

ABSTRACT
Background: Mitochondrial dysfunction plays a key role in PD, but the underlying molecular mechanisms remain unresolved. We hypothesized that the disruption of mitochondrial function in PD is primed by rare, protein‐altering variation in nuclear genes controlling mitochondrial structure and function.
Objective: The objective of this study was to assess whether genetic variation in genes associated with mitochondrial function influences the risk of idiopathic PD.
Methods: We employed whole‐exome sequencing data from 2 independent cohorts of clinically validated idiopathic PD and controls, the Norwegian ParkWest cohort (n = 411) and the North American Parkinson's Progression Markers Initiative (n = 640). We applied burden‐based and variance‐based collapsing methods to assess the enrichment of rare, nonsynonymous, and damaging genetic variants on genes, exome‐wide, and on a comprehensive set of mitochondrial pathways, defined as groups of genes controlling specific mitochondrial functions.
Results: Using the sequence kernel association test, we detected a significant polygenic enrichment of rare, nonsynonymous variants in the gene‐set encoding the pathway of mitochondrial DNA maintenance. Notably, this was the strongest association in both cohorts and survived multiple testing correction (ParkWest P = 6.3 × 10−3, Parkinson's Progression Markers Initiative P = 6.9 × 10−5, metaanalysis P = 3.2 × 10−6).
Conclusions: Our results show that the enrichment of rare inherited variation in the pathway controlling mitochondrial DNA replication and repair influences the risk of PD. We propose that this polygenic enrichment contributes to the impairment of mitochondrial DNA homeostasis, thought to be a key mechanism in the pathogenesis of PD, and explains part of the disorder's “missing heritability.” © 2018 The Authors. Movement Disorders published by Wiley Periodicals, Inc. on behalf of International Parkinson and Movement Disorder Society
Keywords: Parkinson's disease, genetics, neurodegeneration, whole‐exome sequencing, genetic association studies
Parkinson's disease (PD) affects 1.8% of the population aged older than 65 years.1 Although the etiology of PD remains unknown, mitochondrial dysfunction clearly plays a role. Mutations in most of the genes causing familial PD, including SNCA,2 LRRK2,3 PINK1, PRKN,4 PARK7 (DJ‐1),5 and VPS35,6 have been shown to disrupt mitochondrial quality control. Moreover, impaired mitochondrial DNA (mtDNA) maintenance7, 8 and widespread respiratory chain dysfunction9 occur in the brains of individuals with idiopathic PD. The molecular etiology underlying mitochondrial impairment in idiopathic PD remains, however, unresolved.
Pathogenic mutations in several nuclear mitochondrial genes involved in mtDNA homeostasis have been shown to cause severe nigrostriatal degeneration of a similar type as the one that occurs in PD.10, 11, 12, 13 Moreover, inherited variation in POLG 14 and TFAM,15 both of which are essential for mtDNA maintenance, has been associated with an increased risk of idiopathic PD, although the reported effects were generally weak and have not been reproduced by large genome‐wide association studies. Therefore, it remains undetermined whether mitochondrial dysfunction in PD can, to some extent, be attributed to inherited genetic variation.
We hypothesized that the disruption of mitochondrial function in PD is partly caused by synergistic effects of rare genetic variation in nuclear genes controlling mitochondrial function. To test our hypothesis, we employed whole‐exome sequence data from 2 independent, prospectively collected PD cohorts: the Norwegian ParkWest study (ParkWest)16 and the Parkinson's Progression Markers Initiative (PPMI).17
Methods
Cohorts and Sequencing
We sequenced all individuals with clinically validated PD (n = 192) from the Norwegian ParkWest study, a prospective population‐based cohort of idiopathic PD.16 Controls (n = 219) were provided from cohorts of previously sequenced individuals with testis cancer (n = 167) or acoustic neuroma (n = 52) who had been recruited and examined at our hospital and had no clinical signs of neurodegenerative or other neurological disorders. DNA was extracted from blood by routine procedures and sequenced at HudsonAlpha Institute for Biotechnology (Huntsville, Alabama) using Roche‐NimbleGen Sequence Capture EZ Exome v2 (173 controls) and v3 (all PD and 46 controls) kits (Roche, Brussels, Switzerland) and paired‐end 100 bp sequencing on the Illumina HiSeq platform (Illumina, San Diego, USA). The reads were mapped to the hg19 reference genome using BWA v0.6.2,18 polymerase chain reaction duplicates removed with Picard v1.118,19 and the alignment refined using the Genome Analysis Toolkit v3.3.020 applying base quality score recalibration and realignment around indels recommended in the GATK Best Practices workflow.21, 22 Variants were called in all samples using the GATK HaplotypeCaller20 with default parameters. Next, variant quality score recalibration was performed using 99.9% sensitivity threshold.20 The remaining variants were filtered against the intersection of capture targets (v2 and v3) using BEDtools23 and VCFtools.24 Variants with total depth below 10X were marked as unknown genotype (no‐call) using BCFtools.25 Indel calls, which were found to be less reliable than single‐nucleotide variants, were excluded from downstream analyses.
Additional data used in the preparation of this article were obtained from the PPMI database.17 Whole‐exome sequence data was available for 640 individuals (459 cases, 181 controls). Sequencing had been performed using the Illumina Nextera Rapid Capture Expanded Exome Kit and paired‐end 100 bp reads on the Illumina HiSeq 2500 (Illumina, San Diego, USA). Calling and alignment had been performed by the PPMI. Indels were removed prior to variant quality control (QC) using VCFtools.24
Variant Filtering and QC
Whole‐exome sequence data were recoded into binary PLINK input format, and QC of individual and single nucleotide polymorphism (SNP) data was performed on the ParkWest and PPMI datasets individually using PLINK v1.90.26 Individuals were excluded if their genotypic data showed a missing rate >2%, abnormal heterozygosity (±3 standard deviations, calculated for common and rare variants separately), conflicting sex assignment, cryptic relatedness (identity by descent > 0.2), or divergent ancestry (non‐European). Population stratification was studied using multidimensional scaling against the HapMap populations.27
SNPs were excluded as a result of genotyping rate less than 98%, different call rates between cases and controls (P < .02) or departure from the Hardy‐Weinberg equilibrium (P < 10−5). Only autosomes were considered. Monomorphic and multiallelic variants were removed. The transition‐transversion ratio was calculated before and after QC. Principal component analysis was performed using Eigensoft28, 29 with standard filtering settings (5 iterations, 10 principal components, sigma 6). Analysis of variance of the first 10 principal components was performed with significance level cutoff set to P < .01. For ParkWest, there were 2 significant principal components (3 and 6) that were corrected for in downstream analyses. There were no significant principal components for PPMI. All QC analyses were performed using PLINK v1.9026 and R30 unless otherwise specified.
