

Abstract
Progesterone plays an important role in the female reproductive system. However, there is also evidence that gynecologic disorders/diseases such as uterine fibroids and endometriosis are progesterone‐dependent. Steroidal and non‐steroidal selective progesterone receptor modulators (SPRMs) have shown potential for the treatment of such diseases. Steroidal SPRMs, including mifepristone and ulipristal acetate, have proven effective in clinical trials. However, several steroidal SPRMs containing a dimethylamino substituent have been associated with elevated liver enzymes in patients. An earlier drug discovery program identified lonaprisan as a highly selective SPRM that did not show drug‐related change in liver enzyme activity. Building on data obtained from that work, here we describe the research program that culminated in the discovery of a novel steroidal SPRM, vilaprisan, which combines an extremely high potency with very favorable drug metabolism and pharmacokinetic properties. Vilaprisan has entered clinical development and is currently undergoing phase 3 clinical trials.
Keywords: fluorinated ligands, gynecologic therapies, methyl sulfone, selective progesterone receptor modulator, structure–activity relationships
Introduction
Progesterone is one of the key regulators of female reproductive functions, such as uterine and mammary gland development, ovulation, and decidualization of the endometrium. During pregnancy, progesterone plays a key role in implantation and myometrial relaxation. Most of the diverse effects of progesterone on the female reproductive target tissues are mediated via the progesterone receptor (PR).1 Following the discovery of progesterone in the early 1930s, the identification of synthetic progesterone analogues has served as a basis for the development of oral contraceptives.
In the 1980s, the first PR antagonist, mifepristone (RU 486), was discovered by the French company Roussel–Uclaf. Mifepristone is a 19‐nor‐testosterone derivative, which has a 4‐(dimethylamino)phenyl group at the 11β‐position (see Scheme 1). The 11β‐phenyl group was found to be a crucial structural element of all potent steroidal PR antagonists.2 More recently, it has become evident that mifepristone and other compounds of this structural class can exhibit partial agonistic activity in a species‐ and tissue‐selective manner.3 Therefore, these compounds are referred to as selective PR modulators (SPRMs).
Scheme 1.
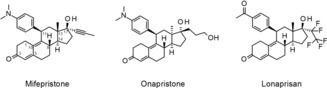
Clinically tested progesterone receptor modulators.
There is clinical evidence that the onset and progression of gynecologic disorders/diseases, such as uterine fibroids, endometriosis and breast cancer, are progesterone‐dependent.4 Therefore, SPRMs appear to be a promising option for the treatment of such diseases.
In addition to its PR antagonistic activity, mifepristone also shows relatively strong antagonistic effects toward the glucocorticoid receptor (GR).5 For long‐term application as for the treatment of chronic conditions, improvement in selectivity is considered to be essential. Therefore, substantial effort has been devoted toward optimizing receptor selectivity of steroidal SPRMs. Several variations of the steroid skeleton have been studied.
Onapristone is characterized by inversion of stereochemistry at C‐13 and C‐17 compared with mifepristone, and a replacement of the propynyl side chain at C‐17 by a 3‐hydroxypropyl group (Scheme 1). This compound shows an improved selectivity toward antiglucocorticoidal effects.6 Onapristone demonstrated efficacy in phase 2 clinical trials for the treatment of advanced breast cancer. However, clinical development was terminated as some patients experienced liver enzyme abnormalities.4f Elevated levels of liver enzymes have also been observed for mifepristone, ulipristal acetate and proellex (CDB‐4124).7 All of these compounds have the 4‐(dimethylamino)phenyl group in common. This structural element is accessible to metabolic demethylations, which may lead to the formation of aniline metabolites that have been reported to cause undesired effects in the liver by the formation of reactive intermediates.8 The hitherto available data from clinical studies with SPRMs lacking the dimethylamino phenyl group, for example, lonaprisan, have provided no evidence of a clinically relevant, drug‐related change in liver enzyme activity.
Investigations have shown that replacement of the dimethylamino group by an acetyl group was tolerated without losing efficacy. However, the 17α side chain was identified as the key factor for selectivity toward other nuclear hormone receptors. This position was therefore chosen for a detailed structure–activity relationship (SAR) analysis to help identify highly potent SPRMs with further decreased endocrine side effects. As a result of an intensive discovery program, lonaprisan (BAY 865044/ZK 230211), which exhibits a unique potency and selectivity, was identified in the late 1990s. Lonaprisan has a 4‐acetylphenyl substituent at C‐11 and a pentafluoroethyl group as a new 17α side chain (Scheme 1).9 The compound has demonstrated much stronger antiproliferative activity10 than mifepristone and onapristone in hormone‐dependent in vitro systems. Lonaprisan has also shown superior antitumor activity in a number of preclinical in vivo models compared with mifepristone and onapristone.11 In two randomized, placebo‐controlled, phase 1 studies in healthy postmenopausal women, lonaprisan was well tolerated without any sign of elevated liver enzyme levels, indicating that potential hepatotoxicity was not a concern with this drug.12 The efficacy of lonaprisan as second‐line endocrine therapy was evaluated in a randomized, phase 2 study of postmenopausal women with stage IV, PR‐positive metastatic breast cancer. Although disease stabilization was observed in some patients for a clinically useful period, the study did not meet its primary endpoint and was therefore terminated.13
The failure of lonaprisan in this clinical oncology trial was a serious setback for the compound class of steroidal SPRMs for the treatment of hormone‐dependent cancer. However, the potential of steroidal SPRMs for the treatment of uterine fibroids and endometriosis has been demonstrated in several clinical trials with different compounds, including mifepristone, ulipristal acetate, asoprisnil, and proellex (CDB‐4124). Development work on these and other steroidal SPRMs, both for clinical indications and as discovery leads, has continued. Ulipristal acetate (ESMYATM), for example, is now indicated for pre‐surgical treatment and repeated intermittent treatment of uterine fibroids in the EU and several other countries.14
Lonaprisan was also considered to be a potential candidate for gynecologic indications, due to its very high potency in combination with its favorable selectivity profile. Three major metabolites of lonaprisan formed by the biodegradation of the 4‐acetyl group were identified in human plasma. These metabolites significantly contribute to the overall activity of the drug candidate (Table 1), as measured by receptor transactivation assays. The presence of pharmacodynamically active metabolites—in particular, those with long half‐lives and thus prolonged circulation in vivo—is considered to be disadvantageous in intermittent treatment regimens. Such intermittent treatment regimens are currently being developed for SPRMs in long‐term use.15 Accordingly, lonaprisan is not considered to be an ideal candidate for the treatment of uterine fibroids and endometriosis.
Table 1.
