hippocampus, and striatum by over 50%.168 Significant neuroprotection was maintained in all regions except CA1/2 hippocampus when cooling was initiated 5.5 h after cerebral ischemia169 but was largely lost when initiated 7.5 h after ischemia, a time at which most fetuses were already experiencing electrophysiologic seizures.170 These results influenced the design of the clinical trials in that inclusion criteria require initiation of hypothermia within 6 h of birth.
Hypothermia has been done on newborn piglets subjected to HI. In the model of severe hypoxia induced by ventilation with 6% oxygen, 3 h of hypothermia initiated immediately after HI provided partial neuroprotection, but the neuroprotection was lost in those subjected to a severe insult.171 When hypothermia was extended to 6 h, hypothermia decreased glutamate in microdialysates of extracellular fluid to levels below baseline and prevented a delayed increase in the ratio of citrulline to arginine, indicative of an increase in NOS activity beyond 3 h.172 The duration of hypothermia was then extended to 24 h with the use of selective head cooling. In piglets sedated with propofol and fentanyl during hypothermia, robust neuroprotection was observed,102 whereas whole body cooling in unanesthetized piglets for 24 h failed to protect the brain.173 The lack of protection in the latter case was attributed to the cold stress and increased cortisol release.173 Replacing fentanyl with dexmedatomidine for sedation during hypothermia can augment cardiovascular instability and exacerbate brain damage, suggesting caution in selecting the choice of sedatives.174
Beneficial effects of 24 h of hypothermia accompanied by fentanyl sedation also have been reported in the piglet model of hypoxia followed by complete asphyxia. In this model, putamen is highly vulnerable and whole body hypothermia provided essentially complete neuroprotection in this region when assessed 11 days after HI (Table 2).175 When the onset of hypothermia was delayed by 3 or 4 h, survival of neurons in putamen decreased to 61% and 45%, respectively (Table 2).152,176 These results showing partial protection with a 3–4 h delay in piglets are consistent with the partial protection seen in fetal sheep with a 5.5-h delay. However, they differ from a study in the piglet model of 45 min of severe hypoxia, where a 3-h delay in hypothermia onset had no significant improvement in the overall brain neuropathology score,177 possibly because the injury is more severe in the model. It is noteworthy that in the early clinical trials, hypothermia was delayed by 3–6 h after birth in the majority of patients, and hypothermia is believed to have been less effective in those that presented with more severe symptoms and in those with a 3–6 h delay compared with those treated within 3 h of birth.178 Because more severe cerebral ischemia can accelerate various forms of cell death signaling, the therapeutic window for initiating hypothermia is not likely to be fixed, but rather varies inversely with the severity of the insult. In line with the animal data, the onset of hypothermia is now attempted to be minimized whenever possible in neonatal clinical practice. Even with a clinically relevant 2-h delay, some of the neurons in cortex that would ordinarily undergo a delayed necrotic form of cell death in the hypoxia + asphyxia piglet model instead undergo apoptosis that is amenable to treatment with a caspase-3 inhibitor.179
The fetal sheep and neonatal piglet models do have some limitations as translation models for therapeutic hypothermia. Cerebral ischemia rather than whole body HI has been the model of choice for the fetal sheep hypothermia studies. Moreover, the fetal sheep recover in the intrauterine environment without undergoing the cardiopulmonary transition that results in increased oxygenation after HI. It should also be noted that selective head cooling produces less cooling of subcortical structures.180 On the other hand, the HI insults in the piglets are performed postpartum and the duration of hypothermia has generally been limited to 24 h because of critical care needs. Results in fetal sheep indicate that 72 h of head cooling may be superior to 48 h of head cooling,181 with no additional benefit of 120 h of head cooling.182,183 Overall, work in fetal sheep and neonatal piglets both supported the testing of targeted temperature management in term newborns with HIE.
White matter injury after hypothermia
In term HIE newborns treated with hypothermia, MRI indicates a decrease in injury in vulnerable gray matter regions, such as basal ganglia and thalamus.184 White matter is particularly vulnerable to HI in preterm newborns, but white matter injury is also present in term human newborns with HIE.32,51 Whereas hypothermia affords some protection of white matter, such as in the posterior limb of the internal capsule,185 white matter injury persists in term HIE neonates.186–188 Persistent white matter injury has also been seen in term monkey fetuses subjected to complete asphyxia189 and in newborn piglets subjected to hypoxia-asphyxia.190 In piglets with a 2-h delay in hypothermia, increases in apoptotic cells, thought to be mainly mature oligodendrocytes, were seen in multiple white matter regions not only after rewarming, but also during hypothermia relative to that seen with normothermic recovery from HI.190 Unexpectedly, piglets that received hypothermia without undergoing HI also displayed an increase in subcortical white matter apoptosis.190 The latter effect may be related to the unfolded protein response as assessed by increased immunostaining of endoplasmic reticulum-to-nucleus signaling-1 protein in oligodendrocytes and astrocytes.191 Thus, damaging side effects of therapeutic hypothermia are an important clinical consideration.
Hypothermia plus adjunct therapy
Because some gray and white matter damage persists following delayed use of hypothermia, several agents have been tested in combination with hypothermia in large animal models. In the piglet model of severe hypoxia, inhalation of 50% xenon for 18 h starting 30 min after HI combined with 12 hypothermia initiated immediately after HI produced additive neuroprotection that was equivalent to that obtained with 24 h of hypothermia alone.103 Combining 18 h of xenon inhalation with 24 h of hypothermia produced robust neuroprotection, with protection ranging from 72%–100% among brain regions.103 Moderate protection was also reported in the carotid occlusion + hypoxia model when xenon and hypothermia treatment was delayed 2 h.111 This collective work in piglets and earlier work in rat pups192 led to a small clinical trial comparing hypothermia with hypothermia plus 30% xenon inhalation.193 The trial demonstrated no significant increase in adverse effects, but efficacy of combined treatment based on short-term MRI outcomes could not be demonstrated. In another study in piglets, the combination of hypothermia and argon inhalation initiated at 2 h appeared to provide more robust protection than the combination with xenon.112
Melatonin possesses antioxidant properties in a variety of pro-oxidant models.194 Administration of melatonin to pregnant ewes was found to decrease hydroxyl radical formation and 4-hydroxynonrenal immunoreactivity in fetal brain after umbilical cord occlusion.195 Administration to preterm fetal sheep after umbilical cord occlusion decreases oxidant stress and inflammation in white matter.196 In the piglet model of carotid occlusion + hypoxia, treatment with melatonin enhanced neuroprotection with hypothermia.110 Here, melatonin was infused from 10 min through 6 h after HI, whereas hypothermia was delayed until 2 h after HI. In the piglet model of hypoxia + asphyxia, administration of a different antioxidant, EUK134, at 30 min failed to augment the protection in putamen afforded by hypothermia initiated at 4 h of reoxygenation (Table 2).176 Properties of antioxidant agents and the timing of administration when used as an adjunct therapy to delayed hypothermia may be critical. Administration of the 20-HETE synthesis inhibitor, HET0016, also reduces oxidative damage when administered at 5 min after hypoxia + asphyxia.153 When administered soon after the insult, HET0016 moderately augmented neuronal survival afforded by a 3-h delay in hypothermic onset: from 61% to 70% in putamen (Table 2), from 57% to 73% in thalamus, and from 71% to 86% in somatosensory cortex.152 Thus, with a clinically relevant delay in hypothermia onset, it is possible to administer agents to augment neuroprotection, but some of these may need to be given rapidly after birth to produce a substantial added benefit.
