

Abstract
P-glycoprotein (PgP) is the major drug efflux pump in human cerebral microvessels. PgP prevents pathogens, toxins and therapeutic drugs from entering the CNS. Understanding the molecular regulation of PgP activity will suggest novel mechanisms to improve CNS drug delivery. Previously, we found that during peripheral inflammatory pain (PIP) (3 h after λ carrageenan injection in the rat paw), PgP traffics to the cortical microvessel endothelial cell plasma membrane concomitant with increased PgP activity. In the current study, we measured the changes in composition of PgP-containing protein complexes after PIP in rat microvessel isolates. We found that a portion of the PgP is contained in a multi-protein complex that also contains the caveolar proteins CAV1, SDPR, PTRF and PRKCDBP. With PIP, total CAV1 bound to PgP was unchanged; however, phosphorylated CAV1 (Y14P-CAV1) in the complex increased. There were few PgP/CAV1 complexes relative to total PgP and CAV1 in the microvessels suggesting CAV1 bound to PgP is unlikely to affect total PgP activity. However, both PgP and CAV1 trafficked away from the nucleus in response to PIP. These data suggest that P-CAV1 bound to PgP potentially regulates PgP trafficking and contributes to the acute PgP activity increase after a PIP stimulus.
Keywords: Blood–brain barrier, caveolin1, pain, p-glycoprotein, protein trafficking
Introduction
The blood–brain barrier (BBB) is a biochemical and physical barrier that selectively regulates passage of substances between the blood and the brain. The physical barrier consists of an extensive network of non-fenestrated capillaries where the endothelial cells surrounding the capillary lumen are tethered to each other via tight junctions.1 Tight junction integrity is regulated by pericytes, which surround the endothelial cells on the abluminal side of the capillaries.2 Signals from the astrocytes, neurons and microglia also contribute to tight junction regulation.1,2 The biochemical barrier consists of drug transporters, particularly the ATP-binding cassette efflux pumps located at the luminal endothelial cell plasma membrane, and metabolic enzymes.3 This combination of barrier properties limits paracellular and transcellular movement of substances from the blood into the brain. The BBB is necessary to prevent pathogens and toxins from entering the brain; however, it is a significant clinical challenge for the delivery of drugs to the brain to treat pathologies with a CNS component.3,4
P-glycoprotein (MDR1/ABCB1, E.C. 3.6.3.44) is the most abundant drug efflux pump in human BBB endothelial cells. P-glycoprotein (PgP) has a wide range of substrates including opioids, the major drug used for the treatment of pain.5,6 The efflux efficiency and abundance of PgP make it a major regulator and controller of CNS drug uptake. Many pathological conditions or treatments with PgP substrates increase PgP activity over basal levels amplifying the clinical challenge.6–8 Agents that directly inhibit PgP have not proven clinically viable because patients die of xenobiotic toxicity and infection.9,10 An alternative strategy with clinical potential is to identify signaling pathways that cause an acute increase or decrease in PgP activity at the BBB.4 Manipulation of these pathways would allow for short-term targeted CNS drug delivery without the toxicity of long-term PgP inhibition.
Two recent reviews summarize the current knowledge of PgP regulation at the BBB.4,6 As indicated by these reviews, conditions and signals that lead to chronic upregulation of PgP are the most well studied; mechanisms and signaling pathways that acutely regulate PgP activity are poorly understood. The Miller and Cannon laboratory has successfully used ex vivo incubation of isolated cortical microvessels to identify signals including vascular endothelial growth factor (VEGF), sphingosine-1P, ceramide 1P and lysophosphatidic acid that acutely regulate basal PgP activity.11–14 Our laboratory has used an in vivo acute peripheral inflammatory pain (PIP) model, 3 h after λ carrageenan injection into the hind paw, to study regulation of BBB integrity in response to a pain stimulus. The acute PIP response is characterized by increased paracellular leak accompanied by alterations in tight junction structure and membrane location.15,16 At 3 h, PgP activity is increased concomitant with decreased accumulation of morphine, an opioid pain therapeutic that is a PgP substrate, in the rat brain.5,17 Administering a nerve block16 or blocking PgP activity with cyclosporine A during the PIP stimulus enhances morphine uptake into the brain.5 The PIP stimulus causes a redistribution of PgP within membrane microdomains17 and trafficking of PgP from the nucleus to the plasma membrane of the endothelial cells.18
Based on the above data, we hypothesized that an acute increase in PgP activity is responsible for the decreased morphine uptake and efficacy we observe in this PIP model. The data suggest potential mechanisms by which PgP could acutely alter CNS morphine uptake in this model including: 1. An increase in the amount of PgP due to increased transcription or translation; 2. Trafficking of existing PgP molecules from intracellular reservoirs to the luminal plasma membrane so that more molecules are in a position to efflux drug into the blood; and 3. Activation of PgP molecules at the plasma membrane by a change in membrane microenvironment or proteins bound to PgP. To determine whether any of these mechanisms could contribute to the acute upregulation of PgP activity, we measured: the amount of PgP protein; components of the PgP microenvironment; PgP trafficking; and, in particular, PgP binding to and co-localization with caveolar proteins after a PIP stimulus because PgP binding to CAV1 and localization in caveolae have been reported by others to regulate PgP activity in in vitro models.19–22 We found that a portion of the PgP binds to caveolar proteins in a multi-protein complex. After a 3-h pain stimulus, the percentage of CAV1 in the complex that is phosphorylated increases. Both PgP and CAV1 decrease in the nucleus suggesting CAV1 may be involved in the PgP trafficking to the plasma membrane after PIP. Identification of the steps involved in PgP trafficking will provide novel drug targets to improve CNS drug delivery for PgP substrates.
Methods
Reagents
OptiPrep was purchased from Accurate Chemical (Westbury, NY). We obtained Criterion TGX gels, tris (2-carboxyethyl) phosphine hydrochloride, 4X Laemmli sample buffer, Precision Plus prestained molecular weight standards and Clarity Western ECL Substrate from Bio-Rad (Hercules, CA). Coomassie plus protein assay reagent and the pierce BCA protein assay kit were purchased from ThermoFisher (Waltham, MA). We obtained autoradiography film from ISC Bioexpress (Kaysville, UT). We used antibodies purchased from the following vendors: P-glycoprotein (MDR1) (sc8313) (Santa Cruz Biotechnology, Dallas, TX); GLUT1 (ab32551), caveolin1 (ab2910) and PTRF (ab48824) from AbCam (Cambridge, MA); P-caveolin1 (tyr14) (3244) and α-tubulin (2144) from Cell Signaling (Danvers, MA); P-caveolin1 (tyr14) (orb14814) from Biorbyt (Cambridge, UK); PRKCDBP (ProteinTech16250-1-AP) and SDPR (ProteinTech12339-1-AP) from ThermoFisher; and HRP-linked goat anti-rabbit and HRP-linked goat anti-mouse from GEHealthcare (Piscataway, NJ). All other chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise noted.
Animals and treatments
All animal protocols were written according to the National Institutes of Health guidelines for animal research and approved by the University of Arizona Institutional Animal Care and Use Committee. Results were reported according to the ARRIVE guidelines to the best of our ability. Female Sprague-Dawley rats (200–250 g; Envigo, Indianapolis, IN) were maintained under standard 12-h light/12-h dark conditions with food and water ad libitum. Three hours prior to sacrifice, 100 µl 0.9% NaCl or ʎ carrageenan (3% in 0.9% NaCl) was injected subcutaneously into the left hind paw. Animals were randomly assigned to treatment groups.