In vitro activity and metabolic stability in liver microsomes of lonaprisan and its human metabolites.
| |||||
|---|---|---|---|---|---|
| Compound | Y | PR transactivation assay | Metabolic stability assay | ||
| IC50 [nm][a] | Efficacy [%] | F max Human [%][a] | F max Rat [%][a] | ||
| lonaprisan |
|
0.02 | 100 | 77 | 83 |
| metabolite 1 |
|
0.14 | 100 | 58 | 81 |
| metabolite 2 |
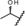
|
0.10 | 100 | 50 | 57 |
| metabolite 3 |
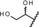
|
3.3 | 100 | 68 | 82 |
[a] Concentration of drug resulting in 50 % inhibition. [b] Maximum oral bioavailability calculated from in vitro hepatic extraction ratio (E H) in liver microsomes, assuming 100 % absorption (F max=1−E H); see the Experimental Section for details.
On the other hand, the surprisingly high potency of some of the metabolites—in particular, compound 1—showed that higher polarity is tolerated at the 4‐position of the 11β‐phenyl group than originally anticipated. Furthermore, the lonaprisan metabolite 1 was found to be about seven times less potent than the parent compound in transactivation assays, although only a threefold higher dose was required for early pregnancy termination tests in rats to achieve a similar efficacy as observed for lonaprisan. This indicates an improved oral bioavailability due to the more favorable physicochemical properties of metabolite 1 (compound 1 exhibits a 10‐fold higher aqueous solubility compared with the parent compound lonaprisan).
Based on these results, a new drug discovery program was initiated aiming to identify compounds with potency and selectivity similar to those of lonaprisan, but with higher metabolic stability to avoid the formation of active circulating metabolites.
Results and Discussion
Discovery of vilaprisan
Because the 17α‐pentafluoroethyl group demonstrated a favorable selectivity profile, this position was left unchanged. Instead, the new program focused on modifications of the 4‐substituent attached to the 11β‐phenyl ring, which was found to be metabolically unstable in lonaprisan. Broad modifications of position 17 have been described in the literature and even the steroid skeleton has been reported with several variations,6, 16 whereas only a relatively low number of 4‐substituents have been characterized in detail. Until the start of our new program, only three different substituents at the 4‐position of the 11β‐phenyl ring had been included in the structure of clinical candidates. Excluding lonaprisan (4‐acetyl) and asoprisnil (4‐hydroxyiminomethyl), another representative of this compound class,16 all other SPRMs that entered clinical trials carried the 4‐dimethylamino group. Our new study included the investigation of a broad range of other groups of compounds. Beyond the variation of 4‐substituent of the 11β‐phenyl ring described in this paper, we also explored the influence of substituents at other positions of the phenyl ring including disubstituted compounds. All these derivatives were found to be not superior compared with the para‐substituted derivatives.
The new derivatives were tested for their PR antagonistic activity in vitro using transactivation assays. However, potency indicated in transactivation assays cannot be directly translated to in vivo efficacy without consideration of the absorption, distribution, metabolism, excretion properties that are relevant to achieve the required exposure of the compound in vivo. Therefore, we decided to consider all compounds for in vivo characterization in a pharmacologic animal model, which had both an IC50 value of 0.5 nm or lower and showed sufficient in vitro metabolic stability when tested in human and rat liver microsomes.
As shown in Table 2, initially the 4‐unsubstituted derivative 4 was synthesized and characterized in vitro. This compound was found to be relatively potent in vitro (IC50=0.17 nm), but interestingly showed only 90 % efficacy. Earlier observations from our previous program (unpublished results) indicated that full inhibitory efficacy was required in the transactivation assay for compounds to show antagonistic effects in vivo. Compounds that reached only 90 % antagonistic efficacy in vitro were found to be full PR agonists in vivo.
Table 2.
Structure–activity relationship exploration and metabolic stability in liver microsomes of alkyl, carboxyl, and carboxamide substituents.
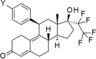
| |||||
|---|---|---|---|---|---|
| Compound | Y[a] | PR transactivation assay | Metabolic stability assay | ||
| IC50 [nm] | Efficacy [%] | F max Human [%] | F max Rat [%] | ||
| 4 | H | 0.17 | 90 | 79 | 88 |
| 5 |

|
0.01 | 99 | 31 | 54 |
| 6 |
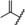
|
0.01 | 100 | 28 | 53 |
| 7 |
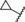
|
0.05 | 100 | 66 | 61 |
| 8 |
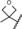
|
0.04 | 100 | 8 | 47 |
| 9 |
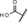
|
25 | 99 | 98 | n.d.[b] |
| 10 |
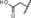
|
14 | 99 | n.d.[b] | n.d.[b] |
| 11 |
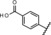
|
0.09 | 100 | 97 | 97 |
| 12 |
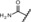
|
0.90 | 100 | 75 | 95 |
| 13 |
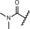
|
1.4 | 99 | 39 | 60 |
[a] Substituent at the 4′ position of the 11β‐phenyl ring. [b] Not determined.
Introduction of a vinyl group at the 4‐position (compound 5) led to almost complete antagonistic efficacy and a significant improvement in potency compared with the unsubstituted derivative (compound 4).
A further increase of the size of the 4‐substituent resulted in some compounds, such as derivatives 6–8, that exhibited full antagonistic efficacy in vitro. However, as these derivatives showed lower metabolic stability in liver microsomes, they were not tested further in vivo.
The pharmacologic activity determined for the lonaprisan metabolites showed that a certain polarity was tolerated at the 4‐position. Thus, several functional groups were introduced at different distances to the steroid skeleton to systematically explore the SAR. Derivatives with hydroxy groups, such as the lonaprisan metabolite 2, in general exhibited high potency (Table 1). However, only low metabolic stability was observed for these compounds.
Introduction of a carboxylic acid function at the same position (compound 9) led to an almost complete loss of activity. Addition of a carboxylic acid was further studied by the introduction of a broad range of spacers of different lengths between the carboxy group and the 11β‐phenyl group, such as in compounds 10 and 11. These compounds yielded variable results. For example, the acid 10 did not show any improvement in potency. By contrast, the biphenyl derivative 11 was unexpectedly found to be very potent. In addition, compound 11 exhibited very high metabolic stability in both rat and human liver microsomes. Therefore, this compound was selected for in vivo characterization.
Replacement of the carboxylic acid in compound 9 by small amides led to an improvement in potency (for example, in compounds 12 and 13), showing IC50 values of around 1 nm, although they did not reach the IC50 values of the most potent derivatives. To further explore the SAR of amide derivatives, we synthesized a library of such compounds, including examples with one or more additional functional groups in the amino part. Some of these more complex amides showed improvements in potency, but none of them could fulfill the desired profile to qualify them for an in‐depth characterization due to, for example, poor metabolic stability.
