

Abstract
Human immunodeficiency virus (HIV) has infected 76 million people and killed an estimated 35 million. During its 40‐year history, remarkable progress has been made on antiretroviral drugs. Progress toward a vaccine has also been made, although this has yet to deliver a licensed product. In 2007, I wrote a review, HIV AIDS Vaccines: 2007. This review, HIV AIDS Vaccines: 2018, focuses on the progress in the past 11 years. I begin with key challenges for the development of an AIDS vaccine and the lessons learned from the six completed efficacy trials, only one of which has met with some success.
Challenges posed by human immunodeficiency virus‐1 for vaccine development
Development of a human immunodeficiency virus (HIV) vaccine faces many challenges that include the virus life cycle favoring the rapid establishment of hard to clear chronic infections, the high diversity and structure of the envelope glycoprotein limiting the ability to elicit broadly neutralizing antibodies (bnAbs), and the tropism of the virus for T helper cells facilitating infection, spread, and persistence (see Box 1).
Box 1. Challenges for HIV vaccine development.
| Prevention, not just control, of infection to limit the establishment of proviral reservoirs |
| Generation of highly mutated Ab with atypical structures to achieve broad neutralization |
| Providing CD4+ T cell help that is not kindling for infection |
| Achieving breadth for the diversity of transmitted viruses |
Ab, antibody; HIV, human immunodeficiency virus.
Upon infection, HIV reverse transcribes its RNA to DNA, which integrates into the host genome as a DNA provirus. The provirus, established within hours of infection, is a chronic infection that can lie latent, unable to be recognized by the immune system or eradicated by drugs. Most vaccines work by memory responses expanding and limiting disease. A protective memory response for HIV may need to prevent the establishment of proviral DNA.
Prevention of infection is best mediated by neutralizing antibody (Ab) blocking the entry of virus into a cell. HIV is a highly diverse virus. Each transmitted virus is a unique virus. For a vaccine to be effective, it will need to recognize and block conserved targets for neutralization that are common to the strains undergoing transmission. Despite years of intensive effort by highly skilled laboratories, broadly neutralizing antibody (bnAb) for the strains of HIV that are undergoing transmission has yet to be achieved in humans.1, 2
Another challenge is the cellular receptor for HIV being CD4. CD4 is the signature surface molecule for the T cells that provide help in the form of cytokines and co‐stimulation for both Ab and T cell responses. Vaccines readily elicit memory CD4+ helper T cells that rapidly migrate to sites of an incoming infection. For most infections, these CD4+ T cells support control of the infection. For HIV, they serve as kindling as well as providing help.3, 4 For a CD4+ T cell to be infected, it also needs to display CCR5, which serves as a co‐receptor for HIV. Not all types of CD4+ T cells display the CCR5 co‐receptor for infection. However, memory cells that localize at mucosal surfaces do display CCR5, providing target cells at mucosal sites of entry. The T follicular helper cells that migrate to the germinal centers of lymph nodes, where they support somatic hypermutation of Abs, also display the CCR5 co‐receptor. Because germinal centers restrict the entry of cytotoxic T cells, the T follicular helper cells not only are kindling for HIV infection but also carry the infection to a sanctuary where it can persist and thrive.5 Thus, vaccination can raise responses that exacerbate as well as control infection.
Logistical considerations have also slowed vaccine development. Challenge studies need to be done in nonhuman primates (NHPs), an expensive model in which limited numbers of animals can be used. Preclinical challenge trials are further complicated by there not being sure correlates for protection—with different vaccines potentially eliciting responses that protect by different mechanisms.
Despite these challenges, there is hope: some candidate vaccines successfully prevent simian immunodeficiency virus and chimeric simian‐human immunodeficiency virus infections in NHPs. In these vaccine trials, success is measured not by delivering a single high dose challenge, but by delivering repeated low dose challenges that infect about 30% of the animals at each dose.6 Hopefully, vaccines that have been successful in NHPs can be translated into humans where transmission is typically much less frequent than once for three exposures.7
Efficacy trials for HIV vaccines
By far, the most significant milestones on the path to an HIV vaccine have been the efficacy trials (Figure 1). The first two trials, one in discordant couples in the United States and one in intravenous drug users in Thailand, tested bivalent subunits of the gp120 receptor binding subunit of Env for the ability to raise protective antibodies.8, 9 Unlike the hepatitis B and papillomavirus vaccines, where protein vaccines raise protective responses, the gp120 HIV protein failed to protect either of the high‐risk populations. In retrospect, this failure to protect seems to have been due in part to multiple boosts (7 total) driving the Ab response to a nonprotective subgroup of immunoglobulin G (IgG; see below).
Figure 1.
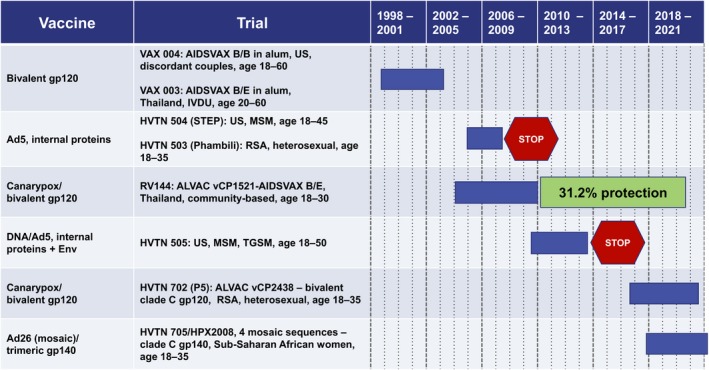
Timeline for human immunodeficiency virus (HIV) vaccine efficacy trials. Years at the top are for 4‐year intervals, individual years are designated by dotted lines. Ad5, adenovirus serotype 5 vector; Ad26, adenovirus serotype 26 vector; ALVAC, canarypox vector; HVTN, HIV Vaccine Trials Network; IVDU, intravenous drug users; MSM, men who have sex with men; RSA, Republic of South Africa; TGSM, male to female transgender individuals who have sex with men; US, United States; VCP, canary pox vector.
The second two completed trials tested adenovirus five (Ad5)‐vectored internal HIV proteins (Gag, Pol, and Nef) for the ability to raise protective cytotoxic CD8+ T cells. These trials, termed STEP and Phambili, were conducted in men who have sex with men (MSM) in the United States and heterosexual couples in South Africa, respectively. Both trials were stopped early by their data safety monitoring boards because of harm: a higher incidences of HIV infection in vaccinated than nonvaccinated participants.10, 11 The most broadly accepted hypothesis for the failure of the STEP and Phambili trials is the elicitation by the Ad5 vector of CD4+ T cells that served as preferential targets for infection.12
The fifth completed trial, RV144, a 16,000‐participant community‐based trial in Thailand, tested a canary poxvirus prime (ALVAC) followed by an ALVAC + bivalent gp120 protein in alum boost. This trial used the same gp120 protein that had been used in the Thai trial testing gp120 protein alone. RV144 was overseen by the US Military and the Thai Ministry of Public Health and, despite controversy about the wisdom of conducting the trial,13 provided the first and only protection (31.2%) achieved by an HIV vaccine.14 Interestingly, the vaccine seemed to have achieved a 60.5% rate of protection during the first 6 months after its peak response. This protection waned with the rapid contraction of a poorly durable Ab response.15 Post hoc analyses of CD4+ T cell responses have revealed that the T cells elicited by the RV144 vaccine had relatively poor susceptibility to HIV infection,12 a phenomenon that likely contributed to its protective potential.