Cell culture
GPNT rat brain endothelial cells23,24 were a kind gift from Dr. Pierre Olivier-Couraud (INSERM, Paris, France). Cells were maintained in a 1:1 mixture of Ham's F12/ MEM alpha medium (Mediatech, Inc., Manassas, VA) with the following supplements: 10% fetal bovine serum, 2 mM L-glutamine, 100 IU penicillin and 100 µg/ml streptomycin (Mediatech, Inc.); 5 µg/ml transferrin, 5 µg/ml insulin and 5 ng/ml selenite (Sigma-Aldrich); and 2 ng basic Fibroblast Growth Factor (BD, San Diego, CA). Cells were grown on collagen I-coated flasks (ThermoFisher) at 37℃ in a humidified CO2 environment. Cells were subcultured every three to four days by incubating with 0.25% trypsin: 0.53 nM EDTA (Gemini Bioproducts, Woodland, CA) and then diluting the resulting suspension into new medium. Cells harvested for immunoprecipitation were ∼85% confluent and between passages 14 and 50.
Microvessel isolation and fractionation
Rat cortical microvessels were isolated and the lysates fractionated as previously described.25,26 Additional details are in supplementary methods.
Microscopy sample preparation and immunofluorescence staining
Formalin-fixed paraffin embedded (FFPE) sections were prepared and stained for fluorescence using the protocol accompanying the Nxgen Decloaking Chamber (Biocare Medical, Concord, CA) as previously described.18 The primary and AlexaFluor-labeled secondary antibodies used for this assay were: PTRF (ab48824) from AbCam; CAV1 (SAB4200216) from Sigma-Aldrich, (ab2910) from AbCam and (BD606100) from BD Biosciences (San Jose, CA); PgP (orb11267) from Biorbyt and (ab3366) from AbCam and AlexaFluor488 goat anti-rabbit (A-11008) and AlexaFLuor568 goat anti-mouse (A-11004) from Life Technologies (Carlsbad, CA). Slides were stored at 4℃ until analysis. Additional details are in supplementary methods.
Proximity ligation assay
FFPE sections were prepared for the proximity ligation assay (PLA) as for immunofluorescence staining. Primary antibodies were sequentially added to the slides and incubated for 1 h at room temperature in a humidified chamber. The PLA was performed using the Duolink In Situ – Fluorescence kit (Sigma-Aldrich) according to the manufacturer's protocol starting with the addition of the PLA probes. For this assay, we used the anti-mouse MINUS and anti-rabbit PLUS probes coupled with the green detection reagents and the in situ mounting medium with DAPI recommended by and purchased from the kit manufacturer. The primary antibodies used for this assay were the same as those used for immunofluorescence staining.
Microscopy
Images for brightfield and widefield fluorescence were acquired on a Leica DMI6000B inverted microscope (Leica Microsystems, Buffalo Grove, IL) equipped with a Leica DFC450 color CCD camera and a 16 bit Hamamatsu Flash 4.0 sCMOS camera (Hamamatsu Corp., Bridgewater, NJ). Images were acquired using Leica LAS X 1.x software (Leica). Co-localization analysis was done on images acquired using a DeltaVisionRT Deconvolution Microscopy System (GEHealthcare, Piscataway, NJ) which includes an inverted Olympus IX 70 microscope and color CCD camera that is coupled to SoftWorx software that controls the image acquisition and deconvolution. Negative control images were collected under the exact conditions used to collect positive images, however, no primary antibody was included in the staining protocol.
Cholesterol assay
Total cholesterol in each gradient fraction was quantified using the Invitrogen Amplex® Red Cholesterol Assay Kit (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. Additional details are in supplementary methods. Cholesterol values per fraction were normalized for the total cholesterol loaded onto each gradient.
Immunoprecipitation
Co-immunoprecipitations were done using the Pierce Co-immunoprecipitation kit (ThermoFisher) according to the manufacturer's protocol. Briefly, 10 µg antibody was linked to 25 µl resin. Samples were lysed in the kit lysis buffer and protein concentration measured using the Pierce BCA Protein Assay kit (ThermoFisher). The sample was pre-cleared using the control agarose resin and the sample incubated with the antibody-linked resin at 4℃ overnight. For comparison of SAL and CAR samples, equal protein was loaded on the antibody-linked resin column. Samples were eluted, protein measured, the proteins separated by SDS-PAGE and the proteins identified by immunoblotting. When SAL and CAR samples were compared, equal eluate protein was loaded on the gels. Additional details are in supplementary methods.
Immunoblots
Gradient fractions or the eluates from the co-immunoprecipitations were separated on SDS-PAGE gels, transferred to membranes and the proteins detected using standard methods.25 Bands were quantitated using the algorithm in FIJI.27 Additional details are in supplementary methods.
Microscopy image processing and analysis
Immunofluorescence images were acquired and analyzed as described in detail previously.18 Additional details are in supplementary methods.
Calculations and statistics
To determine whether more of protein X bound protein Y after treatment, we used the following equation: X(S)/X(C) divided by Y(S)/Y(C), where S = saline injection and C = carrageenan injection. Statistical difference was determined by calculating the 95% confidence interval. If the values between the mean plus and the mean minus the 95% confidence interval value did not contain 1, the means were considered significantly different (p = 0.05). Difference between means for the co-localization analysis was tested with the Student's t-test using the algorithms in Excel (Microsoft, Redmond, WA).
Results
Trafficking of PgP with caveolar proteins during PIP
One potential mechanism by which PgP activity could increase in the cortical microvessels is by increasing the amount of PgP. As shown in Figure 1(a) and (b) (and Supplementary Data), we did not see an increase in the amount of PgP in the microvessel isolates 3 h after a λ carrageenan injection in the paw compared to control animals. These data suggest that post-translational mechanisms are responsible for the observed increase in PgP activity after an acute pain stimulus.
Figure 1.

Two pools of PgP/caveolar proteins occur in membranes from rat brain microvessel isolates. (a) Representative immunoblot of total PgP and α-tubulin in microvessel isolates from saline-injected control (S) and ʎ carrageenan-injected (C) animals. (n = 4 pools of three rats). (b) Quantitation of PgP corrected for α-tubulin as a loading control in microvessel isolates from saline-injected control (SAL) and ʎ carrageenan-injected (CAR) animals. Values are the mean + SEM (n = 4 pools of three rats). (c) Total protein (closed circles), cholesterol (open circles) and density (closed diamonds) profile of the OptiPrep gradient fractions loaded with a sample of rat brain microvessel isolate. Values are the mean +/− SEM (n = 3 pools of three rats). (d) Representative immunoblots indicating the gradient fractions that contain specific proteins from samples from saline-injected control (SAL) and ʎ carrageenan-injected (CAR) animals (n = 3 pools of three rats). PgP: P-glycoprotein; CAV1: caveolin1; PTRF/cavin1: polymerase 1 and transcript release factor; PRKCDBP/cavin3: protein kinase C delta binding protein; SDPR/cavin2: serum deprivation response protein; GLUT1:glucose transporter1.
We previously showed that a portion of the PgP in rat cerebral microvessel endothelial cells moves from a nuclear location to the plasma membrane after a PIP stimulus.18 Within the plasma membrane in naïve animals, a portion of the PgP is located in caveolae, while some of it is not.28 The activity of PgP within the membrane depends in part on the lipid microenvironment. In cells, the presence of cholesterol has been linked to increased PgP activity.19,29 We used density gradients of cerebral microvessel isolates from control rats (SAL) and those that received a ʎ carrageenan injection in the left hind paw (CAR) to determine whether PgP co-localized with the caveolar proteins and cholesterol in the plasma membrane. As shown in Figure 1(c), there were two major pools of PgP in the density gradient fractions. These pools were similar in both control and treated animals. The two main fractions that contain PgP were also the fractions with the highest protein concentration and cholesterol content. Although the amount of cholesterol was the highest in the two fractions with the major amount of PgP, there was some in fractions of lower density where there was also a smaller amount of PgP. These lower density fractions correspond to those that contain lipid raft markers26 where cholesterol is reportedly less concentrated than in the caveolae, but at a higher concentration than in the rest of the plasma membrane. The least dense fractions, which are at the top of the gradient, also contained a small amount of cholesterol. Free cholesterol, not in plasma membrane structures, is expected to fractionate there.
When we probed for a marker of endothelial cell plasma membrane (GLUT1), we found that this protein cofractionated with PgP in microvessel isolates from both the control and ʎ carrageenan-injected animals. This indicates that the main pools of PgP we observe are in the plasma membrane.
