

Summary
Avatrombopag, an oral thrombopoietin receptor agonist, was compared with placebo in a 6‐month, multicentre, randomised, double‐blind, parallel‐group Phase 3 study, with an open‐label extension phase, to assess the efficacy and safety of avatrombopag (20 mg/day) in adults with chronic immune thrombocytopenia (ITP) and a platelet count <30 × 109/l (ClinicalTrials.gov identifier NCT01438840). The primary endpoint was the cumulative number of weeks of platelet response (platelet count ≥50 × 109/l) without rescue therapy for bleeding; secondary endpoints included platelet response rate at day 8 and reductions in the use of concomitant medications. Amongst the 49 patients randomised, avatrombopag (N = 32) was superior to placebo (N = 17) in the median cumulative number of weeks of platelet response (12·4 vs. 0·0 weeks, respectively; P < 0·0001). At day 8, a greater platelet response rate was also observed for patients treated with avatrombopag compared with placebo (65·63% vs. 0·0%; P < 0·0001), and use of concomitant ITP medications was also reduced amongst patients receiving avatrombopag. The safety profile of avatrombopag was consistent with Phase 2 studies; the most common adverse events were headache and contusion. Overall, avatrombopag was well tolerated and efficacious for the treatment of chronic ITP.
Keywords: bleeding disorders, thrombocytopenia, thrombopoietin, platelet count, platelet disorders
Current first‐line treatments for chronic immune thrombocytopenia (ITP) include agents that decrease platelet destruction (e.g., corticosteroids, intravenous gamma globulin and intravenous anti‐RHo[D]) or suppress the production of antiplatelet antibodies (e.g., immunosuppressants) (Provan et al, 2010). However, these drugs have variable, transient efficacy and significant toxicities, and relapse is common upon discontinuation (Provan et al, 2003, 2010).
Two second‐generation thrombopoietin (TPO) receptor agonists, eltrombopag and romiplostim, have been approved for the treatment of ITP in the United States and Europe. Clinical guidelines recommend their use for patients with ITP and a risk of severe bleeding who are not candidates for splenectomy, and who have failed at least one other therapy (Bussel et al, 2007; Kuter et al, 2008, 2010). Eltrombopag, a small molecule TPO receptor agonist with a half‐life of ~12 h, increases platelet counts after 8 days of daily oral dosing, with levels returning to baseline 12 days after the last dose (Jenkins et al, 2007). Romiplostim, a recombinant fusion polypeptide administered weekly via subcutaneous injection, increases platelet counts within 5 to 8 days, with levels returning to baseline after 28 days (Wang et al, 2004). Both TPO receptor agonists are generally well tolerated, with the most common adverse event (AE) observed in 6‐month clinical studies being headache (Kuter et al, 2008; Cheng et al, 2011). Eltrombopag, however, is associated with elevations in alanine aminotransferase and bilirubin, for which it has a boxed warning for the risk of severe and potentially life‐threatening hepatotoxicity, and has important restrictions relative to the timing of specific types of food intake and drug administration (http://pi.amgen.com/united_states/nplate/nplate_pi_hcp_english.pdf; https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/promacta.pdf). Both eltrombopag and romiplostim have also been associated with increases in thromboembolic events (Bussel et al, 2009; Wong et al, 2017); for romiplostim, bone marrow fibrosis has also been reported in some patients (Kuter et al, 2009; Ghanima et al, 2011).
Given the side‐effect profiles and inconvenient dosing requirements and/or route of administration of currently available therapies, there remains an important unmet medical need for novel, orally administered TPO receptor agonists, with improved safety and pharmacokinetic profiles for the treatment of patients with chronic ITP.
Avatrombopag is a small molecule TPO receptor agonist that mimics the biological effects of TPO in vitro and in vivo (Fukushima‐Shintani et al, 2008, 2009). In preclinical studies, avatrombopag stimulated the proliferation of cultured human c‐Mpl–Ba/F3 cells and promoted the differentiation of CD34+ cells from human cord blood to megakaryocytes in a concentration‐dependent manner (Fukushima‐Shintani et al, 2008, 2009). Notably, avatrombopag can be administered orally with food, has no significant hepatotoxicity, and avoids the safety and immunogenic risks of recombinant parenteral agents (Bussel et al, 2014). In Phase 2 studies, oral avatrombopag increased platelet production in patients with chronic ITP and was more effective than placebo in improving the platelet response rate on day 28 (Bussel et al, 2014). Avatrombopag was generally well tolerated, with headache, fatigue and epistaxis being the most frequently reported AEs that occurred at exposure‐adjusted incidence rates comparable with placebo (Bussel et al, 2014).
The aim of the current Phase 3 study was to demonstrate the superiority of avatrombopag to placebo in raising and maintaining platelet counts in patients with chronic ITP within a target range (50–150 × 109/l) over a 6‐month treatment period, and to assess the safety and efficacy of long‐term therapy with avatrombopag.
Methods
Study design
This Phase 3 multicentre, randomised, double‐blind, parallel‐group, placebo‐controlled study included a core study and open‐label extension phase, and evaluated the efficacy and safety of oral avatrombopag versus placebo in patients with chronic ITP (ClinicalTrials.gov identifier: NCT01438840). The study was conducted at 27 study sites in Australia, Belgium, Bulgaria, Czech Republic, New Zealand, The Netherlands, Poland, Singapore, Slovakia, South Africa and Ukraine. Efficacy was evaluated over a 6‐month treatment period, and safety was assessed for up to 76 weeks following completion of treatment. The concurrent use of other standard of care treatments for chronic ITP and rescue therapy was allowed.
The protocol, informed consent form, and other documents were reviewed and approved by the sites’ Institutional Review Board or Independent Ethics Committee. The study was conducted in compliance with the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH), and the provisions of the Declaration of Helsinki, Good Clinical Practice guidelines and local laws and regulations.
The study included the core study and the extension phase (Fig 1). The pre‐randomisation phase included a 4‐week screening period, and the core study consisted of baseline/randomisation (day 1), titration (6 weeks), concomitant ITP medication reduction period (12 weeks), maintenance (8 weeks, including the end‐of‐treatment [EOT] visit), dose tapering (4 weeks) and follow‐up (4 weeks). Dose‐tapering and follow‐up periods were only for patients who did not continue into the extension phase. The extension phase included a 6‐week conversion period, a 90‐week maintenance/concomitant ITP medication reduction period, a dose‐tapering period and follow‐up.
Figure 1.