The sixth trial tested DNA priming followed by Ad5 boosting. This trial, HVTN 505, conducted in the United States in MSM and transgender men who have sex with men, differed from the first Ad5 trial by including immunogens that expressed a secreted form of Env and a DNA prime, which had augmented immunogenicity of recombinant Ad5 vectors.16 Because of the enhanced infection in the first trial testing an Ad5 vaccine, HVTN 505 was closely monitored by its data safety monitoring board and stopped early, not because of harm, but because of futility.17
There are currently two ongoing efficacy trials (HVTN 702 and HVTN 705). HVTN 702 tests conditions used in the Thai RV144 trial, but with appropriately matched clade C immunogens for a South African cohort. An additional boost is being given to help maintain protective Ab responses. The second ongoing trial, HVTN 705, tests four Ad26 vectored mosaic sequences followed by a trimeric gp140 Env boost to protect 18–35‐year‐old sub‐Saharan African women. More information for these ongoing efficacy trials, as well as two trials testing the protective potential of passively transferred bnAb, are presented below (see Table 1). All of the immunogens used to date have been well tolerated with only mild to moderate local reactogenicity and limited systemic side effect.
Table 1.
Ongoing efficacy trials
| Vaccine, phase, sponsor | Designation | Clade | n | Cohort | Start date | Immunogens | Comment |
|---|---|---|---|---|---|---|---|
|
Canarypox/bivalent gp120, phase IIb/III, NIAID |
HVTN 702 | C | n = 5,400: 2,700 treated, 2,700 placebo | South African adults, age 18–35 |
Oct 2016 |
2 primes: ALVAC‐HIV (1 × 107 CCID50 (vCP2438) at weeks 0 and 4 3 boosts: ALVAC plus bivalent protein (100 µg TV1.C gp120 and 100 µg of 1086.C gp120) in MF59 at weeks 12, 24, and 52 |
Repeat of RV144 trial but with sequences for epidemic in South Africa. Will test for prevention of infection over 24 and possibly 36 months |
|
Ad26/gp140 phase IIb, Janssen vaccines and prevention B.V. |
HVTN 705 | All |
n = 2,600: 1,300 treated, 1,300 placebo |
Sub‐Saharan African women, age 18–35 | Nov 2017 |
2 primes: Tetravalent Ad26.Mos4.HIV (5 × 1010 viral particles) at weeks 0 and 12 2 boosts: Ad26.Mos4.HIV + Clade C gp140.HIV (250 µg) in aluminum phosphate at weeks 24 and 52 |
Testing for prevention of acquisition months 7–24 |
|
AMP phase IIb, NIAID |
HVTN 703, HPTN 081 |
All |
n = 1500: 500 for each of 2 treatment groups and 500 placebo |
Women from 7 countries in sub‐Saharan Africa, age 18–50 | May 2016 | Passive transfer: VRC01 bnAb transfused 10 times at 8‐week intervals at 20 or 30 mg/kg | Testing for prevention of acquisition by week 80, observation until week 92 |
|
AMP phase IIb, NIAID |
HVTN 704, HPTN 085 |
All |
n = 2,700: 900 for each of 2 treatment groups plus 900 placebo |
United States, Brazil, Peru, Switzerland MSM, transgender men, age 18–50 |
Apr 2016 | Passive transfer: VRC01 bnAb transfused 10 times at 8‐week intervals at 20 or 30 mg/kg | Testing for prevention of acquisition by week 80, observation until week 92 |
ALVAC, canarypox vector; AMP, antibody‐mediated protection; bnAb, broadly neutralizing antibody; CCID50, cell culture infectious dose for 50% of cultures; HIV, human immunodeficiency virus; HPTN, US National Institutes of Health Sponsored HIV Prevention Trials Network; HVTN, US National Institutes of Health sponsored HIV Vaccine Trials Network; MF59, a squalene‐based emulsion that is used as an adjuvant; MSM, men who have sex with men; NIAID, US National Institutes of Allergy and Infectious Diseases.
Mining of immune responses in RV144 for correlates of risk, protective non‐neutralizing antibody
Just as the achievement of protection in RV144 was not anticipated, the correlates for reduced risk were also unexpected.18 These correlates did not include bnAb or CD8+ T cells, the immune responses that had been most sought by vaccine developers. Rather, case control studies showed that binding Ab, which was not neutralizing, correlated with reduced risk. This protective non‐neutralizing antibody (pnnAb) belonged to two subgroups of IgG: IgG1 and IgG3 with IgG3 being particularly effective.19, 20 In contrast, IgG2, IgG4, and serum IgA did not correlate with reduced risk. Indeed, serum IgA, which had a high response rate, was associated with increased, not reduced risk.
The protective binding Ab had specific targets on Env. Ab to the tip of the highly variable V1V2 loop correlated with reduced risk.18 Sequence analysis revealed that break‐through viruses had been sieved for V1V2 sequences that were not recognized by the binding Ab.21 A second target for pnnAb was the C1‐C2 region of the inner domain of gp120, a region of Env that is a major target for antibody‐dependent cellular cytotoxicity (ADCC).22 Recombinant Ab to C1‐C2 from RV144 participants had high ADCC activity in the presence of low, but not high serum IgA, which seemed to interfere with IgG‐initiated ADCC.23, 24 A third protective target was the V3 loop of Env. This activity did not neutralize virus; but, rather, was associated with ADCC, which, again, was most active in the presence of low serum IgA.23
Two subsets of polyfunctional CD4+ T cells correlated with reduced risk.25 The first of these co‐expressed CD40 ligand, interleukin (IL)‐2, IL‐4, interferon‐ϒ, and tumor necrosis factor‐α; and, the second, CD40 ligand, IL‐2, and IL‐4. It was hypothesized that these CD4 subsets contributed to protection by providing CD4+ T cell help for the elicited pnnAb. The expression of IL‐4 by both subsets is of interest because such is expressed by type 2 CD4+ T cells that do not display the CCR5 coreceptor. Consistent with this, ALVAC‐elicited CD4+ T cells have been found to be relatively resistant to HIV infection.12 Provocatively, type 2 CD4+ T cells promote Ig gene rearrangements to IgA, an Ab response that correlated with increased risk in RV144.18
Antibody mediated protection, differences between bnAb and pnnAb
Neutralizing Ab prevents infection by blocking functional activities of viral proteins that mediate entry into cells. Upon infection, or vaccination, naïve B cells whose Ab receptors bind to proteins exposed on the foreign agent (or vaccine) expand and undergo mutation and selection in lymph nodes for increasingly tight binding to the foreign agent. These mutations are centered in the Fab region of the Ab molecule (Figure 2 a) and for most infections can generate an Ab capable of neutralizing the infection. However, broad neutralization of HIV requires an Ab that can recognize conserved functional regions of Env, which are masked by heavily glycosylated highly variable sequences (Figure 2 b; for reviews, see Refs. 1 and 2). Abs that can block conserved functional regions are broadly neutralizing and can neutralize many isolates, including cross‐clade isolates of HIV. Mutants of these conserved functional regions are generally lethal and, therefore, most do not support escape. Only a fraction of infected people generate such bnAbs26 and these Abs, when they are generated, have atypical structures generated by mutation of as much as 30% of their Fab sequence.
Figure 2.
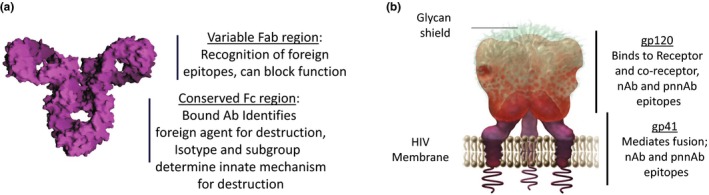
Functional regions of antibody and the human immunodeficiency virus (HIV) envelope. (a) Schematic of an antibody (Ab) showing variable Fab and conserved Fc regions. Fab and Fc fragments are generated by papain cleavage: Fab is the variable antigen binding fragment, and Fc is the crystallizable constant fragment. (b) Schematic of the HIV envelope glycoprotein (Env) with receptor binding (gp120) and transmembrane proteins (gp41) indicated. nAb, neutralizing Ab; pnnAb, protective non‐neutralizing Ab. Schematic of immunoglobulin G (IgG) is reproduced from commons.wikimedia.org by Gareth White and is licensed under CC BY 2.0. Schematic of HIV Env is reproduced with permission from David H. Spach, MD and the National HIV Curriculum.
