

Abstract
X-linked adrenoleukodystrophy (X-ALD) is a rare inherited metabolic disease affecting the nervous system and the adrenal glands. It is caused by a mutation of the ABCD1 gene, resulting in the impaired degradation of very long-chain fatty acids and their subsequent accumulation in several organs and tissues. X-ALD is notable for its high phenotypical variability, that includes isolated adrenocortical insufficiency, slowly progressive myelopathy with paraparesis, ataxia, and peripheral neuropathy to severe childhood cerebral forms.
Here, we describe the case of an X-ALD patient with a p.Gly343Val mutation in ABCD1 gene, who presented in adulthood with a spinal syndrome of mild severity, and later developed a progressive cognitive and behavioral syndrome. Our patient showed a striking correlation between clinical phenotype and neuroimaging, including a brain fluoro-2-deoxy-d-glucose positron emission tomography that displayed an atypical cerebral glucose metabolism.
Keywords: X-linked adrenoleukodystrophy (X-ALD), Missense mutation, Frontal lobe dysfunction, Cortical and subcortical atrophy, Brain FDG-PET
Introduction
X-linked adrenoleukodystrophy (X-ALD) is a rare inherited metabolic disease affecting the nervous system and the adrenal glands. It is a monogenic disorder caused by mutations of the ABCD1 gene, located on chromosome Xq28 [1,2]). The ABCD1 gene codes for the peroxisomal transporter ATP-binding cassette subfamily D member (ABCD1, formerly ALDP) that mediates the import of very long-chain fatty acids (VLCFAs) Coenzyme-A esters across the peroxisomal membrane [3,4], resulting in an impaired degradation of VLCFAs in peroxisomes and subsequent accumulation of various lipid species in several organ and tissues.
This pathogenic pathway may be responsible of different clinical phenotypes, distinguished by age of onset, severity, and affected systems [5,6]. Three main phenotypes have been identified in affected males: a childhood cerebral form, an adrenomyeloneuropathy variant (AMN), and isolated Addison's disease.
-
•
The childhood-onset cerebral form (CCALD) affects 35% of male patients, and typically presents at 4-8 years of age. This form bears an initial resemblance to the syndrome of attention deficit disorder and hyperactivity, but subsequently leads to developmental regression, vision and hearing loss, and disturbances of motor function. It often has a fatal course during the first decade of life.
-
•
"Pure" AMN (40%-45% of affected males) manifests more commonly in the late 20s, with symptoms limited to the spinal cord and the peripheral nerves that lead to slowly progressive paraparesis, sphincter incontinence, and sometimes adrenocortical insufficiency. About 20% of adults with AMN may later develop brain lesions and present progressive symptoms such as cognitive impairment, behavioral and/or speech disorders, spinocerebellar degeneration (adrenoleukomyeloneuropathy).
-
•
Isolated Addison disease (10% of affected males) is characterized by primary adrenocortical insufficiency as its only symptom. This form has an extremely variable age of onset that ranges from 2 years of age to adulthood, but symptoms of AMN may also develop later in life.
Other clinical presentations of X-ALD are described (10% of cases): (1) cerebral juvenile, cerebral adult form (cerebral adrenoleukodystrophy); (2) a cerebello-brainstem form; (3) progressive pure dementia; (4) a variant with dysautonomia; and (5) an asymptomatic variant [7].
In X-ALD, the majority of female carriers remain asymptomatic, albeit approximately 20% of them show a subclinical AMN, with a later onset (age ≥ 35 years) and a milder phenotype than the affected males [8]. The diagnosis of X-ALD is based on clinical examination, instrumental and laboratory data; in particular, a high plasma level of VLCFAs represents the standard diagnostic biomarker of X-ALD [4]. However, genetic analysis is still necessary in order to confirm atypical cases [9,10]. Brain and spine magnetic resonance imaging (MRI) is a helpful tool to better define the X-ALD clinical phenotype. In the last years, brain [18F]-fluoro-2-deoxy-d-glucose positron emission tomography (FDG-PET) has emerged as useful neuroimaging technique able to reveal in X-ALD abnormalities independent from morphologic changes detected by MRI. Brain metabolic pattern seems to be characteristic of the disease, displaying increased uptake within frontal areas and anterior cingulate cortex and decreased uptake in temporal areas and cerebellum [11].