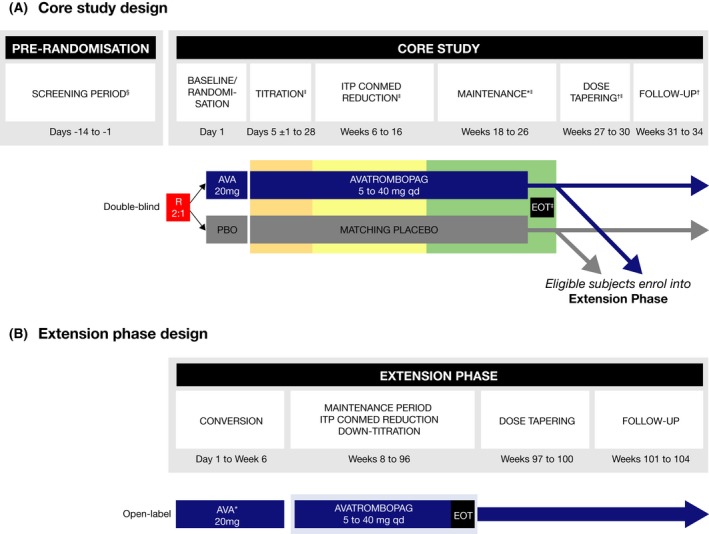
Study design. (A) Core study. (B) Extension phase. AVA, avatrombopag; CONMED, concomitant medication; E, extension; EOT, end‐of‐treatment; ITP, immune thrombocytopenia; PBO, placebo; qd, once‐daily; R, randomised. *At EOT visit (visit 22), patients could enter the extension phase and receive open‐label avatrombopag therapy. Patients who did not continue into the extension phase entered the dose‐tapering and follow‐up phase. †Only for patients who did not enter the extension phase. ‡Optional entry into the open‐label extension phase. §The screening visit and day 1 baseline/randomisation visit platelet counts were averaged to obtain the baseline platelet count value. The two samples were obtained ≥48 h and ≤2 weeks apart and the results were available prior to randomisation. Therefore, an additional screening platelet count may have been required due to issues with scheduling. ‖Patients who discontinued early who met the criteria for a lack of treatment effect may have moved directly into the open‐label extension phase. [Colour figure can be viewed at http://wileyonlinelibrary.com]
Patients
Eligible patients were males and females, ≥18 years of age, with ITP ≥12 months in duration, and an average of two platelet counts <30 × 109/l. Other inclusion criteria included a peripheral blood smear consistent with ITP and previous treatment with one or more therapies for ITP or patients must have had a bone marrow examination consistent with ITP within 3 years to rule out myelodysplastic syndrome or other cause of thrombocytopenia. Key exclusion criteria included secondary ITP, clinically significant arterial or venous thrombosis, cardiovascular disease, chronic active hepatitis, cirrhosis, gastric atrophy, malignant disease, myelodysplastic syndrome, pernicious anaemia or portal hypertension, and use of romiplostim or eltrombopag within 4 weeks of randomisation. All patients gave written informed consent prior to screening.
Pre‐randomisation phase
Screening visit and day 1 baseline/randomisation visit platelet counts (obtained ≥48 h and ≤2 weeks apart) were averaged to obtain the baseline platelet count prior to randomisation.
Randomisation and masking
Patients were centrally stratified by splenectomy status (yes or no), baseline platelet count (≤15 × 109/l or >15 to <30 × 109/l), and the use of concomitant ITP medication (yes or no), and randomised using an interactive voice and web response system to receive either avatrombopag or placebo (2:1) in a double‐blind fashion. During the randomisation phase, patients and all personnel involved with the conduct and interpretation of the study—including the investigators, investigational site personnel and sponsor staff—were blinded to the treatment codes (but not dose levels). Randomisation data were kept strictly confidential, filed securely at each site, and were accessible only to authorised persons until the time of unblinding. A master list of all treatments was maintained in a sealed envelope with the sponsor. Corresponding patient numbers associated with a specific treatment were blinded in the interactive voice and web response system database. The extension phase was not blinded.
Core study
The study drugs were packaged in a double‐blind configuration. Patients received therapy of either avatrombopag at a starting dose of 20 mg or placebo, once‐daily. The dose could be titrated up to a maximum dose of 40 mg or down to a minimum dose of 5 mg, according to pre‐specified protocol thresholds and individual response to treatment. The primary endpoint assessment was made during the maintenance period of the core study, as concomitant ITP medications were to be administered at a stable dose and downward titration of the study medication was not permitted during this period. The overall goal of the dose modifications was to maintain platelet counts ≥50 × 109/l and ≤150 × 109/l, and to reduce the need for concomitant ITP medications.
Extension phase
Patients who completed the maintenance period of the core study or discontinued from the core study early due to lack of treatment effects, and who had no significant safety or tolerability concerns, were eligible to continue into the extension phase. All patients started the extension phase with open‐label avatrombopag 20 mg, once‐daily. During the 90‐week maintenance period of the extension phase, avatrombopag dose titration and downward titration of concomitant ITP medications were allowed. At the end of the extension phase, a 4‐week, dose‐tapering period was followed by a 30‐day follow‐up after the last dose of avatrombopag.
Concomitant therapy
Patients receiving concomitant ITP medication when entering the study were allowed to have this medication down‐titrated and ultimately eliminated during the concomitant ITP medication reduction period of the core study and during the maintenance period/concomitant ITP medication reduction period of the extension phase. Down‐titration of concomitant ITP medication was implemented at the discretion of the investigator and could only be considered if the patient's platelet count remained >150 × 109/l. Patients with a platelet count >150 × 109/l to ≤250 × 109/l could have their concomitant ITP medication down‐titrated by ≤25% of the original dose for 14 days, whereas the dose was reduced by ≤50% in patients with a platelet count >250 × 109/l for 14 days. If a patient was receiving ≥2 concomitant ITP medications, only one medication was down‐titrated at a time.
Study assessments
At each visit, platelet counts were measured and the use of concomitant medication recorded to assess the use of any rescue therapy. Rescue therapy (addition of any new ITP medication) was administered at the discretion of the investigators based on clinical assessment (e.g., due to life‐threatening thrombocytopenia, a major bleeding event or signs/symptoms of a potential bleeding event). The World Health Organization (WHO) bleeding scale was used to assess the incidence and severity of bleeding events, as previously reported (Bussel et al, 2007; Kuter et al, 2008, 2010). Safety was assessed by monitoring all treatment‐emergent adverse events (TEAEs) and serious adverse events (SAEs) throughout the study. In addition, physical examinations, vital signs, electrocardiograms (ECGs), blood chemistries, haematologies, coagulation tests, a panel of gastric biomarkers and urine analyses were regularly conducted. Bone marrow biopsies were required during the study of patients who developed immature or dysplastic cells on peripheral blood smear. In addition, patients could elect to provide optional marrow biopsies during the course of the study.