The fractions that contained the major portion of PgP also contained the caveolar proteins, CAV1, PTRF/cavin1 (polymerase 1 and transcript release factor), SDPR/cavin2 (serum deprivation response protein) and PRKCDBP/cavin3 (protein kinase C delta binding protein). This indicates that there are two caveolar/PgP populations that fractionate at different densities in our cerebral microvessel isolates. We did not see a difference in the location of the caveolar proteins in the gradients 3 h after ʎ carrageenan injection although there was a trend toward more of the caveolar proteins in the higher density PgP-containing fraction after PIP. These data suggest that PgP traffics to cholesterol rich membrane regions after acute pain.
An increase in density in the PgP/caveolar protein complexes is indicative of a change in either the protein or lipid composition of the complexes. We have, in the current study and previously, characterized some of the proteins in these two peaks and found no difference in protein content.25 Peak I contained approximately 18% of the total protein and approximately 18% of the total cholesterol (Figure 1(c)). In contrast, Peak II, which is denser, had approximately 13% of the total protein and 8% of the total cholesterol. The different ratio of protein to cholesterol suggests the lipid composition of the lipoprotein complexes in these two peaks differs which could contribute to the observed difference in densities.
PgP forms protein complexes with the caveolar proteins
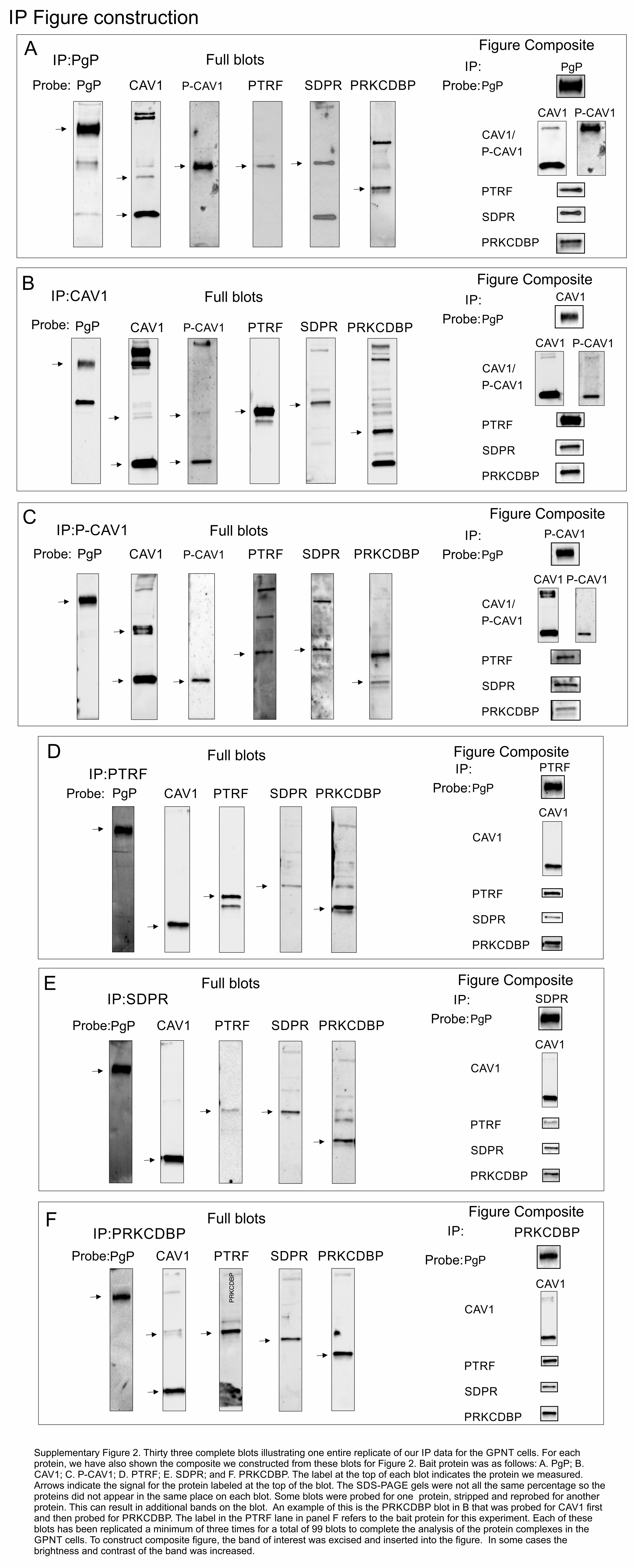
Based on the data that show that caveolar proteins co-fractionate with PgP, we tested whether these proteins could be found in a protein complex with PgP. The first step was to determine whether the caveolar proteins co-immunoprecipitated with PgP in GPNT cells and microvessel isolates. As shown in Figure 2 (Supplementary Figure 2), an immunoprecipitation for PgP in GPNT cells, which are a pure rat brain endothelial cell culture model, resulted in the co-immunoprecipitation of caveolin1, some of which is phosphorylated at Tyr14 (Y14P-CAV1). Using either CAV1 or Y14P-CAV1 as bait resulted in the co-immunoprecipitation of PgP. In the GPNT cells, when PgP was used as bait, most of the CAV1 appeared at ∼21 kDa on the blot, where monomers are expected to appear, with a faint CAV1 immunoreactive band at ∼50 kDa. In other types of cell culture cells, the ∼50 kDa band has been identified as a CAV1 dimer.30,31 When the blots were probed with an antibody for Y14P-CAV1, the majority of the signal was contained in the ∼50 kDa band. When CAV1 was used as the bait, most of the CAV1 appeared at ∼21 kDa. There was a faint ∼50 kDa band. Probing these blots for Y14P-CAV1 indicated that the major P-CAV1 band also appeared at ∼21 kDa. Use of alternate CAV1 and Y14P-CAV1 antibodies yielded the same result indicating that both bands contain CAV1 and Y14P-CAV1 immunoreactive protein. Similar to the data for CAV1, when P-CAV1 was used as bait, most of the CAV1 and P-CAV1 appeared at ∼21 kDa. A long exposure showed a minor band at ∼50 kDa. These data suggest that a higher molecular weight complex containing P-CAV1 is the predominant P-CAV1 form that binds PgP, but that this is only a small portion of the total P-CAV1.
Figure 2.

PgP is a component of a multi-protein complex in GPNT cells and rat brain microvessel isolates. (a) Representative blot of PgP and caveolar protein co-immunoprecipitations from GPNT cells probed for P-glycoprotein (PgP); caveolin1 (CAV1); Y14 phospho-caveolin1 (P-CAV1); polymerase I and transcript release factor (PTRF); serum deprivation-response protein (SDPR); protein kinase C delta binding protein (PRKCDBP). (n = 3 or 4). (b) Representative blot of PgP and caveolar protein co-immunoprecipitations probed for PgP and caveolar proteins in microvessel isolates from control animals. (n ≥ 3 pools of 6 animals (minimum total of 18 animals) per treatment).
Using PgP as bait, we found that other caveolar proteins, PTRF, SDPR and PRKCDBP, co-immunoprecipitated with the PgP. Immunoprecipitation using PTRF, SDPR and PRKCDBP as the bait resulted in the co-immunoprecipitation of PgP (Figure 2, Supplementary Figure 2). Each of the caveolar proteins used as bait co-immunoprecipitated the other caveolar proteins (Figure 2, Supplementary Figure 2). When we probed the blots for CAV1, most of the protein appeared at 21 kDa with a faint band at 50 kDa in the eluates from the SDPR and PRKCDBP co-immunoprecipitation reactions. We were unable to detect P-CAV1 in the eluates from the PTRF, SDPR and PRKCDBP reactions. The ability to use PgP and each of the caveolar proteins as bait to co-immunoprecipitate each of the other proteins suggests that the cells contain a multi-protein complex that contains all these components.
