

Abstract
Neutrophils represent the most abundant population of circulating cytotoxic effector cells. Moreover, their number can be easily increased by treatment with granulocyte‐colony stimulating factor or granulocyte macrophage‐colony stimulating factor, without the need for ex vivo expansion. Because neutrophils express Fc receptors, they have the potential to act as effector cells during monoclonal antibody therapy of cancer. Additionally, as neutrophils play a role in the regulation of adaptive immune responses, exploiting neutrophils in mAb therapy may result in long‐term antitumour immunity. There is limited evidence that neutrophils play a prominent role in current immunoglobulin G‐based immunotherapy. However, as IgA induces neutrophil recruitment, novel therapeutic strategies that aim to target the IgA Fc receptor FcαRI may fully unleash the potential of enlisting neutrophils as cytotoxic effector cells in antibody therapy of cancer.
Keywords: antibody‐dependent cellular cytotoxicity, cancer, FcαRI, IgA, immunotherapy, neutrophils
1. INTRODUCTION
The availability of monoclonal antibodies (mAbs) for cancer therapy has significantly improved the therapeutic options of patients.1, 2, 3 Several mAbs are standard care of treatment nowadays, which has improved clinical outcome in a number of cancers such as B‐cell malignancies, melanoma and subsets of breast cancer. In many types of malignancies, benefits of mAb treatment are however modest, and improvement of therapeutic efficacy is warranted.4 Two different major classes of antibodies can be distinguished. First, antibodies can be directed against the tumour environment. These include mAbs that target angiogenic factors, such as the antivascular endothelial growth factor (VEGF) mAb bevacizumab.3 Additionally, prominent successes in especially melanoma treatment have been recently established by targeting checkpoints, such as cytotoxic T‐lymphocyte‐associated protein 4, programmed death 1 or programmed death ligand 1 on infiltrating immune cells.5 The second class of anticancer mAbs targets tumour cells directly. Once bound, mAbs can initiate multiple different effector functions, which can result in eradication of tumour cells. As mAbs mediate dissimilar mechanisms, depending on the target antigen or the antibody isotype, the main mode(s) of action of most clinical mAbs is still incompletely clear, in spite of an overwhelming number of in vitro, in vivo and patient studies.
mAbs can have direct and indirect mechanisms (Figure 1).6 Direct effects include induction of tumour cell death through cross‐linking of receptors or via blockade of receptor‐ligand interactions (Figure 1A). For instance, the anti‐HER‐2 mAb trastuzumab prevents dimerization and internalization of HER‐2, which hampers induction of intracellular signalling.1, 2, 3 This is only effective when tumour cells overexpress HER‐2. Anti‐epidermal growth factor receptor (EGFR) antibodies inhibit binding of EGF and thereby decrease proliferation. Mutations in signalling pathway downstream of EGFR (eg, in KRAS, BRAF) hamper the effectiveness of anti‐EGFR mAbs, as proliferation signals are still sent irrespective of blocking EGF binding to EGFR. Because treatment with anti‐EGFR mAbs is ineffective in patients with KRAS mutations, the involvement of this direct inhibitory effect on proliferation is supported.7 Complement‐dependent cytotoxicity (CDC) has been proposed as mode of action as well (Figure 1B). The Fc part of IgG can bind to the complement component C1q, which will activate the classical pathway. The terminal components (C5‐C9) of the complement pathway form a membrane attack complex that creates pores in the target cell membrane, resulting in lysis. In some mouse models, an important role for CDC after mAb therapy was found, but not in others.8, 9 Clear evidence in patients is lacking, although it has been reported that polymorphisms in the C1QA gene correlated with clinical responses after rituximab treatment in patients with follicular lymphoma.10
Figure 1.
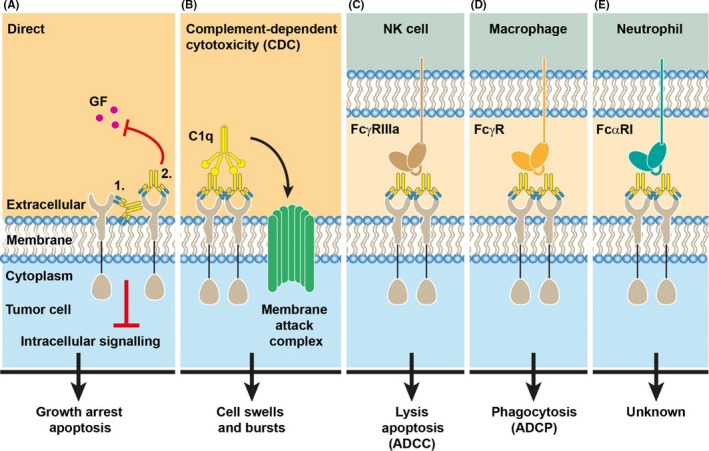
Modes of antibody‐induced killing of tumour cells. A, mAbs can exert direct effects on tumour cell survival or proliferation via induction of apoptosis or blockade of growth factor binding, which inhibits downstream signalling leading to growth arrest. B, IgG binds the complement factor C1q, resulting in activation of the classical complement system that ultimately leads to formation of the membrane attack complex, which induces lysis of the tumour cell. This process is referred to as complement‐dependent cytotoxicity (CDC). C‐E, Antibody‐opsonized tumour cells can be recognized and killed by a large variety of Fc receptor‐expressing immune cells. Among them are (C) NK cells that recognize IgG‐opsonized tumour cells via FcγRIIIa. Interaction induces ADCC and leads to apoptosis in the tumour cell. D, Macrophages express various FcγRs that allows phagocytosis of the opsonized target cell (ADCP). E, The dominant Fc receptor on neutrophils is FcαRI that functionally recognizes IgA‐opsonized tumour targets. In contrast to the well‐established killing modes of NK cells and macrophages, the mechanisms of neutrophil‐induced tumour killing are still under debate
Through interaction with the IgG Fc domain, mAbs furthermore bind to IgG Fc receptors (Fcγ receptors) and in this way bridge tumour cells with immune effector cells. Requirement of Fcγ receptor‐mediated mechanisms for in vivo efficacy was demonstrated for most mAbs. Loss of activating Fcγ receptors abrogated therapeutic efficacy, whereas the absence of the inhibitory receptor FcγRIIb enhanced the eradication of tumour after mAb therapy.11 The importance of Fcγ receptor‐mediated mechanisms was also shown in patient studies, as Fcγ receptor polymorphisms that affect binding affinities to IgG (FcγRIIa‐131H/R or FcγRIIIa‐158V/F) were associated with clinical responses after anti‐CD20, anti‐EGFR or anti‐HER‐2 mAb therapy in lymphoma, colorectal or breast cancer, respectively.1, 2, 3, 4 Two major effector cell populations have been identified. Natural killer (NK) cells induce apoptosis in tumour cells via release of perforins and granzymes or through activation of death receptor pathways, which is referred to as antibody‐dependent cellular cytotoxicity (ADCC) (Figure 1C).12 Additionally, antibody‐dependent phagocytosis (ADCP) by macrophages is an effective mechanism to eradicate tumour cells after mAb therapy (Figure 1D).9, 13, 14, 15 The involvement of neutrophils after mAb therapy is less clear, even though these are potent cytotoxic cells that express Fc receptors (Figure 1E). Moreover, the increasing evidence supports a role in regulating adaptive immune responses. It is now generally accepted that for curative treatment, antibody immunotherapies must not only induce direct tumour cell killing but also initiate potent adaptive immune responses to establish long‐lasting cancer‐specific immunity. As such, neutrophils may represent promising effector cells in mAb therapy of cancer.