Here, we describe the atypical case of an adult patient who presented with a mild form of adrenomyeloneuropathy, that later developed a severely progressive cognitive and behavioral syndrome, with a striking correlation between clinical and neuro-imaging data.
Case presentation
A 46-year-old Caucasian man was referred to our department for the insidious onset of walking difficulties and bladder dysfunction with urinary urgency; he was described by family members as careless. At baseline his neurological examination showed signs of a pyramidal syndrome confined to the lower limbs, with weakness, hyperreflexia, bilateral Babinski sign. The Spastic Paraplegia Rating Scale (SPRS) score was 20/52. Neuropsychological assessments revealed a mild deficit of semantic fluency and a Mini-Mental State Examination score (MMSE) of 27.89/30. In the course of the interview he appeared easily distractible, and apathetic towards his cognitive difficulties (anosodiaphoria). Brain and spine MRI showed subtle T2-weighted hyperintense areas in the periventricular frontal white matter associated with atrophy of the spinal cord. Nerve conduction studies were normal. Endocrine screening detected an initial adrenal insufficiency, affecting both glucocorticoid and mineralocorticoid incretion, and a slight reduction in testosterone levels.
His pedigree comprised several cases of walking difficulties and febrile infant death, as shown in Fig. 1. In particular, the proband's mother (II, 1) was affected by a foot dystonia, his maternal uncle (II, 4) died at age 46 for complications of a spastic paraplegia with onset at 30 years; another maternal uncle (II, 8) died at 2 years of age for an unspecified epileptic encephalopathy; his maternal aunt (II, 3) had a gait disorder and her son (III, 9) died 14 years as a result of a fever. The proband's older brother (III, 2) was affected by spastic paraplegia with childhood onset and died at 46 years of age for complications of his bedridden state; his younger brother (III, 4), 43 years old, reportedly exhibited signs suggestive of psychosis with paranoid ideation, but repeatedly refused medical care. In addition, another brother died at 55 years of age for multiform glioblastoma.
Fig. 1.

Family tree. The presented case is marked with an arrow. Subject II,4; II,8; III,2; III,9 were affected by ALD/AMN; II,1 and II,3 are female carriers. ALD = adrenoleukodystrophy; AMN = adrenomyeloneuropath.
Considering these data, a diagnosis of X-ALD/AMN was suspected. VLCFAs were increased in the plasma. Molecular analysis of the ABCD1 gene detected a rare hemizygous missense mutation c.1028G>T (p.Gly343Val) in the exon 2, thus confirming the diagnosis.
Five years later, the patient was readmitted to our department due to the progression of his behavioral disorder with disinhibition, distractibility, absence of insight, apathy, anhedonia, and impulse control disorder, associated with urinary incontinence. The neurologic examination showed Romberg sign, spastic paraplegia (Spastic Paraplegia Rating Scale score: 22/52), dysmetria at nose-finger test, apallesthesia in the lower limbs, Epstein sign. A second neuropsychological evaluation showed a mild cognitive impairment (MMSE 24.99/30) with a severe deficit in frontal functions (Frontal Assessment Battery: 11.2/18; Frontal Behavioral Inventory: 32).