PgP is contained in caveolar protein complexes in microvessels isolated from rat brains
A CAV1/PgP protein complex has been documented in rat microvessel isolates;11 however, whether a complex containing PgP and the other caveolar proteins exists in rat microvessels or whether this complex changes with PIP is unknown. As shown in Figure 2, using PgP as bait co-immunoprecipitated CAV1, Y14P-CAV1, PTRF, SDPR and PRKCDBP from the microvessel isolate lysates from control animals. Similar to the data from the GPNT cells, most of the co-immunoprecipitated CAV1 appeared at ∼21 kDa; however, there was also a higher molecular weight band, ∼50 kDa, which contained CAV1 immunoreactive protein. Most of the Y14P-CAV1 that immunoprecipitated with PgP was in the higher molecular weight form. Using the caveolar proteins as bait resulted in the co-immunoprecipitation of PgP and each of the other caveolar proteins. These data suggest that a portion of the PgP is contained in a multi-protein complex in isolated microvessels similar to what we observed in GPNT cells in vitro. Similar findings in the microvessel isolates, which are enriched for endothelial cells, but contain multiple cell types, to GPNT cells suggests that the complexes are characteristic of in vivo endothelial cells and that the GPNT cells could provide a model to further investigate these complexes.
Increased P-CAV1 is bound to PgP after a PIP stimulus
To determine whether the PgP-caveolar protein complex was altered after a PIP stimulus, we measured the relative amount of each of the caveolar proteins that co-immunoprecipitated with PgP in control and treated animals. As shown in Figure 3(a), using PgP as bait resulted in the co-immunoprecipitation of the other caveolar proteins in animals 3 h after a PIP stimulus. Using the immunoblot data, we calculated whether more or less of each caveolar protein was contained in the complex with PgP. We first normalized the amount of each caveolar protein to the amount of PgP immunoprecipitated, then calculated the relative amount in the saline- compared to the λ carrageenan-treated animals. We found that there was no change in the amount of the caveolar proteins that co-immunoprecipitated with PgP 3 h after a PIP stimulus (Figure 3(b)). The Y14P-CAV1 immunoreactive protein bound to PgP was in the ∼50 kDa band in samples from both control and treated animals. There was a significant increase in the Y14P-CAV1 bound to PgP in the CAR-treated animals (p ≤ 0.05).
Figure 3.

P-caveolin1 bound to PgP increases with peripheral inflammatory pain. (a) Representative blots indicating the amount of PgP and the caveolar proteins that co-immunoprecipitate with PgP in microvessels isolated from animals 3 h after injection with saline (S) or ʎ carrageenan (C). (b) Quantitation of the amount of caveolar proteins that co-immunoprecipitate with PgP in microvessels isolated from saline control (SAL) and ʎ carrageenan- (CAR) injected animals. (c) Representative blots indicating the amount of PgP and the caveolar proteins that co-immunoprecipitate with CAV1 in microvessels isolated from animals 3 h after injection with saline (S) or ʎ carrageenan (C)(d) Quantitation of the amount of PgP and caveolar proteins that co-immunoprecipitate with CAV1 in microvessels isolated from saline control (SAL) and ʎ carrageenan (CAR) injected animals. For quantitation, values are the absolute value of 1 – mean relative amount of each protein corrected for the amount of bait protein detected in the sample calculated using the formula in the methods (n ≥ 3 pools of six rats – minimum total = 18 rats for saline: n ≥ 3 pools of nine rats – minimum total = 27 rats for carrageenan). Values were considered significant if the 95% confidence interval did not include 1.
Using CAV1 as bait (Figure 3), we co-immunoprecipitated PgP and the other caveolar proteins. We saw no change in the amount of any of the proteins bound to CAV1 with PIP; however, there was a significant increase in Y14P-CAV1 in the ∼21 kDa band after PIP. Taken together, these data suggest that there is no change in the amount of CAV1 bound to PgP after treatment, but a larger portion of the bound CAV1 is phosphorylated in the treated samples. Although we could detect SDPR bound to both PgP and CAV1, the amounts were too low for accurate quantitation; qualitatively, there was no difference in the SDPR signal in microvessel isolates from control and treated animals.
Location of PgP/CAV1 complexes in the microvessel cross section
Our data showing that PIP increased Y14P-CAV1 bound to PgP concomitant with an increase in PgP activity was unexpected. In cell culture cells, binding of Y14P-CAV1 to PgP is correlated with decreased PgP activity.20 In rat cortical microvessels, decreased PgP activity also correlates to increased Y14P-CAV1 bound to PgP after increased VEGF signaling.11 As a first step toward understanding this paradox, we examined the location and frequency of the complexes in the microvessels. The co-immunoprecipitation data indicate that PgP/CAV1 complexes occur in the microvessel isolates which are an enrichment for microvessel endothelial cells, but also contain pericytes and some additional cellular material from the cortex. To identify the subcellular location of these complexes, we used a PLA to detect complexes in FFPE sections of microvessel isolates. Figure 4(a) shows a cross section of a representative H & E-stained microvessel and indicates the structures we identified in the cross section. We saw endothelial cells surrounding a lumen and pericytes surrounding the endothelial cells. Figure 4(b) shows staining of the microvessels for each pair of antibodies we used for the PLA. When we stained the slides for PgP and CAV1, we saw that there was some PgP in the nucleus, some at the endothelial cell luminal membrane and some in the pericytes (Figure 4(b)). Lower intensity staining for PgP was seen in the cytosol. CAV1 was primarily located at the membrane; however, lower intensity staining could also be seen in the nucleus and cytosol. Pericytes also had CAV1 staining, particularly at the membrane. When the channels were merged, there was colocalization of some of the signal particularly at the membrane, but the overlap was not complete and patches of red signal and green signal could still be identified. As a control, we also stained for CAV1 and PTRF proteins which associate with each other in caveolae in cell culture cell models.32 PTRF and CAV1 staining was mostly at the lumen and the overlap of the signals occurred frequently.
Figure 4.