2. NEUTROPHILS AS EFFECTOR CELLS
2.1. Tumoricidal ability of neutrophils
Neutrophils are the most abundant circulating white blood cell population, and their numbers can easily be augmented through treatment with granulocyte colony‐stimulating factor (G‐CSF) or granulocyte‐macrophage colony‐stimulating factor (GM‐CSF).16 Massive accumulation of neutrophils was observed in murine colon carcinoma tumours that had been transduced with the G‐CSF gene, which resulted in elimination.17 The relative ease of obtaining large numbers of effector cells without the need for ex vivo expansion represents a major advantage of recruiting neutrophils for mAb therapy of cancer. Neutrophils can be tumoricidal in the absence of mAbs. For instance, the antitumour effect of Bacillus Calmette‐Guérin (BCG)‐based immunotherapy of bladder cancer was absent when neutrophils had been depleted.18 Nonetheless, the cytotoxic ability of neutrophils is greatly increased after mAb therapy, and it was shown that neutrophils were necessary and sufficient for elimination of subcutaneous tumours after mAb therapy.19 Additionally, selective depletion of neutrophils significantly reduced protective activity of treatment with the anti‐CD52 IgG mAb alemtuzumab (Campath‐1H) in a xenograft mouse model of CD52+ tumours.20 Cotreatment with G‐CSF furthermore augmented therapeutic activity. Similarly, rituximab treatment was less effective in a B‐cell lymphoma model in SCID mice after depletion of neutrophils.21 Efficacy of a cotreatment of an antitumour antibody, recombinant interleukin‐2 with an extended half‐life, anti‐PD‐1 and a T‐cell vaccine, was decreased after depletion of neutrophils, which was comparable to depletion of macrophages or NK cells. Depletion of CD8 cells abrogated therapeutic efficacy of this cocktail most prominently.22
Recruitment of tumoricidal neutrophils was induced when a combination therapy with β‐glucan was given, which increased the protective ability of mAbs in animal models.23, 24 It was recently demonstrated that Imprime PGG (a clinical grade soluble β‐glucan) formed immune complexes with naturally occurring anti‐β glucan antibodies in human blood, which activated complement and primed neutrophils and monocytes via complement receptor 3 and FcγRIIa.25 The combination of Imprime PGG and cetuximab (anti‐EGFR mAb) treatment of patients with stage‐IV KRAS‐mutant colorectal cancer showed modest clinical activity, suggesting that priming of innate myeloid cells occurs in vivo.26 The evidence supporting an active role for neutrophils in current IgG‐based immunotherapies is however limited. Effectiveness of treatment of patients with neuroblastoma with an antiganglioside GD2 mAb (in combination with GM‐CSF) was dependent on a polymorphism in FcγRIIa (H131/R131),27 which may suggest that neutrophils were involved as effector cells. However, as FcγRIIa is widely expressed on immune cells, including neutrophils, monocytes, macrophages and dendritic cells (Figure 2), it cannot be excluded that this combination treatment regime also induces the generation of adaptive immune responses through antigen presentation. Likewise, higher response rates were observed when patients with follicular lymphoma were cotreated with rituximab and GM‐CSF.28 This may be due to increased neutrophil numbers and activity, but a role for other myeloid immune cells cannot be excluded.
Figure 2.
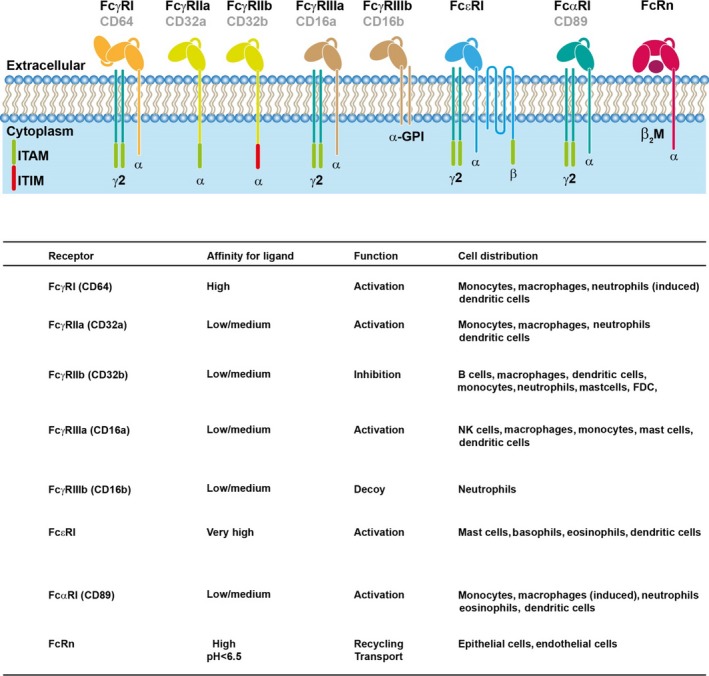
Fc receptors on effector cells. The principal Fc receptors for IgG, IgE and IgA, as well as associated signalling chains, are shown. FcγRI, FcγRIIa, FcγRIIIa, FcεRI and FcαRI are activating receptors. FcγRIIb contains an ITIM motif and is an inhibitory receptor, whereas FcγRIIIb is a GPI‐linked molecule without associated signalling chains. FcRn is involved in recycling of IgG and transport of immune complexes. Affinity, function and global cell distribution are indicated. ITAM: immunoreceptor tyrosine‐based activation motif, ITIM; immunoreceptor tyrosine‐based inhibitory motif
In addition to FcγRIIa, neutrophils constitutively express the low to intermediate affinity IgG Fc receptors FcγRIIIb (CD16), as well as the inhibitory receptor FcγRIIb on subpopulations (Figure 2).29, 30 FcγRIIIb, a glycosyl‐phosphatidylinositol‐anchored receptor, is the most abundant Fcγ receptor on neutrophils but likely not significantly involved in efficient killing of IgG‐opsonized tumour cells.31 It has been hypothesized that FcγRIIIb acts as decoy receptor for Fc‐engineered IgG1 antibodies.32 As such, IgG2 antibodies may represent an interesting antibody isotype for neutrophil recruitment, due to its lower affinity for FcγRIIIb.33
The treatment with IFN‐γ or G‐CSF upregulates the high‐affinity Fc receptor for IgG (FcγRI; CD64). Specific targeting of FcγRI with bispecific antibodies (BsAb) has been employed as strategy to overcome potential inhibition via FcγRIIb. In vitro, FcγRI BsAb proved very effective in recruiting FcγRI‐expressing neutrophils of G‐CSF‐treated patients as effector cells.34 Several FcγRI BsAb directed against a multitude of tumour antigens have been described that target different malignancies.16 Furthermore, in vivo efficacy of FcγRI BsAb in combination with G‐CSF or CpG (an adjuvant derived from bacterial DNA) was demonstrated in FcγRI transgenic mice.35, 36 Subsequently, several phase I/II clinical trials were performed to demonstrate clinical efficacy of FcγRI BsAb in patients with cancer.16, 37 Treatment was generally well‐tolerated, and some partial clinical responses were observed. Combination treatments with G‐CSF, GM‐CSF or IFN‐γ have been tried as well to improve clinical outcome. This did, however, not result in significantly improved outcome, but induced severe side effects in some cases, leading to discontinuation of therapy. Thus, overall efficacy of FcγRI BsAb treatment was disappointing.16, 37 This may have been due to the fact that patients who were included in these trials had advanced disease and were heavily pretreated. FcγRI BsAb furthermore had a short half‐life, which likely contributed to the lack of prominent clinical responses.