A second brain and spine MRI confirmed the bilateral and confluent Flair and T2-weighted hyperintensities of the periventricular frontal, also involving the white parietal matter (Fig. 2A). The anterior cingulate cortex and the corpus callosum were also both damaged, with involvement of the isthmus but especially of the genu of the anterior part of the body, extending to the white matter of the forceps minor (Fig. 2D). The deep frontal white matter was more affected than the parietal, showing also a higher hyperintensity on T2-weighted images and a remarkable hypointensity on T1-weighted images and on the exponential Apparent Diffusion Coefficient map (Fig. 2A and B). This kind of signal is typical of a severe damage and characterized by an increase of the water molecules movements in Diffusion Imaging, correlating with the frontal origin of the disease, also confirmed by the findings of the first MRI. The U fibers were completely spared. A bilateral pyramidal tract demyelination of the frontopontine fibers in the anterior limb of the internal capsule (Fig. 2A) and of the corticospinal tracts in the posterior internal capsule and encephalic trunk was also clearly evident on Flair and T2-weighted images. Some pontine transverse fibers showed the same pathological signal that thickened and symmetrically extended in middle cerebellar peduncles and in the deep hemispheric white matter (Fig. 2C). Both the brainstem and the medulla appeared quite atrophic. Some of these brainstem MR features resembled an olivopontocerebellar atrophy. The superior cerebellar peduncles and the periaqueductal gray substance were partially hyperintense on Flair and T2-weighted images too. The ventricular system and subarachnoid fronto-insular cerebral and cerebellar spaces were enlarged because of the atrophy, greater in the frontal lobes (Fig. 2A). The temporal and the occipital white matter was almost normal. This kind of cerebral and cerebellar involvement does not conform to any of the 5 neuroradiological patterns described by Loes et al because of the presence of both cerebellar and cerebral lesions, with involvement of the periventricular white matter but also of the internal capsule [12]. Even though there were some motion artifacts, the spinal cord appeared clearly atrophic, especially in the thoracic tract (Fig. 3).
Fig. 2.

(A-D) Brain MRI. Axial turbo spin-echo (TSE) T2-weighted image (A) well depicts bilateral and confluent white matter fronto-parietal hyperintensities more extensive and characterized by a higher signal in the frontal lobes. The anterior limb of the internal capsule is also clearly affected, bilaterally. The ventricular system and the fronto-insular subarachnoid spaces are enlarged. Axial diffusion-weighted imaging (DWI) eADC map (B) shows a marked hypointensity of the deep frontal white matter, rather than the parietal one. Axial TSE T2-weighted image at subtentorial level (C) reveals a bilateral hyperintense signal of the middle cerebellar peduncles associated to a subtle hyperintensity of the ventral pons that results quite atrophic. Sagittal TSE T2-weighted image shows thinning of the entire corpus callosum with abnormal hyperintense signal of the isthmus, the genu and the anterior part of the body (D). eADC = exponential Apparent Diffusion Coefficient; MRI = magnetic resonance imaging.