In contrast, essentially all HIV‐infected people generate non‐neutralizing Abs that can recognize and bind nonfunctional regions of Env. These Abs, despite not being neutralizing, can protect by the Fc region of the bound Ab (see Figure 2 a) triggering complement (Cʹ) or innate immune cells, such as macrophages and neutrophils, to engulf or lyse the bound virus or infected cell. The Fc regions of an Ab, in contrast to Fab regions, are conserved (Figure 2 a). These conserved regions are represented by different isotypes, such as IgM, IgG, IgA, and IgE. Within isotypes are defined subgroups, such as the IgG isotype having IgG1, IgG2, IgG3, and IgG4 subgroups. Each of these isotypes and subgroups are specialized for activating distinct but often overlapping phagocytic or lytic responses.
Broadly neutralizing Ab is considered preferable to pnnAb because of its ability to block incoming HIV and prevent establishment of proviral DNA, which can be latent and invisible to the immune response and drugs (Figure 3). In contrast, pnnAb, by acting on infected cells (as well as on incoming virus), limits and even clears cells that underwent the initial infection (Figure 3). Whereas bnAb is like St. George killing the dragon (an HIV virion) with a single well‐aimed thrust of a spear, pnnAb is more like the Lilliputians of Gulliver's Travels where the Fc regions of multiple Abs are required to trigger responses, such as complement (Cʹ)‐mediated lysis, ADCC, and antibody‐dependent phagocytosis to kill an infected cell or inactivate virus.
Figure 3.

Comparison of the activities of broadly neutralizing antibodies (bnAbs) and protective non‐neutralizing antibodies (pnnAbs). Cʹ, complement.
Because pnnAb requires cross‐linking of Cʹ or Fc receptors to activate a protective response, binding of Abs with Fc regions belonging to different isotypes/subgroups can compete with the triggering of a protective innate response. Indeed, such likely explains the inhibitory activity of serum IgA in RV144.24 In retrospect, the multiple inoculations (7 in total) in the failed efficacy trials of bivalent gp120 protein had driven the evolution of Env‐specific Abs from protective IgG1 and IgG3 subgroups to nonprotective IgG4 responses.19, 20, 27, 28 Very high levels of Abs also can be detrimental to pnnAb by competition for binding resulting in the binding of single (not both) Fab arms of an Ab (see Figure 2 a) and the failure of the Fc regions of the bound Ab to cross‐link the Cʹ or Fc receptors that trigger protective innate responses. Such is called a prozone and has been reported for ADCC,29 antibody‐dependent phagocytosis,30 Cʹ mediated lysis, and protection by passive transfer of Ab.31
Forms of Env being explored for the elicitation of protective Ab, tiers of nAb
Multiple forms of Env are being developed and tested for the ability to elicit bnAb as well as the more readily achieved, pnnAb (Figure 4). Two important forms are the native receptor binding form, the mediator for the first step for entry, and the CD4‐induced (CD4i) form, a post‐binding intermediate for virus‐cell fusion.32, 33 Most research has focused on the CD4 binding form of Env, which is a target for both bnAb and pnnAb (for example of dual target, see Ref. 34). However, the CD4i form also can be effective for incoming virus because this form has a half‐life of 30 minutes or longer on the surface of cells, which affords time for pnnAb to bind, cross‐link, and activate innate responses.35, 36 Studies also use forms of Env that are poor mimics of natural Env, but technically easier to produce, such as gp120 monomers and gp41 peptides (Figure 4).
Figure 4.
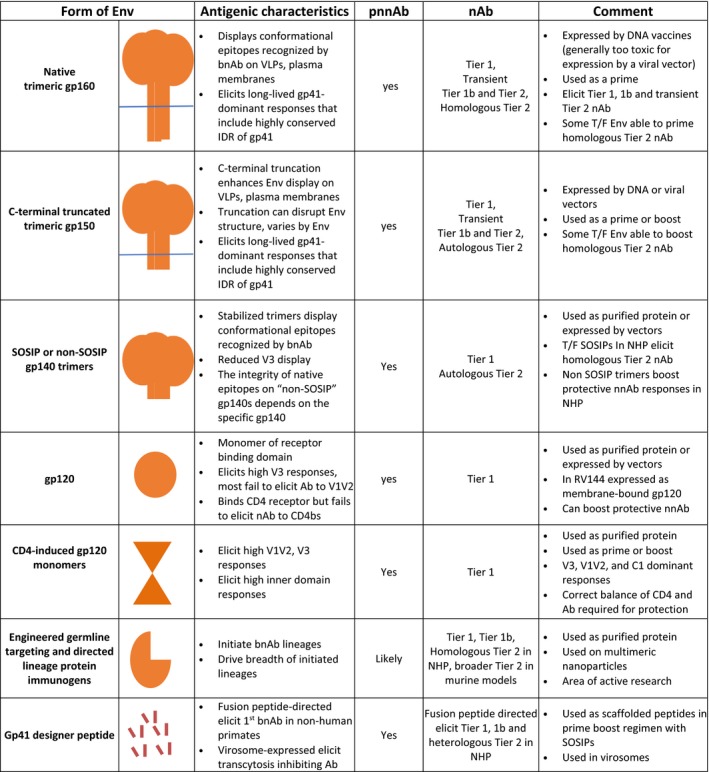
Schematics for forms of the human immunodeficiency virus (HIV) envelope glycoprotein (Env) being developed and used for immunizations. Ab, antibody; bnAb, broadly neutralizing antibody; C1, constant region 1 of gp120; IDR, highly conserved immunodominant region of gp41; nAb, neutralizing antibody; pnnAb, protective non‐neutralizing Ab; SOSIP, disulfide stabilized, cleaved trimeric form of gp140; T/F, transmitted founder virus for an infection; V1V2, variable loops one and two that form the apex of the HIV Env; V3, variable loop 3 of the HIV gp120; VLP, virus‐like particle. Parallel blue line, viral or plasma membrane. See text for more information and references.
Figure 4 presents different forms of Env that are being used in studies for eliciting bnAb and pnnAb. But first, a brief background on tiers of HIV nAb and immune checkpoints for HIV bnAb. Tiers of HIV nAb are ranked according to their potency for neutralization of laboratory‐adapted and primary isolates.37 Tier 1 nAb neutralizes easy to neutralize laboratory‐adapted isolates of HIV; tier 1b are the slightly more difficult to neutralize isolates; and tier 2 are the difficult to neutralize isolates that are undergoing transmission. A characteristic of many bnAbs is polyreactivity for host proteins.38, 39 This means that precursors for bnAb can undergo deletion in the bone marrow because of their recognition of self. Furthermore, responding B cells that emerge from the bone marrow can become anergic at immune tolerance checkpoints.40 This can result in failure to express precursors to bnAb or transient expression of an nAb, rather than further mutational development to a bnAb.