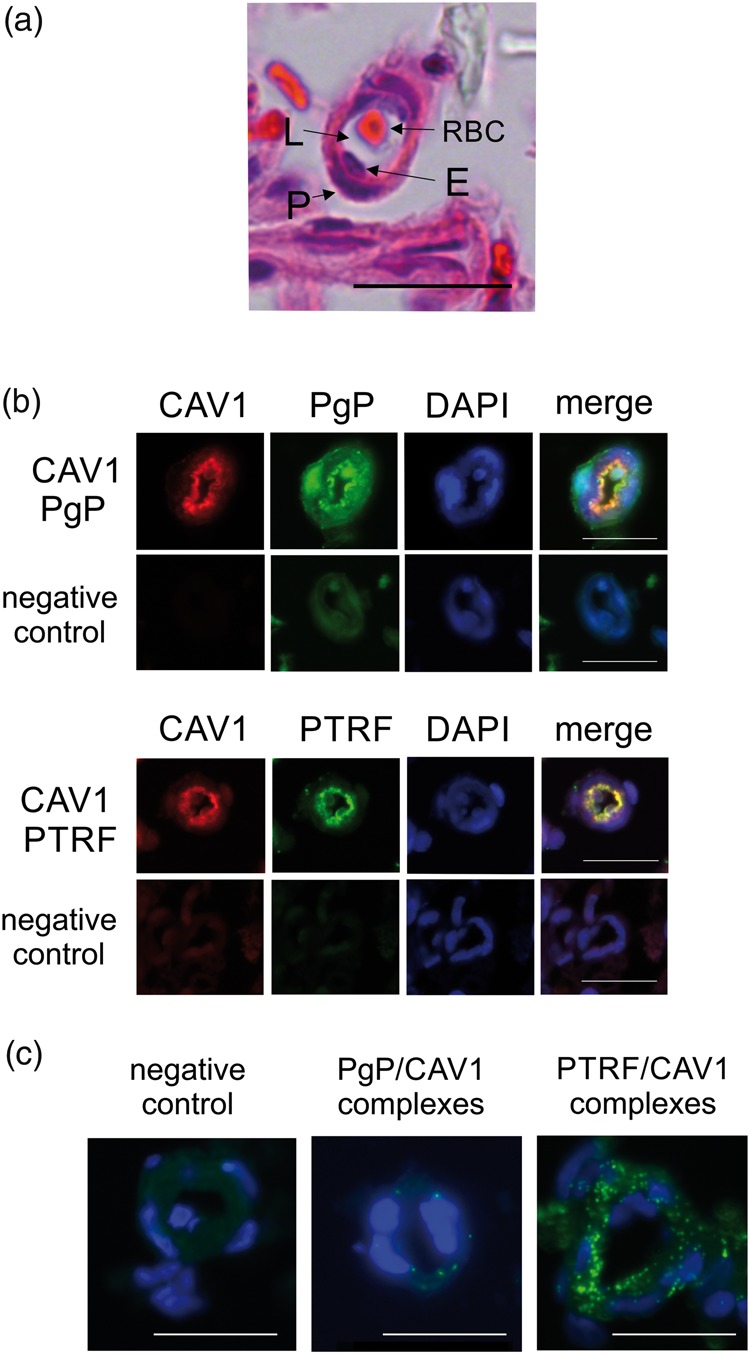
Few P-glycoprotein/caveolin1 complexes occur in rat brain microvessels. (a) Brightfield image of a cross section of a rat brain microvessel stained with H & E showing the endothelial cells (E) surrounding the vessel lumen (L) and pericytes (P) surrounding the endothelial cells. A red blood cell (RBC) sits in the lumen. (b) Representative immunofluorescence images of dual stains for P-glycoprotein (PgP) (green) or PTRF (green) and caveolin1 (CAV1) (red) in cross-sections of rat brain microvessels. The negative control shows fluorescence in the absence of primary antibody. DAPI (blue) counterstain indicates nuclei. (c) Representative proximity ligation assay results for PgP/ CAV1 binding and PTRF/ CAV1 binding (green signals) in cross sections of rat brain microvessels. Blue staining indicates nuclei. The negative control image shows the background reaction of the PLA reagents in the absence of antibodies. The bar represents 20 µm. Images were acquired from at least three pools of three rats.
The PLA showed numerous PTRF/CAV1 complexes, many of them located at the lumen of the microvessel, although there were some elsewhere in the microvessel cross section (Figure 4(c)). We could detect some signal above background for PgP/CAV1 complexes although we were only able to measure a few complexes. Occasionally a complex was located at the vessel lumen, but most of the signal was not at the lumen which could represent nuclear, cytosolic and abluminal complexes. Alternatively, the signal could be from the pericytes. We confirmed our results using a different PgP/CAV1 antibody combination (data not shown). A qualitative assessment indicated that we did not see a different number of complexes in the samples from saline- and ʎ carrageenan-treated animals; however, the paucity of signals precluded meaningful quantitative measurements. Taken together, these data suggest that there is a low percentage of the total PgP and CAV1 bound together in a complex in the microvessels and the complexes are not predominantly luminal.
Trafficking of PgP with CAV1
The paucity of complexes in the microvessels suggests that binding of Y14P-CAV1 to PgP is unlikely to regulate overall PgP activity at the lumen. In addition to caveolar formation, CAV1 regulates trafficking of several proteins in the cell.33–35 Previously, we found that PgP traffics from the nucleus to the plasma membrane with a PIP stimulus.18 We hypothesized that CAV1 binding to PgP could contribute to the PgP trafficking from the nucleus to the plasma membrane. As a first step toward testing this hypothesis, we measured the co-localization of PgP and CAV1 and measured the relative amount of each of these proteins in the nucleus in control and ʎ carrageenan-injected animals. As shown in the deconvolved images of immunostained control microvessels (Figure 5(b)), there was a large amount of PgP in the nucleus. After a PIP stimulus, the PgP decreased in the nucleus (Figure 5(b) and (c)). Most of the caveolin1 was located on the membrane; however, a small amount of staining overlapped with the DAPI (nuclear) signal. After a PIP stimulus, there was a significant loss of the PgP and CAV1 signals from the nucleus. The co-localization of the PgP and CAV1 was approximately 45% before PIP and remained the same after treatment.
Figure 5.
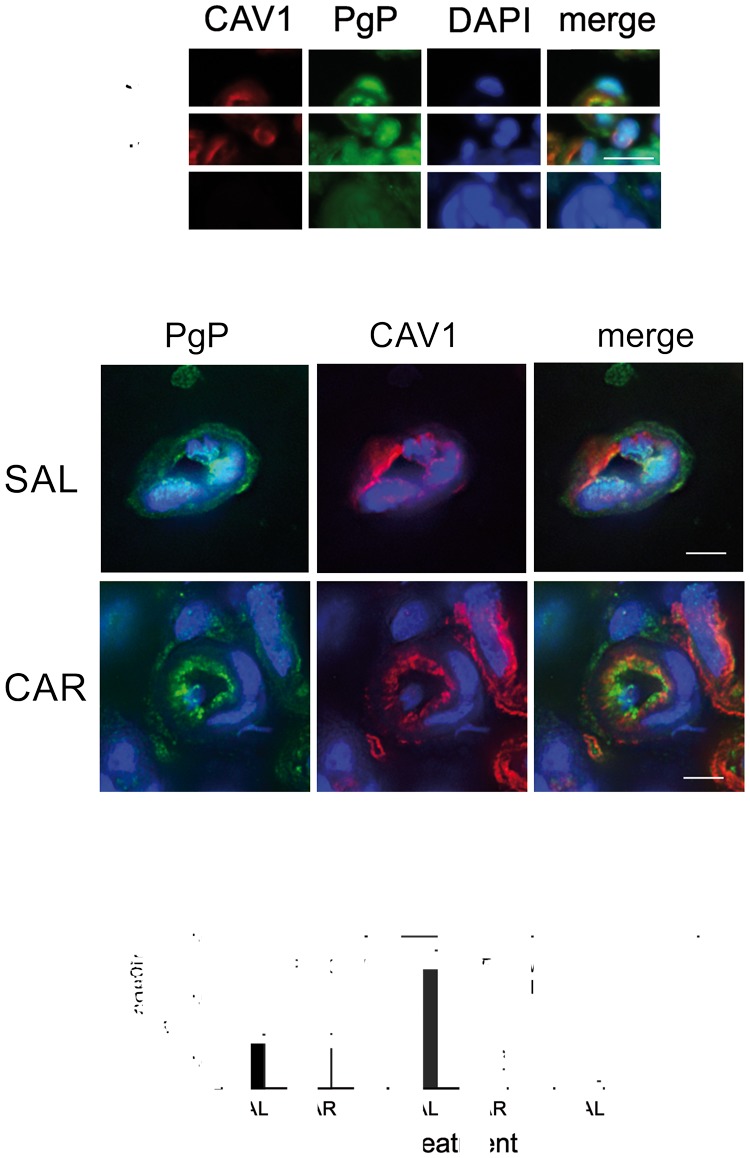
PIP decreases nuclear PgP and CAV1. (a) Representative immunofluorescence images of dual stains for P-glycoprotein (PgP) (green) or and caveolin1 (CAV1) (red) in cross-sections of rat brain microvessels. Negative control shows fluorescence in the absence of primary antibody. DAPI (blue) counterstain indicates nuclei. Bar represents 20 µm. (b) Representative MAX intensity deconvolved images indicating the location of PgP (green) and CAV1 (red) in isolated microvessels from saline-injected (SAL) and ʎ carrageenan-injected (CAR) animals. DAPI (blue) counterstain indicates nuclei. Bar represents 5 µm. (c) Co-localization analysis of PgP with CAV1 and with DAPI (nuclear stain) in control (SAL) and ʎ carrageenan (CAR) injected animals. (n = 9–11 images from three pools of three animals). *Indicates significantly different from controls (p ≤ 0.05).