Fig. 3.
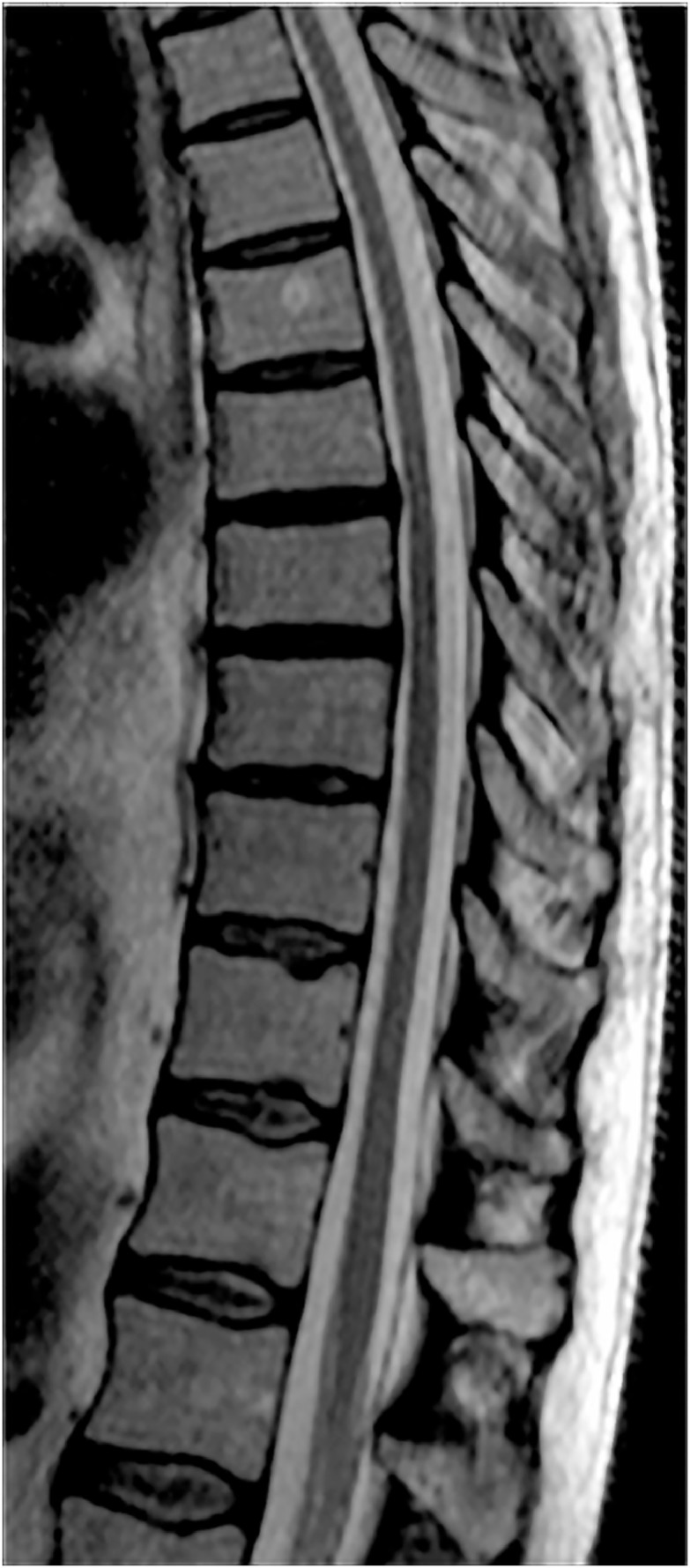
Spine MRI. Sagittal TSE T2-weighted image shows thinning of the dorsal spinal cord, with relative sparing of the conus medullaris. MRI = magnetic resonance imaging.
Brain FDG-PET scan showed a marked cerebral and cerebellar gray matter relative hypometabolism. Cortical FDG uptake was reduced bilaterally in the frontal and the parietal cortices with prevalent and more severe involvement of the entire frontal lobe and the anterior cingulate while the occipital, sensory, and auditory cortices were relatively spared. A decrease in uptake was also bilaterally detected in the striatum, thalamus, and in the hemispheric cerebellar cortex (Fig. 4).
Fig. 4.

Positron emission tomography (PET) transaxial images of [18F]FDG uptake in the patient at 4 different sections (A) show a clear relative hypometabolism involving the associative cerebral cortex, mostly the frontal lobe and the anterior cingulate region, and the cerebellar hemispheres (azure color). To a lesser extent, [18F]FDG uptake appears also reduced in the thalamus and the striatum, bilaterally. These findings are highlighted by the results of single subject statistical parametric mapping (SPM) analysis obtained in our patient in comparison to a group of controls (age range of controls: 27-70, age was considered in the statistical model of SPM as nuisance covariate) and rendered as clusters of reduced relative metabolism (P < .01 uncorrected) onto the sagittal (B), coronal (C) and transaxial (D, E) T1-MRI SPM images spatially normalized to the Montreal Neurological Institute (MNI) space. FDG = fluoro-2-deoxy-d-glucose; MRI = magnetic resonance imaging.
In the following months, the clinical state deteriorated dramatically. The patient became unable to walk, requiring the use of a wheelchair; his cognitive decline progressed to the point of complete aphasia. He also became severely dysphagic, developed cervical dystonia with laterocollis, and experienced several generalized epileptic seizures. These complications were effectively managed through a percutaneous endoscopy gastrostomy, the injection of botulinum toxin in the cervical muscles and with the introduction of antiepileptic therapy (levetiracetam 2000 mg/day), respectively.
Discussion and conclusion
Late-onset cognitive involvement is a rare symptom of X-ALD. In fact, X-ALD presenting with adult-onset dementia has only been described in 2%-3% of cases reported in Western countries [13]. The case that we present is unique in that it shows the development of a severely progressive cognitive syndrome in a patient that had previously been affected only by mild motor and sphincteric symptoms. His neurological history is thus suggestive of a late shift from the AMN phenotype to the cerebral ALD phenotype that essentially took place over the course of 5 years.
Features of this changing neurological profile are apparent in the imaging and functional studies that we performed. Brain and spine MRI images, collected over a 10-year follow-up period, strongly correlate to the clinical course, and highlight the symmetrical progression of the deep white matter damage, which is particularly extensive in the frontal lobes. The severity of the frontal involvement is rare in X-ALD; indeed, the periventricular parietal and occipital white matter is the one predominantly involved in the 85% of the patients. [7,14].
FDG-PET detected cortical and subcortical gray matter hypometabolism, the degree and the pattern of which are again in agreement with the frontal-prevalent cognitive impairment. Even if the typical brain FDG-PET pattern of X-ALD patients is usually characterized by hypermetabolism in frontal areas and anterior cingulate cortex and hypometabolism in temporal areas and cerebellum [11], the different findings in our patient are in line with the topography and the degree of white matter alterations observed at the MRI. Cortical deafferentation, secondary to supratentorial white matter damage and axonal degeneration, might in fact be the primary physiopathological mechanism underlying these cortical metabolic changes. A similar mechanism might also explain the finding of basal ganglia and thalamic hypometabolism. Severe cerebellar hypometabolism, also observed in this patient, might result from direct damage of the cerebellar white matter or from a dysfunction/loss of the cortico/brainstem-cerebellar connections, as reported in rare cases of cerebello–brainstem variant form of adult X-ALD [15]. The degree and extent of these findings allow us to trace a direct line between loss of white matter integrity, altered connectivity, morphological and functional changes, and, finally, the resulting clinical picture. This also underlines how clinical examination, MRI and FDG PET all offer unique contributions to the description of this rare phenotype of X-ALD. Additional features of interest include a mild and not progressive adrenal dysfunction and the absence of peripheral polyneuropathy.
Genetic tests led to the identification of a point mutation in the ABCD1 gene (c.1028G >T p.Gly343Val) in the exon 2. Further molecular and epidemiological studies will be needed to assess whether a definite link exists between this specific mutation and the phenotype presented by this patient.
In conclusion, we believe this case is of interest in that it shows the association of a peculiar cognitive and behavioral phenotype with structural and functional neuroimaging in adult X-ALD/AMN with p.Gly343Val pathogenic mutation of the ABCD1 gene.
Footnotes
Dr Clemente Dato: Study concept and design, acquisition of data, analysis and interpretation of data, drafting the manuscript.
Dr. Guglielmo Capaldo: Study concept and design, acquisition of data, analysis and interpretation of data, drafting the manuscript.
Dr. Chiara Terracciano: Analysis and interpretation of data, revising the manuscript for intellectual content.
Dr. Filomena Napolitano: Analysis and acquisition of data.
Dr. Alessandra D'Amico: Analysis and acquisition of data.
Dr. Sabina Pappatà: Analysis and acquisition of data.
Dr. Filippo Maria Santorelli: Analysis and acquisition of data.
Prof. Giuseppe Di Iorio: Revising the manuscript for intellectual content.
Prof. Simone Sampaolo: Revising the manuscript for intellectual content.
Prof. Mariarosa AB Melone: Study concept and design, study supervision, analysis and interpretation of data, drafting, and revising the manuscript for intellectual content.
Declarations of interest: None.
This study is not industry-sponsored study.
Acknowledgment: S.S. and F.N. thank the Regione Campania (RIS 3—POR FSE CAMPANIA 2014/2020 Asse III, Obiettivo specifico 14), for financial support. C.T. thanks InterUniversity Center for Research in Neurosciences, University of Campania “Luigi Vanvitelli,” Naples, Italy, for financial support.
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.radcr.2018.11.007.