To provide a more realistic scenario of neonatal resuscitation, Juul et al.197 modified the monkey model developed by Ranck and Windle.22 Fetal Macaca nemestrina monkeys were delivered by hysterectomy and the umbilical cord was clamped for 15–18 min before initiating pulmonary ventilation. In this model, the incidence of mortality or persistent moderate or severe neurologic deficit was 43% (6 of 14 monkeys).198 Induction of 72 h of whole body hypothermia over the first 3 h after resuscitation did not improve this outcome (44%; 4 of 9 monkeys). The severity of neurologic deficits correlated with the grade on brainstem histopathology.189 Because erythropoietin has been reported to provide neuroprotection in adult cerebral ischemia models and can act through multiple signaling mechanisms to decrease apoptosis, modulate inflammation, enhance brain repair,199 and is FDA approved for several applications (anemia due to kidney disease and chemotherapy),200 it was chosen as a candidate therapeutic in the newborn monkey model of HI. Remarkably, multi-dose treatment with erythropoietin at 30 min and one, two, and seven days combined with three days of hypothermia reduced the incidence of death and moderate and severe neurologic deficit to 0% (0 of 12 monkeys) and prevented the developmental delay in some behavior tasks, such as climbing up and down a chain and reaching for a toy.198 Combined treatment with erythropoietin and hypothermia also significantly increased myelin staining in hippocampus, although this effect was not significant in other white matter regions. When pooling monkeys with moderate or severe neurologic deficit versus those with mild or no neurologic deficit among groups, fractional anisotropy in internal capsule and corpus callosum and cerebellar cell density and white matter were decreased in those with moderate or severe neurologic impairment.189 These results in monkeys, together with earlier work in rodents, led to a Phase II clinical trial of high-dose erythropoietin as an adjunct therapy with hypothermia.201 Effects on diffusion-weighted lesion volumes at 12 months are encouraging,202 and a large scale Phase III trial is underway.203 Nevertheless, caution is warranted because a variety of hematopoietic and non-hematopoietic cancer stem cells express functional erythropoietin receptors and exhibit potent growth stimulatory effects in its presence.200
Conclusions
The most frequently used large animal models of perinatal HI include partial or complete asphyxia at the time of delivery of fetal macaque monkeys, umbilical cord occlusion and cerebral hypoperfusion at different stages of gestation in instrumented fetal sheep, and severe hypoxia and hypoperfusion in newborn piglets. In monkeys, complete asphyxia with sustained loss of arterial pressure produces neurologic abnormalities symptoms of cerebral palsy with preferential injury to somatosensory, auditory, and vestibular nuclei in brainstem, thalamus, and cerebellum.22,23,189 In models of partial asphyxia in fetal monkeys, fetal sheep, and newborn piglets, preferential injury is produced in primary somatosensory and motor cortices, basal ganglia, and thalamus.43,79,102,122,168 Although both patterns of injury are reported in human autopsy tissue and MRI, the pattern seen with partial asphyxia is more frequent.50 Mechanistically, injury in striatum medium spiny neurons is augmented by activation of NMDA receptors, ASIC 1a, D1 receptors, and adenosine 2A receptors, all of which lead to oxidative and nitrative stress.136,138,142,153,161 Post-insult treatment efficacy of hypothermia in animal models in fetal sheep and newborn piglets is robustly neuroprotective,102,168,175 but white matter injury persists in large animal models,190 thereby replicating that seen on MRI in humans. This experimental work translated to the clinical use of targeted temperature management for term neonatal HI. Research continues to find adjunct therapy to boost the neuroprotection afforded by hypothermia and to ameliorate the residual white matter injury that can result in sustained neurobehavioral deficits throughout childhood.
Acknowledgements
The authors are immensely indebted to Dr. Richard Traystman for providing a rich and vibrant intellectual environment with resources to pursue our investigations of developmental brain injury and for his vision that basic scientists and physician scientists should collaborate to help solve important clinical problems and improve clinical needs for patient care. We also thank Dr. Olga Pletnikova for the access to the human infant brain.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and publication of this article: This work was supported by National Institutes of Health grants R01 NS060703, R21 NS095036, R01 HL139543, R01 NS107417, R01 AG05146, and K08 NS080984.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
References
- 1.Kurinczuk JJ, White-Koning M, Badawi N. Epidemiology of neonatal encephalopathy and hypoxic-ischaemic encephalopathy. Early Hum Dev 2010; 86: 329–338. [DOI] [PubMed] [Google Scholar]
- 2.Shankaran S, Pappas A, McDonald SA, et al. Childhood outcomes after hypothermia for neonatal encephalopathy. N Engl J Med 2012; 366: 2085–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Azzopardi D, Strohm B, Marlow N, et al. Effects of hypothermia for perinatal asphyxia on childhood outcomes. N Engl J Med 2014; 371: 140–149. [DOI] [PubMed] [Google Scholar]
- 4.Shankaran S, Laptook AR, Ehrenkranz RA, et al. Whole-body hypothermia for neonates with hypoxic-ischemic encephalopathy. N Engl J Med 2005; 353: 1574–1584. [DOI] [PubMed] [Google Scholar]
- 5.Gluckman PD, Wyatt JS, Azzopardi D, et al. Selective head cooling with mild systemic hypothermia after neonatal encephalopathy: multicentre randomised trial. Lancet 2005; 365: 663–670. [DOI] [PubMed] [Google Scholar]
- 6.Azzopardi DV, Strohm B, Edwards AD, et al. Moderate hypothermia to treat perinatal asphyxial encephalopathy. N Engl J Med 2009; 361: 1349–1358. [DOI] [PubMed] [Google Scholar]
- 7.Edwards AD, Brocklehurst P, Gunn AJ, et al. Neurological outcomes at 18 months of age after moderate hypothermia for perinatal hypoxic ischaemic encephalopathy: synthesis and meta-analysis of trial data. BMJ 2010; 340: c363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alkharfy TM. Induced hypothermia to treat neonatal hypoxic-ischemic encephalopathy. Review of literature with meta-analysis and development of national protocol. Neurosciences 2013; 18: 18–26. [PubMed] [Google Scholar]
- 9.McAdams RM, Juul SE. Neonatal encephalopathy: update on therapeutic hypothermia and other novel therapeutics. Clin Perinatol 2016; 43: 485–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martinello K, Hart AR, Yap S, et al. Management and investigation of neonatal encephalopathy: 2017 update. Arch Dis Child Fetal Neonatal Ed 2017; 102: F346–F358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Byrom MJ, Bannon PG, White GH, et al. Animal models for the assessment of novel vascular conduits. J Vasc Surg 2010; 52: 176–195. [DOI] [PubMed] [Google Scholar]