It has become clear that targeting Fcγ receptors on neutrophils is far less effective than targeting the Fc receptor for IgA (FcαRI, CD89), which is also constitutively expressed by neutrophils (Figure 2).38, 39 Moreover, immature bone marrow neutrophils were able to kill tumour cells after targeting FcαRI, but were ineffective when FcγRI was triggered.40 The superior ability of FcαRI to trigger neutrophil‐mediated tumour cell killing has now been demonstrated for a plethora of tumour cells and antigens (eg, Ep‐CAM (colon carcinoma), HER‐2 (mamma carcinoma), EGFR (epithelial, colorectal and renal cell carcinoma), carcinoembryonic antigen, CD30 (B‐ and T‐cell lymphoma), HLA class II and CD20 (B‐cell lymphoma)).16, 41, 42, 43, 44 An anti‐EGFR dimeric IgA variant initiated neutrophil‐mediated tumour killing and was also transported by the polymeric Ig receptor that is expressed on epithelial cells, which may be useful when targeting mucosal tumours.45 Interestingly, FcαRI is the only neutrophil Fc receptor that after cross‐linking induces the release of leukotriene B4, which is a potent chemoattractant for neutrophils.46 Migration towards 3‐dimensional tumour colonies was only observed in the presence of FcαRI BsAb or IgA, whereas targeting Fcγ receptors did not result in neutrophil accumulation.40 Recruitment was furthermore enhanced in the presence of an endothelial cell monolayer, which was due to interleukin‐8 production by endothelial cells in response to release of pro‐inflammatory mediators (interleukin‐1β and tumour necrosis factor‐α) by neutrophils (Figure 3).47 Recently, a meta‐analysis of the expression signatures from ∼18 000 human tumours demonstrated that a high neutrophil content in tumours is correlated with poorer prognosis,48 which may be due to their immunosuppressive functions in the tumour microenvironment.49 As granulocytic subsets of myeloid‐derived suppressor cells (gMDSC) express FcαRI (own unpublished data), it would be interesting to investigate whether it is possible to retarget gMDSC into pro‐inflammatory neutrophils. If so, determining the immune landscape of tumours may help to identify patients that have a prominent granulocyte infiltration, and as such may benefit most from therapy with IgA antitumour antibodies.
Figure 3.
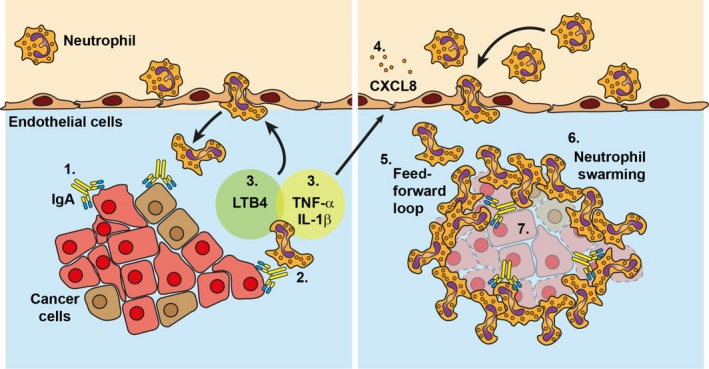
IgA induces neutrophil swarming towards tumour cells. IgA‐opsonized tumour cells elicit a remarkable cascade of activation in neutrophils, resulting in swarming behaviour and killing of the tumour. (1) IgA binds to tumour antigens, which is (2) recognized by neutrophils via their FcαRI. The binding of IgA results in FcαRI cross‐linking and (3) subsequent release of LTB4, TNF‐α and IL‐1β by neutrophils. LTB4 is a chemoattractant and activator for other neutrophils that will amplify the recruitment signal. (4) TNF‐α and IL‐1β induce CXCL8 release by endothelial cells, which further enhances neutrophil migration. (5) This feed‐forward loop of neutrophil recruitment, LTB4 release and subsequent neutrophil activation results in (6) neutrophil swarming behaviour towards IgA‐opsonized tumour cells that ultimately may lead to (7) tumour cell death
In vivo studies with IgA have been hampered due to the lack of suitable animal models. Mice do not express an FcαRI homologue. Although this problem has been addressed by creating a human FcαRI transgenic mouse model in which neutrophils express FcαRI,50 it has only partly solved the problem, as FcαRI only binds human IgA, which has a short half‐life in mice. Treatment with IgA2 anti‐EGFR mAbs successfully reduced the number of peritoneal and lung metastases in FcαRI transgenic mice, although FcαRI+ macrophages, and not neutrophils, were likely the effector cells in this model.51 However, IgA had to be injected daily, as the estimated half‐life of human IgA2 anti‐EGFR antibodies in mice was approximately 15 hours. In comparison, human IgG1 anti‐EGFR antibodies had a half‐life of approximately 4 days in mice,51 due to recycling via the neonatal Fc receptor FcRn (Figure 2). Recently, IgA anti‐HER‐2 antibody variants were generated that contained an albumin‐binding domain in its light chain (IgA‐ABD‐LC), as albumin also interacts with FcRn52 Anti‐HER‐2 IgA‐ABD‐LC antibodies were equally effective in inducing SKBR3 breast cancer cell killing by neutrophils in vitro compared to the parental anti‐HER‐2 IgA. Furthermore, anti‐HER IgA‐ABD‐LC was slightly more effective in reducing tumour burden in a xenograft mouse model, but the relative contribution of neutrophils as effector cells still needs to be established. Similarly, engineering of an IgA anti‐EGFR antibody, which lacked two N‐glycosylation sites and two free cysteines but had stabilized heavy and light chain linkage, has been described.53 This modified IgA showed improved half‐life and was equally effective as IgG1 anti‐EGFR antibodies in reducing tumour outgrowth. Thus, IgA antitumour mAbs have prominent therapeutic efficacy in vivo, especially after modifications have been made to improve half‐life.