Appendix. Supplementary materials
References
- 1.Berger J., Gartner J. X-linked adrenoleukodystrophy: clinical, biochemical and pathogenetic aspects. Biochim Biophys Acta. 2006;1763:1721–1732. doi: 10.1016/j.bbamcr.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 2.Moser H.W., Mahmood A., Raymond G.V. X-linked adrenoleukodystrophy. Nat Clin Pract Neurol. 2007;3:140–151. doi: 10.1038/ncpneuro0421. [DOI] [PubMed] [Google Scholar]
- 3.van Roermund C.W., Visser W.F., Ijlst L., van Cruchten A., Boek M., Kulik W. The human peroxisomal ABC half transporter ALDP functions as a homodimer and accepts acyl-CoA esters. FASEB J. 2008;22:4201–4208. doi: 10.1096/fj.08-110866. [DOI] [PubMed] [Google Scholar]
- 4.Wiesinger C., Kunze M., Regelsberger G., Forss-Petter S., Berger J. Impaired very long-chain acyl-CoA beta-oxidation in human X-linked adrenoleukodystrophy fibroblasts is a direct consequence of ABCD1 transporter dysfunction. J Biol Chem. 2013;288:19269–19279. doi: 10.1074/jbc.M112.445445. doi: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cotrufo R., Melone M.A., Monsurro M.R., Di Iorio G., Carella C., Moser H.W. Phenotype heterogeneity among hemizygotes in a family biochemically screened for adrenoleukodystrophy. Am J Med Genet. 1987;26:833–838. doi: 10.1002/ajmg.1320260410. [DOI] [PubMed] [Google Scholar]
- 6.Moser H.W. Adrenoleukodystrophy: phenotype, genetics, pathogenesis and therapy. Brain. 1997;120:1485–1508. doi: 10.1093/brain/120.8.1485. [DOI] [PubMed] [Google Scholar]
- 7.Steinberg S.J., Moser A.B., Raymond G.V. GeneReviews®[Internet] University of Washington, Seattle; SeattleWA: 1993. X-linked adrenoleukodystrophy. -2016. 1999 Mar 26 [updated 2015 Apr 9] [PubMed] [Google Scholar]
- 8.Engelen M., Barbier M., Dijkstra I.M., Schür R., de Bie R.M., Verhamme C. X-linked adrenoleukodystrophy in women: a cross-sectional cohort study. Brain. 2014;137:693–706. doi: 10.1093/brain/awt361. [DOI] [PubMed] [Google Scholar]
- 9.Wiesinger C., Eichler F.S., Berger J. The genetic landscape of X-linked adrenoleukodystrophy: inheritance, mutations, modifier genes, and diagnosis. Appl Clin Genet. 2015;8:109–121. doi: 10.2147/TACG.S49590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Montagna G., Di Biase A., Cappa M., Melone M.A., Piantadosi C., Colabianchi D. Identification of seven novel mutations in ABCD1 by a DHPLC-based assay in Italian patients with X-linked adrenoleukodystrophy. Hum Mutat. 2005;25:222. doi: 10.1002/humu.9303. [DOI] [PubMed] [Google Scholar]
- 11.Salsano E., Marotta G., Manfredi V., Giovagnoli A.R., Farina L., Savoiardo M. Brain fluorodeoxyglucose PET in adrenoleukodystrophy. Neurology. 2014;83:981–989. doi: 10.1212/WNL.0000000000000770. [DOI] [PubMed] [Google Scholar]
- 12.Loes D.J., Fatemi A., Melhem E.R., Gupte N., Bezman L., Moser H.W. Analysis of MRI patterns aids prediction of progression in X-linked adrenoleukodystrophy. Neurology. 2003;61:369–374. doi: 10.1212/01.WNL.0000079050.91337.83. doi: [DOI] [PubMed] [Google Scholar]
- 13.Inoue S., Terada S., Matsumoto T., Ujike H., Uchitomi Y. A case of adult-onset adrenoleukodystrophy with frontal lobe dysfunction: a novel point mutation in the ABCD1 gene. Intern Med. 2012;51:1403–1406. doi: 10.2169/internalmedicine.51.6899. doi: [DOI] [PubMed] [Google Scholar]
- 14.Volkow N.D., Patchell L., Kulkarni M.V., Reed K., Simmons M. Adrenoleukodystrophy: imaging with CT, MRI, and PET. J Nucl Med. 1987;28:524–527. [PubMed] [Google Scholar]
- 15.Kim J.E., Choi K.G., Jeong J.H., Kang H.J., Kim H.S. Diffuse cortical hypometabolism on (18)F-FDG-PET scan in a case of an adult variant cerebello-brainstem dominant form of ALD manifesting dementia. Parkinsonism Relat Disord. 2012;18:210–212. doi: 10.1016/j.parkreldis.2011.06.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.