

Abstract
Maroteaux–Lamy syndrome (MPS VI) is an autosomal recessive lysosomal storage disorder caused by pathogenic ARSB gene variants, commonly diagnosed through clinical findings and deficiency of the arylsulfatase B (ASB) enzyme. Detection of ARSB pathogenic variants can independently confirm diagnosis and render genetic counseling possible. In this review, we collect and summarize 908 alleles (201 distinct variants, including 3 polymorphisms previously considered as disease‐causing variants) from 478 individuals diagnosed with MPS VI, identified from literature and public databases. Each variant is further analyzed for clinical classification according to American College of Medical Genetics and Genomics (ACMG) guidelines. Results highlight the heterogeneity of ARSB alleles, with most unique variants (59.5%) identified as missense and 31.7% of unique alleles appearing once. Only 18% of distinct variants were previously recorded in public databases with supporting evidence and clinical significance. ACMG recommends publishing clinical and biochemical data that accurately characterize pathogenicity of new variants in association with reporting specific alleles. Variants analyzed were sent to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), and MPS VI locus‐specific database (http://mps6-database.org) where they will be available. High clinical suspicion coupled with diagnostic testing for deficient ASB activity and timely submission and classification of ARSB variants with biochemical and clinical data in public databases is essential for timely diagnosis of MPS VI.
Keywords: ARSB, arylsulfatase B, ASB, databases, lysosomal storage disorder, MPS VI, variants
1. BACKGROUND
Mucopolysaccharidosis type VI (MPS VI, Maroteaux–Lamy syndrome, MIM# 253200) is a rare autosomal recessive disease caused by pathogenic variants in the arylsulfatase B (ARSB) gene, coding for one of the enzymes deputed to the degradation of mucopolysaccharides within lysosomes. Pathogenic ARSB variants result in deficient activity of the lysosomal enzyme N‐acetylgalactosamine 4‐sulfatase (arylsulfatase B, ASB, EC 3.1.6.12), causing excessive lysosomal storage and elevated urinary excretion of ASB enzyme substrates, specifically the glycosaminoglycans (GAGs) dermatan sulfate and chondroitin‐4‐sulfate. Increased lysosomal GAG storage causes cellular injury and can result in a diverse range of clinical manifestations (Jurecka et al., 2011b; Valayannopoulos, Nicely, Harmatz, & Turbeville, 2010; Wang, Bodamer, Watson, & Wilcox, 2011).
The prevalence of MPS VI varies among populations, with estimates ranging from 0.0132 per 100,000 live births in Poland to 8.0 per 100,000 live births in Saudi Arabia to 20.0 per 100,000 live births in Monte Santo, a small town in Brazil (Costa‐Motta et al., 2014; Jurecka, Ługowska, Golda, Czartoryska, & Tylki‐Szymańska, 2015; Moammar, Cheriyan, Mathew, & Al‐Sannaa, 2010; Vairo et al., 2015). While several variables likely influence prevalence rates across populations, it is expected that populations with higher degrees of consanguinity display a higher prevalence of the disease (Baehner et al., 2005; Costa‐Motta et al., 2014). Importantly, disease prevalence is likely underestimated in all populations since these epidemiological studies generally rely on clinical identification of disease rather than prenatal or newborn screening (Valayannopoulos et al., 2010).
MPS VI is clinically heterogeneous. Age of onset, rate of disease progression, and clinical manifestations vary widely among individuals. MPS VI can manifest with growth retardation/short stature, coarse facial features, joint stiffness, dysostosis multiplex, hepatosplenomegaly, corneal clouding, macrocephaly, hearing loss, and cardiac and respiratory compromise. Unlike many other lysosomal storage disorders (LSDs), mental development is generally normal, although other symptoms (such as hearing loss) may partially cause developmental delay in some individuals. MPS VI, like other LSDs, is progressive in nature. Untreated patients will continue to deteriorate as GAGs further accumulate within the cell (Jurecka et al., 2011b; Vairo et al., 2015; Valayannopoulos et al., 2010; Wang et al., 2011).
Historically, individuals with MPS VI have been classified into severity groups based on factors such as age of onset, height, and urinary GAG level. Patients with the classical form of MPS VI (sometimes referred to as severe or rapidly progressing) generally present within the first 3 years of life and display elevated urinary GAG levels (generally >100 μg/mg creatinine). In contrast, patients with non‐classical MPS VI (sometimes referred to as atypical, attenuated, or slowly progressing) present later in life with more subtle disease manifestations, and may demonstrate slightly elevated (generally <100 μg/mg creatinine) or near normal urinary GAG levels (Vairo et al., 2015; Valayannopoulos et al., 2010). Intermediate forms are also observed. Fibroblasts and leukocytes of individuals with MPS VI demonstrate absent or reduced ASB enzyme activity compared with healthy controls (Valayannopoulos et al., 2010). In addition, a non‐classical “cardiac phenotype” has been described in some patients with MPS VI. This form manifests later in adulthood with primarily cardiac features such as valvular disease, cardiomyopathy, and/or acute heart failure, although other symptoms (such as musculoskeletal abnormalities) may also be present (Golda, Jurecka, & Tylki‐Szymanska, 2012; Jurecka, Golda, Opoka‐Winiarska, Piotrowska, & Tylki‐Szymańska, 2011a; Thümler et al., 2012).