Additionally, it was demonstrated that a “cross‐isotype” antibody (referred to as IgGA), in which properties of IgG and IgA1 had been combined, bound to FcγRI, FcγRIIa and FcαRI with similar affinities compared to IgG and IgA1, respectively.54 IgG anti‐HER‐2 antibodies were unable to activate neutrophils, but IgGA anti‐HER‐2 antibodies potently induced killing of SKBR3 breast cancer cells by neutrophils. Comparable results were found with a tandem IgG1/IgA2 antibody format, which activated neutrophils through the incorporated IgA domain, while retaining binding to Fcγ receptors, including FcRn.55 Thus, an IgG/IgA hybrid molecule may enhance therapeutic efficacy through several mechanisms. First, it was demonstrated that simultaneous engagement of FcαRI and FcγRI on neutrophils enhanced killing of antibody‐coated tumour cells.56 Second, the IgG part may recruit NK cells and/or macrophages as effector cells and may induce CDC. And finally, through interaction with FcRn, half‐life will be increased compared to IgA.
2.2. Mechanisms of antibody‐dependent elimination of tumour cells by neutrophils
The mechanism(s) through which neutrophils kill tumour cells remain elusive, although several modes of action have been described that may play a role (Figure 4). Neutrophils produce multiple cytotoxic molecules such as reactive oxygen species (ROS), proteases and defensins, which is consistent with their potent antimicrobial functions.57 Some of these molecules, such as defensins, induced damage to cell membranes, especially in the presence of H2O2 15 (Figure 4A). Myeloperoxidase, which is involved in release of the ROS hypochlorous acid (HOCl), was required for neutrophil‐mediated tumour cell killing in the absence of antibodies. Remarkably, antibody‐mediated tumour cell killing by neutrophils was not dependent on ROS production. Neither ROS scavengers nor inhibitors hampered this process. Similarly, neutrophils from patients with chronic granulomatous disease, which are unable to produce ROS, still effectively mediated tumour cell killing in the presence of mAbs.58 This also questions the absolute necessity of neutrophil extracellular traps (NETs) for induction of tumour cell killing (Figure 4B). NETs are composed of nuclear components such as DNA and histones as well as specific granular and cytoplasmic proteins, including myeloperoxidase and elastase.59 Generally, release of NETs requires ROS production. As such, neutrophils of patients with chronic granulomatous disease cannot form NETs, but are nonetheless capable of mediating antibody‐dependent tumour cell killing. However, as it was shown that NETs induce endothelial and epithelial cell damage and death,60 NETs may contribute to tumour cell death when present. We recently demonstrated that IgA greatly enhances NET formation by neutrophils.61
Figure 4.
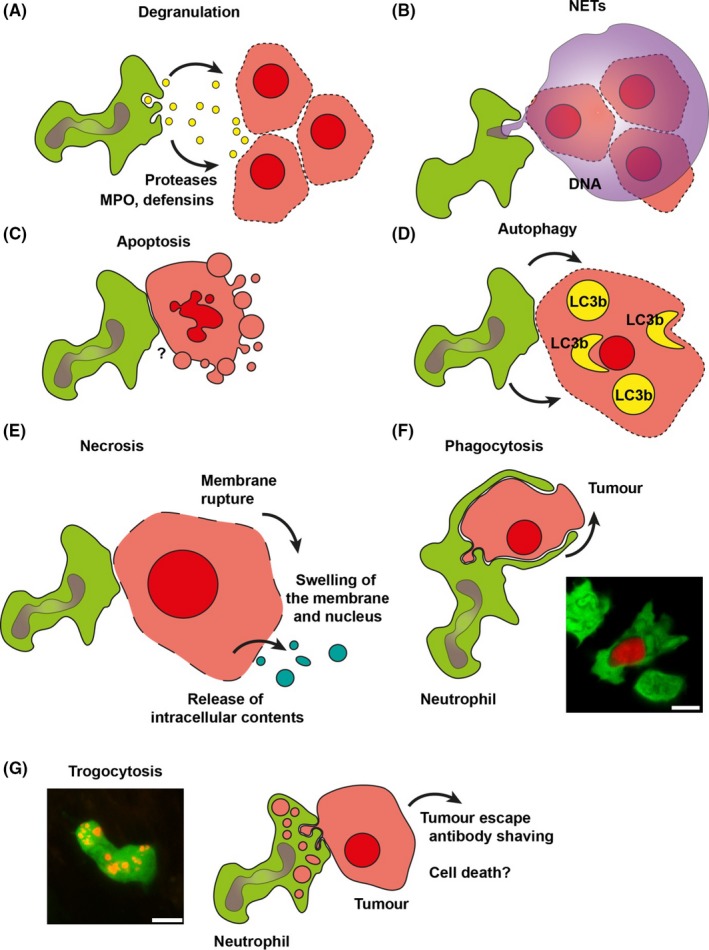
Mechanisms of antibody‐dependent elimination of tumour cells by neutrophils. The mechanisms by which neutrophils kill antibody‐opsonized tumour cells are ill‐understood and sometimes controversial. The mode(s) of action depends among others on the type of antibody, target cell and (density of) tumour epitope. This large number of variables might explain the variety of distinct effector mechanism that has been described in the literature. This includes tumour cell killing via A, degranulation; B, release of neutrophil extracellular traps (NETs); C, apoptosis; D, autophagic cell death; E, necrosis; F, phagocytosis and G, uptake of tumour‐derived membrane resembling trogocytosis. The outcome of the latter process is controversial. Trogocytosis may contribute to killing, but it has also been shown that this process by contrast protects tumour cells by shaving off tumour antigen‐antibody complexes. Pictures in f and g were acquired with a Leica TCS SP8 STED 3X super‐resolution microscope equipped with white light laser and Hybrid photodetectors. Neutrophils (green) and rituximab opsonized human B‐CLL (red) in the spleen of a LysM‐EGFP mice were recorded with a voxel size of 0.38 × 0.38 × 1 μm, a 40× numerical aperture (NA) 1.3 oil immersion objective and a pixel dwell time of 1.96 μs
It has been reported that neutrophils may induce apoptosis in tumour cells (Figure 4C). Incubation of HER‐2‐expressing tumour cells for 20 hours with neutrophils and anti‐HER‐2 mAbs resulted in ADCC (as detected by51 Cr release assays) as well as in induction of apoptosis.58 However, it is unclear how the latter was mediated, as this process was perforin and caspase‐independent, which are both employed by NK cells and cytotoxic T lymphocytes for apoptosis induction. Moreover, the expression of granzymes and perforin in neutrophils is contested.16 Two additional nonapoptotic cell death pathways have been described when neutrophil FcαRI was engaged as target molecule.62 The majority of tumour cells underwent autophagy, as evidenced by formation of LC3b‐positive autophagosomes (Figure 4D). Autophagy is a cell survival mechanism that involves self‐digestion to generate energy. Under disproportionate stress conditions or apoptosis resistance, autophagy may lead to cell death, although it is debatable whether this represents an autophagic cell death pathway or simply a failure of the cell to survive.63 The latter may be reflected by the fact that many tumour cells died through necrosis when incubated with FcαRI BsAb and neutrophils (Figure 4E).62 Phagocytosis of small B‐CLL cells by neutrophils in the presence of (glycoengineered) anti‐CD20 mAbs has also been described (ref 64, and own unpublished data; Figure 4F), although this is likely a very specific situation, as in general most tumour cells are too big to be phagocytosed by neutrophils. It was however shown that neutrophils and tumour cells have intimate contact in the presence of mAbs, resulting in mutual exchange of fluorescent membrane lipid dyes, which resembles trogocytosis (Figure 4G). Whether trogocytosis results in tumour cell death is still under debate. Whereas accumulation of neutrophils with tumour cell‐derived membrane lipids was associated with effective killing of SKBR3 breast cancer cells (ref 58 and Hanke Matlung & Timo van den Berg, personal communication, 2018), trogocytosis of chronic lymphocytic leukaemia B cells (B‐CLL) by neutrophils did not lead to a diminished number of tumour cells.65 In this case, trogocytosis may by contrast result in removal of the antibody from the tumour cell surface, hereby hampering therapeutic efficacy.