- 12.Rice JE, III, Vannucci RC, Brierley JB. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol 1981; 9: 131–141. [DOI] [PubMed] [Google Scholar]
- 13.Mallard C, Vexler ZS. Modeling ischemia in the immature brain: how translational are animal models? Stroke 2015; 46: 3006–3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang L, Zhao F, Qu Y, et al. Animal models of hypoxic-ischemic encephalopathy: optimal choices for the best outcomes. Rev Neurosci 2017; 28: 31–43. [DOI] [PubMed] [Google Scholar]
- 15.Thornton C, Baburamani AA, Kichev A, et al. Oxidative stress and endoplasmic reticulum (ER) stress in the development of neonatal hypoxic-ischaemic brain injury. Biochem Soc Trans 2017; 45: 1067–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thornton C, Jones A, Nair S, et al. Mitochondrial dynamics, mitophagy and biogenesis in neonatal hypoxic-ischaemic brain injury. FEBS Lett 2018; 592: 812–830. [DOI] [PubMed] [Google Scholar]
- 17.Thornton C, Leaw B, Mallard C, et al. Cell death in the developing brain after hypoxia-ischemia. Front Cell Neurosci 2017; 11: 248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Northington FJ, Chavez-Valdez R, Martin LJ. Neuronal cell death in neonatal hypoxia-ischemia. Ann Neurol 2011; 69: 743–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin LJ, Chang Q. DNA damage response and repair, DNA methylation, and cell death in human neurons and experimental animal neurons are different. J Neuropathol Exp Neurol 2018; 77: 636–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martin LJ, Cork LC. The non-human primate striatum undergoes marked prolonged remodeling during postnatal development. Front Cell Neurosci 2014; 8: 294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stroke Therapy Academic Industry R. Recommendations for standards regarding preclinical neuroprotective and restorative drug development. Stroke 1999; 30: 2752–2758. [DOI] [PubMed] [Google Scholar]
- 22.Ranck JB, Jr., Windle WF. Brain damage in the monkey, macaca mulatta, by asphyxia neonatorum. Exp Neurol 1959; 1: 130–154. [DOI] [PubMed] [Google Scholar]
- 23.Faro MD, Windle WF. Transneuronal degeneration in brains of monkeys asphyxiated at birth. Exp Neurol 1969; 24: 38–53. [DOI] [PubMed] [Google Scholar]
- 24.Lawrence DG, Kuypers HG. The functional organization of the motor system in the monkey. I. The effects of bilateral pyramidal lesions. Brain 1968; 91: 1–14. [DOI] [PubMed] [Google Scholar]
- 25.Lawrence DG, Kuypers HG. The functional organization of the motor system in the monkey. II. The effects of lesions of the descending brain-stem pathways. Brain 1968; 91: 15–36. [DOI] [PubMed] [Google Scholar]
- 26.Sukoff MH, Ragatz RE. Cerebellar stimulation for chronic extensor-flexor rigidity and opisthotonus secondary to hypoxia. Report of two cases. J Neurosurg 1980; 53: 391–396. [DOI] [PubMed] [Google Scholar]
- 27.Baker SN. The primate reticulospinal tract, hand function and functional recovery. J Physiol 2011; 589: 5603–5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cahill-Rowley K, Rose J. Etiology of impaired selective motor control: emerging evidence and its implications for research and treatment in cerebral palsy. Dev Med Child Neurol 2014; 56: 522–528. [DOI] [PubMed] [Google Scholar]
- 29.Brandt T, Dieterich M. Vestibular syndromes in the roll plane: topographic diagnosis from brainstem to cortex. Ann Neurol 1994; 36: 337–347. [DOI] [PubMed] [Google Scholar]
- 30.Quattrocchi CC, Fariello G, Longo D. Brainstem tegmental lesions in neonates with hypoxic-ischemic encephalopathy: magnetic resonance diagnosis and clinical outcome. World J Radiol 2016; 8: 117–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schneider H, Ballowitz L, Schachinger H, et al. Anoxic encephalopathy with predominant involvement of basal ganglia, brain stem and spinal cord in the perinatal period. Report on seven newborns. Acta Neuropathol 1975; 32: 287–298. [DOI] [PubMed] [Google Scholar]
- 32.Rutherford M, Pennock J, Schwieso J, et al. Hypoxic-ischaemic encephalopathy: early and late magnetic resonance imaging findings in relation to outcome. Arch Dis Child Fetal Neonatal Ed 1996; 75: F145–F151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Vries LS, Groenendaal F. Patterns of neonatal hypoxic-ischaemic brain injury. Neuroradiology 2010; 52: 555–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sladky JT, Rorke LB. Perinatal hypoxic/ischemic spinal cord injury. Pediatr Pathol 1986; 6: 87–101. [DOI] [PubMed] [Google Scholar]
- 35.Martin E, Barkovich AJ. Magnetic resonance imaging in perinatal asphyxia. Arch Dis Child Fetal Neonatal Ed 1995; 72: F62–F70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schiering IA, de Haan TR, Niermeijer JM, et al. Correlation between clinical and histologic findings in the human neonatal hippocampus after perinatal asphyxia. J Neuropathol Exp Neurol 2014; 73: 324–334. [DOI] [PubMed] [Google Scholar]
- 37.Clancy RR, Sladky JT, Rorke LB. Hypoxic-ischemic spinal cord injury following perinatal asphyxia. Ann Neurol 1989; 25: 185–189. [DOI] [PubMed] [Google Scholar]
- 38.Gilles FH, Murphy SF. Perinatal telencephalic leucoencephalopathy. J Neurol Neurosurg Psychiatry 1969; 32: 404–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arend I, Rafal R, Ward R. Spatial and temporal deficits are regionally dissociable in patients with pulvinar lesions. Brain 2008; 131: 2140–2152. [DOI] [PubMed] [Google Scholar]
- 40.Saxon SV, Ponce CG. Behavioral defects in monkeys asphyxiated during birth. Exp Neurol 1961; 4: 460–469. [DOI] [PubMed] [Google Scholar]
- 41.Saxon SV. Differences in reactivity between asphyxial and normal Rhesus monkeys. J Genet Psychol 1961; 99: 283–287. [DOI] [PubMed] [Google Scholar]
- 42.Sechzer JA. Memory deficit in monkeys brain damaged by asphyxia neonatorum. Exp Neurol 1969; 24: 497–507. [DOI] [PubMed] [Google Scholar]
- 43.Myers RE. Two patterns of perinatal brain damage and their conditions of occurrence. Am J Obstet Gynecol 1972; 112: 246–276. [DOI] [PubMed] [Google Scholar]
- 44.Brann AW, Jr., Myers RE. Central nervous system findings in the newborn monkey following severe in utero partial asphyxia. Neurology 1975; 25: 327–338. [DOI] [PubMed] [Google Scholar]
- 45.Myers RE, Brann AW., Jr Abruptio placentae in rhesus monkey causing brain damage to the fetus. Am J Obstet Gynecol 1976; 126: 1048–1049. [DOI] [PubMed] [Google Scholar]
- 46.Myers RE. Experimental models of perinatal brain damage: relevance to human pathology. In: Gluck L. (ed). Intrauterine asphyxia and the developing fetal brain, Chicago: Year Book Medical, 1977, pp. 37–97. [Google Scholar]
- 47.Larroche JC, Korn G. Brain damage in intrauterine growth retardation. In: Gluck L. (ed.). Intrauterine asphyxia and the developing fetal brain, Chicago: Year Book Medical, 1977, pp. 25–35. [Google Scholar]
- 48.Larroche JC, DeVries L. Fetal and neonatal brain damage. In: Gilbert-Barness E. (ed). Potter's pathology of the fetus and infant 1996; Vol. 2, St. Louis, MO: Mosby-Year Book, Inc., pp. 1099–1131. [Google Scholar]