Discussion
Our data suggest that post-translational mechanisms including protein trafficking and changes in protein complexes are responsible for the acute increase in PgP activity in our PIP model. We found that a portion of the PgP in GPNT cells and rat brain microvessel isolates is contained in a multi-protein complex that includes several caveolar proteins. We see a similar amount of PgP in these complexes 3 h after a PIP stimulus; however, there is a significant increase in the phosphorylation of CAV1 (Y14P) within the complex. Our data also suggest that of the total PgP and CAV1 molecules within the microvessels, only a small percentage is bound in a complex. The complexes are not found in abundance at the luminal surface of the endothelial cells. The scarcity and lack of luminal location of the complexes indicate that CAV1 binding to PgP would have little effect on overall PgP activity. Our co-localization analysis is consistent with an alternative function of the CAV1 bound to PgP, redistribution of PgP within the cells during PIP. We propose a new model (Figure 6) whereby phosphorylation of CAV1 in a PgP/CAV1 complex is a signal for PgP trafficking from the nucleus to the plasma membrane within BBB endothelial cells. Dissociation of CAV1 from (or dephosphorylation of CAV1 within) the complex at the destination would then allow for local regulation of PgP activity by additional signals and microenvironmental factors.
Figure 6.

Model of PgP trafficking in response to PIP. At baseline, PgP is partially distributed in the nucleus, the caveolae and the non-caveolar portions of the membrane. Upon a PIP stimulus, phosphorylation of CAV1 (a) results in the trafficking of PgP in vesicles from the nucleus to the plasma membrane (b). (c) At the plasma membrane, PgP becomes activated (depicted by arrow) and increases drug efflux into the capillaries. Movement of PgP from the caveolae into lipid rafts in the plasma membrane may also occur.
A portion of the PgP in microvessels is located in caveolae. It co-localizes with other caveolar proteins and GLUT1 which is a marker of endothelial cell plasma membranes. This is consistent with electron microscopy images of cortical microvessels from naïve rats that show a portion of the total PgP is in the caveolae.28 We identified two different caveolar populations in the plasma membrane that fractionate at different densities. The different densities of the caveolar protein complexes may be due to different protein components or the lipid composition of the complex. Our cholesterol measurements indicate that a lower percentage of the total cholesterol is contained in the denser fraction that contains caveolar proteins. Previous studies indicate that the lipid composition of the microenvironment containing PgP impacts PgP activity.21,22 In particular, higher cholesterol, which influences membrane fluidity, is correlated with higher activity. Caveolae and lipid rafts both are richer in cholesterol than other portions of the plasma membrane.36 PgP traffics to the endothelial cell plasma membrane in response to PIP.18 The PgP protein in the membrane fractions has the same pattern in control and treated animals suggesting that the PgP is trafficking to caveolae. The PgP could also then distribute to lipid rafts which we would not detect using this analysis.
A portion of the PgP is bound to CAV1. Previous studies have shown that CAV1 directly binds PgP as we see in our study.11,19,20 We also see that other caveolar proteins co-immunoprecipitate with PgP from GPNT cells and the rat microvessel isolates. Co-immunoprecipitation of other caveolar proteins, PTRF, SDPR and PRKCDBP, when PgP is used as bait indicates either that these proteins bind PgP or that they bind another protein that binds to PgP. Each of PTRF, SDPR and PRKCDBP are known to bind directly to CAV1 or to each other in cell culture cells.37 Our immunoprecipitation protocol is detergent-free; therefore, membrane lipids will not be removed from the complexes prior to binding to the protein bait. Some of the interactions between proteins in the complex may be lipid dependent or enhanced.32,37 For example, the amount of PTRF bound to CAV1 depends on the amount and species of membrane lipids present.32,37 Whether each of these proteins bind PgP directly and whether the complex is lipid dependent remains to be determined. However, our ability to co-immunoprecipitate all the other caveolar proteins and PgP no matter which protein is used as bait, suggests that a portion of the PgP in microvessels is bound in a multi-protein complex. This multi-protein complex may be contained within the caveolae or elsewhere in the cells. A multi-protein complex of this type could serve trafficking and regulatory functions.
Of the total PgP and CAV1, our data suggest that only a few PgP/CAV1 complexes exist in the microvessels. Others have estimated that in endothelial cell tissue culture, models between 3 and 5% of the molecules exist in a complex.19,38 Careful analysis of fluorescence and deconvolution microscopy images suggests that there is not particularly good overlap of the signal for these proteins; however, both show overlap with signal for GLUT1 in the plasma membrane.18 Analysis of co-localization by microscopy has limitations; molecules that are bound in a complex may not show signal overlap due to distortion of the tissue, steric hindrance of antibodies and fluorescent labels and image resolution. The PLA assay, which produces a positive signal if the epitopes are within 40 nm of each other, results in a few positive signals. The most likely explanation of our fluorescence data combined with the PLA results is that, although both proteins are in the plasma membrane and even in the caveolae, there are very few of the molecules bound to each other. More advanced analytical techniques are required to measure this more precisely.
Our data indicate that there is an increase in the Y14P-CAV1 bound to PgP after PIP, but no change in the total PgP/CAV1 complexes. In cell culture cells, siRNA-mediated knockdown of CAV1 increased PgP activity.20 Overexpression of CAV1 decreased PgP activity, while overexpression of tyrosine phosphorylation deficient mutant CAV1 did not. Activity changes after these manipulations were modest which is consistent with a low percentage of PgP/CAV1 complexes in the cell. In the complexes we observed in the microvessels, the increased Y14P-CAV1 runs at ∼50 kDa. In cell culture cells, this was identified as a dimer of CAV1 that was more tightly bound and difficult to dissociate.30,31 The higher weight molecular complexes were particularly prevalent in the nucleus. These data fit with the PgP bound to CAV1 being in a difficult to dissociate complex that would inhibit activity. However, since there are few complexes in the microvessels, it is unlikely that this would affect overall PgP activity.
In rat brain microvessels, an increase in Y14P-CAV1 bound to PgP occurs both under conditions where PgP activity increases5,17 and where PgP activity decreases.11 Taken together these data suggest that Y14P-CAV1 bound to PgP is not regulating overall activity. Particularly considering that there are few complexes relative to the overall amount of PgP in the cells. What both the above processes have in common is that the signals cause increased trafficking of PgP.17,18,39 In the case of PIP, the PgP moves to the plasma membrane from internal stores18 and in the case of VEGF signaling, the PgP moves away from the plasma membrane.11 Phosphorylation of CAV1 could be a signal for redistribution of PgP in the microvessels in response to external signals. In the cell culture cells where CAV1 manipulation results in altered PgP activity, PgP trafficking was not measured.20 Modulation of PgP trafficking could contribute to the changes in PgP activity reported in that study.
There is precedent for CAV1 acting in a trafficking capacity. Numerous studies have linked CAV1 to vesicular trafficking and redistribution of other proteins within the cell. The best characterized trafficking pathway is phosphorylation of CAV1 (Y14P) by Src which loosens CAV1 molecules in the caveolar coat, causes constriction of the neck of the caveolae and the internalization of vesicles to increase trafficking away from the caveolae.33,35 Vesicles are subsequently sorted and directed to various intracellular destinations; this pathway is frequently used to internalize membrane-bound receptors. Caveolin-mediated internalization of PgP is consistent with the data by Hawkins et al.11,39 which showed that VEGF decreases PgP activity and proteolytic access to PgP from the luminal plasma membrane side of the microvessels. During PIP, however, there is increased trafficking of PgP and CAV1 away from the nucleus concomitant with increased PgP molecules and activity at the luminal surface of the endothelial cells.5,17,18 Several studies in cell culture cells indicate that CAV1 (particularly Y14P-CAV1) participates in other less well characterized intracellular trafficking events. These include: movement of PgP to caveolae in breast cancer cells;34 endosomal progression, but not internalization, of EGFR;40 targeting of Src to focal adhesions;41,42 long range trafficking of proteins on microtubules;43 and perinuclear accumulation and movement of proteins to and from nuclear membranes.30,43,44 Our data suggest the possibility that CAV1 participates in PgP trafficking away from the nucleus. CAV1 could also be responsible for movement of PgP from the caveolae to other microdomains in the plasma membrane such as lipid rafts. Whether CAV1 participates in PgP trafficking after PIP and whether Src signaling is involved remains to be determined. Our data suggest that CAV1 has a more complex and critical role in PgP trafficking in cortical microvessels than previously assumed.