Although the mechanism(s) of neutrophil‐mediated tumour cell death have not been fully elucidated, it is known that close contact, referred to as the “immunological” or “cytotoxic synapse,” is required. Complement receptor 3 (CR3) was essential for formation of the synapse, as CR3−/− neutrophils were unable to spread on antibody‐coated tumour cells, resulting in absence of tumour cell killing.50, 66 Furthermore, treatment with the antimelanoma mAb TA99 was less effective in CR3−/− mice, although formally it was not demonstrated that therapeutic efficacy was dependent on neutrophils.67 Nonetheless, in vivo occurrence of cytotoxic synapses between neutrophils and tumour cells after mAb therapy was previously reported.68
3. NEUTROPHILS AND REGULATION OF ADAPTIVE IMMUNE RESPONSES
Neutrophils have long been regarded as terminally differentiated cells without prominent biosynthesis ability. It has, however, become clear that neutrophils can modulate immune responses through secretion of inflammatory mediators, cytokines and chemokines as well as via direct cell‐cell contact (Figure 5) (previously extensively reviewed by others69, 70). In an influenza model, neutrophils that migrated to the infection site paved the way for CD8 T cells, by depositing a membranous trail, which contained the chemokine CXCL12 to serve as a chemokine map for migrating CD8 T cells (Figure 5A).71 In addition, it was hypothesized that neutrophils act as danger sensors by communicating the presence of infection to dendritic cells (Figure 5B).72, 73 Moreover, several in vivo studies have demonstrated the ability of neutrophils to transport antigen to lymphoid organs, which indirectly or directly promoted antigen cross‐presentation to T cells (Figure 5C).74, 75 Furthermore, it was shown that neutrophils promote maturation and function of NK cells (Figure 5D).76 In bladder cancer mouse models, depletion of neutrophils led to decreased CD4 T‐cell recruitment and reduced CD8 T‐cell activation, resulting in enhanced tumour outgrowth (Figure 5E).18 Conversely, it was recently shown that neutrophils suppressed CD8 T cells in a mouse breast cancer model and promoted metastasis (Figure 5E).77 Thus, depending on the tumour micro milieu, neutrophils can have opposite effects on (adaptive) immune responses. This may also depend on the stages of tumour development. For instance, it was shown that in early‐stage human lung cancer tumour‐associated neutrophils stimulate T‐cell responses.78, 79 Little is known about the role of neutrophils in inducing adaptive immune responses after antibody therapy of cancer. However, it was demonstrated that neutrophils release pro‐inflammatory cytokines after cross‐linking of FcαRI, suggesting that activated neutrophils may be able to counteract the immunosuppressive tumour environment.47
Figure 5.
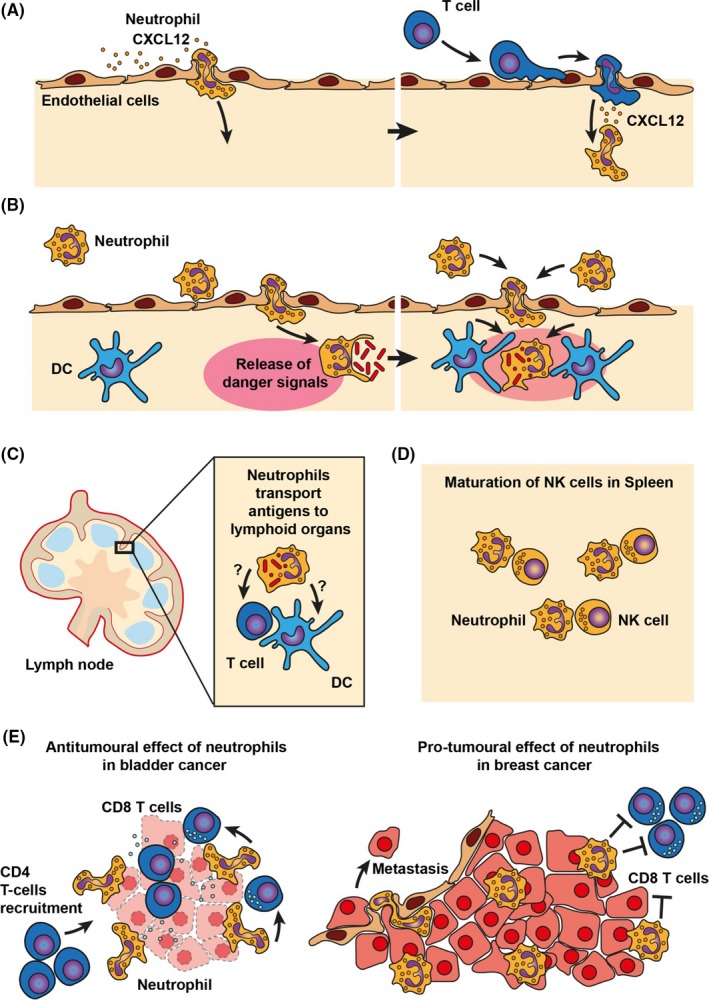
Neutrophils and regulation of adaptive immune responses. Neutrophils modulate immune responses via secretion of inflammatory mediators, cytokines and chemokines as well as via direct cell‐cell contact. A, It was shown that neutrophils can direct T‐cell recruitment via secretion of CXCL12‐rich membrane trails. B, At sites of infection, neutrophils may act as danger sensors by communicating the presence of infection to dendritic cells. C, Neutrophils can transport antigens to lymphoid organs, which directly or indirectly promotes antigen cross‐presentation to T cells. D, In the spleen and lymphoid organs, direct cell‐cell contact between neutrophils and NK cells is required for maturation of NK cells E, Neutrophils can have opposite functions in cancer, depending on the microenvironment. Left panel: In bladder cancer, neutrophils contribute to recruitment of T cells and promote T cell–mediated killing of tumour cells. Right panel: In breast cancer, neutrophils suppress CD8 T cells and promote metastasis
4. CONCLUDING REMARKS AND FUTURE DIRECTIVES
Neutrophils may represent promising effector cells in antibody‐based immunotherapy of cancer for a number of reasons. Firstly, they already represent a formidable army of effector cells, and their numbers can be easily expanded without the need for complicated ex vivo culture or manipulation, which is necessary for, for example, NK cells, dendritic cells or (CAR) T cells. Secondly, neutrophil kills via several nonapoptotic mechanisms, which may be advantageous when tumour cells have mutations in apoptosis pathways. Thirdly, neutrophils can regulate adaptive immune responses, which may lead to in situ generation of long‐term antitumour immunity. The contribution to current IgG‐based immunotherapies is likely limited. However, when the inhibitory SIRPα‐CD47 pathway was simultaneously blocked, killing of breast cancer cells by neutrophils in the presence of IgG anti‐HER‐2 mAbs was significantly enhanced.80 A cotreatment with antagonistic antibodies against CD47 or SIRPα may therefore greatly enhance the contribution of neutrophils as effector cells in IgG‐based antibody therapy. Alternatively, targeting of FcαRI with IgA, or hybrid IgA/IgG antibodies may result in prominent recruitment of neutrophils as cytotoxic effector cells.