- 49.Barkovich AJ, Westmark K, Partridge C, et al. Perinatal asphyxia: MR findings in the first 10 days. Am J Neuroradiol 1995; 16: 427–438. [PMC free article] [PubMed] [Google Scholar]
- 50.Miller SP, Ramaswamy V, Michelson D, et al. Patterns of brain injury in term neonatal encephalopathy. J Pediatr 2005; 146: 453–460. [DOI] [PubMed] [Google Scholar]
- 51.Ferrari F, Todeschini A, Guidotti I, et al. General movements in full-term infants with perinatal asphyxia are related to Basal Ganglia and thalamic lesions. J Pediatr 2011; 158: 904–911. [DOI] [PubMed] [Google Scholar]
- 52.Dobbing J, Sands J. Comparative aspects of the brain growth spurt. Early Hum Dev 1979; 3: 79–83. [DOI] [PubMed] [Google Scholar]
- 53.Gleason CA, Hamm C, Jones MD., Jr Cerebral blood flow, oxygenation, and carbohydrate metabolism in immature fetal sheep in utero. Am J Physiol Regulatory Integrative Comp Physiol 1989; 256: R1264–R1268. [DOI] [PubMed] [Google Scholar]
- 54.Gleason CA, Hamm C, Jones MD., Jr Effect of acute hypoxemia on brain blood flow and oxygen metabolism in immature fetal sheep. Am J Physiol Heart Circ Physiol 1990; 258: H1064–H1069. [DOI] [PubMed] [Google Scholar]
- 55.Jones MD, Jr., Sheldon RE, Peeters LL, et al. Regulation of cerebral blood flow in the ovine fetus. Am J Physiol Heart Circ Physiol 1978; 235: H162–H166. [DOI] [PubMed] [Google Scholar]
- 56.Rosenberg AA, Harris AP, Koehler RC, et al. Role of O2 -hemoglobin affinity in the regulation of cerebral blood flow in fetal sheep. Am J Physiol Heart Circ Physiol 1986; 251: H56–H62. [DOI] [PubMed] [Google Scholar]
- 57.Gleason CA, Jones MD, Jr., Traystman RJ, et al. Fetal cerebral responses to ventilation and oxygenation in utero. Am J Physiol Regul Integr Comp Physiol 1988; 255: R1049–R1054. [DOI] [PubMed] [Google Scholar]
- 58.Ohata H, Gebremedhin D, Narayanan J, et al. Onset of pulmonary ventilation in fetal sheep produces pial arteriolar constriction dependent on cytochrome p450 omega-hydroxylase activity. J Appl Physiol 2010; 109: 412–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jones MD, Jr., Traystman RJ, Simmons MA, et al. Effects of changes in arterial O2 content on cerebral blood flow in the lamb. Am J Physiol Heart Circ Physiol 1981; 240: H209–H215. [DOI] [PubMed] [Google Scholar]
- 60.Jones MD, Jr., Rosenberg AA, Simmons MA, et al. Oxygen delivery to the brain before and after birth. Science 1982; 216: 324–325. [DOI] [PubMed] [Google Scholar]
- 61.Rosenberg AA, Jones MD, Jr., Traystman RJ, et al. Response of cerebral blood flow to changes in PCO2 in fetal, newborn, and adult sheep. Am J Physiol Heart Circ Physiol 1982; 242: H862–H866. [DOI] [PubMed] [Google Scholar]
- 62.Papile LA, Rudolph AM, Heymann MA. Autoregulation of cerebral blood flow in the preterm fetal lamb. Pediatr Res 1985; 19: 159–161. [DOI] [PubMed] [Google Scholar]
- 63.Helou SM, Koehler RC, Gleason CA, et al. Cerebrovascular autoregulation during fetal development in sheep. Am J Physiol Heart Circ Physiol 1994; 266: H1069–H1074. [DOI] [PubMed] [Google Scholar]
- 64.Tweed WA, Cote J, Pash M, et al. Arterial oxygenation determines autoregulation of cerebral blood flow in the fetal lamb. Pediatr Res 1983; 17: 246–249. [DOI] [PubMed] [Google Scholar]
- 65.Szymonowicz W, Walker AM, Yu VY, et al. Regional cerebral blood flow after hemorrhagic hypotension in the preterm, near-term, and newborn lamb. Pediatr Res 1990; 28: 361–366. [DOI] [PubMed] [Google Scholar]
- 66.Ting P, Yamaguchi S, Bacher JD, et al. Hypoxic-ischemic cerebral necrosis in midgestational sheep fetuses: physiopathologic correlations. Exp Neurol 1983; 80: 227–245. [DOI] [PubMed] [Google Scholar]
- 67.Wagner KR, Ting P, Westfall MV, et al. Brain metabolic correlates of hypoxic-ischemic cerebral necrosis in mid-gestational sheep fetuses: significance of hypotension. J Cereb Blood Flow Metab 1986; 6: 425–434. [DOI] [PubMed] [Google Scholar]
- 68.Clapp JF, Peress NS, Wesley M, et al. Brain damage after intermittent partial cord occlusion in the chronically instrumented fetal lamb. Am J Obstet Gynecol 1988; 159: 504–509. [DOI] [PubMed] [Google Scholar]
- 69.de Haan HH, Gunn AJ, Williams CE, et al. Brief repeated umbilical cord occlusions cause sustained cytotoxic cerebral edema and focal infarcts in near-term fetal lambs. Pediatr Res 1997; 41: 96–104. [DOI] [PubMed] [Google Scholar]
- 70.Gunn AJ, Parer JT, Mallard EC, et al. Cerebral histologic and electrocorticographic changes after asphyxia in fetal sheep. Pediatr Res 1992; 31: 486–491. [DOI] [PubMed] [Google Scholar]
- 71.Gunn AJ, Maxwell L, De Haan HH, et al. Delayed hypotension and subendocardial injury after repeated umbilical cord occlusion in near-term fetal lambs. Am J Obstet Gynecol 2000; 183: 1564–1572. [DOI] [PubMed] [Google Scholar]
- 72.de Haan HH, Gunn AJ, Williams CE, et al. Magnesium sulfate therapy during asphyxia in near-term fetal lambs does not compromise the fetus but does not reduce cerebral injury. Am J Obstet Gynecol 1997; 176: 18–27. [DOI] [PubMed] [Google Scholar]
- 73.Mallard EC, Waldvogel HJ, Williams CE, et al. Repeated asphyxia causes loss of striatal projection neurons in the fetal sheep brain. Neuroscience 1995; 65: 827–836. [DOI] [PubMed] [Google Scholar]
- 74.Mallard EC, Gunn AJ, Williams CE, et al. Transient umbilical cord occlusion causes hippocampal damage in the fetal sheep. Am J Obstet Gynecol 1992; 167: 1423–1430. [DOI] [PubMed] [Google Scholar]
- 75.Mallard EC, Williams CE, Johnston BM, et al. Increased vulnerability to neuronal damage after umbilical cord occlusion in fetal sheep with advancing gestation. Am J Obstet Gynecol 1994; 170: 206–214. [DOI] [PubMed] [Google Scholar]
- 76.Davidson JO, Drury PP, Green CR, et al. Connexin hemichannel blockade is neuroprotective after asphyxia in preterm fetal sheep. PLoS One 2014; 9: e96558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.George SA, Barrett RD, Bennet L, et al. Nonadditive neuroprotection with early glutamate receptor blockade and delayed hypothermia after asphyxia in preterm fetal sheep. Stroke 2012; 43: 3114–3117. [DOI] [PubMed] [Google Scholar]
- 78.Furuta A, Martin LJ. Laminar segregation of the cortical plate during corticogenesis is accompanied by changes in glutamate receptor expression. J Neurobiol 1999; 39: 67–80. [PubMed] [Google Scholar]
- 79.Williams CE, Gunn AJ, Mallard C, et al. Outcome after ischemia in the developing sheep brain: an electroencephalographic and histological study. Ann Neurol 1992; 31: 14–21. [DOI] [PubMed] [Google Scholar]
- 80.Williams CE, Gunn AJ, Synek B, et al. Delayed seizures occurring with hypoxic-ischemic encephalopathy in the fetal sheep. Pediatr Res 1990; 27: 561–565. [DOI] [PubMed] [Google Scholar]
- 81.Mallard EC, Williams CE, Gunn AJ, et al. Frequent episodes of brief ischemia sensitize the fetal sheep brain to neuronal loss and induce striatal injury. Pediatr Res 1993; 33: 61–65. [DOI] [PubMed] [Google Scholar]
