

Summary
Actin assembly is important for cell motility. The ability of actin subunits to join or leave filaments via the barbed end is critical to actin dynamics. Capping protein (CP) binds to barbed ends to prevent subunit gain and loss, and is regulated by proteins that include V-1 and CARMIL. V-1 inhibits CP by sterically blocking one binding site for actin. CARMILs bind at a distal site and decrease the affinity of CP for actin, suggested to be caused by conformational changes. We used hydrogen-deuterium exchange with mass spectrometry (HDX-MS) to probe changes in structural dynamics induced by V-1 and CARMIL binding to CP. V-1 and CARMIL induce changes in both proteins’ binding sites on the surface of CP, along with a set of internal residues. Both also affect the conformation of CP’s β-subunit “tentacle,” a second distal actin-binding site. Concerted regulation of actin assembly by CP occurs through allosteric couplings between CP modulator and actin binding sites.
Keywords: allostery, actin regulation, CARMIL, hydrogen/deuterium exchange, mass spectrometry
Introduction
Dynamic assembly and disassembly of the actin cytoskeleton are key determinants of cell shape and movement (Pollard and Cooper, 2009). Actin assembles into filaments with barbed (plus) ends and pointed (minus) ends, with the barbed ends thermodynamically and kinetically favored for polymerization (Pollard and Cooper, 2009). In cells, barbed ends are often oriented towards a membrane, and polymerization from those filament ends can drive movement of that membrane and associated structures, such as organelles and pathogens (Carlier et al., 2015). Cells regulate barbed-end polymerization via different proteins that bind to the barbed end and inhibit or promote the gain or loss of actin subunits at the barbed end.
The α/β heterodimeric actin capping protein (CP) is one such regulator (Cooper and Sept, 2008). CP is widely distributed across eukaryotic organisms and cell types. CP binds tightly to barbed ends and prevents gain and loss of subunits. In cells, the concentration of CP (μM) and the affinity of CP for barbed ends (nM) would be sufficient to cap all barbed ends. To initiate actin assembly and movement, cells activate nucleators of actin polymerization, notably Arp2/3 complex and formins, near membranes (Carlier et al., 2015; Skau and Waterman, 2015). These nucleators create free barbed ends, which polymerize and push on the membrane until the ends become capped. Arp2/3 complex creates branched networks of actin filaments, and capping barbed ends is necessary for actin polymerization and actin-driven motility to exist and proceed continuously at steady state (Loisel et al., 1999). Formins create filaments that are not branched; a formin dimer remains with the growing barbed end and protects it from being capped by CP (Goode and Eck, 2007).
The α and β subunits of CP are extensively intertwined into a conformation that melts in a single irreversible transition (Yamashita et al., 2003; Sizonenko et al., 1996). X-ray crystal structural models show that CP has a pseudo two-fold rotational axis of symmetry, and the two subunits have similar secondary structural elements and tertiary folds (Yamashita et al., 2003) (Figure 1). Overall, the CP structure has the shape of a mushroom. The stalk of the mushroom, known as the N-stalk, is composed of six α helices from the N-termini, three from the α subunit (H1α, H2α, & H3α) and three from the β subunit (H1β, H2β, & H3β) (Figure 1). Above the stalk sits a central β sheet with 10 strands of alternating polarity, five from each subunit. Above the β sheet, a single long α helix from each subunit crosses the surface (H5α and H5β , i.e.”antiparallel helices 5”) (Yamashita et al., 2003), terminating with a shorter C-terminal region often called a “tentacle” (H6α & H6β).
Figure 1. Diagrammatic structure of CP heterodimer.
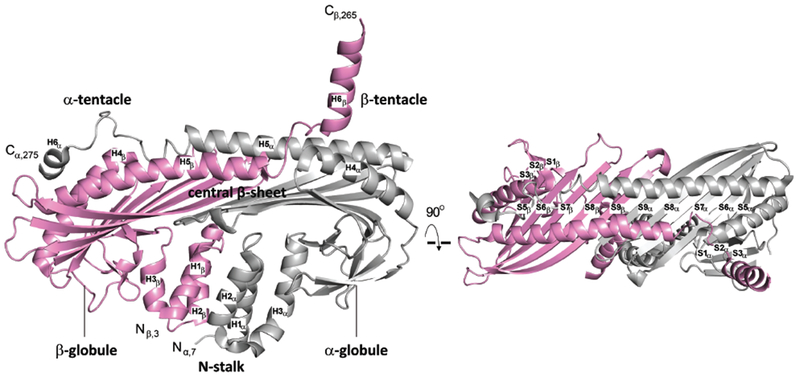
Two views of ribbon representations of two CP crystal structures from PDB 3AAE (Takeda et al., 2010). CP α- and β-subunits are shown in grey and pink, respectively. The N-stalk (H1, H2, H3), central β–sheet (S5-S9), α- and β-globules, antiparallel helices (H5), and α- and β-tentacle (H6) regions of CP are labeled, following the nomenclature of Yamashita and colleagues (Yamashita et al., 2003).
The top surface of the mushroom is one of two major binding sites for the barbed end of the actin filament (Wear et al., 2003; Kim et al., 2010; Narita et al., 2006) (Figure 1). This region includes hydrophilic and charged residues, located on the C-terminal tentacle of the α subunit and on the exposed surfaces of the α helices of the α and β subunits that cross the mushroom top. The second actin-binding site consists of the hydrophobic side of the amphipathic helix that constitutes the C-terminal tentacle of the CP β subunit (Wear et al., 2003; Kim et al., 2010; Narita et al., 2006). The hydrophobic side of this amphipathic helix binds to a hydrophobic groove on the surface of actin, which serves as the binding site for WH2 domains of several actin regulatory proteins (Dominguez, 2016). Structural and biochemical studies led Narita and colleagues to propose a two-step model (Narita et al., 2006) for CP binding to the barbed end. The first step is an electrostatic interaction between the first site described above with the two end protomers of the barbed end. The second step is the hydrophobic interaction of the second site described above with one end protomer. The first step is proposed to primarily influence the association rate while the second step is proposed to primarily influence the dissociation rate (Narita et al., 2006).
CP regulation is critical for proper cytoskeletal function, and the actin-binding activity of CP can be regulated by other proteins, which function in two distinct modes (Takeda et al., 2010). In the first mode, a protein known as V-1 or myotrophin can bind electrostatically to the polar actin-binding site on the top surface of the CP mushroom, competing directly for binding to actin (Jung et al., 2016; Zwolak et al., 2010a; Bhattacharya et al., 2006; Takeda et al., 2010). This steric blocking mechanism is derived from biochemical studies of actin polymerization and structural studies of complexes of V-1 with CP.