ACKNOWLEDGEMENTS
N.H. wrote the paper and designed the illustrations. M.v.E. wrote the paper. This work was supported by Worldwide Cancer Research (grant 15‐1240).
Heemskerk N, van Egmond M. Monoclonal antibody‐mediated killing of tumour cells by neutrophils. Eur J Clin Invest. 2018;48(Suppl. 2):e12962 10.1111/eci.12962
REFERENCES
- 1. Carter PJ, Lazar GA. Next generation antibody drugs: pursuit of the ‘high‐hanging fruit’. Nat Rev Drug Discov. 2018;17:197‐223. [DOI] [PubMed] [Google Scholar]
- 2. Weiner LM, Murray JC, Shuptrine CW. Antibody‐based immunotherapy of cancer. Cell. 2012;148:1081‐1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer. 2012;12:278‐287. [DOI] [PubMed] [Google Scholar]
- 4. Kellner C, Otte A, Cappuzzello E, Klausz K, Peipp M. Modulating cytotoxic effector functions by Fc engineering to improve cancer therapy. Transfus Med Hemother. 2017;44:327‐336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism‐driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16:275‐287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bakema JE, van Egmond M. Fc receptor‐dependent mechanisms of monoclonal antibody therapy of cancer. Curr Top Microbiol Immunol. 2014;382:373‐392. [DOI] [PubMed] [Google Scholar]
- 7. Bibeau F, Lopez‐Crapez E, Di Fiore F, et al. Impact of Fc{gamma}RIIa‐Fc{gamma}RIIIa polymorphisms and KRAS mutations on the clinical outcome of patients with metastatic colorectal cancer treated with cetuximab plus irinotecan. J Clin Oncol. 2009;27:1122‐1129. [DOI] [PubMed] [Google Scholar]
- 8. Di Gaetano N, Cittera E, Nota R, et al. Complement activation determines the therapeutic activity of rituximab in vivo. J Immunol. 2003;171:1581‐1587. [DOI] [PubMed] [Google Scholar]
- 9. Uchida J, Hamaguchi Y, Oliver JA, et al. The innate mononuclear phagocyte network depletes B lymphocytes through Fc receptor‐dependent mechanisms during anti‐CD20 antibody immunotherapy. J Exp Med. 2004;199:1659‐1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Racila E, Link BK, Weng WK, et al. A polymorphism in the complement component C1qA correlates with prolonged response following rituximab therapy of follicular lymphoma. Clin Cancer Res. 2008;14:6697‐6703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med. 2000;6:443‐446. [DOI] [PubMed] [Google Scholar]
- 12. Hatjiharissi E, Xu L, Santos DD, et al. Increased natural killer cell expression of CD16, augmented binding and ADCC activity to rituximab among individuals expressing the Fc{gamma}RIIIa‐158 V/V and V/F polymorphism. Blood. 2007;110:2561‐2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. van der Bij GJ, Bögels M, Otten MA, et al. Experimentally induced liver metastases from colorectal cancer can be prevented by mononuclear phagocyte‐mediated monoclonal antibody therapy. J Hepatol. 2010;53:677‐685. [DOI] [PubMed] [Google Scholar]
- 14. Gül N, Babes L, Siegmund K, et al. Macrophages eliminate circulating tumor cells after monoclonal antibody therapy. J Clin Invest. 2014;124:812‐823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gül N, van Egmond M. Antibody‐dependent phagocytosis of tumor cells by macrophages: a potent effector mechanism of monoclonal antibody therapy of cancer. Cancer Res. 2015;75:5008‐5013. [DOI] [PubMed] [Google Scholar]
- 16. van Egmond M, Bakema JE. Neutrophils as effector cells for antibody‐based immunotherapy of cancer. Semin Cancer Biol. 2013;23:190‐199. [DOI] [PubMed] [Google Scholar]
- 17. Colombo MP, Ferrari G, Stoppacciaro A, et al. Granulocyte colony‐stimulating factor gene transfer suppresses tumorigenicity of a murine adenocarcinoma in vivo. J Exp Med. 1991;173:889‐897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Suttmann H, Riemensberger J, Bentien G, et al. Neutrophil granulocytes are required for effective Bacillus Calmette‐Guérin immunotherapy of bladder cancer and orchestrate local immune responses. Cancer Res. 2006;66:8250‐8257. [DOI] [PubMed] [Google Scholar]
- 19. Albanesi M, Mancardi DA, Jönsson F, et al. Neutrophils mediate antibody‐induced antitumor effects in mice. Blood. 2013;122:3160‐3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Siders WM, Shields J, Garron C, et al. Involvement of neutrophils and natural killer cells in the anti‐tumor activity of alemtuzumab in xenograft tumor models. Leuk Lymphoma. 2010;51:1293‐1304. [DOI] [PubMed] [Google Scholar]
- 21. Hernandez‐Ilizaliturri FJ, Jupudy V, Ostberg J, et al. Neutrophils contribute to the biological antitumor activity of rituximab in a non‐Hodgkin's lymphoma severe combined immunodeficiency mouse model. Clin Cancer Res. 2003;9:5866‐5873. [PubMed] [Google Scholar]
- 22. Moynihan KD, Opel CF, Szeto GL, et al. Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses. Nat Med. 2016;22:1402‐1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hong F, Hansen RD, Yan J, et al. Beta‐glucan functions as an adjuvant for monoclonal antibody immunotherapy by recruiting tumoricidal granulocytes as killer cells. Cancer Res. 2003;63:9023‐9031. [PubMed] [Google Scholar]
- 24. Zhong W, Hansen R, Li B, et al. Effect of yeast‐derived beta‐glucan in conjunction with bevacizumab for the treatment of human lung adenocarcinoma in subcutaneous and orthotopic xenograft models. J Immunother. 2009;32:703‐712. [DOI] [PubMed] [Google Scholar]
- 25. Chan AS, Jonas AB, Qiu X, et al. Imprime PGG‐mediated anti‐cancer immune activation requires immune complex formation. PLoS ONE. 2016;11:e0165909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Segal NH, Gada P, Senzer N, Gargano MA, Patchen ML, Saltz LB. A Phase II efficacy and safety, open‐label, multicenter study of imprime PGG injection in combination with cetuximab in patients with stage IV KRAS‐mutant colorectal cancer. Clin Colorectal Cancer. 2016;15:222‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cheung NK, Sowers R, Vickers AJ, Cheung IY, Kushner BH, Gorlick R. FCGR2A polymorphism is correlated with clinical outcome after immunotherapy of neuroblastoma with anti‐GD2 antibody and granulocyte macrophage colony‐stimulating factor. J Clin Oncol. 2006;24:2885‐2890. [DOI] [PubMed] [Google Scholar]