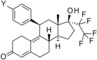
The dimethylamino group at position 4 of the phenyl ring, originally discovered in mifepristone, is one of the most prominent substituents for SPRMs at this position. This functional group is metabolized to aniline‐like derivatives which may be responsible for the hepatotoxicity observed in humans at higher doses with various SPRMs.4f, 7, 8 Accordingly, options to shift the amino function away from the phenyl ring to avoid the metabolic formation of aniline derivatives were evaluated and a library of such compounds was generated by parallel synthesis. The potency of these compounds was compared with the dimethylamino derivative 14, illustrated in Table 3. Surprisingly, and in contrast to other series with different 17‐substituents than pentafluoroethyl, it was observed that replacement of the acetyl group in lonaprisan by a dimethylamino function led to a 25‐fold decrease in potency. Introduction of a methylene group between the dimethylamino group and the phenyl group (compound 15) led to a further decrease in potency by a factor of more than 15. Compound 16, with a methyl piperazine at the benzylic position, and several other derivatives that are not shown here, exhibited potencies that did not qualify them for in vivo characterization. Other compounds in this series exhibited an improvement in potency without overcoming the unfavorable pharmacokinetic (PK) properties.
Table 3.
Structure–activity relationship exploration and metabolic stability in liver microsomes of amines and sulfur‐substituted compounds.
| |||||
|---|---|---|---|---|---|
| Compound | Y | PR transactivation assay | Metabolic stability assay | ||
| IC50 [nm] | Efficacy [%] | F max Human [%] | F max Rat [%] | ||
| 14 |
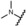
|
0.50 | 100 | n.d. | n.d. |
| 15 |
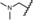
|
8.0 | 100 | 98 | 100 |
| 16 |
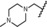
|
3.5 | 100 | 57 | 88 |
| 17 |
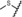
|
0.01 | 100 | 31 | 34 |
| 18 (vilaprisan) |
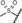
|
0.09 | 100 | 94 | 87 |
| 19 |
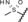
|
0.9 | 100 | 75 | 69 |
Derivatives with sulfur‐containing substituents at the 4‐position of the 11β‐phenyl ring were also synthesized, three of which are shown at the bottom of Table 3. The methylsulfide derivative 17 exhibited high potency in the transactivation assay but showed only low metabolic stability, probably due to oxidation at the sulfur atom. Therefore, the sulfone 18 was synthesized. This compound was slightly less potent in the transactivation assay than lonaprisan, but demonstrated high metabolic stability in human and rat liver microsomes. Therefore, compound 18, later named vilaprisan, was selected for in vivo characterization. The sulfoximine 19 was found to be less potent by a factor of approximately 10 compared with the sulfone and did not fully meet the criteria for further in vivo characterization. However, sulfoximine 19 exhibited an approximately eightfold improvement in aqueous solubility compared with sulfone 18 (nephelometry: 74 mg L−1[19] versus 9 mg L−1[18]). Accordingly, this derivative was also selected for further in vivo characterization. Other sulfur‐containing derivatives (not shown) exhibited properties that made them unsuitable for further characterization, for example, inefficient potency, low metabolic stability or increased lipophilicity.
Compounds 1 (metabolite of lonaprisan), 11, 18 (vilaprisan) and 19 were studied in an early pregnancy test in rats (Table 4), which is a suitable in vivo model to predict PR antagonistic activity.9 Progesterone is essential for an undisturbed pregnancy in mammals. This biologic principle can be used to determine PR antagonistic activity of a compound. In this regard, a PR antagonist competitively blocks the PR in the endometrium, which finally leads to the interruption of an ongoing pregnancy.
Table 4.
Activity of selected compounds in vivo.
| PR antagonist | Dose of full efficacy | Application route[a] |
|---|---|---|
| Termination of early pregnancy in rats (n=6)[b] | ||
| lonaprisan | 0.5 mg kg−1 | p.o. |
| compound 1 | 1.5 mg kg−1 | p.o. |
| compound 11 | 0.5 mg kg−1 | s.c. |
| compound 18 (vilaprisan) | 0.5 mg kg−1 | p.o. |
| compound 19 | 0.5 mg kg−1 | p.o. |
| Endometrial transformation in rabbits (n=5)[c] | ||
| lonaprisan | 1.0 mg kg−1 | p.o. |
| compound 1 | 3.0 mg kg−1 | p.o. |
| compound 11 | 1.0 mg kg−1 | p.o. |
| compound 18 (vilaprisan) | 1.0 mg kg−1 | p.o. |
[a] p.o.: per os; s.c.: subcutaneous. [b] Full efficacy in termination of early pregnancy studies means that all animals of the corresponding dose group lack implantations or have pathologic nidation sites. [c] Full inhibitory efficacy in endometrial transformations studies means that the degree of glandular differentiation in all animals of the corresponding dose group was characterized by a McPhail index of 1 or 1.5.
Besides maintenance of pregnancy, progesterone is responsible for the glandular differentiation and secretory transformation of the endometrium. These actions of progesterone were assessed in juvenile rabbits using the McPhail test, to evaluate PR agonistic and antagonistic effects.9 Priming of the rabbit endometrium with 17β‐estradiol followed by the administration of a PR agonist led to an induction of glandular differentiation. The degree of glandular differentiation was determined by light microscopy (rating grades 1–4; 1=no glandular differentiation, 4=maximal differentiation). Therefore, after priming with 17β‐estradiol, rabbits were either treated with the test compounds alone to show potential PR agonistic effects, or together with the natural hormone progesterone to test the PR antagonistic effects (inhibition of differentiation induced by progesterone). Only a limited number of test compounds that showed favorable PK properties and activity in the early pregnancy test in rats were tested in the McPhail test.
In the early pregnancy test, all compounds were administered orally except for compound 11 (which was given subcutaneously). The doses and corresponding exposures (area under the curve [AUC]) required to achieve full efficacy were compared with that of lonaprisan (Table 4). Compound 1 was found to require doses approximately three times higher compared with vilaprisan to reach full efficacy in this experiment (1.5 mg kg−1 compared with 0.5 mg kg−1), whereas compounds 11, 18 (vilaprisan) and 19 required similar doses to lonaprisan (0.5 mg kg−1). Sulfoximine 19 was partly converted into sulfone vilaprisan (18) in vivo, comprising one‐third of the total exposure of the parent compound. This finding suggests that after administration of sulfoximine 19, vilaprisan contributes significantly to the pharmacologic effect due to its 10‐fold higher potency. Accordingly, compound 19 is considered to exert its high activity, at least in part, as a prodrug of vilaprisan (18).
Compounds 1, 11 and 18 (vilaprisan) were also tested in the endometrial transformation assay in rabbits (Table 4). Compound 1 again demonstrated approximately three times lower potency than lonaprisan, whereas vilaprisan (18) and the biphenylcarboxylic acid 11 were shown to be as potent as lonaprisan. Based on the in vivo results described, vilaprisan (18) and the biphenylcarboxylic acid 11 were selected as the most promising follow‐up candidates.