Gp160
Trimeric gp160, the native form of Env, has been expressed on virus‐like particles (VLPs) and the plasma membranes of vector‐infected or DNA‐transfected cells. It is used to present the conformationally intact receptor binding forms of Env to the immune system.41 The gp160, a transmembrane protein, undergoes denaturation when purified and is sufficiently toxic to eukaryotic cells to not be stable in viral vectors. However, when expressed by DNA it primes heterologous tier 1 nAb as well as mostly transient tier 1b and tier 2 nAbs.41, 42 The binding Ab response to gp160 is largely to gp41, including the immunodominant region (IDR) of gp41,43 a highly conserved target for pnnAb activities.44, 45, 46 The IDR is a desirable target because it does not readily survive mutations that might support escape.47 DNA‐expressed gp160 has advanced through phase IIa testing as a prime for a modified vaccinia Ankara (MVA) boost.48
Gp150
Trimeric gp150, a C‐terminal truncated, transmembrane form of gp160, is the second closest form to the native receptor‐binding form of Env for eliciting bnAb and pnnAb. The truncation removes toxic sequences allowing the production of stable gp150‐expressing viral vectors.49 The truncation also enhances the display of trimeric Env on VLPs and plasma membranes due to the deletion of endocytosis domains.50 However, depending on the Env, the truncation can reduce the integrity of the receptor binding form of Env.51 Like gp160, gp150 elicits heterologous tier 1 nAbs and low titer, but frequently transient, tier 1b and tier 2 nAbs.41, 42 In addition, like the gp160 native form of Env, the predominant binding Ab elicited by gp150 is to gp41 and the highly conserved IDR of gp41. The MVA‐expressed gp150 has advanced through phase IIa trials where it has served as the boost for the VLP DNA prime.48 The IgG elicited by the DNA prime/MVA boost for gp41, including the IDR, is much more durable than IgG to the gp120 subunit, with IgG1 having a longer half‐life than IgG3.43
SOSIPs and sgp140
SOSIPs, which are disulfide‐stabilized cleaved, trimeric forms of gp140, display native‐like receptor binding forms of gp120.52 SOSIPs are exceptionally useful because they represent the one form of purified protein that retains a native‐like CD4 binding form of Env. SOSIPS are produced in cultured cells and purified for use as protein immunogens. Non‐SOSIP gp140s also are used as immunogens. Attempts to stabilize non‐SOSIP gp140 forms of Env by removing the cleavage site between gp120 and gp41 can result in “sprung” Envs that have lost native structures.53 SOSIPs elicit heterologous tier 1 and homologous tier 1b and tier 2 neutralizing antibody whereas non‐SOSIP gp140 trimers primarily boost heterologous tier 1 nAb and pnnAb responses.54 SOSIPS are targeted to enter phase I testing in Q4 2018. A non‐SOSIP gp140 trimer in aluminum phosphate is being used in the ongoing phase IIb HVTN 705 Ad26/gp140 protein boost trial (Figure 1 and Table 1).
Gp120 monomers
The gp120 monomers have elicited tier 1 nAbs directed to the V3 loop in multiple vaccine trials. Despite showing good binding to soluble CD4, gp120 has not been effective at eliciting binding Ab that blocks the CD4bs on intact, native Env. This is hypothesized to reflect the angle of approach to the CD4bs in the monomer being a poor mimic of the angle of approach in the native trimer.55 One of the correlates for reduced risk in RV144 was Ab to the V1V2 loop of gp120, which in this trial seemed to have been elicited primarily by the A244 component of the bivalent gp120.56 Most gp120s do not elicit much V1V2 binding Ab (for example, see Ref. 42). However, gp120 does consistently elicit Abs to the C1 region of Env, which is a target for ADCC.22, 29 Gp120 is generally used as a boost for DNA‐primed or vector‐primed responses (see Figure 1). It has, however, been used as a prime and boost in a preclinical trial of a pentavalent gp120 that showed some protection attributed to pnnAb.57 A canarypox‐expressed membrane‐anchored gp120 displayed on VLPs and bivalent gp120 protein were used in the partially successful RV144 trial and are being used in the ongoing HVTN 702 efficacy trial (Figure 1 and Table 1). The Ab responses to gp120 generally have short half‐lives. In RV144, these underwent a more than 10‐fold contraction from peak responses in 6 months, with protection waning with the waning of the gp120 response.15
CD4i gp120
The CD4i gp120 monomers may be more capable of eliciting pnnAbs then noninduced monomers. The most developed of these, termed IHV01 or full‐length single chain, consists of HIV‐Bal gp120 tethered to domains one and two of CD4.58, 59 In contrast to most gp120s, the CD4i gp120 raises good titers of binding Ab to the V1V2 region of Env, which is exposed in the induced form. Protective responses in trials using simian prototypes of IHV01 have depended on achieving a “sweet spot” for CD4+ T cell and pnnAb responses. Elicitation of too many CD4+ T cells, which are preferred targets for infection,60 and not enough Abs, does not provide protection.3, 4 The CD4i gp120 (IHV01) is being tested in the clinic in ongoing phase I safety trials. The Ab to CD4i gp120, like Ab to gp120, is short‐lived.4
Heavily engineered recombinant Envs
With the identification of each class of bnAb, researchers have hastened to identify the germ‐line Ig sequence that initiated the lineage for the bnAb (for comprehensive review see Ref. 61). As germ line sequences, termed unmutated common ancestors (UCAs), are identified, or inferred, recombinant Env proteins that can stimulate these UCAs are sought. For example, eOD‐GT8, a highly redesigned outer domain of the HXB2g120, and 426C, a modified and truncated gp120 of the primary clade C virus 426, have been developed to stimulate UCAs for bnAb to the CD4 binding site. Another approach to eliciting bnAb to the CD4bs uses serial Envs that co‐evolved with the elicitation of bnAb in an infected patient.62 In patient CH0505 the transmitted/founder (T/F) Env weakly binds to one of the UCAs for bnAb to the CD4bs. The CH0505 T/F Env as well as Envs from major nodes for broadening of the neutralizing Ab response are now being explored for the ability to initiate and direct a bnAb response to the CD4bs. For elicitation of bnAb to the V1V2 apex of Env, both SOSIPs and small scaffolded proteins are being used. Elicitation of bnAb targeting the N332 supersite at the base of the V3 loop is being approached using mutagenized Envs (expressed on the surface of mammalian cells) for binding to inferred germ line Ab. As inferred germ line sequences are identified, additional designer Envs (frequently with mutations in N‐linked glycosylation sites) are tested for their ability to drive the response toward bnAb. The furthest of these highly engineered Envs in clinical development is the eOD‐GT8‐60mer for the CD4bs.63 The eOD‐GT8 is targeted to enter clinical trials in Q2 2018.
Gp41 peptides and mutated gp41
Recently, an epitope‐based vaccine for the fusion domain of gp41 distinguished itself by being the first to raise bnAb in NHP.64 In a tour de force of sequence analyses and X‐ray crystallography, a patient‐derived bnAb to the fusion peptide guided iterative immunizations with fusion peptide scaffolds followed by SOSIPs to generate the fusion‐peptide specific bnAb. The gp41 sequences have also been used for immunizations using virosomes (phospholipid membrane vesicles with virus‐derived proteins that support fusion with target cells).65 In the HVTN 505 DNA/Ad5 clinical trial (Figure 1), a mutated form of gp4166 raised Abs that cross‐reacted with commensal bacteria.67 This cross‐reactivity has not been observed in immunization with gp41 expressed in gp160 and gp150 forms of Env43 and may be specific to the mutant gp41 expressed by the 505 immunogens.