Annotation and Subset Filtering
Datasets were annotated using ANNOVAR31 according to the RefSeq gene transcripts, and 2 variant subsets were extracted for further analyses. These were defined as “nonsynonymous” and “damaging” according to ANNOVAR. Nonsynonymous variants comprised missense changes (Sequence Ontology: 0001583). Damaging variants comprised all single nucleotide changes that had a deleterious score in all of the following 5 prediction algorithms: PolyPhen2 HumDiv, PolyPhen2 HumVar, MutationTaster, SIFT, and LRT. In the ParkWest dataset, rare variants were defined as having a minor allele frequency (MAF) of < 1%. Because the PPMI cohort had a substantially uneven number of cases and controls, rare variants were defined as having a MAF of <1% in either cases or controls to avoid a unidirectional variant inclusion bias.
Pathway Curation
The mitochondrial pathways were defined as groups of genes encoding functionally and/or structurally linked proteins directly involved in mitochondrial function. Pathways were manually curated from Mitocarta v2.0,32 a collection of all known proteins with strong support for mitochondrial localization. The pathways were subsequently expanded using STRING33 to include genes encoding additional proteins that are involved in the pathways, but have no proven mitochondrial localization. Specifically, for each pathway we compiled a list of additional candidate proteins ranked by the number and strength (STRING combined score) of STRING interactions with the original pathway. The resulting candidate lists were manually inspected, and the original pathways were supplemented with additional genes encoding proteins with a known involvement in mitochondrial pathways, but not established mitochondrial localization (Supplemental Table S1).
Genetic Association Analyses
Single‐point association was performed for common variants (MAF > 1%) using the chi‐square test for PPMI and logistic regression with significant principal components as covariates for ParkWest. For collapsing tests, variants were annotated using ANNOVAR and analyzed using a weighted burden34 and the sequence kernel association test (SKAT)35 as implemented by the SKAT R package v1.3.2.36 In these analyses, variants within a specified region (ie, a gene or pathway) were combined to a single genetic score that was subsequently tested for association in a logistic regression framework. The burden test assumes that all variants in a specified region are causal and influence the phenotype in the same direction, that is, either increase or decrease the risk of PD. Consequently, it suffers from a substantial loss of power when these premises are not valid. SKAT aggregates variants within a specified region without considering the direction of effect for individual variants. Therefore, SKAT is better suited for detecting associations where both risk and protective variants and/or numerous noncausal variants are present. For our analyses, standard weights were used,35 and only rare variants were considered. Different methods of per‐hypothesis resampling were applied as described by Lee and colleagues.36 Genes with only 1 variant were excluded from the gene‐based analyses as this would be representative of single SNP associations rather than true enrichment. Using resampling, the minimum achievable P value (MAP) was determined for each gene and pathway.36 Genes/pathways with minimum achievable P values above the Bonferroni‐corrected threshold for statistical significance were excluded to reduce statistical noise. In total, 3,441 gene tests were performed in the ParkWest cohort, and 13,034 in the PPMI cohort (across all tests and subsets). Meta P values for pathways were calculated using the optimally weighted Z test37 as implemented by the metap R package.38
Multiple Testing Correction
Single‐variant gene and pathway meta‐analysis associations were corrected using the Bonferroni method. To control the family‐wise error in individual cohort pathways, the minP/maxT method39 was implemented using 10,000 null‐permuted phenotype samples for ParkWest and 100,000 for PPMI (different number of permutations as a result of the difference in dataset size).
Power Analysis
The average statistical power of the gene‐based SKAT and burden test were calculated using the SKAT R package v1.3.2.36 The disease model assumed a prevalence of PD of 0.015 and used the empirical MAFs of the ParkWest cohort. Power calculations were carried out using different assumptions of the percentage of causal SNPs (10%, 25%, 50%, 75%, and 100%). The original function from the SKAT R package was modified to calculate the average power of the exonic regions instead of the default random genomic regions.
Standard Protocol Approvals, Registrations, and Patient Consents
These studies were approved by the Regional Committee for Medical and Health Research Ethics, Western Norway (REK 131/04). Written, informed consent was obtained from all participants.
Results
After alignment, variant calling and QC the final ParkWest dataset comprised 377 individuals (181 cases and 196 controls). Mean depth per individual was 80.2 and 92% of targeted bases were covered at ≥20‐fold. In the ParkWest dataset we observed 130,500 variants, of which 61.1% (79,763) were rare. We identified 56,429 nonsynonymous variants and 8,646 damaging variants. The transition‐transversion ratio‐ratio for exonic variants was 3.28 (3.23 before QC). The final PPMI dataset comprised 513 individuals (371 cases and 142 controls). We observed 380,423 variants of which 73.6% (280,099) were rare. We identified 107,130 nonsynonymous variants and 16,644 damaging variants. The transition‐transversion ratio for exonic variants was 3.13 (3.02 before QC). Synonymous and nonsynonymous variants had a similar distribution in the 2 cohorts (Supplemental Table S2) as well as between cases and controls (Supplemental Table S3). Singleton variants comprised 37.4% of the ParkWest dataset and 51.1% of the PPMI dataset (Supplemental Table S4).