In the second mode, a diverse set of otherwise unrelated proteins with CP-interacting (CPI) motifs, including CP ARP2/3 myosin I linker (CARMIL), CD2-associated protein (CD2AP), casein kinase 2 interacting protein (CKIP-1) and the Fam21 subunit of WASH- and capping protein–associated protein (Fam21/WASHCAP), bind to the undersurface of the mushroom, around the N-stalk (Hernandez-Valladares et al., 2010; Edwards et al., 2014). This binding appears to induce a conformational change that allosterically affects actin binding, based on biochemical studies of actin polymerization and structural studies of complexes of CPI motifs with CP (Kim et al., 2012; Takeda et al., 2010; Zwolak et al., 2010a; Hernandez-Valladares et al., 2010). The allosteric nature of the interaction imparts CPI motifs with the ability to promote the dissociation of CP from barbed ends of actin filaments that are capped by CP, i.e. “uncap” a barbed end (Yang et al., 2005; Fujiwara et al., 2010). The complex of CP with a CPI motif does retain a lower level of capping activity, and this level appears to be physiologically relevant in cells (Edwards et al., 2015).
The steric blocking mechanism for V-1 implies that the V-1 / CP complex, which exists in cells (Jung et al., 2016), is not able to cap barbed ends. The allosteric effect of the CPI motif weakens the binding of CP for V-1, in addition to affecting actin binding (Fujiwara et al., 2010). The converse, how the binding of V-1 to CP affects the affinity of a CPI motif for CP, is not known. Current data points to coupling between CP-interacting molecules; however, the mechanistic basis of the coupling remains to be established.
This combination of actions led Fujiwara and colleagues to propose a model for actin polymerization in cells in which CPI-motif proteins function locally to enhance the activity of CP by promoting the dissociation of V-1, which diffuses rapidly to function as a global inhibitor (Fujiwara et al., 2014). Support for this model comes from the observation that the phenotype of certain point mutants of CP, which are unable to bind a CPI motif, resembles the loss of CP (Edwards et al., 2015).
In this study, we sought to test the molecular basis for this model by examining how the structural dynamics of CP are affected by the binding of V-1 and CARMIL. We used hydrogen-deuterium exchange with mass spectrometry (HDX-MS) to interrogate the conformational dynamics and solvent accessibility of the backbone and sidechains of CP, free and in complex with V-1 and fragments of CARMIL that include CPI motifs. Our results indicate that the binding of V-1 and CARMIL result in structural dynamic perturbations, involving a common set of residues, which reveals linkage between the conformations of the two binding sites and known actin-binding sites and provides insight into the molecular mechanism of CP regulation.
Results
CP α-tentacle, β-tentacle, and N-stalk are dynamic / solvent-exposed
Rates of amide hydrogen exchange with solvent D2O are determined in part by local fluctuations in protein structure and thereby report on protein conformational dynamics. To determine CP conformational fluctuations, we employed HDX-MS (Chalmers et al., 2011; Pascal et al., 2012) to derive the first peptide-level analysis of solvent accessibility for CP. CP was deuterium-labeled at exchangeable protons (amide protons) for 10, 30, 60, 120, 900, 3600, and 14400 sec (i.e. up to 4 hrs) and digested on-line with pepsin. In a preliminary experiment, the peptic peptides were identified by high resolving power, accurate-mass analysis to cover ~99% of the sequence. Upon incubation in D2O, HDX extent was measured at each time point by subtracting the centroid of the isotopic distribution of the non-deuterated peptide from that of the deuterated peptide. The percentage of deuterium uptake, relative to the maximum measured deuterium incorporation based on the peptide mass for each peptide, was mapped onto a crystal structure of CP (PDB 3AAE (Takeda et al., 2010)). The results are displayed in Figure 2 and Figure S1 with a color scheme superimposed on the structure. Corresponding kinetic curves for individual peptides are shown in Figure S2 and Figure S3.
Figure 2. HDX-MS analysis of solvent accessibility for CP.
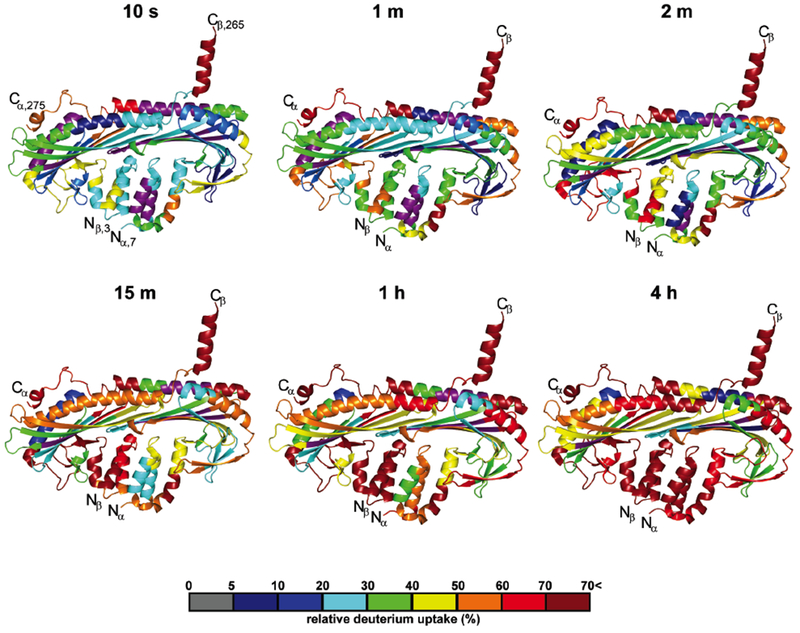
Cartoon representation of a CP crystal structure from PDB 3AAE (Takeda et al., 2010). The percentage of deuterium incorporation for representative peptides is mapped for 6 incubation times (10 sec, 1 min, 2 min, 15 min, 1 hr, and 4 hr). Relative deuterium uptake for representative peptides is displayed as a color gradient (see scale at bottom) and highlighted in the cartoon representation. Data shown are representative of two independent experiments. See also Figure S1-S4 and Table S1.
Monitoring the rate of deuterium incorporation revealed solvent-accessible regions of CP. The C-terminal CP α- and β-tentacles (Figure S1A) exchanged rapidly, with >50% deuterium uptake at the shortest incubation time (10 sec). CP α-subunit C-terminal peptides, corresponding to residues 247-255, 256-267, 266-276, and 278-286, reached maximum deuterium uptake within 15 minutes. In contrast, CP β-subunit C-terminal peptides corresponding to residues 249-264 and 265-272 exchanged more quickly, reaching maximum deuterium uptake within 10 seconds. These results indicate that while both CP α- and β-tentacles are likely solvent exposed, the β-tentacle is relatively more accessible to the solvent than is the α-tentacle. The depicted α-helical character of the β-tentacle may not be applicable to the solution structure. The HDX of the β-tentacle is consistent with an intrinsically disordered region, and NMR studies of CP in solution support this view (Zwolak et al., 2010a). In crystal structures in which the β-tentacle is resolved, two alpha-helical conformations have been observed (Figure S4). In one conformation, the alpha helix extends out from the body of the mushroom; in the other, it is oriented closer toward the surface of the α-subunit’s globule domain (PDB 3AAE, 11ZN,2KZ7, 2KXP) (Takeda et al., 2010; Yamashita et al., 2003; Zwolak et al., 2010a). Structures of CP bound to the actin-like Arp1 filament in dynactin complex, derived from cryoEM, also show the β-tentacle in an α-helical conformation, in contact with actin (Urnavicius et al., 2015; Urnavicius et al., 2018), and in a position consistent with the model for CP binding to actin (Narita et al., 2006).