Binding of PgP in a complex with the other caveolar proteins in addition to CAV1 suggests some other trafficking possibilities. PRKCDBP is the protein responsible for vesicular trafficking to and away from the caveolae.37 We do not see an increase in PgP bound in the caveolar protein complex with PIP. This would be consistent with PgP traveling to locations in the plasma membrane during PIP and then dissociating from the complex. Protein trafficking to and from caveolae is a rapid and dynamic process. We are using a single snapshot at 3 h post-λ carrageenan injection to examine this process. Other timepoints could yield additional information. Recent data using Brefeldin A treatment of ex vivo microvessels to block vesicular trafficking from the endoplasmic reticulum to the Golgi resulted in the inability of a ceramide 1P signal to increase PgP activity at the microvessel lumen.13 The authors did not specifically measure PgP trafficking in this study; however, trafficking of PgP from endoplasmic reticulum stores is one explanation of their data. Trafficking of mature PgP from the endoplasmic reticulum to the microvessel lumen in response to PIP may also occur. This would be below our limits of detection using our experimental protocol because of the greater amount of PgP trafficking from the nucleus. Whether multiple trafficking pathways contribute to the upregulation of PgP activity at the capillary lumen in response to PIP remains to be tested.
Taken together, these data are consistent with Y14P-CAV1 participating in PgP trafficking in response to PIP. An ability to acutely regulate PgP at the plasma membrane such that the basal level remains intact, but the acute increase due to PIP is blocked has clinical application for the treatment of acute pain. For example, post-surgical pain can be difficult to treat resulting in an opioid dose escalation. Blocking the acute activity increase due to the pain would decrease the opioid dose needed to inhibit the post-surgical pain. Identification of the signals responsible for the trafficking of PgP to the plasma membrane will indicate targets for drug development that prevent the acute PgP activity increase. Our finding that Y14P-CAV1 bound to PgP increases with PIP concomitant with increased PgP activity suggests that phosphorylation of CAV1 could be responsible for both the increase in PgP trafficking and subsequent activity at the plasma membrane. These data also suggest a PgP regulatory pathway that has potential as a novel clinical target to modulate PgP activity and enhance CNS drug delivery for the treatment of pathologies with a CNS component.
Supplemental Material
Supplemental material, Supplemental material1 for Acute pain alters P-glycoprotein-containing protein complexes in rat cerebral microvessels: Implications for P-glycoprotein trafficking by Margaret E Tome, Chelsea K Jarvis, Charles P Schaefer, Leigh M Jacobs, Joseph M Herndon, Kristen C Hunn, Nathan B Arkwright, Kathryn L Kellohen, Peyton C Mierau and Thomas P Davis in Journal of Cerebral Blood Flow & Metabolism
Supplemental Material
{kind=link}
Supplemental material, Supplemental material2 for Acute pain alters P-glycoprotein-containing protein complexes in rat cerebral microvessels: Implications for P-glycoprotein trafficking by Margaret E Tome, Chelsea K Jarvis, Charles P Schaefer, Leigh M Jacobs, Joseph M Herndon, Kristen C Hunn, Nathan B Arkwright, Kathryn L Kellohen, Peyton C Mierau and Thomas P Davis in Journal of Cerebral Blood Flow & Metabolism
Supplemental Material
Supplemental material, Supplemental material3 for Acute pain alters P-glycoprotein-containing protein complexes in rat cerebral microvessels: Implications for P-glycoprotein trafficking by Margaret E Tome, Chelsea K Jarvis, Charles P Schaefer, Leigh M Jacobs, Joseph M Herndon, Kristen C Hunn, Nathan B Arkwright, Kathryn L Kellohen, Peyton C Mierau and Thomas P Davis in Journal of Cerebral Blood Flow & Metabolism
Acknowledgements
We thank Doug Cromey, MS, and Monika Schmelz, PhD, for advice and expert assistance with the microscopy; and Dr. Pierre Olivier-Couraud for the kind donation of the GPNT cells.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funding support for this study comes from: DA 011271 and NS 042652 to TPD; and Cancer Center Core Grant (P30-CA23074) support of TACMASR.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors' contributions
Study concept and design (MET, TPD); Data acquisition and analysis (MET, CKJ, JMH, CPS, LMJ, KCH, NBA, KLK, PCM); Data interpretation (MET, JMH, CPS, TPD); wrote the manuscript (MET).
Supplementary material
Supplementary material for this paper can be found at the journal website: http://journals.sagepub.com/home/jcb
References
- 1.Abbott NJ, Patabendige AA, Dolman DE, et al. Structure and function of the blood-brain barrier. Neurobiol Dis 2010; 37: 13–25. [DOI] [PubMed] [Google Scholar]
- 2.Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood-brain barrier. Nat Med 2013; 19: 1584–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Borst P, Elferink RO. Mammalian ABC transporters in health and disease. Annu Rev Biochem 2002; 71: 537–592. [DOI] [PubMed] [Google Scholar]
- 4.Miller DS, Cannon RE. Signaling pathways that regulate basal ABC transporter activity at the blood- brain barrier. Curr Pharm Des 2014; 20: 1463–1471. [DOI] [PubMed] [Google Scholar]
- 5.Seelbach MJ, Brooks TA, Egleton RD, et al. Peripheral inflammatory hyperalgesia modulates morphine delivery to the brain: a role for P-glycoprotein. J Neurochem 2007; 102: 1677–1690. [DOI] [PubMed] [Google Scholar]
- 6.Miller DS. Regulation of P-glycoprotein and other ABC drug transporters at the blood-brain barrier. Trends Pharmacol Sci 2010; 31: 246–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ambudkar SV, Kim IW, Sauna ZE. The power of the pump: mechanisms of action of P-glycoprotein (ABCB1). Eur J Pharm Sci 2006; 27: 392–400. [DOI] [PubMed] [Google Scholar]
- 8.Loo TW, Bartlett MC, Clarke DM. Permanent activation of the human P-glycoprotein by covalent modification of a residue in the drug-binding site. J Biol Chem 2003; 278: 20449–20452. [DOI] [PubMed] [Google Scholar]
- 9.Thomas H, Coley HM. Overcoming multidrug resistance in cancer: an update on the clinical strategy of inhibiting p-glycoprotein. Cancer Control 2003; 10: 159–165. [DOI] [PubMed] [Google Scholar]
- 10.Liang XJ, Aszalos A. Multidrug transporters as drug targets. Curr Drug Targets 2006; 7: 911–921. [DOI] [PubMed] [Google Scholar]
- 11.Hawkins BT, Sykes DB, Miller DS. Rapid, reversible modulation of blood-brain barrier P-glycoprotein transport activity by vascular endothelial growth factor. J Neurosci 2010; 30: 1417–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cannon RE, Peart JC, Hawkins BT, et al. Targeting blood-brain barrier sphingolipid signaling reduces basal P-glycoprotein activity and improves drug delivery to the brain. Proc Natl Acad Sci U S A 2012; 109: 15930–15935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mesev EV, Miller DS, Cannon RE. Ceramide 1-phosphate increases P-glycoprotein transport activity at the blood-brain barrier via prostaglandin E2 signaling. Mol Pharmacol 2017; 91: 373–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Banks DB, Chan GN, Evans RA, et al. Lysophosphatidic acid and amitriptyline signal through LPA1R to reduce P-glycoprotein transport at the blood-brain barrier. J Cereb Blood Flow Metab 2017; 38: 857–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huber JD, Hau VS, Borg L, et al. Blood-brain barrier tight junctions are altered during a 72-h exposure to lambda-carrageenan-induced inflammatory pain. Am J Physiol Heart Circ Physiol 2002; 283: H1531–H1537. [DOI] [PubMed] [Google Scholar]