Prediction of human exposure‐efficacy and dose
For the two compounds 11 and 18 (vilaprisan), the prediction of the efficacious human dose was performed based on the predicted human PK and the exposure‐efficacy relationship established in the animal models, confirming exposure (AUC) as the driver of efficacy. The data were corrected by plasma protein binding in human and animal species. For vilaprisan (18), prediction of human PK was performed by a single species scaling approach using the in vivo clearance obtained from cynomolgus monkeys extrapolated to humans on a body weight basis. The efficacious exposure during the 24 h dosing interval at steady state [AUC(0–24 h)ss] in the rat and rabbit models was 116 μg×h L−1 and 126 μg×h L−1, respectively, translating into a predicted efficacious human daily oral dose of 2.5 mg, considering free fraction in human, rat and rabbit plasma of 5 % in all species. For compound 11, prediction of human PK was performed by a single species scaling using in vivo clearance from the rat combined with a direct scaling approach using in vitro clearance from rat and human liver microsomes. Considering free fractions of 0.6 % in human plasma and 0.7 % in rat plasma and the efficacious AUC(0–24 h)ss in rats of 573 μg×h L−1, this translated into a predicted efficacious human daily oral dose of 11 mg.
The significantly lower human daily dose predicted for vilaprisan (18) compared with compound 11 supported the selection of vilaprisan (18) as a candidate for further development. A comparison of vilaprisan (18) with the previous clinical candidate lonaprisan in terms of drug metabolism and PK in humans is briefly summarized below.
Drug metabolism and pharmacokinetics in humans
In vitro and in vivo experiments show that lonaprisan is rapidly metabolized, with the formation of three major metabolites (compounds 1, 2, 3) in human plasma (see Table 1). They are formed by the biodegradation of the 4‐acetyl group. The exposure in terms of AUC of each of these metabolites significantly exceeded the AUC of the parent compound lonaprisan (see Table 5). The IC50 values of compounds 1 and 2 (see Table 1) in the in vitro transactivation assay were only 5–7‐fold higher compared with the parent compound lonaprisan. Compounds 1 and 2 were therefore expected to significantly contribute to the pharmacologic activity of the drug candidate in humans. The metabolite compound 1 was also found to be highly active in both in vivo models (see Table 4).
Table 5.
Activity of lonaprisan, vilaprisan, and their metabolites in the in vitro transactivation assay and the percentage of AUC(0–∞) of their metabolites in human plasma.
| Compound | PR transactivation assay IC50 [nm] |
% AUC of parent |
|---|---|---|
| lonaprisan | 0.02 | N/A |
| 1 | 0.14 | 285[a] |
| 2 | 0.10 | 312[a] |
| 3 | 3.3 | 920[a] |
| 18 (vilaprisan) | 0.09 | N/A |
| 20 | 5.3 | 24[b] |
| 21 | 610 | 19[b] |
[a] Determined by LC–MS after single oral administration of 200 mg lonaprisan to humans. [b] Determined by LC–MS after multiple oral administration of 5 mg day−1 vilaprisan.
In contrast, the unchanged parent compound vilaprisan (compound 18) was the main component in human plasma.17 Only a few minor metabolites formed by the reduction and oxidation of the steroid skeleton were identified in human plasma, each not exceeding 10 % of the AUC of total drug‐related compounds. This was a consequence of the modification of the 4‐substituent at the 11β‐position, as the 4‐sulfonyl group in vilaprisan was not metabolized. Vilaprisan was metabolized by oxidation reactions at different positions of the steroid skeleton and reduction of the carbonyl group in the 3‐position, as well as a combination of both modifications. The two distinct metabolites of vilaprisan that were identified in human plasma were formed by reduction in the 3‐position (compound 20) and by the combination of reduction and oxidation (compound 21) (see Scheme 2). The AUC of these two metabolites after multiple oral administrations of 5 mg vilaprisan exhibited approximately 20 % of the AUC of unchanged vilaprisan (see Table 5). The metabolites 20 and 21 showed approximately a factor of 60 (compound 20) and a factor >6700 (compound 21) lower potency in vitro in the transactivation assay. Considering the relatively low potency of the two metabolites and the low fraction in exposure compared with vilaprisan in humans,17 neither metabolite contributes to the in vivo activity of vilaprisan.
Scheme 2.
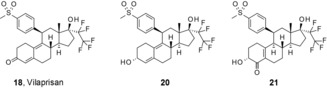
Vilaprisan and its human plasma metabolites.
Syntheses
The synthesis of lonaprisan was described previously.9 Key steps in the synthetic route are the introduction of the 11β‐substituent in a copper‐catalyzed Grignard reaction and the introduction of the 17α‐(pentafluoroethyl) group in a modified Gassman procedure.19
The principal focus of the current drug discovery project was the systematic modification of the 11β‐substituent. Therefore, the order of the reactions was modified by introducing the 17α side chain prior to the 11β‐substituent. This strategy allowed an efficient and broad modification of the 11β‐substituent at the end of the sequence. Only the lonaprisan metabolite 2 was synthesized following the original sequence using a modified building block for the introduction of the 11β‐phenyl group. Introduction of the 17α‐pentafluoro ethyl group19 into the previously published intermediate 22 20 yielded compound 23, which was used for the introduction of the 11β phenyl group. With one exception (the introduction of the oxetane building block in compound 8 required special reaction conditions, as outlined in the Supporting Information), all other 11β‐substituents were introduced in a copper‐catalyzed Grignard reaction, leading to the intermediates 24 a–l as shown in Scheme 3. In some cases, the 4‐substituent Y′ was already the final group Y, whereas in other cases additional reactions were needed to build up the final substituent. The derivatives 1, 3, 4, 5, 6, 8, 14 and 17 were prepared directly from the intermediates (24 a–e, 24 j, 24 k, 24 l) by an acidic cleavage of the protecting group(s) and concomitant elimination of the 5α‐hydroxy group.
Scheme 3.
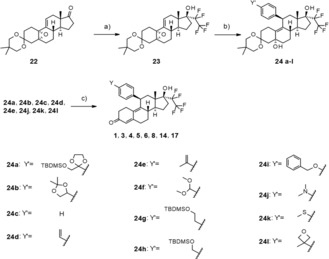
General synthetic approach: a) C2F5I, MeLi‐LiBr, −70 °C then 0 °C; b) phenyl Grignard, cat. CuCl, THF, 0 °C; for compound 24 l: iso‐propylmagnesium chloride, nBuLi, 3‐(4‐bromophenyl)‐3‐methyloxetane, cat. CuCl, THF, 0 °C; c) acidic cleavage.
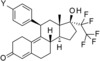
As shown in Scheme 4, the synthesis of compounds 7, 9, 10, 12, 13, 15, 16 and 18 required additional modification of the 4‐substituent. Cyclopropanation of intermediate 24 d yielded compound 25, which was transferred to compound 7 by acidic cleavage of the protecting group. Acidic deprotection of intermediate 24 f produced 26, which was used for the preparation of the carboxylic acid 9. Amide formation of 9 under standard conditions produced the dimethylamide 13. Cleavage of the silyl ether and the 3‐ketal starting from intermediate 24 g produced compound 27, which was oxidized in two steps to the carboxylic acid 10. Cleavage of the silyl ether in intermediate 24 h with tetrabutylammonium fluoride produced compound 28, which was oxidized in two steps to the carboxylic acid 30 and transferred into the amide 32 in two additional standard steps. Again, acidic cleavage yielded compound 12. The aldehyde 26, which was described above, could also be used for the synthesis of compounds 15 and 16 by reductive amination. Finally, vilaprisan (18) could be obtained directly from the 3‐protected sulfide 24 k by oxidation with Oxone®, concomitant cleavage of the protecting group, and elimination of the 5‐hydroxy group during work‐up.