Current goals of research on the Ab component for an AIDS vaccine
At present, the feasible Ab to be elicited by an AIDS vaccine is pnnAb (Box 2). However, work on controlling the subgroup, isotype, and magnitude of pnnAbs is needed to optimize its ability to stimulate protective innate responses. The most protective binding Abs for HIV are IgG3 or IgG1, with IgG3 being more protective than IgG1. Serum IgA can block the protective activity of IgG1 and IgG3.24 Empirically, it has been found that ALVAC priming followed by ALVAC + bivalent gp120 boosting elicits more serum IgA than IgG24; whereas DNA priming and MVA boosting elicits much higher serum IgG than IgA.43 Presumably, this reflects the T cell help that is elicited by these two regimens differentially affecting IgG class switching. Another challenge faced by pnnAb is prozone effects, where high levels of elicited Abs compete for binding, diminishing the activation of protective innate responses.29, 30, 31 Indeed, such could explain the boosting of protective DNA/MVA responses with gp120 subunits resulting in less protection despite higher Abs.68
Box 2. Research priorities for an HIV vaccine.
| Areas of research for the Ab component of an HIV vaccine | |
| pnnAb | Maximize elicitation of protective subgroups of IgG (IgG3 and IgG1) |
| Minimize elicitation of non‐protective isotypes/subgroups of Ab (IgA, IgG2, IgG4) | |
| Understand prozone effects for protective Fc‐initiated innate responses | |
| bnAb | Identification/initiation of responses from unmutated common ancestors |
| Broadening of initiated responses by directed‐lineage immunizations | |
| pnnAb and bnAb | Identification of immunogens/regimens that elicit durable Ab |
| Use of mixtures of immunogens to broaden responses | |
| Areas of research for the T cell component of an HIV vaccine | |
| CD4+ helper T cells | Identification of vectors/regimens that do not elicit susceptible CD4+ T cells |
| Identification of adjuvants that do not enhance susceptible CD4+ T cells | |
| CD8+ cytolytic T cells | Increasing the dominance of conserved element sequences |
| Use of mosaic sequences to broaden responses for transmitted viruses | |
| CMV vectors for eliciting MHC‐II and MHC‐E restricted cytolytic T cells | |
Ab, antibody; pnnAb, protective non‐neutralizing antibody; bnAb, broadly neutralizing antibody; CD4, receptor for HIV and cell surface marker for helper cells; CD8, cell surface marker for cytolytic T cells; CMV, cytomegalovirus; HIV, human immunodeficiency virus; IgG, immunoglobulin; MHC, major histocompatibility complex.
The use of directed lineages to elicit bnAb remains a challenge for even the best and the brightest of laboratories (Box 2). The bnAbs can be elicited in knock‐in mice, which have high frequencies of critical B‐cell precursors; but, with the exception of bnAb to the gp41 fusion protein,64 remains to be achieved in NHPs, and ultimately in humans.61 The challenge is developing immunogens that can initiate and direct responses from the several dozen UCAs that have been identified in the past 11 years. Mixtures of immunogens are also being studied for their ability to broaden nAb responses (for examples, see Refs. 54, 57, 69). To date, such broadening has been achieved for pnnAb and tier 1 nAb (but not bnAb) responses.
A major challenge for both pnnAb and bnAb is identification of antigens, adjuvants, and regimens that elicit durable long‐lived Ab responses (Box 2). In RV144, protection waned concomitant with the rapid waning of Abs. At peak Ab response, protection was 60.5%, not the 31.2% present at 1.5 years.15 In humans, Ab responses to gp41 are more durable than Ab responses to gp120. In the first 6 months post‐peak Ab responses of a DNA/MVA vaccine, Ab to gp41 contracted less than twofold as opposed to the ~10‐fold contraction of Ab to gp120.43 Adjuvants also affect the durability of Ab responses. In humans, MF59 (a squalene emulsion) elicits more durable Abs than aluminum salts, particularly in infants.70 Late gp120 boosts are currently being explored as a method of achieving better durability of gp120 Abs in RV144 participants as well as in participants in DNA/MVA vaccine trials. Excellent boosting has occurred for both regimens.71, 72 Following late boosts of RV144, Ab has undergone additional heavy chain mutations (from 2.9–6.7% of the amino acids being mutated), increased ADCC activity and increased breadth of V2 responses, but has not achieved broad neutralization.71 It remains to be reported how the late boosts affected the isotype and subgroup of boosted Abs that are so critical for pnnAbs.
Passive transfer of Ab to achieve bnAb, antibody‐mediated protection
Given the complexity of the multi‐antigen regimens being developed for the elicitation of bnAb, researchers have turned to directly providing antibody‐mediated protection (AMP) by gene therapy or passive transfer of bnAb. The gene therapy approach uses vectors to express bnAbs in the subject being “vaccinated” (for reviews, see Refs. 73, 74). A major problem faced by gene therapy is recognition of the expressed Abs as foreign and clearance of Ab‐expressing cells by the host.
The passive transfer approach uses repeated transfusions to achieve and maintain protective levels of a bnAb.75 Challenges faced by this approach include: (i) the repetitive transfusions required to maintain protective levels of a bnAb (estimated at 10–20 μg/mL for the VRC01 bnAb); (ii) the cost of manufacturing recombinant Ab; (iii) the need for skilled medical personnel to administer the passive Ab; and (iv) the time required for the recipient to be transfused with a protective dose of Abs. AMP is being actively pursued to assess whether it can provide protection to an at‐risk population; and, if so, the level of Ab needed to provide protection. While these studies are ongoing, Fc region mutations are being introduced to extend the half‐life of the transfused Abs. The most popular of these mutates the neonatal Fc receptor to allow the transfused Abs to be salvaged from lysosomal degradation and recycled to the circulation.76 The breadth and potency of the transfused Abs are also being enhanced by the development of combinations of bnAb, of bispecific recombinant Abs, and most recently of trispecific recombinant Ab.77, 78, 79 The goal is to identify Ab and a regimen that allows trimonthly, or even less frequent, subcutaneous administrations of the protective Abs. Precedence for AMP is found in treatments for multiple autoimmune and allergic diseases and for respiratory syncytial virus and hepatitis B virus infections in infants. Only time will tell whether AMP can become a realistic long‐term prophylactic for prevention of HIV. Its use, however, is likely to be highly beneficial for limiting maternal transmission during breast feeding.
Areas of research for T cell vaccines
The high mutability of HIV and consequent potential for escape pose challenges for the CD8+ cytolytic T cell components of candidate vaccines. Two approaches are being taken to cope with sequence diversity: the use of highly conserved sequences for which mutations are lethal80 and the use of mosaic sequences designed to immunize for the totality of escape mutants (essentially all HIV sequences)81(Box 2). An essential resource for defining conserved sequences, and all viable mutants, is the open access Los Alamos Compendium of >95,000 HIV sequences. Definition of conserved elements is achieved by sequence alignment. Encompassing all possible mutations uses a genetic algorithm to optimize epitope coverage in mosaic cocktails of synthetic sequences.81 Empirical testing of mosaics, consensus sequences, and natural sequences for CD8+ T cell responses demonstrated that the mosaic had the best breadth (coverage of epitopes) and depth (coverage of mutants within an epitope) and ability to recognize circulating strains.82 Inclusion of three or four mosaic sequences optimizes CD8+ T cell coverage.82 A four‐mosaic combination with a natural gp140 boost is currently in phase IIb trials (Table 1).
Another approach to improving the T cell component for HIV vaccines uses cytomegalovirus (CMV) vectors modeled on the 68‐1 rhesus macaque CMV vector. This vector underwent a spontaneous mutation during fibroblast adaptation, resulting in the potential to elicit CD8+ T cells that are restricted by major histocompatibility (MHC)‐II and MHC‐E, and not just MHC‐1.83, 84 This results in very broad targets for CD8+ T cell activity that limit escape and are not affected by prior escape of MHC‐I restricted epitopes. The net result is clearance of virus from 55% of animals that have been vaccinated, challenged, and undergone an initial post‐challenge infection.85 A prototype human CMV vector, “P3,” has been constructed and is anticipated to enter human testing in 2019.
Vaccines and AMP in ongoing efficacy trials
Four ongoing efficacy trials include two for vaccines and two for AMP (Table 1). In the first vaccine trial (HVTN 702), two ALVAC primes followed by three ALVAC + gp120 boosts, test clade C versions of the RV144 immunogens for the ability to protect South African adults. Because of the rapid waning of protection with the waning of Ab in RV144,15 a third protein boost at week 52 has been added to the regimen used in RV144. Other differences from RV144 include the use of MF59 instead of alum as the adjuvant.86 Unfortunately, the gp120 proteins being used in HVTN 702 are not particularly effective at eliciting Abs to V1V2 (a major correlate for reduced risk in RV14418). Such was a fortuitous characteristic of the A244 gp120 in RV144 and not appreciated at the time of the choice and manufacture of the gp120 immunogens for HVTN 702. The trial is sponsored by NIAID and funded by NIAID, the Bill and Melinda Gates Foundation and the republic of South Africa.