Population stratification analysis with HapMap confirmed that both cohorts consisted primarily of individuals of Western and Northern European descent. Quantile‐quantile plots of common variant associations after QC showed no inflation of test statistics, with λ = 1.02 and λ = 1.03 for PPMI and ParkWest, respectively. Multidimensional scaling showed no stratification between cases and controls (Fig. 1). No single variants produced exome‐wide significant associations. The lowest P value was for the SNP rs57859638 (P = 1.2 × 10−4), odds ratio (OR) = 0.47, MAF = 0.219) in IQCF1 for ParkWest, and for rs543304 (P = 1.1 × 10−5), OR = 0.49, MAF = 0.197) in BRCA2 in PPMI. The complete results are available in Supplemental Tables S5 and S6.
Figure 1.
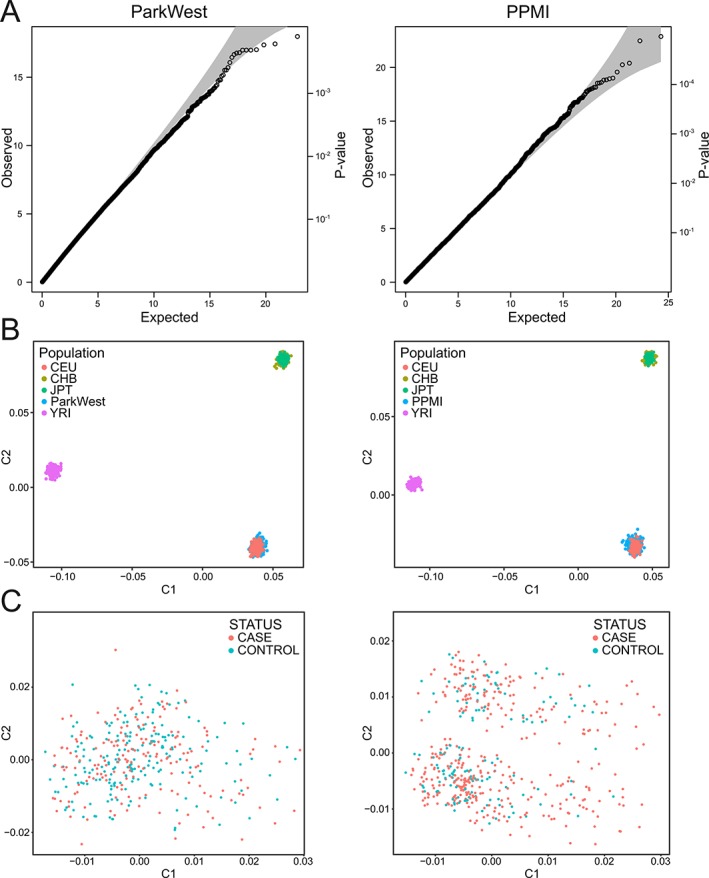
Quality control. (A) Quantile‐quantile‐plots of association for common variants using chi‐square for Parkinson's Progression Markers Initiative and logistic regression with significant principal components (3 and 6) as covariates for ParkWest. (B) Population stratification (multidimensional scaling) against HapMap populations (CEU, Utah residents with Northern and Western European ancestry; CHB, Han Chinese in Beijing, China; JPT, Japanese in Tokyo, Japan; YRI, Yoruba in Ibadan, Nigeria). (C) Multidimensional scaling plots show no stratification between cases and controls in any of the cohorts. All plots are based on data after quality control. C1, first principal component; C2, second principal component.
Gene‐Based Analyses
Gene‐based enrichment analyses for rare nonsynonymous or damaging variants by burden test or SKAT showed no significant associations after Bonferroni correction. Moreover, none of the top 10 nominally significant genes replicated across both the ParkWest and PPMI cohorts (Table 1). The results for all tested genes are shown in Supplemental Tables S7 to S14.
Table 1.
Top single gene results
| Nonsynonymous rare variants | |||||||
|---|---|---|---|---|---|---|---|
| Burden | SKAT | ||||||
| ParkWest | PPMI | ParkWest | PPMI | ||||
| Gene | P value | Gene | P value | Gene | P value | Gene | P value |
| ZNF76 | 3.98 × 10−4 | CHRM2 | 2.46 × 10−4 | TSR1 | 2.57 × 10−4 | CHRM2 | 1.16 × 10−5 |
| KIF20B | 4.74 × 10−4 | UGT1A4 | 4.41 × 10−4 | COL24A1 | 7.87 × 10−4 | MC4R | 2.64 × 10−4 |
| KANK1 | 2.87 × 10−3 | MC4R | 5.27 × 10−4 | WDFY4 | 1.52 × 10−3 | RECQL4 | 2.80 × 10−4 |
| BIRC6 | 4.03 × 10−3 | GLTSCR1 | 6.76 × 10−4 | LRRN2 | 2.99 × 10−3 | CEP131 | 5.22 × 10−4 |
| NUMA1 | 4.75 × 10−3 | TG | 1.05 × 10−3 | ABCC11 | 3.50 × 10−3 | KRTAP10‐7 | 5.52 × 10−4 |
| DNAJC17 | 4.80 × 10−3 | CACNA1H | 1.34 × 10−3 | FAM214A | 5.14 × 10−3 | SETD4 | 6.35 × 10−4 |
| ZNFX1 | 4.88 × 10−3 | HORMAD2 | 1.41 × 10−3 | GEMIN5 | 5.87 × 10−3 | STEAP4 | 6.41 × 10−4 |
| CP | 5.02 × 10−3 | BRPF3 | 1.46 × 10−3 | USP6 | 6.05 × 10−3 | MRPL4 | 7.64 × 10−4 |
| MKI67 | 5.78 × 10−3 | ITGA2B | 1.47 × 10−3 | LRRC75B | 6.52 × 10−3 | FER1L6 | 8.60 × 10−4 |
| CFTR | 6.52 × 10−3 | ADGRG7 | 1.58 × 10−3 | DFNB31 | 7.71 × 10−3 | NDUFA9 | 9.26 × 10−4 |
| Damaging rare variants | |||||||
|---|---|---|---|---|---|---|---|
| Burden | SKAT | ||||||
| ParkWest | PPMI | ParkWest | PPMI | ||||
| Gene | P value | Gene | P value | Gene | P value | Gene | P value |
| DFNB31 | 5.25 × 10−4 | ATP8B4 | 1.75 × 10−3 | DNAH2 | 2.79 × 10−3 | ADAMTS14 | 1.70 × 10−3 |
| ACACB | 4.31 × 10−2 | NRAP | 6.72 × 10−3 | DFNB31 | 2.95 × 10−3 | PPP2R3A | 2.85 × 10−3 |
| LOXHD1 | 6.22 × 10−2 | ATP4A | 1.04 × 10−2 | POLG | 5.02 × 10−3 | HADH | 4.99 × 10−3 |
| CFTR | 6.51 × 10−2 | TEP1 | 1.09 × 10−2 | KIAA0196 | 2.24 × 10−2 | LRP1B | 5.61 × 10−3 |
| LAMA1 | 1.56 × 10−1 | DNAH11 | 1.13 × 10−2 | DNAH3 | 2.63 × 10−2 | PCDHB1 | 7.08 × 10−3 |
| PITRM1 | 1.64 × 10−1 | CSMD1 | 1.47 × 10−2 | ACACB | 3.82 × 10−2 | INADL | 9.07 × 10−3 |
| DNAH2 | 1.73 × 10−1 | PCDHB1 | 1.50 × 10−2 | RELN | 5.96 × 10−2 | CEL | 1.10 × 10−2 |
| FAT2 | 1.92 × 10−1 | BSN | 1.66 × 10−2 | PPFIBP2 | 6.20 × 10−2 | UBXN11 | 1.10 × 10−2 |
| TRIM63 | 2.26 × 10−1 | CPXM1 | 2.08 × 10−2 | SYNE2 | 1.19 × 10−1 | RGS11 | 1.40 × 10−2 |
| QRSL1 | 2.49 × 10−1 | HHAT | 2.10 × 10−2 | MASP2 | 1.19 × 10−1 | HHAT | 1.60 × 10−2 |
Nominal P values given, no single gene association was statistically significant after multiple testing correction. PPMI, Parkinson's Progression Markers Initiative; SKAT, sequence kernel association test.
Power analyses showed that our sample size was underpowered for detecting single gene associations. Assuming that 50% of the nonsynonymous variants in a gene influence the phenotype in any direction (ie, deleterious or protective), our calculations suggest that a sample size of approximately 8,000 for the SKAT and 11,000 for the weighted burden test are required to reach an average power of 80% across all genes at a significance cut‐off of α = 0.05 and surviving Bonferroni correction for 20,000 genes (corrected α = 2.5 × 10−6; Fig. 2).
Figure 2.
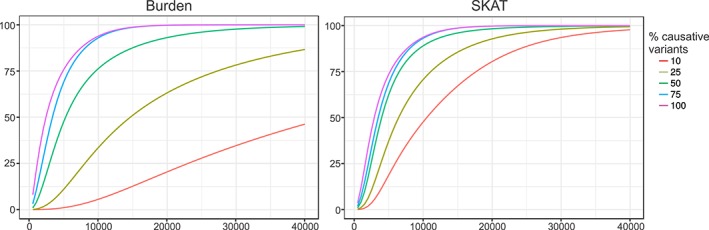
Power analysis. Power estimation for sequence kernel association test (SKAT) and weighted burden test using the rare variant matrix from the ParkWest dataset as basis for the simulation. Genome‐wide significance levels were set to α = 2.5 × 10−6, assuming a total of 20,000 genes. The y‐axis shows the average power across all genes, and the x‐axis shows total sample size. Simulations were made for different percentages of causal variants.
Pathway‐Based Analyses
Starting with Mitocarta, we curated a total of 28 pathways with known molecular function, comprising a total of 868 unique genes (including 100 non‐Mitocarta genes added after expansion with functionally relevant genes lacking evidence of mitochondrial localization). The remaining Mitocarta proteins could not be confidently assigned to a pathway because of limited information regarding their function. The entire noncurated Mitocarta list was also included as a separate pathway. A complete description of the pathways is provided in Supplemental Table S1. After excluding pathways with minimum achievable P value above the threshold for statistical significance, we analyzed 26/18 pathways in ParkWest and 29/26 pathways in PPMI for nonsynonymous/damaging variants, respectively.
Mitochondrial pathway analyses by burden test showed no evidence of enrichment in PD. SKAT detected a significantly skewed distribution of rare, nonsynonymous variants between individuals with PD and controls. The 2 most significant associations were found for the pathways of mtDNA maintenance and mitochondrial calcium homeostasis (Table 2) in both the ParkWest and PPMI cohorts. The strongest association was for mtDNA maintenance (ParkWestP = 6.3 × 10−3, PPMI P = 6.9 × 10−5, meta P = 3.2 × 10−6), which survived multiple testing correction. mtDNA maintenance was also among the top results for damaging variants in both datasets, but did not survive multiple testing correction. None of the other mitochondrial pathways showed significant associations that survived multiple testing correction or had significant nominal P values in both cohorts, except from the meta‐analysis of mitochondrial calcium homeostasis (meta P = 6.7 × 10−5). Detailed pathway results are available in the supplemental data (Supplemental Table S15). Repeating the pathway analyses with rare synonymous variants only yielded no significant results, indicating that the observed enrichment is specific for protein‐altering variation and does not reflect a generally skewed distribution of rare variants in the material.
Table 2.
SKAT analyses of pathways
| Pathway | Nonsynonymous | Damaging | ||||
|---|---|---|---|---|---|---|
| ParkWest | PPMI | Meta | ParkWest | PPMI | Meta | |
| Amino acid metabolism | 3.35 × 10−1 | 6.17 × 10−1 | 4.79 × 10−1 | 5.08 × 10−1 | 5.01 × 10−1 | 5.06 × 10−1 |
| Apoptosis | 5.08 × 10−1 | 5.52 × 10−1 | 5.44 × 10−1 | 5.74 × 10−1 | 7.40 × 10−1 | 7.29 × 10−1 |
| Dopamine metabolism | NA | 4.41 × 10−1 | NA | NA | NA | NA |
| Fatty acid metabolism | 3.88 × 10−1 | 4.18 × 10−1 | 3.66 × 10−1 | 5.91 × 10−1 | 6.41 × 10−2 | 1.58 × 10−1 |
| Glycolysis/gluconeogenesis | 7.43 × 10−1 | 3.92 × 10−1 | 5.86 × 10−1 | 5.65 × 10−1 | 3.65 × 10−1 | 4.38 × 10−1 |
| Heat production | 5.82 × 10−1 | 7.32 × 10−1 | 7.27 × 10−1 | NA | NA | NA |
| Heme metabolism | 8.43 × 10−2 | 8.86 × 10−2 | 2.74 × 10−2 | NA | 7.34 × 10−1 | NA |
| Iron homeostasis | 7.52 × 10−1 | 4.12 × 10−1 | 6.08 × 10−1 | NA | 9.11 × 10−1 | NA |
| Iron‐sulfur cluster building | NA | 7.76 × 10−1 | NA | NA | 6.94 × 10−1 | NA |
| PPAR signaling | 9.60 × 10−2 | 8.68 × 10−2 | 2.99 × 10−2 | 5.57 × 10−1 | 2.17 × 10−1 | 3.08 × 10−1 |
| Krebs cycle | 9.75 × 10−2 | 1.57 × 10−2 | 6.62 × 10−3 | 5.45 × 10−1 | 4.56 × 10−2 | 1.13 × 10−1 |
| Mitocarta | 3.31 × 10−1 | 1.01 × 10−1 | 1.05 × 10−1 | 1.80 × 10−1 | 6.06 × 10−2 | 3.82 × 10−2 |
| Mitochondrial acetylation | 8.67 × 10−1 | 6.26 × 10−1 | 8.33 × 10−1 | NA | 5.75 × 10−1 | NA |
| Mitochondrial calcium homeostasis | 2.44 × 10−3 | 4.41 × 10−3 | 6.67 × 10−5 a | NA | 1.63 × 10−3 | NA |
| Mitochondrial dynamics and quality control | 6.98 × 10−2 | 2.62 × 10−1 | 7.42 × 10−2 | 3.08 × 10−2 | 5.52 × 10−1 | 1.32 × 10−1 |
| Mitochondrial ribosome | 2.97 × 10−2 | 1.56 × 10−1 | 2.30 × 10−2 | 2.40 × 10−2 | 3.87 × 10−1 | 6.62 × 10−2 |
| Mitochondrial transcription | 5.59 × 10−1 | 6.31 × 10−1 | 6.37 × 10−1 | NA | 9.81 × 10−1 | NA |
| Mitochondrial translation | 3.00 × 10−1 | 4.42 × 10−1 | 3.26 × 10−1 | 3.56 × 10−1 | 6.20 × 10−1 | 4.97 × 10−1 |
| Mitochondrial transport | 9.68 × 10−1 | 3.76 × 10−1 | 8.32 × 10−1 | 9.03 × 10−1 | 9.55 × 10−1 | 9.84 × 10−1 |
| Mitochondrial tRNA homeostasis | 2.93 × 10−1 | 1.29 × 10−1 | 1.12 × 10−1 | 2.74 × 10−1 | 5.36 × 10−1 | 3.73 × 10−1 |
| mtDNA maintenance | 6.35 × 10−3 | 6.95 × 10−5 a | 3.17 × 10−6 a | 1.29 × 10−2 | 1.91 × 10−2 | 1.25 × 10−3 |
| NAD metabolism | NA | 6.27 × 10−1 | NA | NA | 4.22 × 10−1 | NA |
| One carbon and folate metabolism | 2.30 × 10−1 | 6.15 × 10−3 | 8.63 × 10−3 | 3.30 × 10−1 | 1.92 × 10−2 | 3.16 × 10−2 |
| Oxidative phosphorylation | 8.24 × 10−1 | 1.47 × 10−2 | 1.47 × 10−1 | 4.24 × 10−1 | 3.03 × 10−3 | 1.36 × 10−2 |
| Pyruvate metabolism | 3.80 × 10−2 | 1.11 × 10−1 | 1.87 × 10−2 | 2.34 × 10−2 | 3.72 × 10−2 | 4.05 × 10−3 |
| ROS metabolism | 4.82 × 10−1 | 2.06 × 10−1 | 2.57 × 10−1 | 4.56 × 10−1 | 6.25 × 10−2 | 1.08 × 10−1 |
| Steroid metabolism | 5.61 × 10−1 | 2.30 × 10−1 | 3.22 × 10−1 | NA | 3.33 × 10−1 | NA |
| Sulfur metabolism | 7.87 × 10−1 | 1.00 | 1.00 | 9.70 × 10−1 | 9.71 × 10−1 | 9.96 × 10−1 |
| Urea cycle | 9.30 × 10−1 | 7.90 × 10−2 | 4.55 × 10−1 | NA | NA | NA |
Nominal P values given. mtDNA, mitochondrial DNA; NA, not analyzed; NAD, nicotinamide adenine dinucleotide; PPAR, peroxisome proliferator‐activated receptor; PPMI, Parkinson's Progression Markers Initiative; ROS, reactive oxygen species; SKAT, sequence kernel association test; tRNA, transfer ribonucleic acid.
Statistically significant after multiple testing correction.
Gene‐based SKAT analysis revealed no specific mono‐ or oligogenic signals sufficient to drive the observed association in both cohorts, suggesting multiple genes contribute to the observed signal (Supplemental Table S16). This is also visualized in Figure 3, which shows the amount of variance contributed by each gene in the pathway. mtDNA maintenance is a complex process requiring a well‐orchestrated synergy of several biological pathways controlled by overlapping, but functionally distinct protein groups. To assess whether the observed association was primarily driven by a particular functional subnetwork, we subdivided the mtDNA maintenance pathway into mtDNA replication, mtDNA repair, and nucleotide homeostasis. None of these subpathways alone was sufficient to drive the overarching pathway signal and observed association with PD (Supplemental Table S17).
Figure 3.
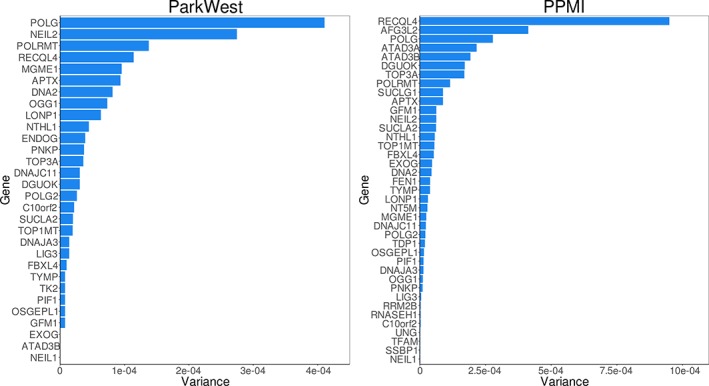
Gene‐based variance in the mitochondrial DNA (mtDNA) maintenance pathway. Sequence kernel association test (SKAT)‐based gene‐based variance for the mtDNA maintenance pathway in the ParkWest and Parkinson's Progression Markers Initiative datasets for rare, nonsynonymous variation. Variance is a measure of SKAT‐based enrichment (ie, the degree of skewed variant distribution between cases and controls) and is defined as the sum of squared difference in minor allele frequency (MAF) between cases and controls for all variants within each gene, . A high variance score for a gene indicates that variants within that gene show a highly uneven distribution between cases and controls. [Color figure can be viewed at http://wileyonlinelibrary.com]
Discussion
We show for the first time that idiopathic PD is associated with a significant enrichment of rare, protein‐altering genetic variants in the pathway of mtDNA replication and repair. This association was significant in both the ParkWest and PPMI cohorts and survived multiple testing correction. Moreover, this association was driven by the combined effects of multiple variants and genes spanning the mtDNA homeostasis network, consistent with a true polygenic signal.
The discrepancy between burden and SKAT in our results suggests that protein‐altering variation in the genes controlling mtDNA maintenance can both be deleterious or protective for PD. Biological pathways generally have a high degree of complexity, such that genetically determined variation in their function may be phenotypically neutral or affect associated disease traits in both directions (ie, increase or decrease the risk). This may particularly apply to late‐onset disorders that are not likely to be affected by selection pressure, such as PD. SKAT is a variance‐component test, which integrates pathway‐level information without consideration of the directionality of single‐variant effects and is therefore particularly powerful in the presence of both protective and risk variants or numerous noncausal variants.
Our results are highly consistent with prior knowledge of mitochondrial involvement in PD. The disruption of mtDNA maintenance as a result of mutations in genes controlling mtDNA replication and repair or mitochondrial nucleotide homeostasis causes nigrostriatal degeneration with or without clinical parkinsonism.11, 12, 13 Moreover, we and others have shown that mtDNA maintenance is impaired in idiopathic PD, resulting in the accumulation of somatic damage and progressive depletion of the wild‐type mtDNA population.7, 40 Based on our results, we propose that this impairment may be partly determined by inherited variation in the genes encoding the pathway of mtDNA homeostasis.
The observation that genetic variation in the mtDNA maintenance pathway appears to influence the risk of PD in both directions is intriguing, but hardly surprising. Maintaining quantitative and qualitative mtDNA integrity in aging neurons requires a balanced interplay of multiple factors including replication processivity, fidelity, nucleotide metabolism, repair mechanisms, and the underlying mtDNA sequence. It is conceivable that variation in the multitude of genes controlling any and each of these processes may inhibit or enhance mtDNA maintenance thus increasing or decreasing the risk of PD, respectively. It is known for instance that mtDNA haplogroup J, which is linked to higher rates of mtDNA replication and transcription, is associated with a decreased risk for PD.41 Intriguingly, even apparently pathogenic mutations may have additional beneficial effects. The murine POLG mutation p.D257A impairs the enzymes proofreading activity, resulting in increased somatic mtDNA mutagenesis and an aged phenotype in mice. At the same time, however, this defect triggers a neuroprotective compensatory mechanism increasing mtDNA copy number in substantia nigra neurons, rendering them resistant to neurodegeneration.42 Moreover, rare protective variants have been shown in other complex disorders including autoimmunity,43 cancer,44 ischemic vascular disease,45 and Alzheimer's disease.46
The pathway of mitochondrial calcium homeostasis also reached nominal significance in both cohorts, but only survived multiple testing correction in the meta‐analysis. Functional evidence suggests that calcium metabolism may be involved in neurodegeneration in PD,47, 48 and this could be partly genetically determined. As this pathway did not survive multiple testing correction in either cohort, however, these results should be interpreted with caution.
Gene‐based tests, by either burden or SKAT, produced no exome‐wide significant associations. Power analyses based on our Norwegian cohort estimated that a substantially larger sample, probably in the order of 8,000 to 15,000, would be required to detect exome‐wide significant genic associations in idiopathic PD by variance‐ or burden‐based tests. Notably, this estimate is based on a clinically homogeneous cohort with validated phenotype, coming from a homogeneous founder population, which is ideal for studying rare variants. Recruiting a PD sample of several thousand individuals will require the combination of multiple cross‐sectional materials across several populations. Such material will unavoidably be more phenotypically and genetically heterogeneous. It is therefore likely that even higher numbers will be required in a real‐life experiment compared to our simulated estimates. A recent study employing a similar rare‐variant collapsing approach in a sample of ∼5,000 schizophrenia cases and controls also failed to produce genic associations at exome‐wide significance.49 Given the fact that schizophrenia has a higher estimated heritability than idiopathic PD, these results corroborate our estimates that samples more than 5,000 individuals will be required to confidently detect gene‐based effects in idiopathic PD (Fig. 2).
In conclusion, our data show that large samples are required to identify rare genetic variation associated with PD at the gene level. In contrast, we show that pathway‐based analysis of rare genetic variation is a powerful tool for deciphering the genetic susceptibility to PD, even in moderately sized samples. Our results suggest that rare, nonsynonymous variation in the genes encoding the complex pathway of mtDNA maintenance influences the risk for idiopathic PD in 2 independent populations. We propose that this variation underlies part of the observed impairment of mtDNA maintenance in the dopaminergic substantia nigra of individuals with PD and may explain part of the disorder's “missing heritability.” The replication of these findings in other sequencing cohorts of idiopathic PD will be essential to further validate these findings.
Author Roles
1) Research Project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript Preparation: A. Writing of the First Draft, B. Review and Critique.
J.J.G.: 1C, 2B, 2C, 3A, 3B
G.S.N.: 1C, 2B, 2C, 3B
P.S.: 1C, 3B
P.M.K.: 1C, 3B
O.D.: 1C, 3B
M.L.‐J.: 1C, 3B
J.M.‐G.: 1C, 3B
G.A.: 1C, 3B
O.‐B.T.: 1C, 3B
S.J.: 1C, 2C, 3B
K.H.: 1A, 1B, 1C, 2C, 3B
C.T.: 1A, 1B, 1C, 2A, 2C, 3A, 3B
Full financial disclosures for the previous 12 months
The authors have no financial disclosures to report for the preceeding 12 months.
Supporting information
Table S1. Pathway_database
Table S2. Distribution_datasets
Table S3. Distribution_case_control
Table S4. Singleton_variants
Table S5. ParkWest_SPA_logistic
Table S6. PPMI_SPA_chisq
Table S7. ParkWest_SGA_nonsynonymous_Burden
Table S8. ParkWest_SGA_nonsynonymous_SKAT
Table S9. ParkWest_SGA_damaging_Burden
Table S10. ParkWest_SGA_damaging_SKAT
Table S11. PPMI_SGA_nonsynonymous_Burden
Table S12. PPMI_SGA_nonsynonymous_SKAT
Table S13. PPMI_SGA_damaging_Burden
Table S14. PPMI_SGA_damaging_SKAT
Table S15. Pathway_results
Table S16. Pathway_gene_analysis
Table S17. Subpathway_analysis
Acknowledgments
We are grateful to the patients and their families for participating in this study. We thank the Parkinson's Progression Markers Initiative for providing access to their exome data. For up‐to‐date information on the study, visit http://www.ppmi-info.org/. We are grateful to Dr Tetyana Zayats for the valuable discussions.
Funding agencies: This work was supported by grants from the Regional Health Authority of Western Norway (Grants 911903 and 911988) and the Research Council of Norway (Grant 240369/F20). Parkinson's Progression Markers Initiative, a public‐private partnership, is funded by the Michael J. Fox Foundation for Parkinson's Research and funding partners, including AbbVie, Avid Radiopharmaceuticals, Biogen Idec, BioLegend, Bristol‐Meyers Squibb, GE Healthcare, Genentech, GlaxoSmithKline, Eli Lilly and Company, Lundbeck, Merck, Meso Scale Discovery, Pfizer Inc., Piramal Imaging, Roche CNS group, Sanofi Genzyme, Servier, Takeda, Teva, UCB, and Golub Capital.
Relevant conflicts of interests/financial disclosures: Nothing to report.
References
- 1. de Rijk MC, Launer LJ, Berger K, et al. Prevalence of Parkinson's disease in Europe: a collaborative study of population‐based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology 2000;54(11 suppl 5):S21‐S23. [PubMed] [Google Scholar]
- 2. Kamp F, Exner N, Lutz AK, et al. Inhibition of mitochondrial fusion by alpha‐synuclein is rescued by PINK1, Parkin and DJ‐1. EMBO J 2010;29(20):3571‐3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang X, Yan MH, Fujioka H, et al. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum Mol Genet 2012;21(9):1931‐1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Park J, Lee G, Chung J. The PINK1‐Parkin pathway is involved in the regulation of mitochondrial remodeling process. Biochem Biophys Res Commun 2009;378(3):518‐523. [DOI] [PubMed] [Google Scholar]
- 5. Thomas KJ, McCoy MK, Blackinton J, et al. DJ‐1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy. Hum Mol Genet 2011;20(1):40‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sugiura A, McLelland GL, Fon EA, McBride HM. A new pathway for mitochondrial quality control: mitochondrial‐derived vesicles. EMBO J 2014;33(19):2142‐2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dolle C, Flones I, Nido GS, et al. Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease. Nat Commun 2016;7:13548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Grunewald A, Rygiel KA, Hepplewhite PD, Morris CM, Picard M, Turnbull DM. Mitochondrial DNA depletion in respiratory chain‐deficient Parkinson disease neurons. Ann Neurol 2016;79(3):366‐378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Flones IH, Fernandez‐Vizarra E, Lykouri M, et al. Neuronal complex I deficiency occurs throughout the Parkinson's disease brain, but is not associated with neurodegeneration or mitochondrial DNA damage. Acta Neuropathol 2018;135:409‐425. [DOI] [PubMed] [Google Scholar]
- 10. Luoma P, Melberg A, Rinne JO, et al. Parkinsonism, premature menopause, and mitochondrial DNA polymerase gamma mutations: clinical and molecular genetic study. Lancet 2004;364(9437):875‐882. [DOI] [PubMed] [Google Scholar]
- 11. Tzoulis C, Schwarzlmuller T, Biermann M, Haugarvoll K, Bindoff LA. Mitochondrial DNA homeostasis is essential for nigrostriatal integrity. Mitochondrion 2016;28:33‐37. [DOI] [PubMed] [Google Scholar]
- 12. Tzoulis C, Tran GT, Schwarzlmuller T, et al. Severe nigrostriatal degeneration without clinical parkinsonism in patients with polymerase gamma mutations. Brain 2013;136(Pt 8):2393‐2404. [DOI] [PubMed] [Google Scholar]
- 13. Palin EJ, Paetau A, Suomalainen A. Mesencephalic complex I deficiency does not correlate with parkinsonism in mitochondrial DNA maintenance disorders. Brain 2013;136(Pt 8):2379‐2392. [DOI] [PubMed] [Google Scholar]
- 14. Balafkan N, Tzoulis C, Muller B, et al. Number of CAG repeats in POLG1 and its association with Parkinson disease in the Norwegian population. Mitochondrion 2012;12(6):640‐643. [DOI] [PubMed] [Google Scholar]
- 15. Gaweda‐Walerych K, Safranow K, Maruszak A, et al. Mitochondrial transcription factor A variants and the risk of Parkinson's disease. Neurosci Lett 2010;469(1):24‐29. [DOI] [PubMed] [Google Scholar]
- 16. Alves G, Muller B, Herlofson K, et al. Incidence of Parkinson's disease in Norway: the Norwegian ParkWest study. J Neurol Neurosurg Psychiatry 2009;80(8):851‐857. [DOI] [PubMed] [Google Scholar]
- 17. Parkinson Progression Marker I . The Parkinson Progression Marker Initiative (PPMI). Prog Neurobiol 2011;95(4):629‐635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li H, Durbin R. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics 2009;25(14):1754‐1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Picard. http://broadinstitute.github.io/picard. Accessed December 2016.
- 20. McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res 2010;20(9):1297‐1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nat Genet 2011;43(5):491‐498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Van der Auwera GA, Carneiro MO, Hartl C, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 2013;43:11 10 11‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Quinlan AR. BEDTools: the Swiss‐Army Tool for Genome Feature Analysis. Curr Protoc Bioinformatics 2014;47:11.12.1‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Danecek P, Auton A, Abecasis G, et al. The variant call format and VCFtools. Bioinformatics 2011;27(15):2156‐2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011;27(21):2987‐2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second‐generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 2015;4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. International HapMap Consortium . The International HapMap Project. Nature 2003;426(6968):789‐796. [DOI] [PubMed] [Google Scholar]
- 28. Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome‐wide association studies. Nat Genet 2006;38(8):904‐909. [DOI] [PubMed] [Google Scholar]
- 29. Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet 2006;2(12):e190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.The R Project for Statistical Computing. https://www.r-project.org. Accessed August 2017.
- 31. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res 2010;38(16):e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Calvo SE, Clauser KR, Mootha VK. MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res 2016;44(D1):D1251‐D1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Szklarczyk D, Franceschini A, Wyder S, et al. STRING v10: protein‐protein interaction networks, integrated over the tree of life. Nucleic Acids Res 2015;43(Database issue):D447‐D452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee S, Emond MJ, Bamshad MJ, et al. Optimal unified approach for rare‐variant association testing with application to small‐sample case‐control whole‐exome sequencing studies. Am J Hum Genet 2012;91(2):224‐237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X. Rare‐variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet 2011;89(1):82‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee S, Fuchsberger C, Kim S, Scott L. An efficient resampling method for calibrating single and gene‐based rare variant association analysis in case‐control studies. Biostatistics 2016;17(1):1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zaykin DV. Optimally weighted Z‐test is a powerful method for combining probabilities in meta‐analysis. J Evol Biol 2011;24(8):1836‐1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dewey M. metap: meta‐analysis of significance values. R package version 082017. https://cran.r-project.org/web/packages/metap/index.html. Accessed August 2017.
- 39. Westfall PH, Young SS. Resampling‐Based Multiple Testing : Examples and Methods for p‐Value Adjustment. Chichester: Wiley; 1993. [Google Scholar]
- 40. Bender A, Krishnan KJ, Morris CM, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet 2006;38(5):515‐517. [DOI] [PubMed] [Google Scholar]
- 41. Hudson G, Nalls M, Evans JR, et al. Two‐stage association study and meta‐analysis of mitochondrial DNA variants in Parkinson disease. Neurology 2013;80(22):2042‐2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Perier C, Bender A, Garcia‐Arumi E, et al. Accumulation of mitochondrial DNA deletions within dopaminergic neurons triggers neuroprotective mechanisms. Brain 2013;136(Pt 8):2369‐2378. [DOI] [PubMed] [Google Scholar]
- 43. Momozawa Y, Mni M, Nakamura K, et al. Resequencing of positional candidates identifies low frequency IL23R coding variants protecting against inflammatory bowel disease. Nat Genet 2011;43(1):43‐47. [DOI] [PubMed] [Google Scholar]
- 44. Fejerman L, Ahmadiyeh N, Hu D, et al. Genome‐wide association study of breast cancer in Latinas identifies novel protective variants on 6q25. Nat Commun 2014;5:5260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jorgensen AB, Frikke‐Schmidt R, Nordestgaard BG, Tybjaerg‐Hansen A. Loss‐of‐function mutations in APOC3 and risk of ischemic vascular disease. N Engl J Med 2014;371(1):32‐41. [DOI] [PubMed] [Google Scholar]
- 46. Jonsson T, Atwal JK, Steinberg S, et al. A mutation in APP protects against Alzheimer's disease and age‐related cognitive decline. Nature 2012;488(7409):96‐99. [DOI] [PubMed] [Google Scholar]
- 47. Guzman JN, Sanchez‐Padilla J, Wokosin D, et al. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ‐1. Nature 2010;468(7324):696‐700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Soman S, Keatinge M, Moein M, et al. Inhibition of the mitochondrial calcium uniporter rescues dopaminergic neurons in pink1‐/‐ zebrafish. Eur J Neurosci 2017;45(4):528‐535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Purcell SM, Moran JL, Fromer M, et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature 2014;506(7487):185‐190. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Pathway_database
Table S2. Distribution_datasets
Table S3. Distribution_case_control
Table S4. Singleton_variants
Table S5. ParkWest_SPA_logistic
Table S6. PPMI_SPA_chisq
Table S7. ParkWest_SGA_nonsynonymous_Burden
Table S8. ParkWest_SGA_nonsynonymous_SKAT
Table S9. ParkWest_SGA_damaging_Burden
Table S10. ParkWest_SGA_damaging_SKAT
Table S11. PPMI_SGA_nonsynonymous_Burden
Table S12. PPMI_SGA_nonsynonymous_SKAT
Table S13. PPMI_SGA_damaging_Burden
Table S14. PPMI_SGA_damaging_SKAT
Table S15. Pathway_results
Table S16. Pathway_gene_analysis
Table S17. Subpathway_analysis