Other regions of CP displayed less deuterium incorporation, compared to the C-terminal regions (Figure 2 and Figure S2 and S3). The CP α-subunit residues 5-128 and bsubunit residues 1-119 comprise the N-stalk and the globule regions, and they showed a different pattern of deuterium uptake than C-terminal tentacles. These regions displayed moderate deuterium uptake (<40%) within 10 seconds (Figure 2 and Figure S1A, S2, and S3) and more extensive deuterium uptake at longer times (Figure 2 and Figure S1B, S2, and S3). The α helices of the CP N-stalk bundle (α 5-69 and β 11-70) displayed significantly slower exchange rates than did the CP C-terminal helices, likely a result of packing and local interactions within the helix bundle. In addition, peptides of the CP central β-sheet, α 129-217 and β 119-179, showed slower exchange than other CP regions, corroborating molecular dynamic simulations (Koike et al., 2016; Lukman et al., 2012) (Figure 2 and Figure S1, S2, and S3). Even at the longest HDX incubation time (14400 seconds), lower levels of deuterium uptake for peptides corresponding to the CP central β-sheet were observed; they represent low exchange most likely due to hydrogen bonding with backbone amide protons of adjacent strands and low solvent accessibility due to protection by the antiparallel helices (Helix 5) along the top of the mushroom-like CP structure (Figure S1B). In contrast, CP helices comprising the CP N-stalk, α- and β-tentacles displayed high relative HDS (>60%) at extended incubation times (>1 h) (Figure S1B). Together, these results indicate that peptides corresponding to the CP N-stalk and the α- and β-tentacles are significantly more accessible and flexible than other protein regions.
Effects of V-1 binding to CP
To characterize the structural dynamics of CP upon V-1 binding and the interaction interface between CP and V-1, we performed HDX-MS on the CP / V-1 complex, with a saturating concentration of V-1. Samples of unbound CP and CP / V-1 complex were evaluated for structural perturbations upon complex formation, as determined by differential HDX rates. These results are shown in Figure 3, Figure S2, and Figure S3. In the CP / V-1 complex, the binding site for V-1 on CP displayed protection from exchange. The CP α subunit antiparallel helix 5 (Yamashita et al., 2003) and the α-tentacle (peptides spanning CP residues α 239-276), were protected >15% from deuterium uptake. This result agrees well with the CP / V-1 interface identified in a crystal structure (PDB 3AAA, (Takeda et al., 2010)) and a solution NMR structure (PDB 2KXP, (Zwolak et al., 2010a)).
Figure 3. HDX-MS analysis of CP, comparing V-1 / CP complex with free CP.

(A) Cartoon representation of a co-crystal structure of V-1 (green surface) and CP (ribbon), based on PDB 3AAA (Takeda et al., 2010). Differences in deuterium uptake induced by V-1 binding to CP are displayed as a color gradient (see scale at bottom) and highlighted in the cartoon representation of CP. Panels B and C show representative comparisons of deuterium uptake curves for free CP (black) with V-1-bound CP (pink) for CP α- and β-subunits, respectively. Data shown are representative of two independent experiments. See also Figure S2 and S3.
Previous studies demonstrated that the binding of CP for V-1 is weakened by the binding of CARMIL at the distal CP N-stalk, suggesting long-range coupling of the V-1 and CARMIL binding sites (Fujiwara et al., 2014). To provide insight into the nature of this coupling, we examined perturbations outside the V-1 direct binding site, looking first at the CARMIL-binding site.
CARMIL contains two known binding sites for CP, in tandem. Structural and biochemical studies identified two motifs: CPI (capping protein interacting) (residues 9711004) and CSI (CARMIL specific interacting) (residues 1021-1035) (Hernandez-Valladares et al., 2010; Takeda et al., 2010; Zwolak et al., 2010b). The CPI and CSI motifs correspond to segments also referred to as CAH3α and CAH3β (Zwolak et al., 2010b). These two motifs wrap around the CP N-stalk at sites distant from the top of the mushroom structure, where the V-1 binding site and one actin-binding site are located. In co-crystal structures, the CPI motif directly contacts CP N-stalk helices H2α, H1β, and H3β (Figure 1), and the CSI motif interacts with the other side of the CP mushroom stalk, lying opposite to the CPI motif, where it contacts CP N-stalk helices H2β and central β-sheet strands S7α, S8 α, S9 α, S8β, and S9 β (Figure 1).
Indeed, our HDX results revealed that V-1 binding to CP led to protection (<20%) of distal CP N-stalk helices, α 5-46 (H1α & H2α) and β 2-28 (H1β & H2β), which contain binding sites for both the CPI motif and the CSI motif (Figure 3 and Figure S3 and S4). In addition, V-1 binding resulted in protection of certain CP central β-sheet residues (β 131-219), those composing the β-sheet strands nearest the α / β inter-subunit interface, β 175-219 (S8β & S9β) and α 193-206 (S8α & S9α). Unexpectedly, we found that V-1 binding also led to moderate protection (5-10%) of the CP β-tentacle, the amphipathic helix for which the hydrophobic side constitutes a second actin-binding site of CP. The β-tentacle is also distal to the direct binding site of V-1 on CP.
In sum, these results indicate that V-1 binding to CP affects deuterium uptake at the direct binding site for V-1, and that V-1 binding allosterically influences the CP α-tentacle, β-tentacle, and N-stalk. V-1 binding increases the solvent accessibility of residues of both the CPI and CSI binding sites, possibly providing a basis for its modulation of CP binding to CARMIL, and of the β-tentacle, which may contribute to V-1 effects on actin binding.
Next, we employed HDX for the V-1 polypeptide binding, comparing V-1 alone with the CP / V-1 complex (Figures 4 and S5). We observed decreased HDX at sites of CP / V-1 complex formation. Direct-binding V-1 residues were defined in previously solved crystal and solution NMR structures of the complex (Takeda et al., 2010; Zwolak et al., 2010a). The residues include the N-terminal residues, Trp8, Lys11, Asn12, Arg36, Asp44, Gln47, His67, Ile69, Tyr77, and Glu78, which bind the CP α-tentacle, and residues Glu5, Trp8, Ala9, Lys11, Asn12, Asp14, Asp16, Glu17, and Tyr41, which bind central β-sheet strands of the CP β-subunit. Comparing CP / V-1 with V-1 alone, we found substantial protection levels (>15%) for V-1 N-terminal helices (residues 2-48 (Figures 4 and S5). Moderate protection was also observed in V-1 helices (residues 49-75), and minimal differential HDX was detected for other V-1 residues 76-118. The observed protection of V-1 N-terminal residues is likely due to decrease solvent exposure and increased H-bonding from the direct binding of CP.
Figure 4. HDX-MS analysis of V-1, comparing V-1 / CP complex with free V-1.
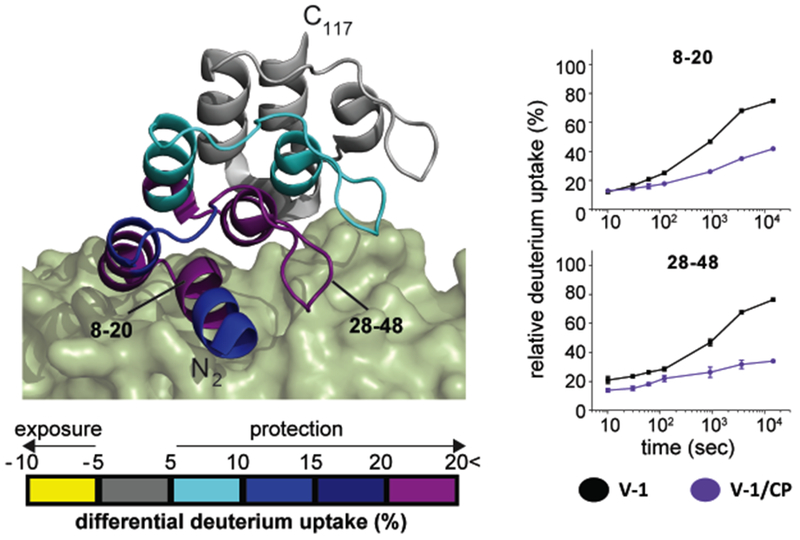
A cartoon representation of V-1 (ribbon) bound to CP (green surface) is shown, based on PDB 3AAA (Takeda et al., 2010). Differences in deuterium uptake induced by CP binding are displayed as a color gradient (see scale at bottom) and highlighted in the cartoon representation of V-1. Representative comparison of deuterium uptake curves of V-1 (black) and CP-bound V-1 (purple) are to the right. Data shown are representative of two independent experiments. See also Figure S5.
Effects of CARMIL binding to CP
In addition to the CPI motif (residues 971-1004) and the CSI motif (residues 10211035), a third region of CARMIL, C-terminal to the CSI motif, has been found to contribute to the biochemical effect of CARMIL on CP, based on actin polymerization studies (Hernandez-Valladares et al., 2010; Zwolak et al., 2010b). This third region of CARMIL has been called CAH3c (Zwolak et al., 2010b). Neither crystallography nor solution NMR studies have provided information about the structure of this region (Hernandez-Valladares et al., 2010; Takeda et al., 2010; Zwolak et al., 2010b), presumably due to its relative lack of order.
Because HDX reports on order and disorder in the structure of a protein, it is particularly well-suited to provide insight into the structural dynamics and function of the different regions of CARMIL, including the unstructured ones. We tested two fragments of CARMIL that differed by the third region. CBR71 is a 71-aa fragment composed of CARMIL residues 964-1034, which includes the CPI motif and the CSI motif; CBR71 corresponds to CAH3a/b. CBR115 is a 115-aa fragment composed of CARMIL residues 964-1078; CBR115 includes CBR71 plus the third C-terminal region (CAH3c).
We evaluated a series of samples, including unbound CP, CP / CARMIL CBR115, and CP / CARMIL CBR71, at saturating levels of CARMIL, for structural perturbations upon complex formation, as determined by differential HDX rates. CARMIL CBR115 binding caused differential HDX of CP α- and β-subunits (Figure 5 and Figure S6 and S7). CP N-stalk helices (α 5-38 and β 2-70) showed the highest levels of protection, at > 15%. These results are consistent with the direct CARMIL binding site determined previously by X-ray crystal and solution NMR structures of CARMIL-bound CP (Hernandez-Valladares et al., 2010; Takeda et al., 2010; Zwolak et al., 2010b).
Figure 5. HDX-MS analysis of CP, comparing CARMIL CBR115-bound CP with free CP.

A cartoon representation of CARMIL CBR115-bound CP is shown, based on PDB 3LK3 (Hernandez-Valladares et al., 2010). CARMIL CBR115 is shown as green surface, with its unresolved residues 1005-1020 between CPI and CSI motifs modeled and C-terminal residues 1034-1078 indicated as a dashed line. Differences in deuterium uptake induced by CARMIL CBR115 binding are displayed as a color gradient (see scale at bottom). Representative comparison of deuterium uptake curves of CP (black) and CARMIL CBR115-bound CP (blue) are shown on the right. These data are representative of two independent experiments. See also Figure S6 and S7.
Perturbations were also observed for regions away from the direct CARMIL binding site. CP α-subunit peptides 239-246, 247-255, and 256-267 were significantly protected (>20%) upon CARMIL binding; these peptides contain residues involved in V-1 binding at the top of the mushroom-like CP structure (Figure 5 and Figure S6 and S7). In addition, CP central β-sheet peptides, β 175-179 and β 176-196, near the V-1 binding site were protected >5% from HDX upon CARMIL binding. Unexpectedly, the highly dynamic CP β-tentacle was also protected (>25%) in the CARMIL-bound state. Only minimal perturbations of HDX rates were observed for the globules of the CP α and β subunits. These results provide experimental evidence to support CARMIL-induced allosteric modulation of conformation of the CP central β-sheet and C-terminal tentacles. Moreover, these results include perturbations similar to the effects seen for CP binding to V-1, comparing V-1 / CP complex with CP (Figure 3).
We compared the effects of the two CARMIL fragments, CBR115 and CBR71, to search for a role for the third region, CAH3c, distal to the CPI and CSI motifs. At longer times (>15 min) of incubation, no significant differences in perturbations were observed between the CP/CBR115 and CP/CBR71 complexes (Figure S6 and S7). However, at short time points (10 sec) HDX differences were observed in the CP α- and β-tentacles, comparing CBR115 with CBR71 (Figure S6 and S7). Residues 256-286 of the α-tentacle and 249-272 of the β-tentacle displayed significantly more protection with CBR115 than with CBR71. Both sets of residues are involved in binding to actin, and the α-tentacle residues involve binding to V-1. The results provide structural dynamic evidence in support of a regulatory role for CARMIL residues 1035-1078, C-terminal to the CSI motif, i.e. CAH3c.
Next, we examined and compared the differential deuterium uptake of CARMIL CBR115 and CARMIL CBR71 peptides upon CP binding (Figure 6). CBR115 and CBR71 peptides in isolation were highly dynamic and underwent rapid HDX (>50%) even at the shortest incubation time (10 sec). CP-bound CARMIL CBR115 and CBR71 showed overall protection (>10%) at incubation times between 10 sec and 15 min. Of note, the CARMIL peptide 1071-1078, which is present in CBR115 but absent in CBR71, was significantly protected (>20%) upon CP binding (Figure 6), providing additional support for a regulatory role for the C-terminal region beyond the CSI motif, i.e. CAH3c.
Figure 6. HDX-MS analysis of CARMIL CBR71 and CARMIL CBR115, comparing CP complexes with free CARMIL fragments.
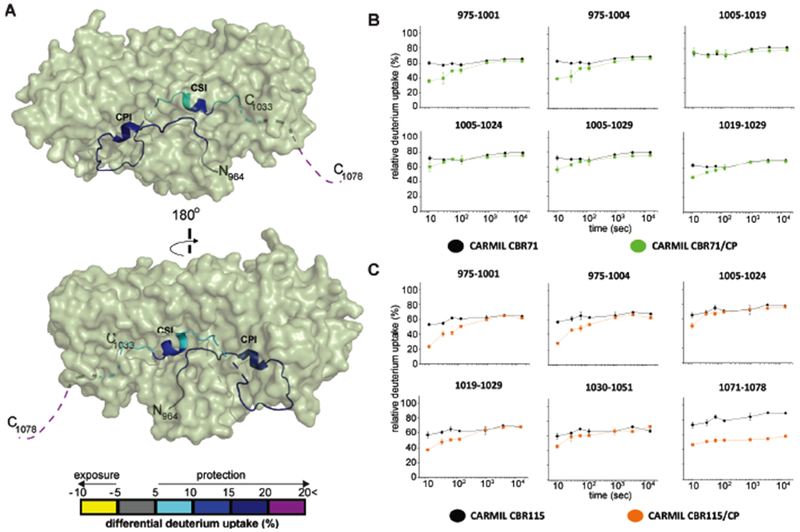
(A) Two views of a cartoon representation of a co-crystal structure of CARMIL CBR115 peptide (ribbon) and CP (green surface), based on PDB 3LK3 (Hernandez-Valladares et al., 2010). Unresolved CARMIL CBR115 residues 1005-1020 between CPI and CSI motifs and C-terminal residues 1034-1078 (CAH3c) are indicated as dashed lines. Differences in deuterium uptake induced by CP binding to CARMIL CBR115 are displayed as a color gradient (see scale at bottom) and highlighted in the ribbon representation of CARMIL CBR115. (B) Deuterium uptake curves for all CARMIL CBR71 peptides are shown for free (black) and CP-bound (green) states. (C) Deuterium uptake curves for CARMIL CBR115 peptides are shown for free (black) and CP-bound (orange) states. Data are representative of two independent experiments.
To compare further the interactions of CBR115 and CBR71 with CP, we measured the binding affinities with fluorescence anisotropy (Figure 7). We measured the fluorescence anisotropy of a fluorescent-labeled peptide, corresponding to a fragment of CARMIL with the CPI motif, in complex with CP. Increasing concentrations of unlabeled CBR115 and CBR71 were added, to compete with and release the fluorescent CPI-motif fragment, decreasing the anisotropy. The results were fit well by a direct-competition model with 1:1 binding stoichiometry. The fitted Kd for CBR115 was 180 ± 120 pM, and the value for CBR71 was 470 ± 130 pM. This difference between CBR115 and CBR71 is consistent with previous comparisons of the two CARMIL fragments in assays of actin capping activity for CP (Hernandez-Valladares et al., 2010; Takeda et al., 2010; Zwolak et al., 2010b). We also performed this experiment using CP for which the C-terminal tentacle region of the β subunit was truncated, ending at L243. The binding affinity was also tight, and no significant differences were found comparing truncated CP with full-length CP, consistent with previous results (Lukman et al., 2012; Koike et al., 2016; Kim et al., 2012).
Figure 7. Binding of CARMIL CBR71 and CARMIL CBR115 to CP by fluorescence anisotropy titration.
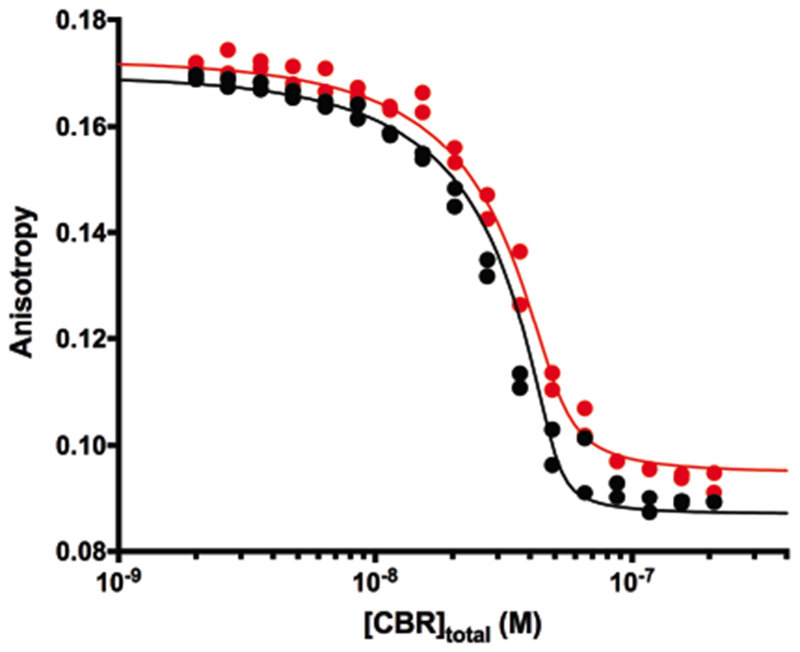
The fluorescence anisotropy of a CPI-motif-containing peptide with a fluorescent label, bound to CP, is plotted vs the total concentration of unlabeled CBR115 (black) or CBR71 (red). The points represent data values from experiments in duplicate, and the lines are the best fits to a competition model with 1:1 stoichiometry. The concentration of CP was 50 nM, and the concentration of the CPI-motif peptide was 20 nM. The fitted value for the binding constant of the CPI-motif peptide to CP was 21.5 nM, which is within the range of experimental values determined from independent direct-binding experiments.
Discussion
CARMIL and V-1 are two regulators that bind to and inhibit the actin capping activity of CP via allosteric and steric mechanisms, respectively. Structural models for CP / CARMIL and CP / V-1 complexes suggest that CARMIL proteins allosterically inhibit the interaction of CP with actin by associating with sites on the N-stalk, distal to the actin-binding site; whereas, V-1 sterically blocks association of CP with actin filament barbed ends by binding to a major actin-binding site at the C-terminal region of the α-subunit. The binding sites for actin and V-1 on CP overlap, so the allosteric effect of CARMIL leads to inhibition of binding for both actin and V-1. The molecular basis of the allosteric cross-talk and the nature of changes in structural dynamics induced by binding of CARMIL and V-1 regulators has been unclear.
To investigate this question, we utilized HDX-MS to provide the first peptide resolution analysis of conformational dynamics for CP alone and in complexes with CARMIL and V-1. We discovered that the CP N-stalk and the C-terminal α- and β-tentacles regions are solvent-exposed and dynamic, relative to the buried and protected central β-sheet, consistent with predictions from molecular dynamics simulations and biochemical studies. Furthermore, we discovered that CARMIL binding results in long-range conformational changes in CP, along the top of the mushroom-like CP structure, which includes the V-1 and actin binding sites, as predicted by biochemistry and molecular dynamics (Lukman et al., 2012; Koike et al., 2016; Kim et al., 2012). In particular, our discoveries provide both an explanation at the molecular level for previous observations that the allosteric effect of CARMIL weakens the binding of CP for V-1 and a foundation for a cellular model for actin polymerization in which CPI-motif proteins work locally and enhance the activity of CP by promoting the dissociation of the rapidly and freely diffusing inhibitor V-1 (Fujiwara et al., 2014).
In addition, we discovered a set of structural perturbations in CP induced by V-1 binding that were similar to those induced by CARMIL. V-1 binding affected HDX at its direct binding site on CP, and it allosterically influenced the CP N-stalk, which contains the CARMIL binding site. Together, perturbations resulting from the binding of V-1 and CARMIL were found in a shared set of CP residues comprising V-1 and CARMIL direct binding sites, as well as the CP inter-subunit interface. The results provide evidence for long-distance communication between V-1 and CARMIL binding sites.
One striking and unexpected discovery ensuing from our results consists of effects on the C-terminal CP β-tentacle, which is proposed to act as a flexible helix, extending away from the body of CP in order to bind actin. The β-tentacle is critical for CP function, in that C-terminal truncations and point mutations decrease the binding affinity of CP for barbed ends (Narita et al., 2006; Wear et al., 2003; Kim et al., 2010). We found that the CP β-tentacle displayed rapid HDX, as expected for regions that are solvent-exposed and highly dynamic. Unexpectedly, we found that the rate of HDX decreased when CP was bound to either V-1 or CARMIL. Previously, an NMR study found that the chemical shifts of the β-tentacle of CP were not affected by V-1 binding (Zwolak et al., 2010a), and an X-ray crystal study found that the base of the β-tentacle in the V-1 / CP complex had a structure similar to that of free CP (Takeda, et al., 2010). An NMR study also found only modest changes in three residues (248-250) of the CP β-subunit upon CARMIL binding (Zwolak et al., 2010b).
Thus, our HDX results that V-1 and CARMIL binding significantly reduce the rate of deuterium incorporation at the distant and mobile CP β-tentacle provide insight into the molecular basis for how CP regulators affect capping of actin filaments. Both V-1 binding and CARMIL binding modulate both of the two actin binding sites of CP – the one found on the CP β-tentacle and the one involving the α-tentacle, where V-1 binds.
Another discovery emerging from our study highlights the roles of the different regions of the CP-binding domain of CARMIL. CARMILs contain a CPI motif (residues 971-1004, aka CAH3a) and a CSI motif (residues 1021-1035, aka CAH3b), along with a C-terminal third region, known as CAH3c (Zwolak et al., 2010b). Here, in a comparison of CARMIL fragment CBR115, which contains all three regions, with a shorter fragment, CBR71, which contains only the first two, we determined that CBR115 binds more tightly to CP than does CBR71, in a fluorescence anisotropy binding assay; this result is consistent with previous assays of the relative ability of CBR115 and CBR71 to inhibit the actin capping activity of CP (Zwolak et al., 2010b; Hernandez-Valladares et al., 2010).The molecular determinants and structural basis of CARMIL CBR115 binding to CP have not been established. Our study provides structural evidence for interaction of this third region, based on differences between how CBR115 and CBR71 interact with CP. Moreover, we found that a CARMIL peptide corresponding to residues 1071-1078, which is present in CBR115 but absent in CBR71, was significantly protected upon CP binding, providing structural evidence for interaction of this C-terminal region.
In sum, these results indicate that the binding of either V-1 or CARMIL to CP affects HDX at the known direct-binding sites for both of the ligands – near the N-stalk and near the actin-binding CP α-tentacle. Unexpectedly, both ligands also affect conformation near the actin-binding β-tentacle, which may contribute further to their ability to inhibit actin binding. Both ligands affect a set of interior beta-sheet residues, suggesting a mechanistic path for the allosteric connection between the two direct-binding sites. The results also support a role for residues of CARMIL distal to the CPI and CSI motifs, in the CAH3c region. Future studies will be required to define the physical basis for the allosteric coupling observed here and to delineate the linkage between distinct sites on CARMIL that appear to regulate cooperatively cytoskeletal dynamics through CP and actin.
Experimental Procedures
Protein Expression & Purification
His-tagged-α1 and β2 subunits of mouse CP (pRSFDuet-1, pBJ 2041) were coexpressed in E. coli BL21 (DE3) pRIL. For CP lacking the β-tentacle, a premature stop codon was introduced, so that the C-terminal residue of the mouse β2 subunit was L243 instead of C272 (pBJ 1891). The fusion proteins were affinity purified on Ni Sepharose 6 Fast Flow (GE Healthcare). The CP eluates were bound to ceramic hydroxyapatite type I, 40 μm (Bio-Rad) in 20 mM Tris-HCl, 20 mM NaCl, 2 mM NaH2PO4, 125 μM CaCl2, 1 mM NaN3, 5 mM DTT, pH 7.5, and eluted with a gradient to 200 mM NaH2PO4, 20 mM NaCl, 125 μM CaCl2, 1 mM NaN3, 5 mM DTT, pH 7.6. CPs were chromatographed on Sephacryl S-200 HR (GE Healthcare) in 20 mM 3-(N-morpholino)propanesulfonic acid (MOPS), 100 mM KCl, 1 mM Tris(2-carboxyethyl)phosphine (TCEP), 1 mM NaN3, pH 7.2 and stored at −70°C.