The second vaccine tests priming at weeks 0 and 12 with a tetravalent mosaic sequence expressed by Ad26 followed by boosting with the tetravalent Ad26 and a clade C gp140 in aluminum phosphate at weeks 24 and 52. This trial (HVTN 705) is being conducted in young women in South Africa, Zambia, and Zimbabwe, a sub‐Saharan population at particularly high risk for HIV infection. The regimen being used has shown good protection in macaques against serial rectal challenges with a pathogenic clade B simian‐human immunodeficiency virus.87 This protection correlated with pnnAb. The vaccine is anticipated to have cross‐clade CD8+ T cell responses due to its use of mosaic sequences. This vaccine is sponsored by Janssen and Janssen B.V. and funded by the NIAID, Janssen and Janssen, and the Ragon Institute. Other vaccines, for example, DNA priming and MVA‐boosting with VLP‐expressing vaccines,48 are progressing toward efficacy testing.
The first two efficacy trials for AMP test VRC01, a broadly neutralizing recombinant Ab to the CD4bs, for the ability to prevent infection in sub‐Saharan women and in MSM and transgender women who have sex with men in the United States, Brazil, Peru, and Switzerland. The target population in South Africa has an estimated infection rate of 5% per year. In the Americas and Switzerland, the estimated infection rate is 3% per year. Both AMP trials are sponsored by the NIAID and both are being conducted jointly by the HVTN and the HIV Prevention Trial Network (HPTN). Both test 20 or 30 mg/kg of VRC01 transfused 10 times at 8‐week intervals. The primary test for efficacy is prevention of acquisition by week 80 (the time of the last transfusion).75 Goals for the AMP trials are to learn whether passive transfer can prevent HIV infection in homosexual and heterosexual partners and, if it can prevent infection, define the level of Ab that is needed to protect these cohorts.
Young age and pre‐existing Ab, potential openings for HIV vaccination?
It has long been known that young age (under 1 year) and advancing age (over 50 years) affect the efficacy of vaccination. Provocatively, immunization of infants (but not those advanced in age) may be more effective than immunization of young adults for eliciting both bnAb and pnnAb.88, 89 Studies in infants also have suggested that passive Abs from an infected mother may enhance the efficiency of vaccination.89 Vaccination of infants, potentially in the presence of passive Abs, a regimen that is used for hepatitis B virus needs to be further explored to determine if it might provide a much‐needed end run to vaccination for HIV.
Concluding challenges
Despite the progress that HIV vaccines have made in the last 11 years,90 the field continues to face enormous challenges. Broadly neutralizing Ab has been sufficiently difficult to raise, that passive transfer of bnAb is being considered as a viable alternative to vaccination. Can modern technology enable routine passive transfer of a cocktail of bnAb to millions of individuals? We should not despair, but this is not a trivial challenge. Meanwhile, the more achievable vaccines that rely on pnnAb have the challenges of Ab durability and vaccine elicited CD4+ T cells that co‐express the CCR5 co‐receptor for HIV serving as kindling for infection.4 Will adjuvants that support better durability of elicited pnnAb also create more target CD4+ T cells? Will continual boosting to maintain Ab titers drive subgroups of IgG to nonprotective IgG2 and IgG4 responses as they did in VAX 003 and VAX 004? New and better assays for the susceptibility of elicited CD4+ T cells to HIV infection are needed. Methods to boost Ab responses without driving their rearrangement to nonprotective subgroups and isotypes are needed. In the next 10 years, we will have the results of at least two more vaccine efficacy trials and two AMP trials. These will guide the path for HIV vaccines and it is hoped will lead to the exhilaration of achieved protection!
Funding
National Institutes of Health/National Institutes of Health and Infectious Diseases Grant Number 5R43AI106422—SBIR phase I, and Grant Number 2R44AI106422—SBIR phase II.
Conflict of Interest
H.L. Robinson is a developer of a DNA/MVA vaccine that has progressed through phase IIa trials and been licensed by GeoVax. She also is an employee of GeoVax and holds a minor equity interest in GeoVax.
Acknowledgments
I am indebted to Dr. J.K. McNicholl and M. Reynolds for critical review of the manuscript and S. Reuland for administrative assistance.
References
- 1. Mayr, LM & Zolla‐Pazner, S . Antibodies targeting the envelope of HIV‐1. Microbiol. Spectr. 3, AID‐0025‐2014 (2015). [DOI] [PubMed] [Google Scholar]
- 2. Burton, D.R. & Hangartner, L. Broadly neutralizing antibodies to HIV and their role in vaccine design. Annu. Rev. Immunol. 34, 635–659 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fouts, T.R. et al Balance of cellular and humoral immunity determines the level of protection by HIV vaccines in rhesus macaque models of HIV infection. Proc. Natl. Acad. Sci. USA 112, E992–E999 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lewis, G.K. , DeVico, A.L. & Gallo, R.C. Antibody persistence and T‐cell balance: two key factors confronting HIV vaccine development. Proc. Natl. Acad. Sci. USA 111, 15614–15621 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Connick, E. et al CTL fail to accumulate at sites of HIV‐1 replication in lymphoid tissue. J. Immunol. 178, 6975–6983 (2007). [DOI] [PubMed] [Google Scholar]
- 6. Keele, B.F. et al Low‐dose rectal inoculation of rhesus macaques by SIVsmE660 or SIVmac251 recapitulates human mucosal infection by HIV‐1. J. Exp. Med. 206, 1117–1134 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Boily, M.C. et al Heterosexual risk of HIV‐1 infection per sexual act: systematic review and meta‐analysis of observational studies. Lancet Infect. Dis. 9, 118–129 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gilbert, P.B. et al Correlation between immunologic responses to a recombinant glycoprotein 120 vaccine and incidence of HIV‐1 infection in a phase 3 HIV‐1 preventive vaccine trial. J. Infect. Dis. 191, 666–677 (2005). [DOI] [PubMed] [Google Scholar]
- 9. Pitisuttithum, P. et al Randomized, double‐blind, placebo‐controlled efficacy trial of a bivalent recombinant glycoprotein 120 HIV‐1 vaccine among injection drug users in Bangkok, Thailand. J. Infect. Dis. 194, 1661–1671 (2006). [DOI] [PubMed] [Google Scholar]
- 10. Buchbinder, S.P. et al Efficacy assessment of a cell‐mediated immunity HIV‐1 vaccine (the Step Study): a double‐blind, randomised, placebo‐controlled, test‐of‐concept trial. Lancet 372, 1881–1893 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gray, G.E. et al Safety and efficacy of the HVTN 503/Phambili study of a clade‐B‐based HIV‐1 vaccine in South Africa: a double‐blind, randomised, placebo‐controlled test‐of‐concept phase 2b study. Lancet Infect. Dis. 11, 507–515 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Auclair, S. et al Distinct susceptibility of HIV vaccine vector‐induced CD4 T cells to HIV infection. PLoS Pathog. 14, e1006888 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Burton, D.R. et al Public health. A sound rationale needed for phase III HIV‐1 vaccine trials. Science 303, 316 (2004). [DOI] [PubMed] [Google Scholar]
- 14. Rerks‐Ngarm, S. et al Vaccination with ALVAC and AIDSVAX to prevent HIV‐1 infection in Thailand. N. Engl. J. Med. 361, 2209–2220 (2009). [DOI] [PubMed] [Google Scholar]