Scheme 4.
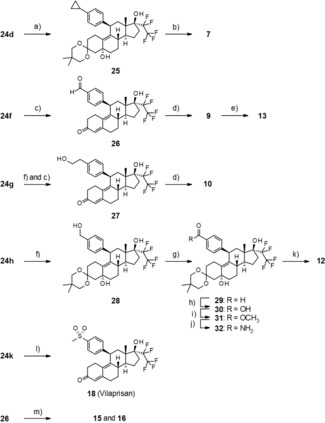
Synthesis of derivatives that require additional modification of Y′: a) diazomethane, Pd(OAc)2, Et2O, 0 °C; b) 2 n HCl, acetone, RT; c) acetic acid, 35 °C; d) Jones reagent, 0 °C; e) TBTU, methylamine, RT; f) TBAF, THF, RT; g) TPAP, NMO, CH2Cl2, molecular sieves, RT; h) 2‐methyl‐2‐butene, THF, tBuOH, NaOCl, 0 °C; i) (trimethylsilyl)diazomethane, THF, RT; j) NH3 in methanol, 85 °C; k) sulfuric acid, methanol, RT; l) oxone, 0 °C, m) amine, sodium triacetoxyborohydride, CH2Cl2, RT.
The synthesis of the biphenylcarboxylic acid 11 is shown in Scheme 5. Starting with intermediate 24 i, the benzyl ether was cleaved by Pd‐catalyzed hydrogenation. The phenol 33 was transferred into the nonaflate 34. A palladium‐catalyzed coupling reaction provided the biphenyl ester 35. Cleavage of the 3‐ketal under acidic conditions followed by saponification yielded compound 11.
Scheme 5.
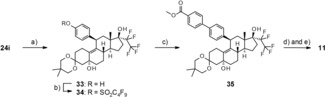
Synthesis of the biphenylcarboxylic acid 11: a) ammonium formate, Pd/C, methanol, RT; b) C4F9SO2F, nBuLi, THF, 0 °C; c) [4‐(methoxycarbonyl)phenyl]boronic acid, Pd(PPh3)4, toluene/ethanol, reflux; d) acetic acid, 35 °C; e) LiOH, THF, 90 °C.
The sulfoximine 19 as mixture of epimers was synthesized according to Scheme 6. Treatment of intermediate 24 k with chloramine‐T trihydrate® yielded compound 36, which was oxidized to 37 by treatment with hydrogen peroxide. Cleavage of the ketal under standard conditions gave compound 38. The release of the sulfoximine 19 required stronger acidic conditions and was achieved by treatment with concentrated sulfuric acid.
Scheme 6.
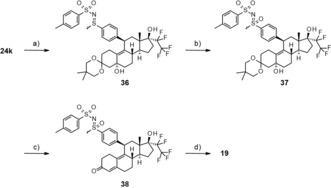
Synthesis of sulfoximine 19 as a mixture of epimers: a) chloramine‐T trihydrate, RT; b) H2O2, ethanol, CH3CN, RT; c) sulfuric acid, methanol, RT; d) conc. sulfuric acid, CHCl3, 0 °C.
Conclusions
The first SPRMs became available more than 25 years ago. Historically, this compound class has been considered for a number of indications with high unmet medical need, such as uterine fibroids and endometriosis. The chronic nature of these conditions implicates the requirement for long‐term medical treatment. However, the first SPRMs demonstrated relatively strong GR antagonistic side effects and therefore were not considered to be appropriate for long‐term application. The SPRM onapristone that succeeded in the development, failed in clinical trials due to unexpected liver intolerance with long‐term use. Later, the SPRM lonaprisan was discovered, which exhibited a favorable selectivity profile and was tolerated without any signs of liver toxicity. However, investigations into its use in the treatment of metastatic breast cancer failed as it did not reach its primary clinical endpoint. In clinical trials, it was found that lonaprisan formed a cocktail of active metabolites that significantly contributed to its overall activity. Therefore, the compound was considered suboptimal for gynecologic indications.
Here, we describe a drug discovery program that focused on the improvement of the PK properties of lonaprisan. Extensive modifications were attempted to stabilize the 4‐substituent of the 11β‐phenyl ring while maintaining the favorable potency, selectivity and safety profile of former candidates. Several potent derivatives were identified and comprehensive characterization of the new derivatives in vitro and, for the most promising compounds, then in vivo, led to the discovery of vilaprisan. This compound combines an extremely high selectivity toward the PR compared with other nuclear receptors8 with favorable tolerability, and significantly improved PK profile compared with lonaprisan. After successful profiling in preclinical development, investigations of the compound have progressed to clinical trials, and phase 121, 22 and phase 223 clinical trials have been completed. Recently, the compound entered phase 3 clinical development to examine the efficacy and safety of vilaprisan in the treatment of symptoms associated with uterine fibroids—measured by decreases in heavy menstrual bleeding, reductions in fibroid size, and improvements in health‐related quality of life of patients. Additionally, a phase 2b study for the treatment of women suffering from endometriosis has been initiated.
Herein we have described a research program that culminated in the discovery of a novel SPRM with a favorable safety profile and improved PK properties. Based on its high potency, high selectivity for the PR, and PK with absence of biologically active metabolites, vilaprisan represents a SPRM optimized for long‐term, intermittent clinical use in pre‐menopausal women.
Experimental Section
Chemistry
Materials and methods: All commercially available starting materials and solvents were purchased and used without further purification. Flash column chromatography was performed using prepacked flash chromatography columns PF‐15‐SIHP purchased from Interchim or KP‐Sil purchased from Biotage using a Biotage Isolera separation system. 1H NMR spectra were recorded at room temperature on Bruker Avance spectrometers operating at 300 or 400 MHz. NMR signal multiplicities are reported as they appeared, without considering higher‐order effects. Chemical shifts (δ) are given in parts per million (ppm) with the residual solvent signal used as a reference (CDCl3: singlet [s], 7.26 ppm; [D6]DMSO: quintet, 2.50 ppm). Liquid chromatography mass spectrometry (LC–MS) spectra were recorded on a Waters Acquity ultra performance liquid chromatography (UPLC)–MS SQD 3001 spectrometer, using an Acquity UPLC BEH C18 1.7 50×2.1 mm column, with acetonitrile and water +0.1 % formic acid as eluents at 60 °C, a flow rate of 0.8 mL min−1, an injection volume of 2 μL, with diode‐array detector scan at 210–400 nm, ELSD. All tested compounds were at least 95 % pure, as determined by 1H NMR spectroscopy.