- 28. Cartron G1, Zhao‐Yang L, Baudard M, et al. Granulocyte‐macrophage colony‐stimulating factor potentiates rituximab in patients with relapsed follicular lymphoma: results of a phase II study. J Clin Oncol. 2008;26:2725‐2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nimmerjahn F, Ravetch JV. Antibodies, Fc receptors and cancer. Curr Opin Immunol. 2007;19:239‐245. [DOI] [PubMed] [Google Scholar]
- 30. Bruhns P, Jönsson F. Mouse and human FcR effector functions. Immunol Rev. 2015;268:25‐51. [DOI] [PubMed] [Google Scholar]
- 31. Valerius T, Wurflein D, Stockmeyer B, Repp R, Kalden JR, Gramatzki M. Activated neutrophils as effector cells for bispecific antibodies. Cancer Immunol Immunother. 1997;45:142‐145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Peipp M, Lammerts van Bueren JJ, Schneider‐Merck T, et al. . Antibody fucosylation differentially impacts cytotoxicity mediated by NK and PMN effector cells. Blood. 2008;112:2390‐2399. [DOI] [PubMed] [Google Scholar]
- 33. Schneider‐Merck T, Lammerts van Bueren JJ, Berger S, et al. . Human IgG2 antibodies against epidermal growth factor receptor effectively trigger antibody‐dependent cellular cytotoxicity but, in contrast to IgG1, only by cells of myeloid lineage. J Immunol. 2010;184:512‐520. [DOI] [PubMed] [Google Scholar]
- 34. Valerius T, Repp R, de Wit TP, et al. Involvement of the high‐affinity receptor for IgG (Fc gamma RI; CD64) in enhanced tumor cell cytotoxicity of neutrophils during granulocyte colony‐stimulating factor therapy. Blood. 1993;82:931‐939. [PubMed] [Google Scholar]
- 35. Honeychurch J, Tutt AL, Valerius T, Heijnen IA, Van De Winkel JG, Glennie MJ. Therapeutic efficacy of FcgammaRI/CD64‐directed bispecific antibodies in B‐cell lymphoma. Blood. 2000;96:3544‐3552. [PubMed] [Google Scholar]
- 36. van Ojik HH, Bevaart L, Dahle CE, et al. CpG‐A and B oligodeoxynucleotides enhance the efficacy of antibody therapy by activating different effector cell populations. Cancer Res. 2003;63:5595‐5600. [PubMed] [Google Scholar]
- 37. Curnow RT. Clinical experience with CD64‐directed immunotherapy. An overview. Cancer Immunol Immunother. 1997;45:210‐215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Valerius T, Stockmeyer B, van Spriel AB, et al. FcalphaRI (CD89) as a novel trigger molecule for bispecific antibody therapy. Blood. 1997;90:4485‐4492. [PubMed] [Google Scholar]
- 39. Deo YM, Sundarapandiyan K, Keler T, Wallace PK, Graziano RF. Bispecific molecules directed to the Fc receptor for IgA (Fc alpha RI, CD89) and tumor antigens efficiently promote cell‐mediated cytotoxicity of tumor targets in whole blood. J Immunol. 1998;160:1677‐1686. [PubMed] [Google Scholar]
- 40. Otten MA, Rudolph E, Dechant M, et al. Immature neutrophils mediate tumor cell killing via IgA but not IgG Fc receptors. J Immunol. 2005;174:5472‐5480. [DOI] [PubMed] [Google Scholar]
- 41. Dechant M, Valerius T. IgA antibodies for cancer therapy. Crit Rev Oncol Hematol. 2001;39:69‐77. [DOI] [PubMed] [Google Scholar]
- 42. Guettinger Y1, Barbin K, Peipp M, et al. A recombinant bispecific single‐chain fragment variable specific for HLA class II and Fc alpha RI (CD89) recruits polymorphonuclear neutrophils for efficient lysis of malignant B lymphoid cells. J Immunol. 2010;184:1210‐1217. [DOI] [PubMed] [Google Scholar]
- 43. Aleyd E, Heineke MH, van Egmond M. The era of the immunoglobulin A Fc receptor FcαRI; its function and potential as target in disease. Immunol Rev. 2015;268:123‐138. [DOI] [PubMed] [Google Scholar]
- 44. Lohse S, Loew S, Kretschmer A, et al. Effector mechanisms of IgA antibodies against CD20 include recruitment of myeloid cells for antibody‐dependent cell‐mediated cytotoxicity and complement‐dependent cytotoxicity. Br J Haematol. 2017;181:413‐417. [DOI] [PubMed] [Google Scholar]
- 45. Lohse S, Derer S, Beyer T, et al. Recombinant dimeric IgA antibodies against the epidermal growth factor receptor mediate effective tumor cell killing. J Immunol. 2011;186:3770‐3778. [DOI] [PubMed] [Google Scholar]
- 46. van der Steen L, Tuk CW, Bakema JE, et al. Immunoglobulin A: Fc(alpha)RI interactions induce neutrophil migration through release of leukotriene B4. Gastroenterology. 2009;137:2018‐2029. [DOI] [PubMed] [Google Scholar]
- 47. Otten MA, Bakema JE, Tuk CW, et al. Enhanced FcαRI‐mediated neutrophil migration towards tumor colonies in the presence of endothelial cells. Eur J Immunol. 2012;42:1815‐1821. [DOI] [PubMed] [Google Scholar]
- 48. Gentles AJ, Newman AM, Liu CL, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21:938‐945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Coffelt SB, Wellenstein MD, de Visser KE. Neutrophils in cancer: neutral no more. Nat Rev Cancer. 2016;16:431‐446. [DOI] [PubMed] [Google Scholar]
- 50. van Egmond M, van Vuuren AJ, Morton HC, et al. Human immunoglobulin A receptor (FcalphaRI, CD89) function in transgenic mice requires both FcR gamma chain and CR3 (CD11b/CD18). Blood. 1999;93:4387‐4394. [PubMed] [Google Scholar]
- 51. Boross P, Lohse S, Nederend M, et al. IgA EGFR antibodies mediate tumor killing in vivo. EMBO Mol Med. 2013;5:1213‐1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Meyer S, Nederend M, Jansen JH, et al. Improved in vivo anti‐tumor effects of IgA‐Her2 antibodies through half‐life extension and serum exposure enhancement by FcRn targeting. MAbs. 2016;8:87‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lohse S, Meyer S, Meulenbroek LA, et al. An anti‐EGFR IgA that displays improved pharmacokinetics and myeloid effector cell engagement in vivo. Cancer Res. 2016;76:403‐417. [DOI] [PubMed] [Google Scholar]