- 15. Robb, M.L. et al Risk behaviour and time as covariates for efficacy of the HIV vaccine regimen ALVAC‐HIV (vCP1521) and AIDSVAX B/E: a post‐hoc analysis of the Thai phase 3 efficacy trial RV 144. Lancet Infect. Dis. 12, 531–537 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Koup, R.A. et al Priming immunization with DNA augments immunogenicity of recombinant adenoviral vectors for both HIV‐1 specific antibody and T‐cell responses. PLoS One 5, e9015 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hammer, S.M. et al Efficacy trial of a DNA/rAd5 HIV‐1 preventive vaccine. N. Engl. J. Med. 369, 2083–2092 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Haynes, B.F. et al Immune‐correlates analysis of an HIV‐1 vaccine efficacy trial. N. Engl. J. Med. 366, 1275–1286 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chung, A.W. et al Polyfunctional Fc‐effector profiles mediated by IgG subclass selection distinguish RV144 and VAX003 vaccines. Sci. Transl. Med. 6, 228ra38 (2014). [DOI] [PubMed] [Google Scholar]
- 20. Yates, N.L. et al Vaccine‐induced Env V1‐V2 IgG3 correlates with lower HIV‐1 infection risk and declines soon after vaccination. Sci. Transl. Med. 6, 228ra39 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rolland, M. et al Increased HIV‐1 vaccine efficacy against viruses with genetic signatures in Env V2. Nature 490, 417–420 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ferrari, G. et al An HIV‐1 gp120 envelope human monoclonal antibody that recognizes a C1 conformational epitope mediates potent antibody‐dependent cellular cytotoxicity (ADCC) activity and defines a common ADCC epitope in human HIV‐1 serum. J. Virol. 85, 7029–7036 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bonsignori, M. et al Antibody‐dependent cellular cytotoxicity‐mediating antibodies from an HIV‐1 vaccine efficacy trial target multiple epitopes and preferentially use the VH1 gene family. J. Virol. 86, 11521–11532 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tomaras, G.D. et al Vaccine‐induced plasma IgA specific for the C1 region of the HIV‐1 envelope blocks binding and effector function of IgG. Proc. Natl. Acad. Sci. USA 110, 9019–9024 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lin, L. et al COMPASS identifies T‐cell subsets correlated with clinical outcomes. Nat. Biotechnol. 33, 610–616 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hraber, P. , Seaman, M.S. , Bailer, R.T. , Mascola, J.R. , Montefiori, D.C. & Korber, B.T. Prevalence of broadly neutralizing antibody responses during chronic HIV‐1 infection. AIDS 28, 163–169 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Balasubramanian, P. et al Functional antibody response against V1V2 and V3 of HIV gp120 in the VAX003 and VAX004 vaccine trials. Sci. Rep. 8, 542 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Karnasuta, C. et al Comparison of antibody responses induced by RV144, VAX003, and VAX004 vaccination regimens. AIDS Res. Hum. Retroviruses 33, 410–423 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pollara, J. et al High‐throughput quantitative analysis of HIV‐1 and SIV‐specific ADCC‐mediating antibody responses. Cytometry A. 79, 603–612 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Macura, N. , Zhang, T. & Casadevall, A. Dependence of macrophage phagocytic efficacy on antibody concentration. Infect. Immun. 75, 1904–1915 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Taborda, C.P. , Rivera, J. , Zaragoza, O. & Casadevall, A. More is not necessarily better: prozone‐like effects in passive immunization with IgG. J. Immunol. 170, 3621–3630 (2003). [DOI] [PubMed] [Google Scholar]
- 32. Guan, Y. et al Diverse specificity and effector function among human antibodies to HIV‐1 envelope glycoprotein epitopes exposed by CD4 binding. Proc. Natl. Acad. Sci. USA 110, E69–E78 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pancera, M. et al Structure of HIV‐1 gp120 with gp41‐interactive region reveals layered envelope architecture and basis of conformational mobility. Proc. Natl. Acad. Sci. USA 107, 1166–1171 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hessell, A.J. et al Fc receptor but not complement binding is important in antibody protection against HIV. Nature 449, 101–104 (2007). [DOI] [PubMed] [Google Scholar]
- 35. Mengistu, M. , Ray, K. , Lewis, G.K. & DeVico, A.L. Antigenic properties of the human immunodeficiency virus envelope glycoprotein gp120 on virions bound to target cells. PLoS Pathog. 11, e1004772 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fernandez‐Larsson, R. , Srivastava, K.K. , Lu, S. & Robinson, H.L. Replication of patient isolates of human immunodeficiency virus type 1 in T cells: a spectrum of rates and efficiencies of entry. Proc. Natl. Acad. Sci. USA 89, 2223–2226 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Seaman, M.S. et al Tiered categorization of a diverse panel of HIV‐1 Env pseudoviruses for assessment of neutralizing antibodies. J. Virol. 84, 1439–1452 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mouquet, H. et al Polyreactivity increases the apparent affinity of anti‐HIV antibodies by heteroligation. Nature 467, 591–595 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Verkoczy, L. & Diaz, M. Autoreactivity in HIV‐1 broadly neutralizing antibodies: implications for their function and induction by vaccination. Curr. Opin. HIV AIDS 9, 224–234 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Haynes, B.F. , Kelsoe, G. , Harrison, S.C. & Kepler, T.B. B‐cell‐lineage immunogen design in vaccine development with HIV‐1 as a case study. Nat. Biotechnol. 30, 423–433 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McCurley, N.P. et al HIV transmitted/founder vaccines elicit autologous tier 2 neutralizing antibodies for the CD4 binding site. PLoS One 12, e0177863 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shen, X. et al HIV‐1 gp120 and modified vaccinia virus Ankara (MVA) gp140 boost immunogens increase immunogenicity of a DNA/MVA HIV‐1 vaccine. J. Virol. 91, e01077‐17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Buchbinder, S.P. et al Immunogenicity of a novel Clade B HIV‐1 vaccine combination: results of phase 1 randomized placebo controlled trial of an HIV‐1 GM‐CSF‐expressing DNA prime with a modified vaccinia Ankara vaccine boost in healthy HIV‐1 uninfected adults. PLoS One 12, e0179597 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu, P. et al Infectious virion capture by HIV‐1 gp120‐specific IgG from RV144 vaccinees. J. Virol. 87, 7828–7836 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Moog, C. et al Protective effect of vaginal application of neutralizing and nonneutralizing inhibitory antibodies against vaginal SHIV challenge in macaques. Mucosal Immunol. 7, 46–56 (2014). [DOI] [PubMed] [Google Scholar]
- 46. Burton, D.R. et al Limited or no protection by weakly or nonneutralizing antibodies against vaginal SHIV challenge of macaques compared with a strongly neutralizing antibody. Proc. Natl. Acad. Sci. USA 108, 11181–11186 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lewis, G.K. , Pazgier, M. & DeVico, A.L. Survivors remorse: antibody‐mediated protection against HIV‐1. Immunol. Rev. 275, 271–284 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Goepfert, P.A. et al Specificity and 6‐month durability of immune responses induced by DNA and recombinant modified vaccinia Ankara vaccines expressing HIV‐1 virus‐like particles. J. Infect. Dis. 210, 99–110 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wyatt, L.S. , Belyakov, I.M. , Earl, P.L. , Berzofsky, J.A. & Moss, B. Enhanced cell surface expression, immunogenicity and genetic stability resulting from a spontaneous truncation of HIV Env expressed by a recombinant MVA. Virology 372, 260–272 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Egan, M.A. , Carruth, L.M. , Rowell, J.F. , Yu, X. & Siliciano, R.F. Human immunodeficiency virus type 1 envelope protein endocytosis mediated by a highly conserved intrinsic internalization signal in the cytoplasmic domain of gp41 is suppressed in the presence of the Pr55gag precursor protein. J. Virol. 70, 6547–6556 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Edwards, T.G. et al Truncation of the cytoplasmic domain induces exposure of conserved regions in the ectodomain of human immunodeficiency virus type 1 envelope protein. J. Virol. 76, 2683–2691 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sanders, R.W. & Moore, J.P. Native‐like Env trimers as a platform for HIV‐1 vaccine design. Immunol. Rev. 275, 161–182 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ringe, R.P. et al Cleavage strongly influences whether soluble HIV‐1 envelope glycoprotein trimers adopt a native‐like conformation. Proc. Natl. Acad. Sci. USA 110, 18256–18261 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bricault, C.A. et al Neutralizing antibody responses following long‐term vaccination with HIV‐1 Env gp140 in guinea pigs. J. Virol. 92, e00369‐18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chen, L. et al Structural basis of immune evasion at the site of CD4 attachment on HIV‐1 gp120. Science 326, 1123–1127 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Alam, S.M. et al Antigenicity and immunogenicity of RV144 vaccine AIDSVAX clade E envelope immunogen is enhanced by a gp120 N‐terminal deletion. J. Virol. 87, 1554–1568 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bradley, T. et al Pentavalent HIV‐1 vaccine protects against simian‐human immunodeficiency virus challenge. Nat. Commun. 8, 15711 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Fouts, T.R. , Lewis, G.K. & Hone, D.M. Construction and characterization of a Salmonella typhi‐based human immunodeficiency virus type 1 vector vaccine. Vaccine 13, 561–569 (1995). [DOI] [PubMed] [Google Scholar]
- 59. Fouts, T.R. et al Expression and characterization of a single‐chain polypeptide analogue of the human immunodeficiency virus type 1 gp120‐CD4 receptor complex. J. Virol. 74, 11427–11436 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Douek, D.C. et al HIV preferentially infects HIV‐specific CD4+ T cells. Nature 417, 95–98 (2002). [DOI] [PubMed] [Google Scholar]
- 61. Stamatatos, L. , Pancera, M. & McGuire, A.T. Germline‐targeting immunogens. Immunol. Rev. 275, 203–216 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Liao, H.X. et al Co‐evolution of a broadly neutralizing HIV‐1 antibody and founder virus. Nature 496, 8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jardine, J. et al Rational HIV immunogen design to target specific germline B cell receptors. Science 340, 711–716 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Xu, K. et al Epitope‐based vaccine design yields fusion peptide‐directed antibodies that neutralize diverse strains of HIV‐1. Nat. Med. 24, 857–867 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bomsel, M. et al Immunization with HIV‐1 gp41 subunit virosomes induces mucosal antibodies protecting nonhuman primates against vaginal SHIV challenges. Immunity 34, 269–280 (2011). [DOI] [PubMed] [Google Scholar]
- 66. Chakrabarti, B.K. et al Modifications of the human immunodeficiency virus envelope glycoprotein enhance immunogenicity for genetic immunization. J. Virol. 76, 5357–5368 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Williams, W.B. , Han, Q. & Haynes, B.F. Cross‐reactivity of HIV vaccine responses and the microbiome. Curr. Opin. HIV AIDS 13, 9–14 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Buge, S.L. et al Gp120‐alum boosting of a Gag‐Pol‐Env DNA/MVA AIDS vaccine: poorer control of a pathogenic viral challenge. AIDS Res. Hum. Retroviruses 19, 891–900 (2003). [DOI] [PubMed] [Google Scholar]
- 69. Cho, M.W. et al Polyvalent envelope glycoprotein vaccine elicits a broader neutralizing antibody response but is unable to provide sterilizing protection against heterologous simian/human immunodeficiency virus infection in pigtailed macaques. J. Virol. 75, 2224–2234 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Martinez, D.R. , Permar, S.R. & Fouda, G.G. Contrasting adult and infant immune responses to HIV infection and vaccination. Clin. Vaccine Immunol. 23, 84–94 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Easterhoff, D. et al Boosting of HIV envelope CD4 binding site antibodies with long variable heavy third complementarity determining region in the randomized double blind RV305 HIV‐1 vaccine trial. PLoS Pathog. 13, e1006182 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Joachim, A. et al Three‐year durability of immune responses induced by HIV‐DNA and HIV‐modified vaccinia virus Ankara and effect of a late HIV‐modified vaccinia virus Ankara Boost in Tanzanian volunteers. AIDS Res. Hum. Retroviruses 33, 880–888 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Fuchs, S.P. & Desrosiers, R.C. Promise and problems associated with the use of recombinant AAV for the delivery of anti‐HIV antibodies. Mol. Ther. Methods Clin. Dev. 3, 16068 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Brady, J.M. , Baltimore, D. & Balazs, A.B. Antibody gene transfer with adeno‐associated viral vectors as a method for HIV prevention. Immunol. Rev. 275, 324–333 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Gilbert, P.B. et al Basis and statistical design of the passive HIV‐1 antibody mediated prevention (AMP) test‐of‐concept efficacy trials. Stat. Commun. Infect. Dis. 9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Saxena, A. & Wu, D. Advances in therapeutic Fc engineering – modulation of IgG‐associated effector functions and serum half‐life. Front. Immunol. 7, 580 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Julg, B. et al Protection against a mixed SHIV challenge by a broadly neutralizing antibody cocktail. Sci. Transl. Med. 9, eaao4235 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wagh, K. et al Potential of conventional & bispecific broadly neutralizing antibodies for prevention of HIV‐1 subtype A, C & D infections. PLoS Pathog. 14, e1006860 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Xu, L. et al Trispecific broadly neutralizing HIV antibodies mediate potent SHIV protection in macaques. Science 358, 85–90 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kulkarni, V. et al Altered response hierarchy and increased T‐cell breadth upon HIV‐1 conserved element DNA vaccination in macaques. PLoS One 9, e86254 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Fischer, W. et al Polyvalent vaccines for optimal coverage of potential T‐cell epitopes in global HIV‐1 variants. Nat. Med. 13, 100–106 (2007). [DOI] [PubMed] [Google Scholar]
- 82. Barouch, D.H. et al Mosaic HIV‐1 vaccines expand the breadth and depth of cellular immune responses in rhesus monkeys. Nat. Med. 16, 319–323 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Bintu, L. et al Dynamics of epigenetic regulation at the single‐cell level. Science 351, 720–724 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Hansen, S.G. et al Cytomegalovirus vectors violate CD8+ T cell epitope recognition paradigms. Science 340, 1237874 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hansen, S.G. et al Profound early control of highly pathogenic SIV by an effector memory T‐cell vaccine. Nature. 473, 523–527 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Karasavvas, N. et al IgG antibody responses to recombinant gp120 proteins, gp70V1/V2 scaffolds, and a cyclicV2 peptide in Thai phase I/II vaccine trials using different vaccine regimens. AIDS Res. Hum. Retroviruses 31, 1178–1186 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Barouch, D.H. et al Evaluation of a mosaic HIV‐1 vaccine in a multicentre, randomised, double‐blind, placebo‐controlled, phase 1/2a clinical trial (APPROACH) and in rhesus monkeys (NHP 13‐19). Lancet 392, 232–243 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Goo, L. , Chohan, V. , Nduati, R. & Overbaugh, J. Early development of broadly neutralizing antibodies in HIV‐1‐infected infants. Nat. Med. 20, 655–658 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Fouda, G.G. et al Infant HIV type 1 gp120 vaccination elicits robust and durable anti‐V1V2 immunoglobulin G responses and only rare envelope‐specific immunoglobulin A responses. J. Infect. Dis. 211, 508–517 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Robinson, H.L. HIV/AIDS vaccines: 2007. Clin. Pharmacol. Ther. 82, 686–693 (2007). [DOI] [PubMed] [Google Scholar]