Lack of treatment effect was defined as platelet counts <30 × 109/l after more than 3 weeks of treatment at the maximum dose of 40 mg. Patients could be discontinued from the core study if the investigator considered platelet counts to be dangerously low after 7 days of treatment at the maximum dose, if patients required rescue therapy more than 3 times, or if they required continuous rescue therapy for more than 3 weeks.
Efficacy endpoints
Core study
The primary efficacy endpoint was the cumulative number of weeks of platelet response, defined as a platelet count ≥50 × 109/l in the absence of rescue therapy over 6 months of once‐daily treatment in adults with ITP.
Secondary efficacy endpoints were the proportion of patients with a platelet response at day 8 and the proportion of patients with a reduction in the use of concomitant ITP medications from baseline. Exploratory efficacy endpoints included: durable platelet response rate (defined as the proportion of patients who had a platelet response for ≥6 of the last 8 weeks of treatment), the incidence and severity of bleeding events, and the proportion of patients receiving rescue therapy. The safety of avatrombopag compared with placebo was also assessed.
Extension phase
In the extension phase, the efficacy and safety of long‐term avatrombopag therapy was assessed by measuring platelet response rate, bleeding, and the use of rescue therapy.
Changes to efficacy endpoints
The study protocol was amended in March 2013, changing the primary endpoint from durable platelet response to the cumulative number of weeks of platelet response over 6 months of treatment in order to support a planned regulatory filing of avatrombopag in Japan. Platelet response at day 8 was also changed from a secondary to an exploratory endpoint, and the anticipated sample size was reduced from 100 to 45 subjects.
Determination of sample size
A resampling method, based on results from a previous 4‐week study that randomised 64 patients to placebo or avatrombopag (Bussel et al, 2014) was used to determine sample size. The proportion of placebo‐ and avatrombopag‐treated patients with cumulative number of weeks of a platelet response in that study were as follows: 0 weeks, 80% vs. 7%; 1 week, 20% vs. 0%; 2 weeks, 0% vs. 7%; 3 weeks, 0% vs. 7%; and 4 weeks, 0% vs. 80%, respectively. It was assumed that a similar treatment difference would be observed in the Phase 3 study with a longer treatment duration, and that there would be a 15% dropout rate, with all dropout patients being considered to have 0 weeks of platelet response. A sample size of 45 patients (15 placebo, 30 avatrombopag) was projected to provide at least 95% power to reject the null hypothesis of no treatment difference between avatrombopag and placebo in the cumulative number of weeks of platelet response during the core study using the Wilcoxon rank sum test (a nonparametric test) at a 0·05 significance level (2‐sided).
Statistical analysis
The full analysis set (FAS) included all randomised patients, and the safety analysis set (SAS) included all patients who received at least one dose of study medication and had a post‐dose safety assessment in either the core study or extension phase. All patients who received study medication and who provided at least one platelet count and corresponding efficacy assessment during the extension phase were included in the modified full analysis set (mFAS). Baseline demographics and characteristics of the FAS and SAS were summarised for each group using descriptive statistics and categorical variables summarised by treatment group and by frequency distribution.
Role of funding source
This study was funded by Eisai Inc. (Woodcliff Lake, NJ, USA). The corresponding author had full access to all of the data in the study and had final responsibility for the decision to submit for publication.
Results
The study was conducted between 6 February 2012 and 9 April 2015. Of 100 screened patients, 49 met eligibility criteria, were randomised to study medication (avatrombopag, N = 32; placebo, N = 17), and were included in the FAS and SAS. Twenty‐two patients (68·8%) in the avatrombopag treatment group and one (5·9%) in the placebo treatment group completed the core study (Fig 2). The most frequent reason for discontinuation from the core study was lack of treatment effect (avatrombopag, N = 7; placebo, N = 15). Thirty‐nine patients who entered the extension phase were included in the mFAS with 29 patients (74·4%) completing the extension phase and nine (23·1%) discontinuing; one patient was lost to follow‐up.
Figure 2.

Subject disposition. (A) Core study. (B) Extension phase. N/n, number of patients. [Colour figure can be viewed at http://wileyonlinelibrary.com]
Baseline demographics and characteristics were generally balanced across the avatrombopag and placebo treatment groups (Table 1), except that there was a higher percentage of avatrombopag‐treated females (71·9%) compared with placebo (47·1%; subsequent analysis suggested that the primary efficacy outcome was similar across male and female subgroups). Most patients were in the low baseline platelet count category (≤15 × 109/l; 57·1%), not splenectomised (67·3%) and were not using concomitant ITP medication (55·1%). Approximately 30% of patients had previously received five or more ITP medications.
Table 1.
Baseline demographics and patient characteristics (FAS)
| Placebo (N = 17) | Avatrombopag (N = 32) | Total (N = 49) | |
|---|---|---|---|
| Age (years), mean (SD) | 41·2 (14·7) | 46·4 (14·2) | 44·6 (14·4) |
| <65 years, N (%) | 16 (94·1) | 29 (90·6) | 45 (91·8) |
| Female, N (%) | 8 (47·1) | 23 (71·9) | 31 (63·3) |
| Race, N (%) | |||
| White | 15 (88·2) | 31 (96·9) | 46 (93·9) |
| Black or African American | 1 (5·9) | 0 | 1 (2·0) |
| Asian | 1 (5·9) | 1 (3·1) | 2 (4·1) |
| Weight (kg), mean (SD) | 84·97 (20·48) | 81·90 (22·71) | 82·97 (21·79) |
| Height (cm), mean (SD) | 170·53 (7·46) | 167·89 (8·00) | 168·81 (7·84) |
| BMI (kg/m2), mean (SD) | 29·24 (6·64) | 28·99 (7·32) | 29·08 (7·02) |
| Baseline platelet count, N (%) | |||
| ≤15 × 109/l | 10 (58·8) | 18 (56·3) | 28 (57·1) |
| 15–30 × 109/l | 7 (41·2) | 13 (40·6) | 20 (40·8) |
| ≥30 × 109/l | 0 | 1 (3·1) | 1 (2·0) |
| Splenectomy, N (%) | 5 (29·4) | 11 (34·4) | 16 (32·7) |
| Use of concomitant ITP medication at baseline, N (%) | 7 (41·2) | 15 (46·9) | 22 (44·9) |
BMI, body mass index; FAS, full analysis set; ITP, immune thrombocytopenia; N, number of patients; SD, standard deviation.