- 54. Kelton W, Mehta N, Charab W, et al. IgGA: a “cross‐isotype” engineered human Fc antibody domain that displays both IgG‐like and IgA‐like effector functions. Chem Biol. 2014;21:1603‐1609. [DOI] [PubMed] [Google Scholar]
- 55. Borrok MJ, Luheshi NM, Beyaz N, et al. Enhancement of antibody‐dependent cell‐mediated cytotoxicity by endowing IgG with FcαRI (CD89) binding. MAbs. 2015;7:743‐751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. van Egmond M, van Spriel AB, Vermeulen H, Huls G, van Garderen E, van de Winkel JG. Enhancement of polymorphonuclear cell‐mediated tumor cell killing on simultaneous engagement of fcgammaRI (CD64) and fcalphaRI (CD89). Cancer Res. 2001;61:4055‐4060. [PubMed] [Google Scholar]
- 57. Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. Neutrophil function: from mechanisms to disease. Annu Rev Immunol. 2012;30:459‐489. [DOI] [PubMed] [Google Scholar]
- 58. Horner H, Frank C, Dechant C, et al. Intimate cell conjugate formation and exchange of membrane lipids precede apoptosis induction in target cells during antibody‐dependent, granulocyte‐mediated cytotoxicity. J Immunol. 2007;179:337‐345. [DOI] [PubMed] [Google Scholar]
- 59. Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532‐1535. [DOI] [PubMed] [Google Scholar]
- 60. Saffarzadeh M1, Juenemann C, Queisser MA, et al. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS ONE. 2012;7:e32366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Aleyd E, van Hout MW, Ganzevles SH, et al. IgA enhances NETosis and release of neutrophil extracellular traps by polymorphonuclear cells via Fcα receptor I. J Immunol. 2014;192:2374‐2383. [DOI] [PubMed] [Google Scholar]
- 62. Bakema JE, Ganzevles SH, Fluitsma DM, et al. Targeting FcαRI on polymorphonuclear cells induces tumor cell killing through autophagy. J Immunol. 2011;187:726‐732. [DOI] [PubMed] [Google Scholar]
- 63. Galluzzi L, Vitale I, Aaronson SA, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486‐541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Bologna L, Gotti E, Da Roit F, et al. Ofatumumab is more efficient than rituximab in lysing B chronic lymphocytic leukemia cells in whole blood and in combination with chemotherapy. J Immunol. 2013;190:231‐239. [DOI] [PubMed] [Google Scholar]
- 65. Valgardsdottir R, Cattaneo I, Klein C, Introna M, Figliuzzi M, Golay J. Human neutrophils mediate trogocytosis rather than phagocytosis of CLL B cells opsonized with anti‐CD20 antibodies. Blood. 2017;129:2636‐2644. [DOI] [PubMed] [Google Scholar]
- 66. van Spriel AB, Leusen JH, van Egmond M, et al. Mac‐1 (CD11b/CD18) is essential for Fc receptor‐mediated neutrophil cytotoxicity and immunologic synapse formation. Blood. 2001;97:2478‐2486. [DOI] [PubMed] [Google Scholar]
- 67. van Spriel AB, van Ojik HH, Bakker A, Jansen MJ, van de Winkel JG. Mac‐1 (CD11b/CD18) is crucial for effective Fc receptor‐mediated immunity to melanoma. Blood. 2003;101:253‐258. [DOI] [PubMed] [Google Scholar]
- 68. Hubert P, Heitzmann A, Viel S, et al. Antibody‐dependent cell cytotoxicity synapses form in mice during tumor‐specific antibody immunotherapy. Cancer Res. 2011;71:5134‐5143. [DOI] [PubMed] [Google Scholar]
- 69. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol. 2011;11:519‐531. [DOI] [PubMed] [Google Scholar]
- 70. Deniset JF, Kubes P. Recent advances in understanding neutrophils. F1000Res. 2016;5:2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lim K, Hyun YM, Lambert‐Emo K, et al. Neutrophil trails guide influenza‐specific CD8⁺ T cells in the airways. Science. 2015;349:aaa4352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. van Gisbergen KP, Sanchez‐Hernandez M, Geijtenbeek TB, van Kooyk Y. Neutrophils mediate immune modulation of dendritic cells through glycosylation‐dependent interactions between Mac‐1 and DC‐SIGN. J Exp Med. 2005;201:1281‐1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Breedveld A, Groot Kormelink T, van Egmond M, de Jong EC. Granulocytes as modulators of dendritic cell function. J Leukoc Biol. 2017;102:1003‐1016. [DOI] [PubMed] [Google Scholar]
- 74. Duffy D, Perrin H, Abadie V, et al. Neutrophils transport antigen from the dermis to the bone marrow, initiating a source of memory CD8+ T cells. Immunity. 2012;37:917‐929. [DOI] [PubMed] [Google Scholar]
- 75. Hampton HR, Bailey J, Tomura M, Brink R, Chtanova T. Microbe‐dependent lymphatic migration of neutrophils modulates lymphocyte proliferation in lymph nodes. Nat Commun. 2015;6:7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Jaeger BN, Donadieu J, Cognet C, et al. Neutrophil depletion impairs natural killer cell maturation, function, and homeostasis. J Exp Med. 2012;209:565‐580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Coffelt SB, Kersten K, Doornebal CW, et al. IL‐17‐producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature. 2015;522:345‐348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Eruslanov EB, Bhojnagarwala PS, Quatromoni JG, et al. Tumor‐associated neutrophils stimulate T cell responses in early‐stage human lung cancer. J Clin Invest. 2014;124:5466‐5480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Eruslanov EB. Phenotype and function of tumor‐associated neutrophils and their subsets in early‐stage human lung cancer. Cancer Immunol Immunother. 2017;66:997‐1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zhao XW, van Beek EM, Schornagel K, et al. CD47‐signal regulatory protein‐α (SIRPα) interactions form a barrier for antibody‐mediated tumor cell destruction. Proc Natl Acad Sci USA. 2011;108:18342‐18347. [DOI] [PMC free article] [PubMed] [Google Scholar]