- 82.Tan WK, Williams CE, Gunn AJ, et al. Suppression of postischemic epileptiform activity with MK-801 improves neural outcome in fetal sheep. Ann Neurol 1992; 32: 677–682. [DOI] [PubMed] [Google Scholar]
- 83.Guan J, Bennet TL, George S, et al. Selective neuroprotective effects with insulin-like growth factor-1 in phenotypic striatal neurons following ischemic brain injury in fetal sheep. Neuroscience 2000; 95: 831–839. [DOI] [PubMed] [Google Scholar]
- 84.Davidson JO, Green CR, Nicholson LF, et al. Connexin hemichannel blockade is neuroprotective after, but not during, global cerebral ischemia in near-term fetal sheep. Exp Neurol 2013; 248: 301–308. [DOI] [PubMed] [Google Scholar]
- 85.Back SA, Luo NL, Borenstein NS, et al. Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J Neurosci 2001; 21: 1302–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Back SA, Luo NL, Mallinson RA, et al. Selective vulnerability of preterm white matter to oxidative damage defined by F2-isoprostanes. Ann Neurol 2005; 58: 108–120. [DOI] [PubMed] [Google Scholar]
- 87.Back SA, Riddle A, Dean J, et al. The instrumented fetal sheep as a model of cerebral white matter injury in the premature infant. Neurotherapeutics 2012; 9: 359–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Back SA, Riddle A, Hohimer AR. Role of instrumented fetal sheep preparations in defining the pathogenesis of human periventricular white-matter injury. J Child Neurol 2006; 21: 582–589. [DOI] [PubMed] [Google Scholar]
- 89.Riddle A, Luo NL, Manese M, et al. Spatial heterogeneity in oligodendrocyte lineage maturation and not cerebral blood flow predicts fetal ovine periventricular white matter injury. J Neurosci 2006; 26: 3045–3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Riddle A, Maire J, Gong X, et al. Differential susceptibility to axonopathy in necrotic and non-necrotic perinatal white matter injury. Stroke 2012; 43: 178–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.McClure MM, Riddle A, Manese M, et al. Cerebral blood flow heterogeneity in preterm sheep: lack of physiologic support for vascular boundary zones in fetal cerebral white matter. J Cereb Blood Flow Metab 2008; 28: 995–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Reddy K, Mallard C, Guan J, et al. Maturational change in the cortical response to hypoperfusion injury in the fetal sheep. Pediatr Res 1998; 43: 674–682. [DOI] [PubMed] [Google Scholar]
- 93.Buser JR, Segovia KN, Dean JM, et al. Timing of appearance of late oligodendrocyte progenitors coincides with enhanced susceptibility of preterm rabbit cerebral white matter to hypoxia-ischemia. J Cereb Blood Flow Metab 2010; 30: 1053–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Riddle A, Dean J, Buser JR, et al. Histopathological correlates of magnetic resonance imaging-defined chronic perinatal white matter injury. Ann Neurol 2011; 70: 493–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Buser JR, Maire J, Riddle A, et al. Arrested preoligodendrocyte maturation contributes to myelination failure in premature infants. Ann Neurol 2012; 71: 93–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Conrad MS, Dilger RN, Johnson RW. Brain growth of the domestic pig (Sus scrofa) from 2 to 24 weeks of age: a longitudinal MRI study. Dev Neurosci 2012; 34: 291–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Conrad MS, Johnson RW. The domestic piglet: an important model for investigating the neurodevelopmental consequences of early life insults. Annu Rev Anim Biosci 2015; 3: 245–264. [DOI] [PubMed] [Google Scholar]
- 98.Dickerson JW, Dobbing J. Prenatal and postnatal growth and development of the central nervous system of the pig. Proc R Soc Lond B Biol Sci 1967; 166: 384–395. [DOI] [PubMed] [Google Scholar]
- 99.Goureau A, Garrigues A, Tosser-Klopp G, et al. Conserved synteny and gene order difference between human chromosome 12 and pig chromosome 5. Cytogenet Cell Genet 2001; 94: 49–54. [DOI] [PubMed] [Google Scholar]
- 100.Schachtschneider KM, Madsen O, Park C, et al. Adult porcine genome-wide DNA methylation patterns support pigs as a biomedical model. BMC Genom 2015; 16: 743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Thoresen M, Haaland K, Loberg EM, et al. A piglet survival model of posthypoxic encephalopathy. Pediatr Res 1996; 40: 738–748. [DOI] [PubMed] [Google Scholar]
- 102.Tooley JR, Satas S, Porter H, et al. Head cooling with mild systemic hypothermia in anesthetized piglets is neuroprotective. Ann Neurol 2003; 53: 65–72. [DOI] [PubMed] [Google Scholar]
- 103.Chakkarapani E, Dingley J, Liu X, et al. Xenon enhances hypothermic neuroprotection in asphyxiated newborn pigs. Ann Neurol 2010; 68: 330–341. [DOI] [PubMed] [Google Scholar]
- 104.Edwards AD, Yue X, Squier MV, et al. Specific inhibition of apoptosis after cerebral hypoxia-ischaemia by moderate post-insult hypothermia. Biochem Biophys Res Commun 1995; 217: 1193–1199. [DOI] [PubMed] [Google Scholar]
- 105.Thoresen M, Penrice J, Lorek A, et al. Mild hypothermia after severe transient hypoxia-ischemia ameliorates delayed cerebral energy failure in the newborn piglet. Pediatr Res 1995; 37: 667–670. [DOI] [PubMed] [Google Scholar]
- 106.Robertson NJ, Kato T, Bainbridge A, et al. Methyl-isobutyl amiloride reduces brain Lac/NAA, cell death and microglial activation in a perinatal asphyxia model. J Neurochem 2013; 124: 645–657. [DOI] [PubMed] [Google Scholar]
- 107.Peeters-Scholte C, Koster J, Veldhuis W, et al. Neuroprotection by selective nitric oxide synthase inhibition at 24 hours after perinatal hypoxia-ischemia. Stroke 2002; 33: 2304–2310. [DOI] [PubMed] [Google Scholar]
- 108.Peeters-Scholte C, Koster J, van den Tweel E, et al. Effects of selective nitric oxide synthase inhibition on IGF-1, caspases and cytokines in a newborn piglet model of perinatal hypoxia-ischaemia. Dev Neurosci 2002; 24: 396–404. [DOI] [PubMed] [Google Scholar]
- 109.Peeters-Scholte C, Braun K, Koster J, et al. Effects of allopurinol and deferoxamine on reperfusion injury of the brain in newborn piglets after neonatal hypoxia-ischemia. Pediatr Res 2003; 54: 516–522. [DOI] [PubMed] [Google Scholar]
- 110.Robertson NJ, Faulkner S, Fleiss B, et al. Melatonin augments hypothermic neuroprotection in a perinatal asphyxia model. Brain 2013; 136: 90–105. [DOI] [PubMed] [Google Scholar]
- 111.Faulkner S, Bainbridge A, Kato T, et al. Xenon augmented hypothermia reduces early lactate/N-acetylaspartate and cell death in perinatal asphyxia. Ann Neurol 2011; 70: 133–150. [DOI] [PubMed] [Google Scholar]
- 112.Broad KD, Fierens I, Fleiss B, et al. Inhaled 45-50% argon augments hypothermic brain protection in a piglet model of perinatal asphyxia. Neurobiol Dis 2016; 87: 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Alvarez FJ, Lafuente H, Rey-Santano MC, et al. Neuroprotective effects of the nonpsychoactive cannabinoid cannabidiol in hypoxic-ischemic newborn piglets. Pediatr Res 2008; 64: 653–658. [DOI] [PubMed] [Google Scholar]
- 114.Lafuente H, Alvarez FJ, Pazos MR, et al. Cannabidiol reduces brain damage and improves functional recovery after acute hypoxia-ischemia in newborn pigs. Pediatr Res 2011; 70: 272–277. [DOI] [PubMed] [Google Scholar]
- 115.Pazos MR, Mohammed N, Lafuente H, et al. Mechanisms of cannabidiol neuroprotection in hypoxic-ischemic newborn pigs: role of 5HT(1A) and CB2 receptors. Neuropharmacology 2013; 71: 282–291. [DOI] [PubMed] [Google Scholar]
- 116.Rocha-Ferreira E, Rudge B, Hughes MP, et al. Immediate remote ischemic postconditioning reduces brain nitrotyrosine formation in a piglet asphyxia model. Oxid Med Cell Longev 2016; 2016: 5763743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ezzati M, Bainbridge A, Broad KD, et al. Immediate remote ischemic postconditioning after hypoxia ischemia in piglets protects cerebral white matter but not grey matter. J Cereb Blood Flow Metab 2016; 36: 1396–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Brambrink AM, Martin LJ, Hanley DF, et al. Effects of the AMPA receptor antagonist NBQX on outcome of newborn pigs after asphyxic cardiac arrest. J Cereb Blood Flow Metab 1999; 19: 927–938. [DOI] [PubMed] [Google Scholar]
- 119.Brambrink AM, Ichord RN, Martin LJ, et al. Poor outcome after hypoxia-ischemia in newborns is associated with physiological abnormalities during early recovery. Possible relevance to secondary brain injury after head trauma in infants. ExpToxicol Pathol 1999; 51: 151–162. [DOI] [PubMed] [Google Scholar]
- 120.Maller AI, Hankins LL, Yeakley JW, et al. Rolandic type cerebral palsy in children as a pattern of hypoxic-ischemic injury in the full-term neonate. J Child Neurol 1998; 13: 313–321. [DOI] [PubMed] [Google Scholar]
- 121.Johnston MV. Selective vulnerability in the neonatal brain. Ann Neurol 1998; 44: 155–156. [DOI] [PubMed] [Google Scholar]
- 122.Martin LJ, Brambrink A, Koehler RC, et al. Primary sensory and forebrain motor systems in the newborn brain are preferentially damaged by hypoxia-ischemia. J Comp Neurol 1997; 377: 262–285. [DOI] [PubMed] [Google Scholar]
- 123.Woolsey CN, Fairman D. Contralateral, ipsilateral, and bilateral representation of cutaneous receptors in somatic areas I and II of the cerebral cortex of pig, sheep, and other mammals. Surgery 1946; 19: 684–702. [PubMed] [Google Scholar]
- 124.Craner SL, Ray RH. Somatosensory cortex of the neonatal pig: I. Topographic organization of the primary somatosensory cortex (SI). J Comp Neurol 1991; 306: 24–38. [DOI] [PubMed] [Google Scholar]
- 125.Jones EG, Coulter JD, Burton H, et al. Cells of origin and terminal distribution of corticostriatal fibers arising in the sensory-motor cortex of monkeys. J Comp Neurol 1977; 173: 53–80. [DOI] [PubMed] [Google Scholar]
- 126.Martin LJ, Brambrink A, Koehler RC, et al. Neonatal asphyxic brain injury is neural system preferential and targets sensory-motor networks. In: Stevenson DK, Sunshine P. (eds). Fetal and neonatal brain injury: mechanisms, management and the risks of practice, Oxford: Oxford University Press, 1997, pp. 374–399. [Google Scholar]
- 127.Low JA, Robertson DM, Simpson LL. Temporal relationships of neuropathologic conditions caused by perinatal asphyxia. Am J Obstet Gynecol 1989; 160: 608–614. [DOI] [PubMed] [Google Scholar]
- 128.Roland EH, Poskitt K, Rodriguez E, et al. Perinatal hypoxic-ischemic thalamic injury: clinical features and neuroimaging. Ann Neurol 1998; 44: 161–166. [DOI] [PubMed] [Google Scholar]
- 129.Martin LJ, Brambrink AM, Price AC, et al. Neuronal death in newborn striatum after hypoxia-ischemia is necrosis and evolves with oxidative stress. Neurobiol Dis 2000; 7: 169–191. [DOI] [PubMed] [Google Scholar]
- 130.Nakajima W, Ishida A, Lange MS, et al. Apoptosis has a prolonged role in the neurodegeneration after hypoxic ischemia in the newborn rat. J Neurosci 2000; 20: 7994–8004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Northington FJ, Zelaya ME, O'Riordan DP, et al. Failure to complete apoptosis following neonatal hypoxia-ischemia manifests as “continuum” phenotype of cell death and occurs with multiple manifestations of mitochondrial dysfunction in rodent forebrain. Neuroscience 2007; 149: 822–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Golden WC, Brambrink AM, Traystman RJ, et al. Failure to sustain recovery of Na,K-ATPase function is a possible mechanism for striatal neurodegeneration in hypoxic-ischemic newborn piglets. Brain Res Mol Brain Res 2001; 88: 94–102. [DOI] [PubMed] [Google Scholar]
- 133.Mueller-Burke D, Koehler RC, Martin LJ. Rapid NMDA receptor phosphorylation and oxidative stress precede striatal neurodegeneration after hypoxic ischemia in newborn piglets and are attenuated with hypothermia. Int J Dev Neurosci 2008; 26: 67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cui D, Sun D, Wang X, et al. Impaired autophagosome clearance contributes to neuronal death in a piglet model of neonatal hypoxic-ischemic encephalopathy. Cell Death Dis 2017; 8: e2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Guerguerian AM, Brambrink AM, Traystman RJ, et al. Altered expression and phosphorylation of N-methyl-D-aspartate receptors in piglet striatum after hypoxia-ischemia. Brain Res Mol Brain Res 2002; 104: 66–80. [DOI] [PubMed] [Google Scholar]
- 136.Yang ZJ, Torbey M, Li X, et al. Dopamine receptor modulation of hypoxic-ischemic neuronal injury in striatum of newborn piglets. J Cereb Blood Flow Metab 2007; 27: 1339–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Martin LJ, Brambrink AM, Lehmann C, et al. Hypoxia-ischemia causes abnormalities in glutamate transporters and death of astroglia and neurons in newborn striatum. Ann Neurol 1997; 42: 335–348. [DOI] [PubMed] [Google Scholar]
- 138.Yang ZJ, Ni X, Carter EL, et al. Neuroprotective effect of acid-sensing ion channel inhibitor psalmotoxin-1 after hypoxia-ischemia in newborn piglet striatum. Neurobiol Dis 2011; 43: 446–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Greengard P. The neurobiology of slow synaptic transmission. Science 2001; 294: 1024–1030. [DOI] [PubMed] [Google Scholar]
- 140.Pastuszko A, Saadat-Lajevardi N, Chen J, et al. Effects of graded levels of tissue oxygen pressure on dopamine metabolism in the striatum of newborn piglets. J Neurochem 1993; 60: 161–166. [DOI] [PubMed] [Google Scholar]
- 141.Park TS, Van Wylen DG, Rubio R, et al. Increased brain interstitial fluid adenosine concentration during hypoxia in newborn piglet. J Cereb Blood Flow Metab 1987; 7: 178–183. [DOI] [PubMed] [Google Scholar]
- 142.Yang ZJ, Wang B, Kwansa H, et al. Adenosine 2A receptor contributes to ischemic brain damage in newborn piglet. J Cereb Blood Flow Metab 2013; 33: 1612–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Svenningsson P, Lindskog M, Rognoni F, et al. Activation of adenosine A2A and dopamine D1 receptors stimulates cyclic AMP-dependent phosphorylation of DARPP-32 in distinct populations of striatal projection neurons. Neuroscience 1998; 84: 223–228. [DOI] [PubMed] [Google Scholar]
- 144.Svenningsson P, Nishi A, Fisone G, et al. DARPP-32: an integrator of neurotransmission. Annu Rev Pharmacol Toxicol 2004; 44: 269–296. [DOI] [PubMed] [Google Scholar]
- 145.Snyder GL, Fienberg AA, Huganir RL, et al. A dopamine/D1 receptor/protein kinase A/dopamine- and cAMP-regulated phosphoprotein (M r 32 kDa)/protein phosphatase-1 pathway regulates dephosphorylation of the NMDA receptor. J Neurosci 1998; 18: 10297–10303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Cheng XJ, Hoog JO, Nairn AC, et al. Regulation of rat Na +, K + -ATPase activity by PKC is modulated by state of phosphorylation of Ser-943 by PKA. Am J Physiol Cell Physiol 1997; 273: C1981–C1986. [DOI] [PubMed] [Google Scholar]
- 147.Harder DR, Narayanan J, Birks EK, et al. Identification of a putative microvascular oxygen sensor. Circ Res 1996; 79: 54–61. [DOI] [PubMed] [Google Scholar]
- 148.Renic M, Klaus JA, Omura T, et al. Effect of 20-HETE inhibition on infarct volume and cerebral blood flow after transient middle cerebral artery occlusion. J Cereb Blood Flow Metab 2009; 29: 629–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Dunn KM, Renic M, Flasch AK, et al. Elevated production of 20-HETE in the cerebral vasculature contributes to severity of ischemic stroke and oxidative stress in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol 2008; 295: H2455–H2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zhang H, Falck JR, Roman RJ, et al. Upregulation of 20-HETE synthetic cytochrome P450 isoforms by oxygen-glucose deprivation in cortical neurons. Cell Mol Neurobiol 2017; 37: 1279–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Renic M, Kumar SN, Gebremedhin D, et al. Protective effect of 20-HETE inhibition in a model of oxygen-glucose deprivation in hippocampal slice cultures. Am J Physiol Heart Circ Physiol 2012; 302: H1285–H1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zhu J, Wang B, Lee JH, et al. Additive neuroprotection of a 20-HETE inhibitor with delayed therapeutic hypothermia after hypoxia-ischemia in neonatal piglets. Dev Neurosci 2015; 37: 376–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Yang ZJ, Carter EL, Kibler KK, et al. Attenuation of neonatal ischemic brain damage using a 20-HETE synthesis inhibitor. J Neurochem 2012; 121: 168–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Yermolaieva O, Leonard AS, Schnizler MK, et al. Extracellular acidosis increases neuronal cell calcium by activating acid-sensing ion channel 1a. Proc Natl Acad Sci USA 2004; 101: 6752–6757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Bhardwaj A, Sawada M, London ED, et al. Potent sigma 1 -receptor ligand 4-phenyl-1-(4-phenylbutyl) piperidine modulates basal and N -methyl-D-aspartate-evoked nitric oxide production in vivo. Stroke 1998; 29: 2404–2411. [PubMed] [Google Scholar]
- 156.Goyagi T, Goto S, Bhardwaj A, et al. Neuroprotective effect of sigma 1-receptor ligand 4-phenyl-1-(4-phenylbutyl) piperidine (PPBP) is linked to reduced neuronal nitric oxide production. Stroke 2001; 32: 1613–1620. [DOI] [PubMed] [Google Scholar]
- 157.Takahashi H, Kirsch JR, Hashimoto K, et al. PPBP [4-phenyl-1-(4-phenylbutyl) piperidine, a potent sigma-receptor ligand, decreases brain injury following transient focal ischemia in cats. Stroke 1995; 26: 1676–1682. [DOI] [PubMed] [Google Scholar]
- 158.Takahashi H, Kirsch JR, Hashimoto K, et al. PPBP[4-phenyl-1-(4-phenylbutyl)piperidine] decreases brain injury after transient focal ischemia in rats. Stroke 1996; 27: 2120–2123. [DOI] [PubMed] [Google Scholar]
- 159.Harukuni I, Bhardwaj A, Shaivitz AB, et al. Sigma 1 -receptor ligand 4-phenyl-1-(4-phenylbutyl)-piperidine affords neuroprotection from focal ischemia with prolonged reperfusion. Stroke 2000; 31: 976–982. [DOI] [PubMed] [Google Scholar]
- 160.Wegleiter K, Hermann M, Posod A, et al. The sigma-1 receptor agonist 4-phenyl-1-(4-phenylbutyl) piperidine (PPBP) protects against newborn excitotoxic brain injury by stabilizing the mitochondrial membrane potential in vitro and inhibiting microglial activation in vivo. Exp Neurol 2014; 261: 501–509. [DOI] [PubMed] [Google Scholar]
- 161.Yang ZJ, Carter EL, Torbey MT, et al. Sigma receptor ligand 4-phenyl-1-(4-phenylbutyl)-piperidine modulates neuronal nitric oxide synthase/postsynaptic density-95 coupling mechanisms and protects against neonatal ischemic degeneration of striatal neurons. Exp Neurol 2010; 221: 166–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Ni X, Yang ZJ, Carter EL, et al. Striatal neuroprotection from neonatal hypoxia-ischemia in piglets by antioxidant treatment with EUK-134 or edaravone. Dev Neurosci 2011; 33: 299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Karnatovskaia LV, Wartenberg KE, Freeman WD. Therapeutic hypothermia for neuroprotection: history, mechanisms, risks, and clinical applications. Neurohospitalist 2014; 4: 153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Trescher WH, Ishiwa S, Johnston MV. Brief post-hypoxic-ischemic hypothermia markedly delays neonatal brain injury. Brain Dev 1997; 19: 326–338. [DOI] [PubMed] [Google Scholar]
- 165.Bona E, Hagberg H, Loberg EM, et al. Protective effects of moderate hypothermia after neonatal hypoxia- ischemia: short- and long-term outcome. Pediatr Res 1998; 43: 738–745. [DOI] [PubMed] [Google Scholar]
- 166.Thoresen M, Bagenholm R, Loberg EM, et al. Posthypoxic cooling of neonatal rats provides protection against brain injury. Arch Dis Child Fetal Neonatal Ed 1996; 74: F3–F9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Wagner BP, Nedelcu J, Martin E. Delayed postischemic hypothermia improves long-term behavioral outcome after cerebral hypoxia-ischemia in neonatal rats. Pediatr Res 2002; 51: 354–360. [DOI] [PubMed] [Google Scholar]
- 168.Gunn AJ, Gunn TR, de Haan HH, et al. Dramatic neuronal rescue with prolonged selective head cooling after ischemia in fetal lambs. J Clin Invest 1997; 99: 248–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Gunn AJ, Gunn TR, Gunning MI, et al. Neuroprotection with prolonged head cooling started before postischemic seizures in fetal sheep. Pediatrics 1998; 102: 1098–1106. [DOI] [PubMed] [Google Scholar]
- 170.Gunn AJ, Bennet L, Gunning MI, et al. Cerebral hypothermia is not neuroprotective when started after postischemic seizures in fetal sheep. Pediatr Res 1999; 46: 274–280. [DOI] [PubMed] [Google Scholar]
- 171.Haaland K, Loberg EM, Steen PA, et al. Posthypoxic hypothermia in newborn piglets. Pediatr Res 1997; 41: 505–512. [DOI] [PubMed] [Google Scholar]
- 172.Thoresen M, Satas S, Puka-Sundvall M, et al. Post-hypoxic hypothermia reduces cerebrocortical release of NO and excitotoxins. Neuroreport 1997; 8: 3359–3362. [DOI] [PubMed] [Google Scholar]
- 173.Thoresen M, Satas S, Loberg EM, et al. Twenty-four hours of mild hypothermia in unsedated newborn pigs starting after a severe global hypoxic-ischemic insult is not neuroprotective. Pediatr Res 2001; 50: 405–411. [DOI] [PubMed] [Google Scholar]
- 174.Ezzati M, Kawano G, Rocha-Ferreira E, et al. Dexmedetomidine combined with therapeutic hypothermia is associated with cardiovascular instability and neurotoxicity in a piglet model of perinatal asphyxia. Dev Neurosci 2017; 39: 156–170. [DOI] [PubMed] [Google Scholar]
- 175.Agnew DM, Koehler RC, Guerguerian AM, et al. Hypothermia for 24 hours after asphyxic cardiac arrest in piglets provides striatal neuroprotection that is sustained 10 days after rewarming. Pediatr Res 2003; 54: 253–262. [DOI] [PubMed] [Google Scholar]
- 176.Ni X, Yang ZJ, Wang B, et al. Early antioxidant treatment and delayed hypothermia after hypoxia-ischemia have no additive neuroprotection in newborn pigs. Anesth Analg 2012; 115: 627–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Karlsson M, Tooley JR, Satas S, et al. Delayed hypothermia as selective head cooling or whole body cooling does not protect brain or body in newborn pig subjected to hypoxia-ischemia. Pediatr Res 2008; 64: 74–78. [DOI] [PubMed] [Google Scholar]
- 178.Thoresen M, Tooley J, Liu X, et al. Time is brain: starting therapeutic hypothermia within three hours after birth improves motor outcome in asphyxiated newborns. Neonatology 2013; 104: 228–233. [DOI] [PubMed] [Google Scholar]
- 179.Wang B, Armstrong JS, Lee JH, et al. Rewarming from therapeutic hypothermia induces cortical neuron apoptosis in a swine model of neonatal hypoxic-ischemic encephalopathy. J Cereb Blood Flow Metab 2015; 35: 781–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Tooley JR, Eagle RC, Satas S, et al. Significant head cooling can be achieved while maintaining normothermia in the newborn piglet. Arch Dis Child Fetal Neonatal Ed 2005; 90: F262F266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Davidson JO, Draghi V, Whitham S, et al. How long is sufficient for optimal neuroprotection with cerebral cooling after ischemia in fetal sheep? J Cereb Blood Flow Metab 2018; 38: 1047–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Davidson JO, Wassink G, Yuill CA, et al. How long is too long for cerebral cooling after ischemia in fetal sheep? J Cereb Blood Flow Metab 2015; 35: 751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Davidson JO, Yuill CA, Zhang FG, et al. Extending the duration of hypothermia does not further improve white matter protection after ischemia in term-equivalent fetal sheep. Sci Rep 2016; 6: 25178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Rutherford MA, Azzopardi D, Whitelaw A, et al. Mild hypothermia and the distribution of cerebral lesions in neonates with hypoxic-ischemic encephalopathy. Pediatrics 2005; 116: 1001–1006. [DOI] [PubMed] [Google Scholar]
- 185.Rutherford M, Ramenghi LA, Edwards AD, et al. Assessment of brain tissue injury after moderate hypothermia in neonates with hypoxic-ischaemic encephalopathy: a nested substudy of a randomised controlled trial. Lancet Neurol 2010; 9: 39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Howlett JA, Northington FJ, Gilmore MM, et al. Cerebrovascular autoregulation and neurologic injury in neonatal hypoxic-ischemic encephalopathy. Pediatr Res 2013; 74: 525–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Lee JK, Poretti A, Perin J, et al. Optimizing cerebral autoregulation may decrease neonatal regional hypoxic-ischemic brain injury. Dev Neurosci 2017; 39: 248–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Tekes A, Poretti A, Scheurkogel MM, et al. Apparent diffusion coefficient scalars correlate with near-infrared spectroscopy markers of cerebrovascular autoregulation in neonates cooled for perinatal hypoxic-ischemic injury. Am J Neuroradiol 2015; 36: 188–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.McAdams RM, Fleiss B, Traudt C, et al. Long-Term neuropathological changes associated with cerebral palsy in a nonhuman primate model of hypoxic-ischemic encephalopathy. Dev Neurosci 2017; 39: 124–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Wang B, Armstrong JS, Reyes M, et al. White matter apoptosis is increased by delayed hypothermia and rewarming in a neonatal piglet model of hypoxic ischemic encephalopathy. Neuroscience 2016; 316: 296–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Lee JK, Wang B, Reyes M, et al. Hypothermia and rewarming activate a macroglial unfolded protein response independent of hypoxic-ischemic brain injury in neonatal piglets. Dev Neurosci 2016; 38: 277–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Thoresen M, Hobbs CE, Wood T, et al. Cooling combined with immediate or delayed xenon inhalation provides equivalent long-term neuroprotection after neonatal hypoxia-ischemia. J Cereb Blood Flow Metab 2009; 29: 707–714. [DOI] [PubMed] [Google Scholar]
- 193.Azzopardi D, Robertson NJ, Bainbridge A, et al. Moderate hypothermia within 6 h of birth plus inhaled xenon versus moderate hypothermia alone after birth asphyxia (TOBY-Xe): a proof-of-concept, open-label, randomised controlled trial. Lancet Neurol 2016; 15: 145–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Tan DX, Manchester LC, Terron MP, et al. One molecule, many derivatives: a never-ending interaction of melatonin with reactive oxygen and nitrogen species? J Pineal Res 2007; 42: 28–42. [DOI] [PubMed] [Google Scholar]
- 195.Miller SL, Yan EB, Castillo-Melendez M, et al. Melatonin provides neuroprotection in the late-gestation fetal sheep brain in response to umbilical cord occlusion. Dev Neurosci 2005; 27: 200–210. [DOI] [PubMed] [Google Scholar]
- 196.Welin AK, Svedin P, Lapatto R, et al. Melatonin reduces inflammation and cell death in white matter in the mid-gestation fetal sheep following umbilical cord occlusion. Pediatr Res 2007; 61: 153–158. [DOI] [PubMed] [Google Scholar]
- 197.Juul SE, Aylward E, Richards T, et al. Prenatal cord clamping in newborn Macaca nemestrina: a model of perinatal asphyxia. Dev Neurosci 2007; 29: 311–320. [DOI] [PubMed] [Google Scholar]
- 198.Traudt CM, McPherson RJ, Bauer LA, et al. Concurrent erythropoietin and hypothermia treatment improve outcomes in a term nonhuman primate model of perinatal asphyxia. Dev Neurosci 2013; 35: 491–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.van der Kooij MA, Groenendaal F, Kavelaars A, et al. Neuroprotective properties and mechanisms of erythropoietin in in vitro and in vivo experimental models for hypoxia/ischemia. Brain Res Rev 2008; 59: 22–33. [DOI] [PubMed] [Google Scholar]
- 200.Debeljak N, Solar P, Sytkowski AJ. Erythropoietin and cancer: the unintended consequences of anemia correction. Front Immunol 2014; 5: 563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Wu YW, Mathur AM, Chang T, et al. High-dose erythropoietin and hypothermia for hypoxic-ischemic encephalopathy: a phase II trial. Pediatrics 2016; 137: pii: e20160191. [DOI] [PubMed] [Google Scholar]
- 202.Mulkey SB, Ramakrishnaiah RH, McKinstry RC, et al. Erythropoietin and brain magnetic resonance imaging findings in hypoxic-ischemic encephalopathy: volume of acute brain injury and 1-year neurodevelopmental outcome. J Pediatr 2017; 186: 196–199. [DOI] [PubMed] [Google Scholar]
- 203.Juul SE, Comstock BA, Heagerty PJ, et al. High-dose erythropoietin for asphyxia and encephalopathy (HEAL): a randomized controlled trial - background, aims, and study protocol. Neonatology 2018; 113: 331–338. [DOI] [PMC free article] [PubMed] [Google Scholar]