1) (5′R,8′S,10′R,13′S,14′S,17′S)‐5,5,13′‐trimethyl‐17′‐(pentafluoroethyl)‐1′,2′,7′,8′,12′,13′,14′,15′,16′,17′‐decahydro‐6′H‐spiro[1,3‐dioxane‐2,3′‐[5,10]epoxycyclopenta[a]phenanthren]‐17′‐ol (23): At −30 °C, 1,1,1,2,2‐pentafluoro‐2‐iodoethane (116 g, 471.7 mmol) was condensed. A solution of (5′R,8′S,10′R,13′S,14′S)‐5,5,13′‐trimethyl‐1′,2′,6′,7′,8′,12′,13′,14′,15′,16′‐decahydro‐17′H‐spiro[1,3‐dioxane‐2,3′‐[5,10]epoxycyclopenta[a]phenanthren]‐17′‐one (50 g, 134.2 mmol, synthesis described previously20) in 500 mL dry toluene was added at −70 °C. A 1.5 m solution of methyllithium–lithium bromide complex in diethyl ether (290 mL, 435 mmol) was added slowly at −70 °C. Afterward, it was stirred for 1 h at 0 °C. The reaction mixture was then poured into saturated aqueous ammonium chloride solution. The aqueous layer was extracted with ethyl acetate three times. The combined organic layers were washed with brine, dried over sodium sulfate, filtered and evaporated. The crude product was dissolved in 80 mL toluene (at 70 °C). This was then diluted with 250 mL hexane. The resultant suspension was stirred for 1 h at 0 °C and was then filtered and dried in vacuo. The obtained product (51.57 g, 104.7 mmol, 78 % yield) was used without further purification. 1H NMR (300 MHz, CDCl3): δ=5.90–6.14 (multiplet [m], 1 H) 3.60 (doublet [d], J=11.49 Hz, 1 H) 3.43–3.50 (m, 1 H) 3.33–3.43 (m, 2 H) 2.54–2.69 (m, 1 H) 2.25–2.44 (m, 2 H) 2.05–2.23 (m, 4 H) 1.94–2.03 (m, 2 H) 1.59–1.89 (m, 6 H) 1.31–1.51 (m, 3 H) 1.20 (broad signal [br] d, J=3.96 Hz, 1 H) 1.05 (s, 3 H) 0.93 (s, 3 H) 0.85 ppm (s, 3 H).
2) General procedure for the introduction of the 11β‐phenyl substituent (derivatives 24a–24k): Dibromomethane (5 μL) was added to 5 mmol magnesium turnings in 0.5 mL dry tetrahydrofuran (to activate the magnesium turnings). A solution of the bromophenyl building block (5.1 mmol) in 6 mL dry tetrahydrofuran was added to this suspension slowly so that the reaction temperature did not exceed 55 °C. The solution was then stirred for 1 h. After stirring, the reaction mixture was cooled to 0 °C and 0.15 mmol copper(I) chloride was added. Stirring was continued for an additional 15 minutes. Afterward, a solution of 1 mmol of compound 23 in 5 mL dry tetrahydrofuran was added at 0 °C. The reaction mixture was allowed to warm up to 23 °C over a 3 h period and stirred for additional 10 h at this temperature. The reaction mixture was then poured into ice‐cold saturated aqueous ammonium chloride solution. After stirring for 30 minutes, it was extracted with ethyl acetate three times. The combined organic layers were washed with brine, dried over sodium sulfate, filtered and evaporated. The crude product was purified by chromatography over silica gel using hexane/ethyl acetate.
3) General procedure for the cleavage of the protecting groups in the final step under acidic conditions:
a) Using hydrochloric acid (applied for the preparation of compounds 5–8): 0.14 mmol of the protected precursor was dissolved in 5 mL acetone. Hydrochloric acid (0.7 mL, 4 n) was added and the reaction mixture was stirred for 1 h at 25 °C. The reaction mixture was then poured into saturated aqueous sodium hydrogen carbonate solution. It was extracted with ethyl acetate. The organic layers were washed with brine and the crude product was purified by column chromatography.
b) Using diluted sulfuric acid (applied for the preparation of compounds 1, 2, 3, 4, 12, 14, 17): 0.43 mmol of the protected precursor was dissolved in 5 mL methanol. Aqueous sulfuric acid (5 mL, 50 %) was added. The reaction mixture was stirred for 2.5 h at 25 °C. The reaction mixture was then poured into saturated aqueous sodium hydrogen carbonate solution. It was extracted with ethyl acetate. The organic layers were washed with brine and the crude product was purified by column chromatography.
c) Using acetic acid (applied for the preparation of compound 11): 0.51 mmol of the protected precursor was dissolved in acetic acid (3 mL, 70 %). The reaction mixture was stirred for 2 h at 25 °C and for another 2.5 h at 35 °C. The reaction mixture was then poured into ice water and stirred for 2 h. The formed precipitate was filtered and purified by column chromatography.
4) (11β,17α)‐20,20,21,21,21‐Pentafluoro‐17‐hydroxy‐11‐[4‐(methylsulfonyl)phenyl]‐19‐norpregna‐4,9‐dien‐3‐one (18, vilaprisan): Compound 24 k (5 g, 8.1 mmol) was dissolved in a mixture of 140 mL tetrahydrofuran and 140 mL methanol. The reaction flask was put in an ice bath. A solution of 20 g Oxone® in 94 mL water was slowly added (the temperature inside the flask increased up to 12 °C). The reaction mixture was kept in the ice bath and stirred for 3.5 h. It was then diluted with water and dichloromethane. The phases were separated and the aqueous layer was extracted with dichloromethane. The combined organic layers were washed with brine and the crude product was purified by column chromatography. The product (3.8 g, 86 %) was obtained as a white solid. 1H NMR (400 MHz, CDCl3) δ=7.86 (d, J=8.62 Hz, 2 H) 7.41 (d, J=8.11 Hz, 2 H) 5.81 (s, 1 H) 4.52 (br d, J=6.84 Hz, 1 H) 3.06 (s, 3 H) 2.68–2.77 (m, 1 H) 2.31–2.65 (m, 8 H) 2.20–2.30 (m, 1 H) 2.18 (s, 1 H) 2.08 (s, 1 H) 1.74–1.88 (m, 3 H) 1.42–1.56 (m, 2 H) 0.53 ppm (s, 3 H); LC–MS (ESI+): t R=1.22 min, m/z 545.2 [M+H]+.
Pharmacology
PR transactivation assay: The transactivation assay was carried out in SK‐N‐MC cells transfected with human PR‐B (pRChPR‐B‐neo) and the mouse mammary tumor virus promotor linked to the luciferase reporter gene. Cells were grown for 24 h either in the absence (negative control) or presence of increasing concentrations of the test compound (0.01 nmol L−1 to 1 mmol L−1). For the determination of antagonistic activity, cells were treated with 0.1 nmol L−1 promegestone, as well as with increasing concentrations of the test compound (0.01 nmol L−1 to 1 mmol L−1). Mifepristone was used as the positive control.