Glutathione S-transferase (GST)-tagged CBR115 fragment of human CARMIL1a (residues Glu964 to Ser1078, pGEX-6P-3, pBJ 2411) and GST-tagged CBR71 fragment of human CARMIL1a (residues Glu964 to Val1034, pGEX-6P-3, pBJ 2052) were each expressed in E. coli BL21 Star (DE3). The fusion proteins were affinity purified on Glutathione Sepharose 4 Fast Flow (GE Healthcare), followed by cleavage with PreScission Protease (GE Healthcare). The cleaved CBR peptides were bound to POROS GoPure XS (Applied Biosystems) in 20 mM NaH2PO4, 100 μM EDTA, 1 mM NaN3, 5 mM DTT, pH 7.5 and eluted with a gradient to 1 M NaCl in the same buffer. Trace GST was removed by re-incubating with Glutathione Sepharose 4 Fast Flow. Purified CBR peptides were dialyzed into 20 mM MOPS, 100 mM KCl, 1 mM TCEP, 1 mM NaN3, 0.005 % TWEEN 20, pH 7.2 and stored at 70°C.
GST-tagged human V-1 (pGEX-KG, pBJ 2438) was expressed in E. coli BL21 Star (DE3). The fusion protein was affinity purified on Glutathione Sepharose 4 Fast Flow, followed by cleavage with bovine thrombin (MP Biomedicals). V-1 was chromatographed on Sephacryl S-200 HR in 20 mM MOPS, 100 mM KCl, 1 mM TCEP, 1 mM NaN3, 0.005% TWEEN 20, pH 7.2. Trace GST was removed by re-incubating with Glutathione Sepharose 4 Fast Flow. Purified V-1 was stored at −70°C.
Hydrogen deuterium exchange mass spectrometry (HDX-MS)
CP, CP / CARMIL CBR115, CP / CARMIL CBR71, and CP / V-1 samples were buffer-exchanged with 1× phosphate saline buffer (PBS), pH 7.4. HDX was initiated by diluting samples (25 μM, 2 μL) 10-fold with 1XPBS prepared in D2O, or 1XPBS H2O buffer for samples measured for no-deuterium control. At different time intervals (10, 30, 60, 120, 360, 900, 3600, and 14400 s), the labeling reaction was quenched by rapidly decreasing the pH to 2.5 with 30 μL of quench buffer (3 M urea, 1% trifluoroacetic acid, H2O) at 4°C. The protein mixture was immediately injected into a custom-built HDX sample-handling device that enabled in-column digestion with resin-immobilized pepsin (2 mm • 20 mm) at a flow rate of 100 μL / min in 0.1% formic acid. The resulting peptic peptides were captured on a ZORBAX Eclipse XDB C8 column (2.1 mm • 15 mm, Agilent) for desalting (3 min). The C8 column was then switched in-line with a Hypersil Gold C18 column (2.1 mm • 50 mm, Thermo Fisher), and a linear gradient (4-40% acetonitrile, 0.1 % formic acid, 50 μL/min flow rate, over 5 min) was used to separate the peptides and direct them to a LTQ-FTICR mass spectrometer (Thermo Fisher) equipped with an electrospray ionization source. Valves, columns, and tubing for protein digestion and peptide separation were immersed in an ice-water bath to minimize back-exchange.
To map the peptic peptides, the digest, in the absence of HDX, was submitted to accurate mass analysis by LC–MS/MS with the LTQ-FTICR, and the peptic peptides identified using Mascot (Matrix Science). Raw HDX spectra and peptide sets also were submitted to HDX Workbench (Pascal et al., 2012) for calculation and data visualization in a fully automated fashion. Peptides for each run were assessed based on relative representation and statistical validation. Only the top six peptides from each MS scan were used in the final analysis. The extent of HDX at each time point was calculated by subtracting the centroid of the isotopic distribution of the non-deuterated peptide from that of the deuterated peptide. The relative deuterium uptake was plotted versus the labeling time to yield kinetic curves. For comparison between apo states and the complexes, differences in HDX for all incubation time points were calculated. Absolute differences in perturbation values larger than 5% D were considered significant. The 15-min time point was mapped onto the protein threedimensional (3D) structure for data visualization. Peptide digestions were optimized under HDX assay conditions and the mass calculations included accommodation for back exchange with solvent.
Fluorescence Anisotropy Binding Titrations
CBR stock solutions were dialyzed into PBS pH 7.4 with 0.005 % TWEEN 20, and protein concentrations were determined by absorbance at 205 nm (Anthis and Clore, 2013) (CBR115 ε205nm = 409870 M−1 cm−1, CBR71 ε205nm = 245320 M−1 cm−1). A fluorescent CARMIL1 peptide comprised of only the CPI motif (G969-A1005, Ncarboxytetramethylrhodamine (TAMRA), C-amide) was purchased from Watsonbio Sciences; labeled CPI concentration was determined by absorbance at 554 nm (TAMRA ε554nm = 80000 M−1cm−1). CP concentration was determined by absorbance at 280 nm (CP ε280nm = 78935 M1-1 cm
Fluorescence anisotropy experiments were performed on a one-channel L-format QM2000 spectrofluorometer using FelixGX software (Photon Technology International). Polarized excitation was at 552 nm; polarized emission was measured at 582 nm. Experiments were performed at 25°C in 20 mM MOPS, 100 mM KCl, 1 mM TCEP, 1 mM NaN3, 0.005 % TWEEN 20, pH 7.2. CBRs were titrated into a solution containing 20 nM labeled CPI and 50 nM CP, with a two-minute incubation after each addition. For each anisotropy experimental value, 1 measurement was recorded every 5 seconds and averaged over 120 seconds.
The data were analyzed using a competitive binding model, assuming that both the CPI and the CBRs compete for the same binding site on CP. The competition model assumes that labeled, X* (CPI), and unlabeled, X (CBR), peptides bind to the same site on CP, defined by two equilibrium dissociation constants, Kd*=[X*][CP]/[X*CP] and Kd=[X][CP]/[XCP], where [X*] , [X] and [CP] are free concentrations of the species.
The observed anisotropy is defined as in eq. (1a)
| (1a) |
where Ao is the anisotropy of free labeled peptide X*, Amax is the anisotropy of the X*CP complex and v* is the fraction of labeled peptide bound to CP:
| (1b) |
In equation (1b), the free concentrations of [X*] and [CP] can be found numerically by solving the system of mass conservation equations (2a), (2b), (2c) using Scientist software (MicroMath Scientific Software):
| (2a) |
| (2b) |
| (2c) |
Using equations (1) and (2a), (2b), (2c) we fit the experimentally observed anisotropy change (A) as a function of three independent variables [X*]tot, [X]tot and [CP]tot to determine the values of four parameters: Ao, Amax, Kd and Kd*.
Information on all reagents and software used in this study are listed in Supplemental Table S1.
Supplementary Material
Acknowledgments
We thank Ms. S. Smith and Ms. M. Torres for general support and coordination. We are grateful to Drs. Daisy Leung, Adam Zwolak, and Benjamin Stark for advice, assistance and discussions. We acknowledge support by the National Institutes of Health (grants P01AI120943 and R01AI123926 to G.K.A. and M.G., R35GM118171 to J.A.C., R01GM030498 to T.M.L., and P41GM103422 to M.G.) and by the Department of the Defense (Defense Threat Reduction Agency grant HDTRA1-16-1-0033 (C. Basler PI) to G.K.A.). The mass spectrometry facilities were supported by P41GM103422 to M.L.G. The content of the information does not necessarily reflect the position or the policy of the federal government, and no official endorsement should be inferred.