The duration of exposure for the core study was longer in the avatrombopag treatment group, with 26/32 (81·3%) patients receiving avatrombopag over 18 weeks, 17/32 (53·1%) over 26 weeks and 2/32 (6·3%) over 30 weeks. In the placebo treatment group, only 3/17 (17·6%) patients were exposed to study medication over 18 weeks, and only one was exposed to study medication for over 26 weeks.
Avatrombopag was shown to be superior to placebo in the cumulative number of weeks of platelet response, the primary study endpoint, with a significantly longer duration of a platelet count ≥50 × 109/l in the absence of rescue therapy with avatrombopag than with placebo (median: 12·4 vs. 0·0 weeks; mean: 12·0 vs. 0·1 weeks; P < 0·0001). In addition, more patients in the avatrombopag treatment group (21/32; 65·6%) had a platelet response at day 8 than in the placebo‐treated group (0/17; 0·0%; P < 0·0001; Table 2). Further, 5/15 (33·3%) patients in the avatrombopag treatment group reduced their use of concomitant ITP medication from baseline compared with 0/7 placebo‐treated patients (33·3% vs. 0%, respectively; 95% confidence interval, 9·48, 57·19) (Table 2), although this treatment difference did not reach statistical significance (N = 22; P = 0·1348) due to the small number of patients using ITP medications at baseline.
Table 2.
Summary of core study efficacy endpoints (FAS)
| Placebo (N = 17) | Avatrombopag (N = 32) | |
|---|---|---|
| Cumulative number of weeks of platelet responsea | ||
| Mean (SD) | 0·1 (0·49) | 12·0 (8·75) |
| Median | 0·0 | 12·4 |
| Min, max | 0, 2 | 0, 25 |
| P‐value of Wilcoxon rank sum test | <0·0001 | |
| Platelet count ≥50 × 109/l at day 8b | ||
| Yes (%, 95% CI) | 0·0 (‐,‐) | 65·6 (49·17, 82·08) |
| No (%) | 100·0 | 34·4 |
| Difference of response rate (95% CI)c | 65·63 (49·17, 82·08) | |
| P‐value of Fisher's exact test | <0·0001 | |
| Reduction in use of concomitant ITP medications from baselined | ||
|---|---|---|
| Placebo (N = 7) | Avatrombopag (N = 15) | |
| Yes (%, 95% CI) | 0·0 (‐,‐) | 33·3 (9·48, 57·19) |
| No (%) | 100·0 | 66·7 |
| Difference of rate of reduction (95% CI)e | 33·33 (9·48, 57·19) | |
| P‐value of Fisher's exact test | 0·1348 | |
CI, confidence interval; FAS, full analysis set; ITP, immune thrombocytopenia; N, number of patients; SD, standard deviation.
Cumulative number of weeks of platelet response is defined as the total number of weeks in which platelet count is ≥50 × 109/l during the core study in the absence of rescue therapy.
Patients with platelet response (≥50 × 109/l) at day 8 in the absence of rescue therapy on or before day 8.
Difference of response rate = platelet response rate at day 8 of avatrombopag – platelet response rate at day 8 of placebo, 95% CI is calculated based on normal approximation.
Only patients with use of concomitant ITP medications at baseline were included in this analysis.
Difference of rate reduction = rate of reduction in use of concomitant ITP medications from baseline of avatrombopag – rate of reduction in use of concomitant ITP medications from baseline of placebo, 95% CI is calculated based on normal approximation.
The durable platelet response rate was significantly greater in avatrombopag‐treated patients compared with those receiving placebo (34·4% vs. 0·0%; P = 0·009), with a treatment difference of 34·4% (95% confidence interval, 17·92, 50·83). The median platelet count by visit in avatrombopag‐treated patients was consistently higher than that of the placebo treatment group starting at day 8 (80·5 × 109/l vs. 8 × 109/l; Fig 3A), while the median platelet count for the placebo treatment group remained unchanged throughout the core study.
Figure 3.

Median (Q1, Q3) platelet count over time. (A) Core study (FAS). (B) Extension phase (modified FAS). FAS, full analysis set; ITP, immune thrombocytopenia; Med, medication; n, number of patients; Q1/3, quartile 1/3. [Colour figure can be viewed at http://wileyonlinelibrary.com]
The overall platelet response rate observed in the core study was generally maintained throughout the extension phase until around week 36 (Fig 3B). Beyond week 38, platelet response was lower and more variable, but the small number of patients (N < 15) at these time points limits further interpretation.
The incidence of any bleeding event during the core study was not statistically different between the avatrombopag and placebo treatment groups (43·8% vs. 52·9%, respectively; P = 0·5394), and was lower for avatrombopag when adjusted for the 2·6‐fold longer mean exposure time for avatrombopag‐treated patients. All bleeding events were WHO Grade 1, except for three patients in the avatrombopag treatment group who experienced Grade 2 (N = 2) or Grade 3 (N = 1) bleeding events. The WHO Grade 3 bleeding event (epistaxis) was also reported as an AE of special interest (AESI).
Although rescue therapy was required by 21·9% of avatrombopag‐treated patients and 11·8% of those who received placebo, there was no statistically significant difference in the use of rescue therapy between these groups (P = 0·4668 using Fisher's exact test). The lower use of rescue therapy by patients in the placebo treatment group is probably artefactual due to the 2·6‐fold shorter period of exposure in placebo‐treated individuals, resulting from the high rate of early discontinuations due to lack of treatment effect.
As per International Working Group criteria, a complete platelet response was defined as a platelet count ≥100 × 109/l and an absence of bleeding (Rodeghiero et al, 2009). Similarly, a platelet response was defined as a platelet count ≥30 × 109/l, with at least a two‐fold increase in platelet count from baseline and an absence of bleeding (Rodeghiero et al, 2009). According to these criteria, 28·1% and 56·3% of avatrombopag‐treated patients achieved a complete platelet response and platelet response, respectively, on day 8, compared with 0% of those treated with placebo.
The mean duration of exposure to avatrombopag (22·8 weeks) was approximately 2·6‐fold longer than that of placebo (8·9 weeks) in the core study, and the median, (26.0 weeks and 6.0 weeks, respectively) 4.3 fold longer. In the combined core study and extension phase, the mean duration of exposure to avatrombopag was 43·9 weeks; the longest exposure to avatrombopag was 75·7 weeks. Throughout the core study and extension phase, 72·3% of patients were exposed to avatrombopag for at least 32 weeks, and 29·8% received avatrombopag for at least 52 weeks.
Assessments of vital signs, ECGs, clinical laboratory evaluations and a panel of gastric biomarkers identified no safety signal.
While the overall incidence of TEAEs in the core study was higher in the avatrombopag treatment group compared with the placebo treatment group (96·9% vs. 58·8%, respectively) (Table 3), there were no clinically important differences in the exposure‐adjusted incidence rates (4·3% per patient‐week vs. 6·6% per patient‐week, respectively); the higher overall incidence of TEAEs in avatrombopag‐treated patients was probably due to the greater mean (2.6 fold) and median (4.3‐fold) duration of exposure. Similarly, the overall incidence of SAEs in the core study was higher in the avatrombopag treatment group compared with placebo‐treated patients (28·1% vs. 5·9%), again, probably due to the increased duration of exposure to avatrombopag. Importantly, nearly all SAEs in avatrombopag‐treated patients occurred in only single patients. Again, exposure‐adjusted incidence rates for SAEs were comparable in the core study — 1·2% per patient‐week and 0·7% per patient‐week for the avatrombopag and placebo treatment groups, respectively (Table 3).
Table 3.
Overview of TEAEs during the core study and extension phase (SAS)
| Core study | Core + extension phase | |||||
|---|---|---|---|---|---|---|
| Incidence | Exposure‐adjusted incidence ratea | Incidence | Exposure‐adjusted incidence ratea | |||
| Placebo (N = 17) N (%) | Avatrombopag (N = 32) N (%) | Placebo (N = 17) % | Avatrombopag (N = 32) % | Avatrombopag (N = 47) N (%) | Avatrombopag (N = 47) % | |
| TEAEs, N (%) | 10 (58·8) | 31 (96·9) | 6·6 | 4·3 | 45 (95·7) | 2·2 |
| Treatment‐related TEAEsb, N (%) | 3 (17·6) | 20 (62·5) | 2·0 | 2·7 | 31 (66·0) | 1·5 |
| TEAE with CTCAE Grade 3 or 4, N (%) | 0 | 6 (18·8) | 0 | 0·8 | 14 (29·8) | 0·7 |
| SAEs, N (%) | 1 (5·9) | 9 (28·1) | 0·7 | 1·2 | 15 (31·9) | 0·7 |
| Deaths | 0 | 0 | 0 | 0 | 0 | 0 |
| Other SAEs | 1 (5·9) | 9 (28·1) | 0·7 | 1·2 | 15 (31·9) | 0·7 |
| TEAEs leading to study‐drug dose adjustment, N (%) | 0 | 5 (15·6) | 0 | 0·7 | 11 (23·4) | 0·5 |
| Withdrawal | 0 | 3 (9·4) | 0 | 0·4 | 6 (12·8) | 0·3 |
| Dose increase | 0 | 1 (3·1) | 0 | 0·1 | 3 (6·4) | 0·1 |
| Dose reduction | 0 | 1 (3·1) | 0 | 0·1 | 2 (4·3) | 0·1 |
| Dose interruption | 0 | 0 | 0 | 0 | 0 | 0 |
AE, adverse event; CTCAE, Common Terminology Criteria for Adverse Events; N, number of patients; SAE, serious adverse event; SAS, safety analysis set; TEAE, treatment‐emergent adverse event. A TEAE (during core study) was defined as an AE that had an onset date (if not present pre‐treatment) or date of worsening in severity relative to pre‐treatment state, on or after the first dose of study medication, up to 30 days after the last dose of study medication during the core study (and before the first dose of extension phase if the patient continued into the extension phase). For each row category, a patient with 2 or more AEs in that category was counted only once.
Exposure‐adjusted incidence rate = number of events/total patient‐weeks exposure × 100%.
Includes TEAEs considered by the investigator to be possibly or probably related to study drug or TEAEs with missing causality.
The same pattern regarding overall incidence and exposure‐adjusted incidence rates was observed for treatment‐related TEAEs, Grade 3/4 AEs and TEAEs leading to study medication dose adjustment (Table 3). Four patients who received avatrombopag experienced Grade 3 TEAEs, which included single SAEs of epistaxis, petechiae, headache and platelet count reduction, which were considered related to study medication. There were only two Grade 4 TEAEs in the core study; one patient with a cerebrovascular accident who discontinued study medication, and the other with worsening ITP that was considered not related to study medication. There were no deaths during the study in any treatment group. The exposure‐adjusted incidence rates for all TEAE categories were comparable in the extension phase of the study.
The most commonly reported TEAEs in the avatrombopag treatment group in the core study were headache, contusion, upper respiratory tract infection, arthralgia, epistaxis, fatigue, gingival bleeding and petechiae, with exposure‐adjusted incidence rates that were all comparable with, or lower than, placebo (Table 4). The only additional TEAEs reported in the open‐label extension phase were a low incidence of thrombocytopenia, hypertension, pharyngitis and nasopharyngitis. With the exception of the two SAEs of vomiting and headache, which were reported in two (6·3%) patients each, all other SAEs in avatrombopag‐treated patients in the core study occurred in only individual patients (Table 4); no safety signal was identified. The only additional SAE reported in the extension phase was thrombocytopenia/platelet count decreased.
Table 4.
Most frequent TEAEs and SAEs during core study and extension phase (SAS)
| Core study | Core + extension phase | |||||
|---|---|---|---|---|---|---|
| Incidence | Exposure‐adjusted incidence ratea | Incidence | Exposure‐adjusted incidence ratea | |||
| Placebo (N = 17) N (%) | Avatrombopag (N = 32) N (%) | Placebo (N = 17) % | Avatrombopag (N = 32) % | Avatrombopag (N = 47) N (%) | Avatrombopag (N = 47) % | |
| Any TEAE | 10 (58·8) | 31 (96·9) | 6·6 | 4·3 | 45 (95·7) | 2·2 |
| Headache | 2 (11·8) | 12 (37·5) | 1·3 | 1·6 | 14 (29·8) | 0·7 |
| Contusion | 4 (23·5) | 10 (31·3) | 2·6 | 1·4 | 19 (40·4) | 0·9 |
| Upper respiratory tract infection | 1 (5·9) | 6 (18·8) | 0·7 | 0·8 | 11 (23·4) | 0·5 |
| Arthralgia | 0 (0) | 4 (12·5) | 0 | 0·5 | 5 (10·6) | 0·2 |
| Epistaxis | 3 (17·6) | 4 (12·5) | 2·0 | 0·5 | 8 (17·0) | 0·4 |
| Fatigue | 1 (5·9) | 4 (12·5) | 0·7 | 0·5 | 7 (14·9) | 0·3 |
| Gingival bleeding | 0 (0) | 4 (12·5) | 0 | 0·5 | 8 (17·0) | 0·4 |
| Petechiae | 1 (5·9) | 4 (12·5) | 0·7 | 0·5 | 7 (14·9) | 0·3 |
| Thrombocytopenia | 0 (0) | 2 (6·3) | 0 | 0·3 | 9 (19·1) | 0·4 |
| Pharyngitis | 1 (5·9) | 0 (0) | 0·7 | 0 | 6 (12·8) | 0·3 |
| Hypertension | 1 (5·9) | 2 (6·3) | 0·7 | 0·3 | 5 (10·6) | 0·2 |
| Nasopharyngitis | 0 (0) | 3 (9·4) | 0 | 0·4 | 5 (10·6) | 0·2 |
| Any SAE | 1 (5·9) | 9 (28·1) | 0·7 | 1·2 | 15 (31·9) | 0·7 |
| Headache | 0 (0) | 2 (6·3) | 0 | 0·3 | 2 (4·3) | 0·1 |
| Vomiting | 0 | 2 (6·3) | 0 | 0·3 | 2 (4·3) | 0·1 |
| Platelet count decreased | 0 | 1 (3·1) | 0 | 0·1 | 2 (4·3) | 0·1 |
AE, adverse event; N, number of patients; SAE, serious adverse event; SAS, safety analysis set; TEAE, treatment‐emergent adverse event.
A TEAE (during core study and extension phase) was defined as an AE that had an onset date (if not present pre‐treatment) or date of worsening in severity relative to pre‐treatment state, on or after the first dose of study medication, up to 30 days after the last dose of study medication during the core study and extension phase. A SAE was defined as any untoward medical occurrence that at any dose resulted in death, was life‐threatening, required inpatient hospitalisation or prolongation of existing hospitalisation, resulted in persistent or significant disability/incapacity, or caused a congenital anomaly/birth defect in the child of an exposed parent. Patients with ≥2 AEs in the same preferred terms was counted only once for that preferred term.
Exposure‐adjusted incidence rate = number of events/total patient‐weeks exposure × 100%.
AESIs were prospectively defined and collected, including the recurrence of thrombocytopenia (defined as a platelet count <10 × 109/l and >10 × 109/l below baseline), thromboembolic events, bleeding events (WHO Grade 3 or 4), neoplastic events, gastric atrophy events, bone marrow pathology and clinically significant liver tests. No AESIs were reported in placebo‐treated patients although the mean duration of treatment in this treatment group was very short (8·9 weeks). No avatrombopag‐treated patients had gastric atrophy events or bone marrow pathology. One patient treated with 40 mg avatrombopag had an AE Grade 3 increase in liver tests (according to the Common Terminology Criteria for Adverse Events, v. 4·03; https://www.eortc.be/services/doc/ctc/CTCAE_4?03_2010-06-14_QuickReference_5x7.pdf) in the core study on day 82 that required no dose adjustment or treatment, and returned to normal on continued avatrombopag dosing; this patient had a history of fatty liver, hepatitis C, obesity, gallstones and past liver function test elevations, and reported the use of alcohol at the time the liver function abnormalities developed. The investigator considered this event unrelated to treatment with avatrombopag. Recurrence of thrombocytopenia, neoplastic events, and bleeding events (epistaxis) were each reported in one avatrombopag‐treated patient (3·1%) over the 26‐week core study. Three patients (9·4%) reported a thromboembolic event in the core study: a deep vein thrombosis (day 8), an asymptomatic pulmonary embolism (day 154) and a cerebrovascular event (day 89). One additional thromboembolic event of jugular vein thrombosis (day 335) was reported in the open‐label extension phase. Of the four patients who reported thromboembolic events, three had multiple risk factors for thromboembolic disease and the reported events were associated with platelet counts from 39–271 × 109/l and were associated with doses of avatrombopag ranging from 10 to 40 mg.
Discussion
Avatrombopag, a novel TPO receptor agonist currently under development for the treatment of chronic ITP, demonstrated efficacy and safety in Phase 2 studies (Bussel et al, 2014; Terrault et al, 2014). These Phase 3 data confirm the safety and efficacy of avatrombopag, which was shown to be superior to placebo as measured by the cumulative number of weeks of platelet response (platelet count ≥50 × 109/l) over 6 months (26 weeks) of once‐daily treatment in adult patients with chronic ITP. Avatrombopag treatment resulted in a median of 12·4 cumulative weeks of platelet response during the core study and was also shown to be superior to placebo (0 weeks), with a greater platelet response rate in avatrombopag‐treated patients at day (65.6 versus 0%, respectively).
Direct stimulation of the TPO signalling pathway to increase platelet production with TPO receptor agonists has proven efficacious in treating patients with chronic ITP (Bussel et al, 2007; Kuter et al, 2008, 2010), supporting their use as second‐line therapies for patients unresponsive to initial treatments (Wang et al, 2016). Efficacy data on the cumulative number of weeks with a platelet count ≥50 × 109/l and the durable platelet response have been reported for eltrombopag and romiplostim (Cheng et al, 2011; Kuter et al, 2008; http://pi.amgen.com/united_states/nplate/nplate_pi_hcp_english.pdf; Amgen Inc., 2017; https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/promacta.pdf). However, some concerns remain over the long‐term use of TPO receptor agonists (Bussel et al, 2009; Kuter et al, 2009; Ghanima et al, 2011; Wong et al, 2017).
In this Phase 3 study, avatrombopag was shown to be well tolerated with a safety profile that was similar to placebo when adjusted for the duration of treatment exposure. Safety findings were consistent with Phase 2 studies and with 6‐month studies with other TPO receptor agonists, with the most frequently reported AE being headache (Bussel et al, 2007, 2014; Kuter et al, 2008, 2010; Cheng et al, 2011). Although eltrombopag and romiplostim are generally considered to be well tolerated, eltrombopag has a boxed warning for the risk of severe and potentially life‐threatening hepatotoxicity, requires routine monitoring of liver function and has restrictions relative to dosing, while romiplostim must be administered parenterally and may increase the risk for development/progression of bone marrow reticulin fibre formation. In contrast, avatrombopag can be administered orally without regard to food type and, importantly, has not been shown to be associated with significant hepatotoxicity.
There was a low incidence of AESI reported with avatrombopag in the Phase 3 core study, with only one avatrombopag‐treated patient reporting a bleeding event (WHO Grade 3 or higher) or recurrence of thrombocytopenia. The three thromboembolic events reported in the core study occurred in single patients and were heterogeneous in nature (one deep vein thrombosis, one pulmonary embolism and one cerebrovascular accident). However, the study population was not large enough to rule out an increased incidence of thromboembolic events with avatrombopag; this will need to be closely monitored in ongoing and future studies.
These Phase 3 data extend the previously reported safety and efficacy findings from prior Phase 2 studies (Bussel et al, 2014) and, taken together, support that long‐term exposure to avatrombopag is well tolerated and that once‐daily oral treatment is effective for the treatment of thrombocytopenia in adult patients with chronic ITP. A US regulatory submission for the approval of avatrombopag for the treatment of patients with chronic ITP based on these data is planned in 2018. Phase 3 studies are ongoing to evaluate the efficacy and safety of avatrombopag in patients with thrombocytopenia scheduled for high‐risk surgical procedures requiring platelet counts of at least 100 × 109/l (NCT03326843), and patients with ovarian, non‐small cell lung and bladder cancer who develop chemotherapy‐induced thrombocytopenia (NCT03471078).
Study limitations
While this Phase 3 study demonstrated the superiority of avatrombopag to placebo in the treatment of patients with chronic ITP, its placebo‐controlled design limits the conclusions regarding the relative efficacy of avatrombopag compared with other approved TPO receptor agonists; although the high statistical significance of both primary and secondary endpoints (P < 0·0001) supports the robustness of the effect of avatrombopag treatment in this patient population. In addition, while 33·3% of patients in the avatrombopag treatment group reduced their use of concomitant ITP medications compared with 0% of placebo‐treated patients, the relatively small number of patients using concomitant medication at baseline is another potential limitation of this study. Another limitation is the low cumulative exposure in the placebo treatment group, due to a higher early discontinuation rate resulting from the lack of efficacy. The significantly higher mean (2.6‐fold) and median (4.3‐fold) exposure duration in the avatrombopag treatment group needs to be considered when evaluating the safety results and limits their interpretation.
In conclusion, this Phase 3 study showed that avatrombopag was superior to placebo in increasing platelet counts in patients with chronic ITP over a 6‐month treatment period. Avatrombopag was generally well tolerated, with exposure‐adjusted incidence rates of TEAEs that were comparable with placebo, and a safety profile that was consistent with that observed in Phase 2 avatrombopag studies and studies of other TPO receptor agonists in patients with ITP.
Conflict of interest disclosure
W.J. has received research funding from Eisai during the conduct of the study. WJ has also received grants from AbbVie, Acerta, Celgene, Celtrion, Gilead, Janssen, Sandoz‐Novartis, Takeda and TG Therapeutics. KC has received personal fees from Baxalta/Shire and NovoNordisk. J.M. and his institution have received research funding from Dova Pharmaceuticals. K.K. received research funding from Eisai during the conduct of the study. B.D.J. is an employee of Dova Pharmaceuticals. W.T. is an employee of Dova Pharmaceuticals. L.F.A. is an employee of Dova Pharmaceuticals.
Authorship contribution
W.J. contributed to the concept and study design, provided patient‐level data, analysed and interpreted the data, contributed to critical revision of the manuscript and approved the final draft. K.C., J.M., K.K., B.D.J. and L.F.A. analysed and interpreted the data, contributed to critical revision of the manuscript and approved the final draft. W.T. analysed the data, contributed to critical revision of the manuscript and approved the final draft.
Acknowledgements
This study was funded by Eisai Inc. The authors would like to thank the sponsor's responsible medical officer Francesco Bibbiani, MD, for his assistance in conducting the study. Medical writing support, under the direction of the authors, was provided by Paul O'Neill, PhD, and Gemma McGregor, PhD, of CMC AFFINITY, a division of Complete Medical Communications Ltd, Glasgow, UK, funded by Dova Pharmaceuticals, Durham, NC, USA, in accordance with Good Publication Practice (GPP3) guidelines.
References
- Amgen Inc. (2017) Nplate® (romiplostim) Prescribing Information. http://pi.amgen.com/united_states/nplate/nplate_pi_hcp_english.pdf (accessed 10 May 2018).
- Bussel, J.B. , Cheng, G. , Saleh, M.N. , Psaila, B. , Kovaleva, L. , Meddeb, B. , Kloczko, J. , Hassani, H. , Mayer, B. , Stone, N.L. , Arning, M. , Provan, D. & Jenkins, J.M. (2007) Eltrombopag for the treatment of chronic idiopathic thrombocytopenic purpura. New England Journal of Medicine, 357, 2237–2247. [DOI] [PubMed] [Google Scholar]
- Bussel, J.B. , Kuter, D.J. , Pullarkat, V. , Lyons, R.M. , Guo, M. & Nichol, J.L. (2009) Safety and efficacy of long‐term treatment with romiplostim in thrombocytopenic patients with chronic ITP. Blood, 113, 2161–2171. [DOI] [PubMed] [Google Scholar]
- Bussel, J.B. , Kuter, D.J. , Aledort, L.M. , Kessler, C.M. , Cuker, A. , Pendergrass, K.B. , Tang, S. & McIntosh, J. (2014) A randomized trial of avatrombopag, an investigational thrombopoietin‐receptor agonist, in persistent and chronic immune thrombocytopenia. Blood, 123, 3887–3894. [DOI] [PubMed] [Google Scholar]
- Cheng, G. , Saleh, M.N. , Marcher, C. , Vasey, S. , Mayer, B. , Aivado, M. , Arning, M. , Stone, N.L. & Bussel, J.B. (2011) Eltrombopag for management of chronic immune thrombocytopenia (RAISE): a 6‐month, randomised, phase 3 study. Lancet, 377, 393–402. [DOI] [PubMed] [Google Scholar]
- Fukushima‐Shintani, M. , Suzuki, K. , Iwatsuki, Y. , Abe, M. , Sugasawa, K. , Hirayama, F. & Kawasaki, T. (2008) AKR‐501 (YM477) in combination with thrombopoietin enhances human megakaryocytopoiesis. Experimental Hematology, 36, 1337–1342. [DOI] [PubMed] [Google Scholar]
- Fukushima‐Shintani, M. , Suzuki, K. , Iwatsuki, Y. , Abe, M. , Sugasawa, K. , Hirayama, F. , Kawasaki, T. & Nakahata, T. (2009) AKR‐501 (YM477) a novel orally‐active thrombopoietin receptor agonist. European Journal of Haematology, 82, 247–254. [DOI] [PubMed] [Google Scholar]
- Ghanima, W. , Junker, P. , Hasselbalch, H.C. , Boiocchi, L. , Geyer, J.T. , Feng, X. , Gudbrandsdottir, S. , Orazi, A. & Bussel, J.B. (2011) Fibroproliferative activity in patients with immune thrombocytopenia (ITP) treated with thrombopoietic agents. British Journal of Haematology, 155, 248–255. [DOI] [PubMed] [Google Scholar]
- Jenkins, J.M. , Williams, D. , Deng, Y. , Uhl, J. , Kitchen, V. , Collins, D. & Erickson‐Miller, C.L. (2007) Phase 1 clinical study of eltrombopag, an oral, nonpeptide thrombopoietin receptor agonist. Blood, 109, 4739–4741. [DOI] [PubMed] [Google Scholar]
- Kuter, D.J. , Bussel, J.B. , Lyons, R.M. , Pullarkat, V. , Gernsheimer, T.B. , Senecal, F.M. , Aledort, L.M. , George, J.N. , Kessler, C.M. , Sanz, M.A. , Liebman, H.A. , Slovick, F.T. , de Wolf, J.T. , Bourgeois, E. , Guthrie, T.H. Jr , Newland, A. , Wasser, J.S. , Hamburg, S.I. , Grande, C. , Lefrere, F. , Lichtin, A.E. , Tarantino, M.D. , Terebelo, H.R. , Viallard, J.F. , Cuevas, F.J. , Go, R.S. , Henry, D.H. , Redner, R.L. , Rice, L. , Schipperus, M.R. , Guo, D.M. & Nichol, J.L. (2008) Efficacy of romiplostim in patients with chronic immune thrombocytopenic purpura: a double‐blind randomised controlled trial. Lancet, 371, 395–403. [DOI] [PubMed] [Google Scholar]
- Kuter, D.J. , Mufti, G.J. , Bain, B.J. , Hasserjian, R.P. , Davis, W. & Rutstein, M. (2009) Evaluation of bone marrow reticulin formation in chronic immune thrombocytopenia patients treated with romiplostim. Blood, 114, 3748–3756. [DOI] [PubMed] [Google Scholar]
- Kuter, D.J. , Rummel, M. , Boccia, R. , Macik, B.G. , Pabinger, I. , Selleslag, D. , Rodeghiero, F. , Chong, B.H. , Wang, X. & Berger, D.P. (2010) Romiplostim or standard of care in patients with immune thrombocytopenia. New England Journal of Medicine, 363, 1889–1899. [DOI] [PubMed] [Google Scholar]
- Novartis (2017) ‘Promacta® (eltrombopag) Prescribing Information’. Available at: https://www.pharma.us.novartis.com/sites/www.pharma.us.novartis.com/files/promacta.pdf (accessed 10 May 2018).
- Provan, D. , Newland, A. & Bolton‐Maggs, P. on behalf of the British Committee for Standards in Haematology General Haematology Task Force . (2003) Guidelines for the investigation and management of idiopathic thrombocytopenic purpura in adults, children and in pregnancy. British Journal of Haematology, 120, 574–596. [DOI] [PubMed] [Google Scholar]
- Provan, D. , Stasi, R. , Newland, A.C. , Blanchette, V.S. , Bolton‐Maggs, P. , Bussel, J.B. , Chong, B.H. , Cines, D.B. , Gernsheimer, T.B. , Godeau, B. , Grainger, J. , Greer, I. , Hunt, B.J. , Imbach, P.A. , Lyons, G. , McMillan, R. , Rodeghiero, F. , Sanz, M.A. , Tarantino, M. , Watson, S. , Young, J. & Kuter, D.J. (2010) International consensus report on the investigation and management of primary immune thrombocytopenia. Blood, 115, 168–186. [DOI] [PubMed] [Google Scholar]
- Rodeghiero, F. , Stasi, R. , Gernsheimer, T. , Michel, M. , Provan, D. , Arnold, D.M. , Bussel, J.B. , Cines, D.B. , Chong, B.H. , Cooper, N. , Godeau, B. , Lechner, K. , Mazzucconi, M.G. , McMillan, R. , Sanz, M.A. , Imbach, P. , Blanchette, V. , Kuhne, T. , Ruggeri, M. & George, J.N. (2009) Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood, 113, 2386–2393. [DOI] [PubMed] [Google Scholar]
- Terrault, N.A. , Hassanein, T. , Howell, C.D. , Joshi, S. , Lake, J. , Sher, L. , Vargas, H. , McIntosh, J. , Tang, S. & Jenkins, T.M. (2014) Phase II study of avatrombopag in thrombocytopenic patients with cirrhosis undergoing an elective procedure. Journal of Hepatology, 61, 1253–1259. [DOI] [PubMed] [Google Scholar]
- Wang, B. , Nichol, J.L. & Sullivan, J.T. (2004) Pharmacodynamics and pharmacokinetics of AMG 531, a novel thrombopoietin receptor ligand. Clinical Pharmacology and Therapeutics, 76, 628–638. [DOI] [PubMed] [Google Scholar]
- Wang, L. , Gao, Z. , Chen, X.P. , Zhang, H.Y. , Yang, N. , Wang, F.Y. , Guan, L.X. , Gu, Z.Y. , Zhao, S.S. , Luo, L. , Wei, H.P. & Gao, C.J. (2016) Efficacy and safety of thrombopoietin receptor agonists in patients with primary immune thrombocytopenia: a systematic review and meta‐analysis. Scientific Reports, 6, 39003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, R.S.M. , Saleh, M.N. , Khelif, A. , Salama, A. , Portella, M.S.O. , Burgess, P. & Bussel, J.B. (2017) Safety and efficacy of long‐term treatment of chronic/persistent ITP with eltrombopag: final results of the EXTEND study. Blood, 130, 2527–2536. [DOI] [PubMed] [Google Scholar]