In vivo assays
Animals: Rats and rabbits were obtained from Charles River (Germany). Wistar rats (100–230 g) and New Zealand white rabbits (35–42 days old, 900–1300 g) had access to food and water ad libitum. All animals were housed according to institutional guidelines at a 12 h/12 h light–dark cycle. Rats were maintained under standard conditions (20–22 °C; 50–70 % humidity). The rats were housed in Makrolon cages type IV, five animals per cage, and fed a pelleted diet (Sniff, Germany). Rabbits were kept at 19±1 °C and 35–65 % humidity. They were conventionally housed in air‐conditioned rooms in Scanbur cages (standard I) and fed Altromin 2023 diet (Altromin, Germany). All animal studies were performed according to the German Animal Welfare Act and were approved by the competent regional authorities. All animal studies were performed in accordance with the ethical guidelines of Bayer AG.
Test for PR activity during early pregnancy (rat) and endometrial transformation (rabbit): The early pregnancy test was carried out as described previously.9, 20 In brief, 5–7 days post‐coitum, rats (n=6) were randomized to receive the test compound in different doses (s.c. or p.o.) or vehicle (benzylbenzoate/castor oil; 1+4 v/v, respectively 85 mg polyoxyethylene‐(50)‐stearate in 100 mL 0.9 % w/v NaCl). Autopsy was performed on Day 9; the absence of implantation sites was assessed as complete interruption of pregnancy and the presence of regressed and/or hemorrhagic implantation sites were defined as pathologic nidation sites. Full efficacy in termination of early pregnancy studies indicated that all animals of the corresponding dose group lacked implantations or had pathologic nidation sites.
PR activities of the test compounds on endometrial glands were tested in juvenile rabbits (900–1300 g). From Days 1 to 4, rabbits were primed with 5.0 mg kg−1 day−1 17β‐estradiol (s.c.). In order to test the anti‐progestational activity, animals were randomized (n=4) to receive 0.2 mg kg−1 day−1 progesterone (s.c.) and the test compound (p.o.) from Days 7 to 10. Progestogenic activity was tested by the test compound (p.o.) alone. Autopsy was performed on Day 11. As the parameter for progestogenic (induction of glandular differentiation) or anti‐progestational activities (inhibition of induction of glandular differentiation), the McPhail index (degree of glandular differentiation) was determined by light microscopy (rating grades 1–4; 1=no glandular differentiation, 4=maximal differentiation). Full efficacy in endometrial transformation studies indicated that the degree of glandular differentiation in all animals of the corresponding dose group was characterized by a McPhail index of 1 or 1.5.
Drug metabolism and pharmacokinetics
Metabolic stability in liver microsomes: The compounds were incubated in human or rat liver microsome suspensions at a final concentration of 0.3–3 μm. The incubations were stopped after 1 h and analyzed by high‐performance liquid chromatography using MS/MS or ultraviolet detection to determine the rate of degradation. This provided the in vitro intrinsic clearance according to Lau et al.24 based on the amount of microsomal protein and specific liver weight. Predicted in vivo clearance (CL H) was calculated based on the “well‐stirred” liver model based on rat or human liver blood flow (Q H) resulting in the hepatic extraction ratio E H according to E H=CL H /Q H. Maximum bioavailability (F max [%]) was calculated from the calculated extraction ratio according to: F max [%]=1−E H assuming 100 % absorption.
Plasma protein binding: Determination of plasma protein binding of test compounds in human and animal plasma was performed according to Schuhmacher et al.25
Determination of metabolites of lonaprisan in human serum: Human serum samples were obtained from volunteers after single oral administration of 200 mg lonaprisan (clinical study ME301781). The concentration–time profiles of the three metabolites (compounds 1, 2 and 3) and lonaprisan were determined by an LC–MS/MS method with external calibration using authentic reference compounds. The PK parameters of lonaprisan and compounds 1, 2 and 3 were calculated by appropriate methods. The product of hydroxylation and reduction in the 11β‐position (compound 3) is the major metabolite of lonaprisan in human serum (AUC=57 % of sum of drug‐related compounds or 920 % of AUC of lonaprisan). The exposures of compound 1 (hydroxylation) and compound 2 (reduction) and lonaprisan were found to be 18 %, 19 % and 6 % of sum of drug‐related compounds, respectively. Accordingly, compound 1 and compound 2 exhibited 285 % and 312 % of the AUC of lonaprisan.
Determination of the metabolites of vilaprisan in human plasma: The exposure of metabolites of vilaprisan (compound 18) in human plasma was investigated following multiple oral administration of 5 mg vilaprisan in healthy volunteers. The concentration–time profiles and the AUC of the two metabolites (compounds 20, 21) and vilaprisan were determined by a LC–MS/MS method with external calibration using authentic reference compounds. The PK parameters of vilaprisan and compounds 21 and 22 were calculated by appropriate methods. The parent compound vilaprisan accounted for the major part of the total drug‐related components in human plasma [AUC(0–24 h)ss ≈438 μg×h L−1 at steady state after daily oral administration of 5 mg].
Metabolite 20, formed by reduction of the 3‐keto group resulting in formation of the 3α‐hydroxy derivative, accounted for an AUC(0–24 h)ss ≈97.8 μg×h L−1 at steady state after daily oral administration of 5 mg vilaprisan corresponding to 22.3 % of the AUC(0–24 h)ss of unchanged vilaprisan. Metabolite 21, formed by reduction at the 3‐keto group and oxidation, accounted for an AUC(0–24 h)ss ≈77.1 μg×h L−1 at steady state after daily oral administration of 5 mg vilaprisan corresponding to 17.6 % of AUC(0–24 h)ss of unchanged vilaprisan.
Conflict of interest
All authors are current or former employees of Bayer AG.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Preclinical and clinical studies presented in this manuscript were funded by Bayer AG.
C. Möller, W. Bone, A. Cleve, U. Klar, A. Rotgeri, A. Rottmann, M.-H. Schultze-Mosgau, A. Wagenfeld, W. Schwede, ChemMedChem 2018, 13, 2271.