- 16.McCaffrey G, Seelbach MJ, Staatz WD, et al. Occludin oligomeric assembly at tight junctions of the blood-brain barrier is disrupted by peripheral inflammatory hyperalgesia. J Neurochem 2008; 106: 2395–2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCaffrey G, Staatz WD, Sanchez-Covarrubias L, et al. P-glycoprotein trafficking at the blood-brain barrier altered by peripheral inflammatory hyperalgesia. J Neurochem 2012; 122: 962–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tome ME, Herndon JM, Schaefer CP, et al. P-glycoprotein traffics from the nucleus to the plasma membrane in rat brain endothelium during inflammatory pain. J Cereb Blood Flow Metab 2016; 36: 1913–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jodoin J, Demeule M, Fenart L, et al. P-glycoprotein in blood-brain barrier endothelial cells: interaction and oligomerization with caveolins. J Neurochem 2003; 87: 1010–1023. [DOI] [PubMed] [Google Scholar]
- 20.Barakat S, Demeule M, Pilorget A, et al. Modulation of p-glycoprotein function by caveolin-1 phosphorylation. J Neurochem 2007; 101: 1–8. [DOI] [PubMed] [Google Scholar]
- 21.Vilas-Boas V, Silva R, Nunes C, et al. Mechanisms of P-gp inhibition and effects on membrane fluidity of a new rifampicin derivative, 1,8-dibenzoyl-rifampicin. Toxicol Lett 2013; 220: 259–266. [DOI] [PubMed] [Google Scholar]
- 22.Troost J, Lindenmaier H, Haefeli WE, et al. Modulation of cellular cholesterol alters P-glycoprotein activity in multidrug-resistant cells. Mol Pharmacol 2004; 66: 1332–1339. [DOI] [PubMed] [Google Scholar]
- 23.Greenwood J, Pryce G, Devine L, et al. SV40 large T immortalised cell lines of the rat blood-brain and blood-retinal barriers retain their phenotypic and immunological characteristics. J Neuroimmunol 1996; 71: 51–63. [DOI] [PubMed] [Google Scholar]
- 24.Demeuse P, Fragner P, Leroy-Noury C, et al. Puromycin selectively increases mdr1a expression in immortalized rat brain endothelial cell lines. J Neurochem 2004; 88: 23–31. [DOI] [PubMed] [Google Scholar]
- 25.Tome ME, Schaefer CP, Jacobs LM, et al. Identification of P-glycoprotein co-fractionating proteins and specific binding partners in rat brain microvessels. J Neurochem 2015; 134: 200–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McCaffrey G, Staatz WD, Quigley CA, et al. Tight junctions contain oligomeric protein assembly critical for maintaining blood-brain barrier integrity in vivo. J Neurochem 2007; 103: 2540–2555. [DOI] [PubMed] [Google Scholar]
- 27.Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 2012; 9: 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bendayan R, Ronaldson PT, Gingras D, et al. In situ localization of P-glycoprotein (ABCB1) in human and rat brain. J Histochem Cytochem 2006; 54: 1159–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang X, Liu T, Bai Y, et al. Polymerase I and transcript release factor acts as an essential modulator of glioblastoma chemoresistance. PLoS One 2014; 9: e93439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanna E, Miotti S, Mazzi M, et al. Binding of nuclear caveolin-1 to promoter elements of growth-associated genes in ovarian carcinoma cells. Exp Cell Res 2007; 313: 1307–1317. [DOI] [PubMed] [Google Scholar]
- 31.Ludwig A, Howard G, Mendoza-Topaz C, et al. Molecular composition and ultrastructure of the caveolar coat complex. PLoS Biol 2013; 11: e1001640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hill MM, Bastiani M, Luetterforst R, et al. PTRF-Cavin, a conserved cytoplasmic protein required for caveola formation and function. Cell 2008; 132: 113–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zimnicka AM, Husain YS, Shajahan AN, et al. Src-dependent phosphorylation of caveolin-1 Tyr-14 promotes swelling and release of caveolae. Mol Biol Cell 2016; 27: 2090–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y, Qu X, Teng Y, et al. Cbl-b inhibits P-gp transporter function by preventing its translocation into caveolae in multiple drug-resistant gastric and breast cancers. Oncotarget 2015; 6: 6737–6748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Minshall RD, Tiruppathi C, Vogel SM, et al. Vesicle formation and trafficking in endothelial cells and regulation of endothelial barrier function. Histochem Cell Biol 2002; 117: 105–112. [DOI] [PubMed] [Google Scholar]
- 36.de LA, Donovan L, Arcaro A. Lipid rafts and caveolae in signaling by growth factor receptors. Open Biochem J 2007; 1: 12–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mohan J, Moren B, Larsson E, et al. Cavin3 interacts with cavin1 and caveolin1 to increase surface dynamics of caveolae. J Cell Sci 2015; 128: 979–991. [DOI] [PubMed] [Google Scholar]
- 38.Radeva G, Perabo J, Sharom FJ. P-Glycoprotein is localized in intermediate-density membrane microdomains distinct from classical lipid rafts and caveolar domains. FEBS J 2005; 272: 4924–4937. [DOI] [PubMed] [Google Scholar]
- 39.Hawkins BT, Rigor RR, Miller DS. Rapid loss of blood-brain barrier P-glycoprotein activity through transporter internalization demonstrated using a novel in situ proteolysis protection assay. J Cereb Blood Flow Metab 2010; 30: 1593–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmidt-Glenewinkel H, Reinz E, Bulashevska S, et al. Multiparametric image analysis reveals role of Caveolin1 in endosomal progression rather than internalization of EGFR. FEBS Lett 2012; 586: 1179–1189. [DOI] [PubMed] [Google Scholar]
- 41.Nethe M, Hordijk PL. A model for phospho-caveolin-1-driven turnover of focal adhesions. Cell Adh Migr 2011; 5: 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gottlieb-Abraham E, Shvartsman DE, Donaldson JC, et al. Src-mediated caveolin-1 phosphorylation affects the targeting of active Src to specific membrane sites. Mol Biol Cell 2013; 24: 3881–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cai T, Wang H, Chen Y, et al. Regulation of caveolin-1 membrane trafficking by the Na/K-ATPase. J Cell Biol 2008; 182: 1153–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gobeil F, Jr., Bernier SG, Vazquez-Tello A, et al. Modulation of pro-inflammatory gene expression by nuclear lysophosphatidic acid receptor type-1. J Biol Chem 2003; 278: 38875–38883. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, Supplemental material1 for Acute pain alters P-glycoprotein-containing protein complexes in rat cerebral microvessels: Implications for P-glycoprotein trafficking by Margaret E Tome, Chelsea K Jarvis, Charles P Schaefer, Leigh M Jacobs, Joseph M Herndon, Kristen C Hunn, Nathan B Arkwright, Kathryn L Kellohen, Peyton C Mierau and Thomas P Davis in Journal of Cerebral Blood Flow & Metabolism
Supplemental material, Supplemental material2 for Acute pain alters P-glycoprotein-containing protein complexes in rat cerebral microvessels: Implications for P-glycoprotein trafficking by Margaret E Tome, Chelsea K Jarvis, Charles P Schaefer, Leigh M Jacobs, Joseph M Herndon, Kristen C Hunn, Nathan B Arkwright, Kathryn L Kellohen, Peyton C Mierau and Thomas P Davis in Journal of Cerebral Blood Flow & Metabolism
Supplemental material, Supplemental material3 for Acute pain alters P-glycoprotein-containing protein complexes in rat cerebral microvessels: Implications for P-glycoprotein trafficking by Margaret E Tome, Chelsea K Jarvis, Charles P Schaefer, Leigh M Jacobs, Joseph M Herndon, Kristen C Hunn, Nathan B Arkwright, Kathryn L Kellohen, Peyton C Mierau and Thomas P Davis in Journal of Cerebral Blood Flow & Metabolism