Footnotes
Declaration of interests The authors declare no competing interests.
References
- Anthis NJ, and Clore GM (2013) Sequence-specific determination of protein and peptide concentrations by absorbance at 205 nm. Protein Science, 22, 851–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya N, Ghosh S, Sept D, and Cooper JA (2006) Binding of Myotrophin/V-1 to Actincapping Protein: Implications for How Capping Protein binds to the Filament Barbed End. J. Biol. Chem, 281, 31021–31030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlier MF, Pernier J, Montaville P, Shekhar S, and Kuhn S (2015) Control of polarized assembly of actin filaments in cell motility. Cell Mol Life Sci, 72, 3051–3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalmers MJ, Busby SA, Pascal BD, West GM, and Griffin PR (2011) Differential hydrogen/deuterium exchange mass spectrometry analysis of protein-ligand interactions. Expert Rev Proteomics, 8, 43–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JA, and Sept D (2008) New insights into mechanism and regulation of actin capping protein. Int Rev Cell Mol Biol, 267, 183–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez R (2016) The WH2 Domain and Actin Nucleation: Necessary but Insufficient. Trends Biochem. Sci, 41, 478–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards M, McConnell P, Schafer DA, and Cooper JA (2015) CPI motif interaction is necessary for capping protein function in cells. Nat Commun, 6, 8415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards M, Zwolak A, Schafer DA, Sept D, Dominguez R, and Cooper JA (2014) Capping protein regulators fine-tune actin assembly dynamics. Nat. Rev. Mol. Cell Biol, 15, 677–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara I, Remmert K, and Hammer JA (2010) Direct observation of the uncapping of capping protein-capped actin filaments by CARMIL homology domain 3. J. Biol. Chem, 285, 2707–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara I, Remmert K, Piszczek G, and Hammer JA (2014) Capping protein regulatory cycle driven by CARMIL and V-1 may promote actin network assembly at protruding edges. Proc. Natl. Acad. Sci. U S A, 111, E1970–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goode BL, and Eck MJ (2007) Mechanism and Function of Formins in the Control of Actin Assembly. Annu. Rev. Biochem, 76, 593–627. [DOI] [PubMed] [Google Scholar]
- Hernandez-Valladares M, Kim T, Kannan B, Tung A, Aguda AH, Larsson M, Cooper JA, and Robinson RC (2010) Structural characterization of a capping protein interaction motif defines a family of actin filament regulators. Nat Struct Mol Biol, 17, 497–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung G, Alexander CJ, Wu XS, Piszczek G, Chen BC, Betzig E, and Hammer JA (2016) V-1 regulates capping protein activity in vivo. Proc. Natl. Acad. Sci. U S A, 113, E6610–E6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, Cooper JA, and Sept D (2010) The Interaction of Capping Protein with the Barbed End of the Actin Filament. J. Mol. Biol, 404, 794–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, Ravilious GE, Sept D, and Cooper JA (2012) Mechanism for CARMIL protein inhibition of heterodimeric actin-capping protein. J. Biol. Chem, 287, 15251–15262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike R, Takeda S, Maeda Y, and Ota M (2016) Comprehensive analysis of motions in molecular dynamics trajectories of the actin capping protein and its inhibitor complexes. Proteins, 84, 948–956. [DOI] [PubMed] [Google Scholar]
- Loisel TP, Boujemaa R, Pantaloni D, and Carlier MF (1999) Reconstitution of actinbased motility of Listeria and Shigella using pure proteins. Nature, 401, 613–616. [DOI] [PubMed] [Google Scholar]
- Lukman S, Robinson RC, Wales D, and Verma CS (2012) Conformational dynamics of capping protein and interaction partners: simulation studies. Proteins, 80, 1066–1077. [DOI] [PubMed] [Google Scholar]
- Narita A, Takeda S, Yamashita A, and Maeda Y (2006) Structural basis of actin filament capping at the barbed-end: a cryo-electron microscopy study. EMBO J, 25, 5626–5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascal BD, Willis S, Lauer JL, Landgraf RR, West GM, Marciano D, Novick S, Goswami D, Chalmers MJ, and Griffin PR (2012) HDX workbench: software for the analysis of H/D exchange MS data. J Am Soc Mass Spectrom, 23, 1512–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard TD, and Cooper JA (2009) Actin, a central player in cell shape and movement. Science, 326, 1208–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sizonenko GI, Karpova TS, Gattermeir DJ, and Cooper JA (1996) Mutational analysis of capping protein function in Saccharomyces cerevisiae. Mol. Biol. Cell, 7, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skau CT, and Waterman CM (2015) Specification of Architecture and Function of Actin Structures by Actin Nucleation Factors. Annu Rev Biophys, 44, 285–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda S, Minakata S, Koike R, Kawahata I, Narita A, Kitazawa M, Ota M, Yamakuni T, Maeda Y, and Nitanai Y (2010) Two distinct mechanisms for actin capping protein regulation--steric and allosteric inhibition. PLoS Biol, 8, e1000416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urnavicius L, Lau CK, Elshenawy MM, Morales-Rios E, Motz C, Yildiz A, and Carter AP (2018) Cryo-EM shows how dynactin recruits two dyneins for faster movement. Nature, 554, 202–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urnavicius L, Zhang K, Diamant AG, Motz C, Schlager MA, Yu M, Patel NA, Robinson CV, and Carter AP (2015) The structure of the dynactin complex and its interaction with dynein. Science, 347, 1441–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wear MA, Yamashita A, Kim K, Maeda Y, and Cooper JA (2003) How capping protein binds the barbed end of the actin filament. Curr. Biol, 13, 1531–1537. [DOI] [PubMed] [Google Scholar]
- Yamashita A, Maeda K, and Maéda Y (2003) Crystal structure of CapZ: structural basis for actin filament barbed end capping. EMBO J, 22, 1529–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Pring M, Wear MA, Huang M, Cooper JA, Svitkina TM, and Zigmond SH (2005) Mammalian CARMIL inhibits actin filament capping by capping protein. Dev Cell, 9, 209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwolak A, Fujiwara I, Hammer JA, and Tjandra N (2010a) Structural basis for capping protein sequestration by myotrophin (V-1). J. Biol. Chem, 285, 25767–25781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwolak A, Uruno T, Piszczek G, Hammer JA, and Tjandra N (2010b) Molecular basis for barbed end uncapping by CARMIL homology domain 3 of mouse CARMIL-1. J. Biol. Chem, 285, 29014–29026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.