References
- 1. Möller C., Hoffmann J., Kirkland T. A., Schwede W., Expert Opin. Invest. Drugs 2008, 17, 469–479. [DOI] [PubMed] [Google Scholar]
- 2. Bélanger A., Philibert D., Teutsch G., Steroids 1981, 37, 361–382. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Han S. J., Tsai S. Y., Tsai M. J., O′Malley B., Endocrinology 2007, 148, 2471–2486; [DOI] [PubMed] [Google Scholar]
- 3b. Afhüppe W., Sommer A., Müller J., Schwede W., Fuhrmann U., Möller C., J. Steroid Biochem. Mol. Biol. 2009, 113, 105–115. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Orihuela P. A., Curr. Opin. Investig. Drugs 2007, 8, 859–866; [PubMed] [Google Scholar]
- 4b. Engman M., Granberg S., Williams A. R. W., Meng C. X., Lalitkumar P. G. L., Gemzell-Danielsson K., Hum. Reprod. 2009, 24, 1870–1879; [DOI] [PubMed] [Google Scholar]
- 4c. Donnez J., Tomaszewski J., Vázquez F., Bouchard P., Lemieszczuk B., Baró F., Nouri K., Selvaggi L., Sodowski K., Bestel E., et al., N. Engl. J. Med. 2012, 366, 421–432; [DOI] [PubMed] [Google Scholar]
- 4d. Kettel L. M., Murphy A. A., Mortola J. F., Fertil. Steril. 1991, 56, 402–407; [DOI] [PubMed] [Google Scholar]
- 4e. Kettel L. M., Murphy A. A., Morales A. J., Ulmann A., Baulieu E. E., Yen S. S., Fertil. Steril. 1996, 65, 23–28; [DOI] [PubMed] [Google Scholar]
- 4f. Robertson J. F., Willsher P. C., Winterbottom L., Blamey R. W., Thorpe S., Eur. J. Cancer 1999, 35, 214–218; [DOI] [PubMed] [Google Scholar]
- 4g. Klijn J. G., Setyono-Han B., Sander H. J., Lamberts S. W. J., de Jong F. H., Deckers G. H., Foekens J. A., Hum. Reprod. 1994, 9, 181–189. [DOI] [PubMed] [Google Scholar]
- 5. Neef G., Beier S., Elger W., Anderson D., Wiechert R., Steroids 1984, 44, 349–372. [DOI] [PubMed] [Google Scholar]
- 6. Wiechert R., Neef G., J. Steroid Biochem. 1987, 27, 851–858. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Murphy A. A., Kettel L. M., Morales A. J., Roberts V. J., Yen S. S., J. Clin. Endocrinol. Metab. 1993, 73, 513–517; [DOI] [PubMed] [Google Scholar]
- 7b. Nieman K. L., Blocker W., Nansel T., Mahoney S., Reynolds J., Blithe D., Wesley R., Armstrong A., Fertil. Steril. 2011, 95, 767–772; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7c.Repros Therapeutics Inc., press release July 17, 2017: http://ir.reprosrx.com/static-files/05b4fe8a-7766-4452-8ccd-d4aeff965fd6 (accessed September 2018);
- 7d. Wagenfeld A., Bone W., Schwede W., Fritsch M., Fischer O. M., Möller C., Hum. Reprod. 2013, 28, 2253–2264; [DOI] [PubMed] [Google Scholar]
- 7e. Kapur A., Angomchanu R, Dey M., J. Obstet. Gynaecol. India 2016, 66, 494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu R., Yu X., Wallqvist A., J. Cheminf. 2015, 7, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fuhrmann U., Hess-Stumpp H., Cleve A., Neef G., Schwede W., Hoffmann J., Fritzemeier K.-H., Chwalisz K., J. Med. Chem. 2000, 43, 5010–5016. [DOI] [PubMed] [Google Scholar]
- 10. Afhüppe W., Beekman J. M., Otto C., Korr D., Hoffmann J., Fuhrmann U., Möller C., J. Steroid Biochem. Mol. Biol. 2010, 119, 45–55. [DOI] [PubMed] [Google Scholar]
- 11. Hoffmann J., Sommer A., J. Steroid Biochem. Mol. Biol. 2005, 93, 191–200. [DOI] [PubMed] [Google Scholar]
- 12.R. Maibauer, C. Zurth, M.-H. Schultze-Mosgau, San Antonio Breast Cancer Symposium, San Antonio, TX (USA); December 14–17, 2006, Poster Number 4074.
- 13. Jonat W., Bachelot T., Ruhstaller T., Kuss I., Reimann U., Robertson J. F. R., Ann. Oncol. 2013, 24, 2543–2548. [DOI] [PubMed] [Google Scholar]
- 14.European Medicines Agency (EMA), summary of opinion, April 23, 2015; EMA/CHMP/245897/2015, WC500186173: http://www.ema.europa.eu/docs/en_GB/document_library/Summary_of_opinion/human/002041/WC500186173.pdf (accessed November 2017).
- 15.European Medicines Agency (EMA), assessment report, April 23, 2015; EMA/CHMP/84021/2015: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Assessment_Report_-_Variation/human/002041/WC500189366.pdf (accessed November 2017).
- 16.
- 16a. Cleve A., Neef G., Ottow E., Scholz S., Schwede W., Tetrahedron 1995, 51, 5563–5572; [Google Scholar]
- 16b. Ottow E., Neef G., Wiechert R., Angew. Chem. Int. Ed. Engl. 1989, 28, 773–776; [Google Scholar]; Angew. Chem. 1989, 101, 776–778; [Google Scholar]
- 16c. Cleve A., Fritzemeier K.-H., Heinrich N., Klar U., Müller-Fahrnow A., Neef G., Ottow E., Schwede W., Tetrahedron 1996, 52, 1529–1542; [Google Scholar]
- 16d. Kloosterboer H. J., Deckers G. H., de Gooyer M. E., Dijkema R., Orlemans E. O. M., Schoonen G. E. J., Ann. N. Y. Acad. Sci. 1995, 761, 192–201. [DOI] [PubMed] [Google Scholar]
- 17. Schultze-Mosgau M.-H., Höchel J., Prien O., Zimmermann T., Brooks A., Bush J., Rottmann A., Clin. Pharmacokinet. 2018, 57, 1001–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.
- 18a. Elger W., Bartley J., Schneider B., Steroids 2000, 65, 713–723; [DOI] [PubMed] [Google Scholar]
- 18b. Chwalisz K., Larsen L., Mattia-Goldberg C., Edmonds A., Elger W., Winkel C. A., Fertil. Steril. 2007, 87, 1399–1412. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Gassman P. G., O′Reilly N. J., Tetrahedron Lett. 1985, 26, 5243–5246; [Google Scholar]
- 19b. Gassman P. G., O′Reilly N. J., J. Org. Chem. 1987, 52, 2481–2490. [Google Scholar]
- 20. Rohde R., Neef G., Sauer G., Wiechert R., Tetrahedron Lett. 1985, 26, 2069–2072. [Google Scholar]
- 21. Schultze-Mosgau M.-H., Schütt B., Hafner F.-T., Zollmann F., Kaiser A., Hoechel J., Rohde B., Int. J. Clin. Pharmacol. Ther. 2017, 55, 16–24. [DOI] [PubMed] [Google Scholar]
- 22. Schütt B., Kaiser A., Schultze-Mosgau M.-H., Seitz C., Bell D., Koch M., Rohde B., Hum. Reprod. 2016, 31, 1703–1712. [DOI] [PubMed] [Google Scholar]
- 23.L. Bradley, X. Ren, E. Groettrup-Wolfers, K. Petersdorf, C. Seitz, ASRM Scientific Congress 2016, Salt Lake City, UT (USA), O-235.
- 24. Lau Y., Sapidou E., Cui X., White R., Cheng K.-C., Drug Metab. Dispos. 2002, 30, 1446–1454. [DOI] [PubMed] [Google Scholar]
- 25. Schuhmacher J., Kohlsdorfer C., Bühner K., Brandenburger T., Kruk R., J. Pharm. Sci. 2004, 93, 816–830. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary