

Abstract
Drug transporter and cytochrome P450 expression is regulated by shared nuclear receptors and, hence, an inducer should induce both, although the magnitude may differ. The objective of this study was to establish relative induction relationships between CYP3A and drug transporters (P‐glycoprotein (P‐gp), organic anion transporting polypeptide (OATP), and breast cancer resistance protein (BCRP)) or other P450s (CYP2C9 and CYP1A2) using ascending doses of the prototypical pregnane xenobiotic receptor (PXR) agonist, rifampin, to elicit weak, moderate, and strong PXR agonism. Healthy subjects received dabigatran etexilate, pravastatin, rosuvastatin, and a midazolam/tolbutamide/caffeine cocktail before and after rifampin 2, 10, 75, or 600 mg q.d. Unlike CYP3A, only moderate induction of P‐gp, OATP, and CYP2C9 was observed and dose‐dependent induction of P‐gp, OATP, and CYP2C9 was always one drug–drug interaction category lower than observed for CYP3A, even when correcting for probe drug sensitivity. Data from this study establish proof‐of‐concept that P450 induction data can be leveraged to inform on the effect on transporters.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ There are limited clinical data on induction of drug transporters that often lead to potentially unnecessary DDI studies and/or conservative assumptions of parity between CYP3A and drug transporter induction in product labeling.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ This study tested the hypothesis that a relationship in induction potential exists between P450 and drug transporters by examining the magnitude of induction of P‐gp, OATP, BCRP, CYP2C9, and CYP1A2 across a range of CYP3A induction magnitude.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ The study identified quantitative relationships between induction of CYP3A and P‐gp, OATP, or CYP2C9. It demonstrated that, unlike CYP3A, strong induction of P‐gp, OATP, and CYP2C9 are unlikely to occur due to PXR agonism and will be one DDI category lower than the level of CYP3A induction.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ The study provides proof‐of‐concept and actionable results to leverage knowledge on effect of an inducer on CYP3A to inform its induction liability for other P450s/transporters, allowing for more focused DDI assessments and reducing the number of clinical studies that are conducted to support development of an NCE.
Both cytochrome P450 isozymes and drug transporters play a major role in the elimination of drugs from the body. Evidence suggests that cytochrome P450 3A (CYP3A) and the drug efflux transporter, P‐glycoprotein (P‐gp), act in functional collaboration during first‐pass elimination of drug.1 P‐gp in the intestine acts through efflux of drug from the enterocyte back into the intestinal lumen, increasing drug residence time in the lumen and decreasing average enterocyte drug concentrations. Decreased drug concentration allows intestinal CYP3A to metabolize drug more efficiently.1 Expression of CYP3A and P‐gp, and ultimately detoxification of many xenobiotics during oral absorption, is regulated by pregnane xenobiotic receptor (PXR). PXR is a highly promiscuous nuclear receptor that also plays a role in the regulation of expression of many other P450s and transporters, including cytochrome P450 2C9 (CYP2C9), breast cancer resistance protein (BCRP), and, potentially, organic anion transporting polypeptide 1B1 and 1B3 (OATP1B).1, 2, 3, 4, 5
Induction of P450s via PXR agonism has been extensively examined and clear regulatory guidance is available for classification of weak (treatment/control AUC ratio (AUCR) of a sensitive probe substrate of 0.8–0.5), moderate (AUCR of 0.5–0.2), and strong (AUCR of <0.2) induction potency in assigning drug–drug interaction (DDI) liability.6, 7 Conversely, the magnitude of inducer effect on drug transporters in vivo is poorly studied. As such, data from P450 induction studies are often utilized to predict transporter induction, e.g., a strong inducer of CYP3A is assumed to also be a strong inducer of P‐gp. However, it is not clear if this assumed parity is appropriate; the little in vivo data available suggests that transporters may be less inducible than P450s, such as CYP3A. For example, rifampin (RIF, a prototypical PXR agonist) coadministration decreased midazolam exposure (MDZ, a CYP3A probe drug) to a greater extent than dabigatran etexilate (DE, a P‐gp sensitive probe drug) exposure (MDZ and total dabigatran AUCR of 0.05–0.10 and 0.33, respectively), suggesting that P‐gp is significantly less inducible than CYP3A.8, 9, 10 Assuming parity in CYP3A and transporter induction may lead to overly conservative recommendations on the use of known enzyme inducers with drugs that are substrates of transporters.
Coregulation by PXR and few clinical observations suggests that it may be possible to establish quantitative relationships between induction of P450s and drug transporters. The primary objective of this study was to characterize the magnitude of induction of P‐gp, OATP, BCRP, CYP2C9, and CYP1A2 across a range of CYP3A induction (weak, moderate, and strong) using ascending dose levels of RIF and to establish their induction relationship to CYP3A. The results of this study seek to establish a proof‐of‐concept that the effect of an inducer on CYP3A can predict its induction liability for other P450s/transporters and generate testable hypotheses towards better leveraging of CYP3A induction data.
RESULTS
Subject demographics
All 40 subjects received all doses of study drugs. Administration of probe cassette was generally safe and well tolerated when administered alone or in combination with RIF 2, 10, 75, or 600 mg to healthy subjects. Overall, half the subjects were male (n = 20; 50%), the majority were white (n = 35; 87.5%), and some were Hispanic or Latino with a mean age of 36 years (range: 20–45 years). The mean body mass index (BMI) at baseline was 26.8 kg/m2 (range: 21.8–29.6 kg/m2). The mean estimated creatinine clearance at baseline was 129.1 mL/min (range: 95.1–195.3 mL/min).
Characterization of rifampin dose‐dependent transporter/P450 induction
Probe drug plasma concentration–time profiles before and after RIF coadminstration are presented in Figure 1 . All probe drug plasma concentration profiles decreased in an RIF dose‐dependent manner compared to when the probe cassette was administered alone. Likewise, a dose‐dependent decrease in area under the curve from time zero to infinity (AUCinf) and maximum plasma concentration (Cmax) after RIF coadministration was observed for all probes (Figure 2 , Table 1). Tabulated probe drug AUCinf, AUClast, and Cmax values, with and without RIF coadministration, are provided in the Supplementary Materials (Tables S1, S2). Strong induction (AUCR <0.2) was only observed for MDZ (AUCR GMR ≈ 0.09 at RIF doses ≥75 mg and Emax = 15) (Figure 2 b). Coadministration of the maximum RIF dose studied (600 mg) resulted in moderate induction (0.2 < AUCR < 0.5) of total dabigatran (TDAB, the sum of conjugated and unconjugated active species of DE), pravastatin (PRA), rosuvastatin (ROS), and tolbutamide (TOL) (Figure 2 a,c–d, respectively). Lack of an additional decrease in ROS AUCR when compared to PRA AUCR (Figure 2 c,e) suggests that only OATP, and not BCRP, is induced by RIF. Rifampin elicited only weak induction (0.5 < AUCR < 0.8) of caffeine (CAF) (Figure 2 f). These results were expected, as CYP1A2 is primarily regulated by aryl hydrocarbon receptor11 and not PXR. Induction of BCRP and CYP1A2 will not be discussed further. Modest within‐subject correlation was observed between PRA and ROS (R = 0.41) as well as ROS and TOL (R = 0.28) ED50 values, whereas correlation between all other ED50 value pairings were weak (R ≤ 0.20, data not shown).
Figure 1.
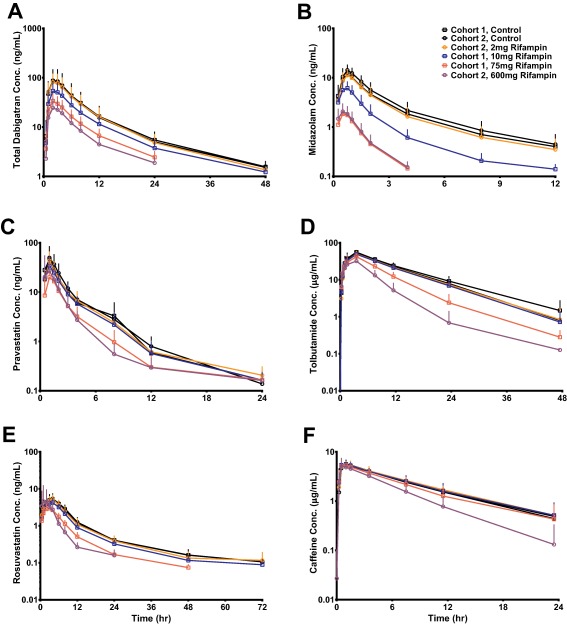
Probe drug mean plasma concentration vs. time curves before and after RIF coadministration. Points and error bars are means and standard deviations, respectively.
Figure 2.
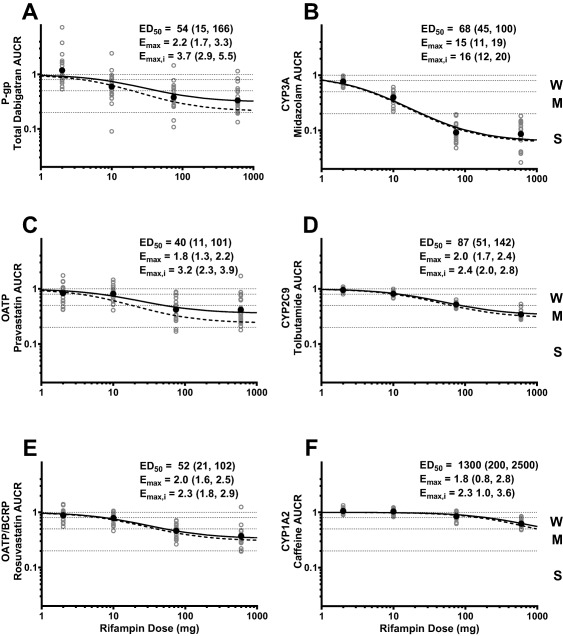
Probe drug mean and individual AUCR as a function of RIF dose. Geometric mean (•) and individual (○) AUCR are depicted. Solid and dashed lines represent estimated and intrinsic induction, respectively. The estimated mean (95% CI) ED50, Emax, and Emax,i for each probe is inset. Weak, moderate, and strong induction classification is denoted as W, M, and S, respectively.
Table 1.
Probe treatment/control geometric mean AUC and Cmax ratio (90% CI) for after coadministration with ascending‐dose RIF
| Probe | RIF q.d. | ||||
|---|---|---|---|---|---|
| 2 mg | 10 mg | 75 mg | 600 mg | ||
| TDAB (P‐gp) | AUCinf | 1.19 (0.93, 1.53) | 0.594 (0.460, 0.768) | 0.380 (0.304, 0.475) | 0.334 (0.269, 0.413) |
| Cmax | 1.07 (0.82, 1.40) | 0.568 (0.423, 0.762) | 0.384 (0.308, 0.479) | 0.305 (0.241, 0.385) | |
| PRA (OATP) | AUCinf | 0.833 (0.716, 0.970) | 0.813 (0.713, 0.926) | 0.421 (0.350, 0.505) | 0.416 (0.333, 0.521) |
| Cmax | 0.856 (0.690, 1.063) | 0.788 (0.660, 0.939) | 0.407 (0.325, 0.511) | 0.473 (0.361, 0.621) | |
| ROS (OATP/BCRP) | AUCinf | 0.882 (0.804, 0.968) | 0.788 (0.716, 0.867) | 0.462 (0.415, 0.514) | 0.371 (0.317, 0.435) |
| Cmax | 0.970 (0.873, 1.076) | 0.846 (0.761, 0.940) | 0.585 (0.520, 0.659) | 0.702 (0.571, 0.864) | |
| MDZ (CYP3A) | AUCinf | 0.769 (0.730, 0.810) | 0.398 (0.359, 0.441) | 0.0916 (0.0795, 0.1055) | 0.0859 (0.0695, 0.1061) |
| Cmax | 0.790 (0.738, 0.846) | 0.516 (0.460, 0.579) | 0.149 (0.130, 0.171) | 0.139 (0.112, 0.172) | |
| TOL (CYP2C9) | AUCinf | 0.955 (0.928, 0.983) | 0.815 (0.786, 0.845) | 0.521 (0.500, 0.543) | 0.345 (0.321, 0.371) |
| Cmax | 0.981 (0.919, 1.047) | 0.894 (0.852, 0.938) | 0.781 (0.736, 0.830) | 0.655 (0.604, 0.711) | |
| CAF (CYP1A2) | AUCinf | 1.06 (1.02, 1.10) | 1.04 (0.99, 1.09) | 0.849 (0.805, 0.896) | 0.623 (0.584, 0.664) |
| Cmax | 1.05 (1.02, 1.09) | 1.05 (1.00, 1.10) | 0.991 (0.939, 1.046) | 0.935 (0.869, 1.005) | |
TDAB, total dabigatran; PRA, pravastatin; ROS, rosuvastatin; MDZ, midazolam; TOL, tolbutamide; CAF, caffeine; RIF, rifampin.
Only moderate induction of P‐gp and OATP probe drug exposure was observed. When adjusted for probe sensitivity (ft of 0.59 and 0.56 for DE and PRA, respectively), the intrinsic induction of transporter activity (Emax,i) was increased, but still did not reach strong categorization (Figure 2 a,c, dashed lines). Overall, the results indicate that strong induction of P‐gp, OATP, CYP2C9, and CYP1A2 is unlikely to be observed with PXR agonism only.
Simulated transporter/P450 induction relationships
Since the induction Emax and ED50 may vary across different P450s/transporters, changes in relationship curves across various scenarios are illustrated in Figure 3 . Only when the induction magnitude of both P450s/transporters is identical will the relationship be linear and approximate the line of unity (Example 2). Example 2 demonstrates when the ED50 for each probe is very different, but Emax is similar. Nonlinear relationships occur when induction capacity is different. Example 3 demonstrates a theoretically likely scenario of when induction capacity differs between each transporter/P450 (Emax,x ≠ Emax,y), but ED50 values are similar due to shared sites of induction (i.e., presystemic and/or systemic).
Figure 3.
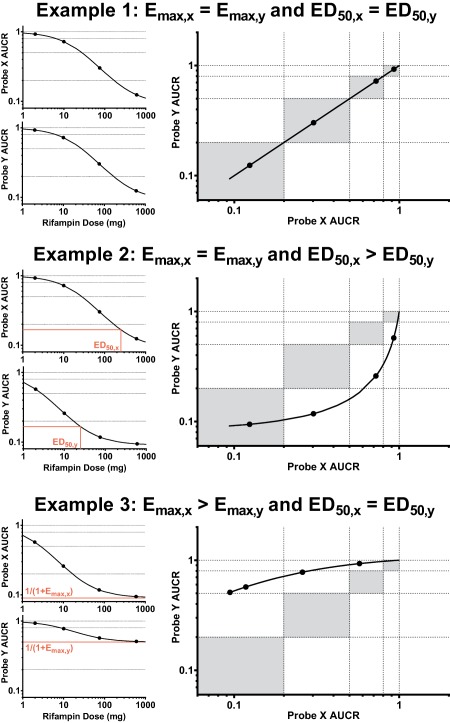
Example rifampin dose vs. probe drug mean AUCR curves and resulting probe clearance induction relationship curve with interpretation. Gray areas represent induction magnitude parity between probes, i.e., similar weak/weak, moderate/moderate, and strong/strong induction of probes X and Y.
Observed transporter/P450 induction relationships
Induction of DE clearance is always one DDI category weaker than that of MDZ clearance across the achievable range of PXR agonism (Figure 4 a, solid line) and, after accounting for differences in probe sensitivity, P‐gp induction remains as one category weaker than CYP3A (Figure 4 a, dashed line). These data suggest that, similar to P‐gp, PXR‐dependent CYP3A induction will always be greater than that of OATP (as evidenced by PRA or ROS AUCR) and CYP2C9 (Figure 4 b–d, respectively).
Figure 4.
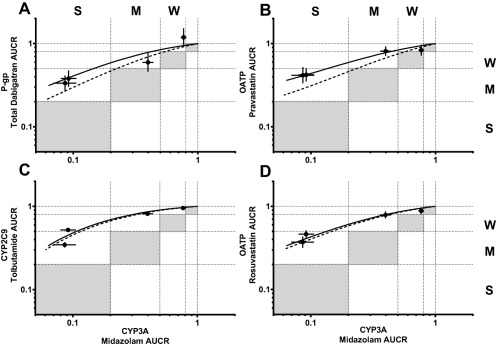
Rifampin dependent induction relationship between CYP3A and P‐gp, OATP, or CYP2C9. The black points and error bars are observed AUCR mean and 90% CI, respectively, for after RIF coadministration. Solid and dashed lines represent estimated and intrinsic induction, respectively. Gray areas represent induction magnitude parity between probes. Weak, moderate, and strong induction classification is denoted as W, M, and S, respectively.
The induction relationships between P‐gp, OATP, and CYP2C9 demonstrate that both the probe drug observed exposure (Figure 5, solid lines) and intrinsic transporter/P450 (Figure 5, dashed lines) induction relationships approximate the line of unity, indicating parity between the induction of P‐gp, OATP (as evidenced by PRA or ROS AUCR), and CYP2C9.
Figure 5.
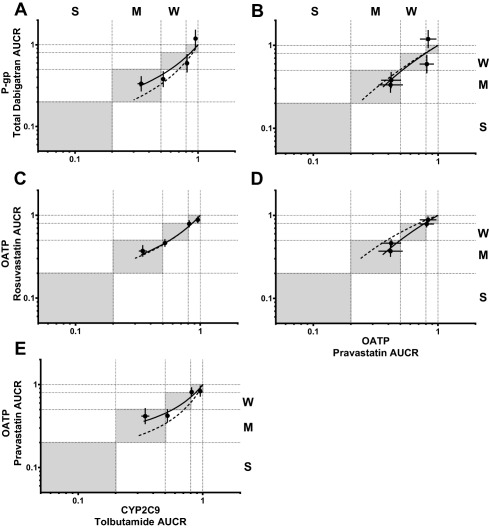
Rifampin dependent induction relationships between P‐gp, OATP, and CYP2C9. The black points and error bars are observed AUCR mean and 90% CI, respectively, for after RIF coadministration. Solid and dashed lines represent estimated and intrinsic induction, respectively. Gray areas represent induction magnitude parity between probes. Weak, moderate, and strong induction classification is denoted as W, M, and S, respectively.
DISCUSSION
This study demonstrated that doses of RIF can be titrated to represent weak, moderate, and strong PXR‐dependent induction during phase I DDI studies. Consistent use of RIF at dose levels less than 600 mg allowed for reliable comparison of induction magnitudes between P450s and transporters. As presented in Figure 2 and Table 1, RIF, the prototypical PXR agonist, elicits dose‐dependent induction of CYP3A, P‐gp, OATP, and CYP2C9. Furthermore, these data demonstrate that, unlike CYP3A, strong induction of P‐gp, OATP, or CYP2C9 is unlikely to occur due to PXR agonism. Furthermore, quantitative induction relationships between CYP3A and P‐gp, OATP, or CYP2C9 demonstrate that induction of P‐gp, OATP, and CYP2C9 will be at least one DDI classification less than that of CYP3A at all points across the achievable range of PXR agonism (Figure 4).
With the induction relationships derived in this study, comparable ED50 values (40–87 mg) were estimated for P‐gp, OATP, CYP3A, and CYP2C9 (Figure 2). This suggests comparable driving force concentrations at the site of induction (e.g., presystemic vs. systemic) of each P450 and transporter. In contrast to the observations noted for ED50, the Emax,i value for CYP3A was much higher than that determined for P‐gp, OATP, and CYP2C9 (16 vs. 2.3–3.7). Collectively, these data indicate that differences in induction magnitude are a result of differences in induction capacity (i.e., maximum fold increase in protein expression) of each transporter or P450 and not driving concentration of the inducer.
There are conflicting literature data on the potential for induction of OATP via PXR agonism. In this study, RIF coadministration decreased PRA and ROS exposure. The exposure of PRA or ROS is not only affected by changes in OATP activity, but also by changes in renal secretion and possibly CYP2C9 activity (specifically for ROS, as it is minimally metabolized (∼10%) by CYP2C9).12 Our analysis shows that RIF did not alter the renal clearance of PRA or ROS (treatment/control renal clearance ratio for after 600 mg RIF coadministration of 1.03 and 1.07, respectively, data not shown), and that observed changes in CYP2C9 (measured with TOL) are modest and would minimally contribute to the observed changes in ROS exposure. Thus, we believe it is OATP induction driving the changes we observed with PRA and ROS (Figure 2 , Table 1) and this is supported by RIF increasing OATP mRNA in vitro.13, 14
While nuclear receptor coregulation may suggest a relationship between induction of PXR coregulated transporters/P450s, direct in vivo evidence was lacking. Induction relationships established with RIF in this study indicate that PXR‐mediated induction of P‐gp, OATP, and CYP2C9 can be predicted based on the magnitude of CYP3A induction; induction of P‐gp, OATP, and CYP2C9 will be at least one DDI classification less than that of CYP3A at all points across the achievable range of PXR agonism (Figure 4). By extension, PXR agonism will elicit the same induction DDI category for P‐gp, OATP, and CYP2C9. These observations allow for useful and simple in vivo DDI liability prediction where limited in vivo induction data may be available but now can be leveraged from the knowledge of one PXR‐regulated transporter/P450 to another. Transporter and P450 induction can be simultaneously assessed using a probe drug cocktail,15, 16 and this method is useful for proactive determination of P‐gp, OATP, CYP3A, and/or CYP2C9 induction by a new chemical entity (NCE). Often, only CYP3A data are available for the inducer NCE or the NCE is not an inducer, but a substrate of P‐gp, OATP, or CYP2C9. In these scenarios, application of the relative induction relationships detailed in this study may be advantageous: Observed CYP3A induction data by the inducer NCE can be leveraged to inform potential exposure decreases of marketed drugs that are P‐gp, OATP, or CYP2C9 substrates. In a similar manner, literature data on marketed CYP3A inducers can inform on potential exposure decreases of the P‐gp, OATP, or CYP2C9 substrate NCE.
The induction relationships between P‐gp, OATP, or CYP2C9 and CYP3A, observed using RIF, are expected to be applicable to any PXR agonist. This hypothesis was tested in vivo with rifabutin and carbamazepine as test inducers against the same P450/transporter probes used in this study; the results will be presented in a subsequent publication.17 The purpose of clinical pharmacology programs in drug development are to inform on a wide variety of questions, one of which is DDI liability prediction. Historically, this has involved the conduct of many DDI studies, each focusing on a single precipitant‐object pairing to provide information on a specific mechanism (e.g., CYP3A). The induction relationships established in this study should allow for decreasing the number of DDI studies conducted, much in the way that DDI probe studies can be extrapolated to other drugs that are sensitive to the mechanism being probed. Application of these induction relationships should provide for more informed labeling recommendations for new chemical entities that are transporter substrates or PXR agonists.
METHODS
Study population
This was a phase I open‐label, multiple‐dose, single‐center study. Eligible subjects were healthy male and nonpregnant, nonlactating female subjects of 18 to 45 years of age with a BMI between 19 and 30 kg/m2. The study protocol and informed consent were approved by the study center's Institutional Review Board, and subjects provided written consent before study participation. Major inclusion criteria included healthy subjects based on medical history/physical examinations/laboratory evaluations, normal 12‐lead electrocardiogram, estimated creatinine clearance (Cockroft‐Gault) >80 mL/min, no evidence of HIV, hepatitis B virus, or hepatitis C virus infection, and use of at least two forms of contraception, including an effective barrier method. Exclusion criteria included plasma and blood donation within 7 and 56 days of study enrollment, respectively, active medical illness, use of prescription drugs within 28 days of study drug dosing (except vitamins, acetaminophen, ibuprofen, and/or hormonal contraceptives).
Study design
Induction was assessed in two cohorts: RIF 10 mg then 75 mg q.d. (N = 20) and RIF 2 mg then 600 mg q.d. (N = 20). Rifampin was administered at night for 10 days of each indicated dose level prior to and then continued through probe drug assessment. A cassette of six probe drugs were orally administered in the morning under fasted conditions, before and after inducer administration (2 days washout between doses of probe drugs, 7 days total probing): 75 mg DE, 20 mg PRA, 10 mg ROS, and a simultaneously administered cocktail of 2 mg MDZ, 500 mg TOL, and 200 mg CAF. These drugs were utilized to assess the activity of P‐gp, OATP, OATP/BCRP, CYP3A, CYP2C9, and CYP1A2, respectively.16, 18, 19, 20 DE was administered as the probe drug, but changes in plasma TDAB (free dabigatran + glucuronides) concentrations were measured (see Bioanalytical procedures, below) for P‐gp activity assessment. DE is rapidly converted to dabigatran after oral absorption, resulting in very low DE plasma exposure,21 necessitating measurement of the active TDAB species. Previous DDI studies indicate that DE (as measured by plasma TDAB) is a sensitive in vivo P‐gp probe8, 18 and is listed as such in the European Medicines Agency (EMA) and US Food and Drug Administration (FDA) DDI guidances.6, 7 The fraction transported (ft) by OATP for PRA and ROS are similar22, 23 and, hence, it can be assumed that any further induction of ROS, relative to PRA, can be attributed to BCRP induction. The MDZ/TOL/CAF cocktail was previously validated to ensure its suitability for simultaneous P450 activity probing.16 The study design is presented in Table 2.
Table 2.
Study design
| Days 1–8 | 9–18 | 19–26 | 27–36 | 37–44 | |
|---|---|---|---|---|---|
| Cassette | Cassette | Cassette | |||
| Cohort 1, N = 20 | RIF 10 mg q.d. | RIF 75 mg q.d. | |||
| Cohort 2, N = 20 | RIF 2 mg q.d. | RIF 600 mg q.d. | |||
| Probe drug cassette | Dose | Abbreviation | P450/transporter | Cassette day | |
| Dabigatran etexilate | 75 mg | DE | P‐gp | 1 | |
| Pravastatin | 20 mg | PRA | OATP | 3 | |
| Rosuvastatin | 10 mg | ROS | OATP/BCRP | 5 | |
| Cocktail | Midazolam | 2 mg | MDZ | CYP3A | 7 |
| Tolbutamide | 500 mg | TOL | CYP2C9 | ||
| Caffeine | 200 mg | CAF | CYP1A2 | ||
*DE was analyzed as total dabigatran (TDAB), the sum of conjugated and unconjugated active species.
Pharmacokinetic (PK) evaluation
Serial blood samples were collected after DE administration on Days 1, 19, and 37: predose (<5 minutes), 0.5, 1, 2, 3, 4, 6, 8, 12, 24, and 48 hours postdose; after PRA administration on Days 3, 21, and 39: predose (<5 minutes), 0.5, 1, 1.5, 2, 3, 4, 8, 12, and 24 hours postdose; after ROS administration on Days 5, 23, and 41: predose (<5 minutes), 0.5, 1, 2, 3, 4, 6, 8, 12, 24, 48, and 72 hours postdose; and after MDZ/TOL/CAF administration on days 7, 25, and 43: predose (<5 minutes), 0.25, 0.5, 0.75, 1, 1.5, 2, 4, 8, 12, 24, and 48 hours postdose.
Bioanalytical procedures
Bioanalysis was conducted at PPD Laboratories (Middleton, WI) and QPS (Newark, DE) using fully validated high‐performance liquid chromatography‐tandem mass spectroscopy (LC/MS/MS) bioanalytical methods. To determine the concentrations of TDAB, PRA, ROS, MDZ, TOL, and CAF in plasma samples, a 50–300 μL aliquot of plasma was spiked with isotopically labeled internal standard (2H and/or 13C). The sample was then processed by protein precipitation, liquid‐liquid, or solid phase extraction, followed by evaporation of the organic solvent. An aliquot of the reconstituted sample extract was injected onto the LC‐MS/MS system. The calibrated ranges of the method were 1.00–500 ng/mL for TDAB, 0.100–100 ng/mL for PRA, 0.0500–50.0 ng/mL for ROS, 0.100–100 ng/mL for MDZ, 100–100,000 ng/mL for TOL, and 20.0–20,000 ng/mL for CAF. For all analytes, precision (%CV) was <15% (<20% at lower limit of quantitation (LLOQ)) and assay accuracy (% relative error) values were within ± 15% of 100% (±20% of 100% at limit of quantitation). All samples were analyzed in the timeframe supported by frozen stability storage data.
PK analyses
The AUC from time zero to infinity (AUCinf) and maximum plasma concentration (Cmax) were determined via noncompartmental analysis using Phoenix (v. 6, Certara USA, Princeton, NJ) for all analytes. For this analysis, actual plasma sampling times were utilized and a minimum of 3 points were used to define the terminal phase. All plasma concentrations <LLOQ and before the first observable timepoint were inputted as zero, whereas those after the first observable concentration were considered missing. Geometric mean treatment/control Cmax and AUCinf ratio (AUC GMR or AUCR) with 90% confidence interval (CI) was calculated.
Characterization of rifampin dose dependent transporter/P450 induction
The RIF dose‐dependent effect on probe AUCR was estimated using R (v. 3.2.2, R Foundation, http://www.r-project.org). A multivariate nonlinear mixed effects model was used to accommodate all six analytes. The mean maximum induction (Emax) and RIF dose (DRIF) at half maximal induction (ED50) were defined using the equation:
| (1) |
To account for within‐subject correlation, a multivariate normal likelihood function with unstructured covariance matrix was used for the response variable in a mixed effects model. Parameter estimation was achieved through a Bayesian approach and Markov Chain Monte Carlo (MCMC) simulation. A normal prior was assigned to the parameter Emax and ED50, and an inverse‐Wishart prior was assigned to the covariance matrix.
Due to differences in probe sensitivity, the maximum intrinsic induction (Emax,i) for the respective transporter/P450 was calculated via correcting Emax based on probe drug fraction metabolized or fraction transported (fm or ft, respectively) by the P450/transporter of interest, a measure of probe sensitivity:
| (2) |
Literature data on increase in probe exposure in the presence of strong selective transporter/P450 inhibition or genetic polymorphism were used to calculate fm or ft values for DE, PRA, ROS, MDZ, TOL, CAF: 0.59, 0.56, 0.86, 0.95, 0.85, and 0.80, respectively.18, 20, 22, 24, 25, 26 Conservative estimates of fm and ft were used to limit underestimation of Emax,i.
Characterization of induction relationships between P450s and transporters
Relative induction between probes was determined via rearrangement of the reference probe (probe X) AUCR equation (Equation 1) to solve for DRIF (Equation 3) and substitution into the object probe (probe Y) AUCR equation (Equation 4) to determine probe Y AUCR as a function of probe X AUCR:
| (3) |
| (4) |
Example induction relationship curves were simulated using Equations 3 and 4; Emax values of 1 and 10 as well as ED50 values of 25 mg and 250 mg were used for these calculations.
Statistical analyses
For each cohort and analyte, a parametric (normal theory) mixed‐effects analysis of variance (ANOVA) model was fitted to the natural log‐transformed values of the single‐dose AUCinf under evaluation using SAS PROC MIXED (Cary, NC). The ratio of geometric least‐squares means (GLSMs) for test vs. reference treatments was calculated, as well as the associated 90% CI. The 2012 Food and Drug Administration (FDA) Drug‐Drug Interaction (DDI) Guidance defines the severity of a given DDI as weak, moderate, or strong based on the mean treatment/control AUCinf ratio range of 0.5–0.8, 0.2–0.5, and <0.2, respectively.6, 7
FUNDING
This study was funded by Gilead Sciences, Inc.
CONFLICT OF INTEREST
The authors declare no competing interests for this work.
AUTHOR CONTRIBUTIONS
J.D.L, B.J.K., and A.M. wrote the article; J.D.L, B.J.K., Q.S., B.M., A.W., B.P.K., and A.M. designed the research; A.W. performed the research; J.D.L, B.J.K., L.W., Q.S., J.L., and B.M. analyzed the data.
Supporting information
Table S1: Mean (%CV) Probe Drug PK Parameters Following Administration Alone or Coadministration with RIF 10 or 75 mg
Table S2: Mean (%CV) Probe Drug PK Parameters Following Administration Alone or Coadministration with RIF 2 or 600 mg
ACKNOWLEDGMENTS
The authors thank Sunila Reddy, PharmD, for help in preparation of the article.
References
- 1. Christians, U. , Schmitz, V. & Haschke, M. Functional interactions between P‐glycoprotein and CYP3A in drug metabolism. Expert Opin. Drug Metab. Toxicol. 1, 641–654 (2005). [DOI] [PubMed] [Google Scholar]
- 2. Francis, G.A. , Fayard, E. , Picard, F. & Auwerx, J. Nuclear receptors and the control of metabolism. Annu. Rev. Physiol. 65, 261–311 (2003). [DOI] [PubMed] [Google Scholar]
- 3. Gerbal‐Chaloin, S. et al Induction of CYP2C genes in human hepatocytes in primary culture. Drug Metab. Dispos. 29, 242–251 (2001). [PubMed] [Google Scholar]
- 4. Lemmen, J. , Tozakidis, I.E. & Galla, H.J. Pregnane X receptor upregulates ABC‐transporter Abcg2 and Abcb1 at the blood‐brain barrier. Brain Res. 1491, 1–13 (2013). [DOI] [PubMed] [Google Scholar]
- 5. Synold, T.W. , Dussault, L. & Forman, B.M. The orphan nuclear receptor SXR coordinately regulates drug metabolism and efflux. Nat. Med. 7, 584–590 (2001). [DOI] [PubMed] [Google Scholar]
- 6. Food & Drug Administration (FDA) . Clinical Drug Interaction Studies — Study Design, Data Analysis, and Clinical Implications — Guidance for Industry. October, 2017.
- 7. European Medicines Agency . Concept paper on a revision of the guideline on the investigation of drug interactions. EMEA/CHMP/694687/2016. March, 2017.
- 8. Hartter, S . et al Decrease in the oral bioavailability of dabigatran etexilate after co‐medication with rifampicin. Br. J. Clin. Pharmacol. 74, 490–500 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kharasch, E.D , Francis, A. , London, A. , Frey, K. , Kim, T. & Blood J. Sensitivity of intravenous and oral alfentanil and pupillary miosis as minimal and noninvasive probes for hepatic and first‐pass CYP3A induction. Clin. Pharmacol. Ther. 90, 100–108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kirby, B.J , Collier, A.C. , Kharasch, E.D. , Whittington, D. , Thummel, K.E. & Unadkat, J.D. Complex drug interactions of HIV protease inhibitors 1: inactivation, induction, and inhibition of cytochrome P450 3A by ritonavir or nelfinavir. Drug Metab. Dispos. 39, 1070–1078 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhou, S.F. , Wang, B. , Yang, L.P. & Liu, J.P. Structure, function, regulation and polymorphism and the clinical significance of human cytochrome P450 1A2. Drug Metab. Rev. 4, 268–354 (2010). [DOI] [PubMed] [Google Scholar]
- 12. Martin, P.D. et al Metabolism, excretion, and pharmacokinetics of rosuvastatin in healthy adult male volunteers. Clin. Ther. 25, 2822–2835 (2003). [DOI] [PubMed] [Google Scholar]
- 13. Jigorel, E. et al Differential regulation of sinusoidal and canalicular hepatic drug transporter expression by xenobiotics activating drug‐sensing receptors in primary human hepatocytes. Drug Metab. Dispos. 34, 1756–1763 (2006). [DOI] [PubMed] [Google Scholar]
- 14. Sahi, J. et al Metabolism and transporter‐mediated drug‐drug interactions of the endothelin‐A receptor antagonist CI‐1034. Chem. Biol. Interact. 159, 156–168 (2006). [DOI] [PubMed] [Google Scholar]
- 15. Prueksaritanont, T. et al Validation of a microdose probe drug cocktail for clinical drug interaction assessments for drug transporters and CYP3A. Clin. Pharmacol. Ther. 101, 519–530 (2017). [DOI] [PubMed] [Google Scholar]
- 16. Blakey, G.E. et al Pharmacokinetic and pharmacodynamic assessment of a five‐probe metabolic cocktail for CYPs 1A2, 3A4, 2C9, 2D6 and 2E1. Br. J. Clin. Pharmacol. 57, 162–169 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lutz, J.D. et al Cytochrome P450 3A induction predicts p‐glycoprotein induction; Part 2: prediction of decreased substrate exposure after rifabutin or carbamazepine. Clin. Pharmacol. Ther. (2018). [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hartter, S. et al Oral bioavailability of dabigatran etexilate (Pradaxa®) after co‐medication with verapamil in healthy subjects. Br. J. Clin. Pharmacol. 75, 1053–1062 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Maeda, K. et al Identification of the rate‐determining process in the hepatic clearance of atorvastatin in a clinical cassette microdosing study. Clin. Pharmacol. Ther. 90, 575–581 (2011). [DOI] [PubMed] [Google Scholar]
- 20. Simonson, S.G. et al Rosuvastatin pharmacokinetics in heart transplant recipients administered an antirejection regimen including cyclosporine. Clin. Pharmacol. Ther. 76, 167–177 (2004). [DOI] [PubMed] [Google Scholar]
- 21. Stangier, J. Clinical pharmacokinetics and pharmacodynamics of the oral direct thrombin inhibitor dabigatran etexilate. Clin. Pharmacokinet. 47, 285–295 (2008). [DOI] [PubMed] [Google Scholar]
- 22. Deng, S. et al Effects of a concomitant single oral dose of rifampicin on the pharmacokinetics of pravastatin in a two‐phase, randomized, single‐blind, placebo‐controlled, crossover study in healthy Chinese male subjects. Clin. Ther. 31, 1256–1263 (2009). [DOI] [PubMed] [Google Scholar]
- 23. Prueksaritanont, T. et al Pitavastatin is a more sensitive and selective organic anion‐transporting polypeptide 1B clinical probe than rosuvastatin. Br. J. Clin. Pharmacol. 78, 587–598 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Christensen, M. et al Low daily 10‐mg and 20‐mg doses of fluvoxamine inhibit the metabolism of both caffeine (cytochrome P4501A2) and omeprazole (cytochrome P4502C19). Clin. Pharmacol. Ther. 71, 141–152 (2002). [DOI] [PubMed] [Google Scholar]
- 25. Kirchheiner, J. et al Impact of CYP2C9 and CYP2C19 polymorphisms on tolbutamide kinetics and the insulin and glucose response in healthy volunteers. Pharmacogenetics 12, 101‐109 (2002). [DOI] [PubMed] [Google Scholar]
- 26. Mathias, A.A. et al Pharmacokinetics and pharmacodynamics of GS–9350: a novel pharmacokinetic enhancer without anti‐HIV activity. Clin. Pharmacol. Ther. 87, 322–329 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Mean (%CV) Probe Drug PK Parameters Following Administration Alone or Coadministration with RIF 10 or 75 mg
Table S2: Mean (%CV) Probe Drug PK Parameters Following Administration Alone or Coadministration with RIF 2 or 600 mg