MPS VI is diagnosed through a combination of clinical findings and demonstration of deficient or absent ASB enzyme activity, in conjunction with normal activity of control enzymes, mainly sulfatases, to exclude a multiple sulfatase deficiency. Detection of pathogenic ARSB variants in the proband and in both parents (trio analysis) independently confirms diagnosis and renders genetic counseling possible. However, this approach may be problematic due to the high proportion of private mutations in MPS VI, thus considering the possibility to identify gene variants that are not yet associated with a clinical phenotype.
Guidelines from the American College of Medical Genetics and Genomics (ACMG) recommend that gene variants be published in association with clinical classification and validating data (Richards et al., 2015). In this study, we collected and analyzed all the ARSB variants reported in the literature and in public databases. Of the 201 unique ARSB variants summarized here, only 18% had been previously recorded in public databases (ClinVar, EmvClass, ClinVitae) in association with supporting evidence/clinical classification. The present update illustrates the heterogeneity of alleles associated with MPS VI, the need of a critical evaluation and correct classification of the identified variants, and the lack of prior representation in publicly available variant databases of characterized ARSB alleles. Variants analyzed in the present paper have been sent to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), where they will be publicly available.
We emphasize the importance to maintain high clinical suspicion during MPS VI diagnosis, as many ARSB alleles identified via molecular testing alone may have yet to be formally classified as MPS VI‐associated and cannot be considered diagnostic per se. Timely submission and classification of ARSB variants in public databases, in association with biochemical and clinical data, will help facilitate timely future diagnoses of MPS VI.
2. METHODOLOGY
To identify all reported pathogenic ARSB variants, a literature search was performed in Embase on October 26, 2017 using the population, intervention and comparators, outcome (PICO) framework where the population was MPS VI (and relevant synonyms) and the outcome was genetic variant (and relevant synonyms); intervention and comparators were not applicable. Results were limited to studies in humans and conference abstracts were excluded. As case reports containing variant data are often not indexed to reflect the presence of this information, a targeted MPS VI case report search was also performed in Embase on September 13, 2017 to supplement the results. The search strategies are described in detail in Supp. Mat. S1. Manual searching of relevant journals not indexed in Embase was also performed. Each identified publication was screened by a single reviewer for information regarding ARSB variants. Reports of individuals with ARSB variants, where variants were reported as linked to MPS VI, were extracted and assessed for redundant reporting to the extent possible.
Public databases were accessed via searches for “ARSB” in ClinVar, EmvClass, ClinVitae, and Human Gene Mutation Database (HGMD) (Hart et al., 2014; Stenson et al., 2003). Identified variants were compared with those from the literature search.
For the three‐dimensional visualization of arylsulfatase B, an image was created using the PyMOL Molecular Graphics System, Version 1.8 Schrödinger, LLC with atomic coordinates from Brookhaven Protein Data Bank accession number 1FSU (Bond et al., 1997).
All variants were described according to the NCBI Reference Sequence NM_000046.4 and the corresponding protein sequence NP_000037.2. In cases where the reported variant did not reference a particular sequence, coordinates were mapped to NM_000046.4 [e.g., “deletion of exon 4” to “c.(690+1_691‐1)_(898+1_899‐1)del”]. Variants cDNA and protein annotations were further checked through “'Name checker'” tool (https://mutalyzer.nl/) according to the HGVS nomenclature v15.11 (https://varnomen.hgvs.org/) and “VariantValidator” tool (https://variantvalidator.org), and corrected if necessary. In case of discrepancies in amino acid sequence impact of coding variants, the variant was simulated and translated in silico using NM_000046.4. All discrepancies and other misreported variants found in our literature analysis were collected in Table 1.
Table 1.
Summary of misreported variants and corrections
| Paper Annotation | Correct Annotation | Type of Error | Error Source | Notes | ||
|---|---|---|---|---|---|---|
| c.208_215del | p.(P70fs*123) | c.208_215del | p.(Pro70Glyfs*54) | Amino acid position | Kantaputra et al. (2014) | |
| c.213insGCCGCAC | p.(Leu72Alafs*57) | c.207_213dup | p.(Leu72Alafs*57) | c.DNA position | Sandberg et al. (2008) | |
| c.247_248delC | p.(D83Qfs*43) | c.247_248del | p.(Asp83Glnfs*43) | c.DNA position | Jurecka et al. (2014b) | |
| c.262_263insA | p.(Pro89Alafs*38) | c.263dup | p.(Pro89Alafs*38) | c.DNA position | Jurecka et al. (2014b) | |
| c.306_312delCTACCAG+146del | p.(Y103SfsX9) | c.306_312+146del | p.(Tyr103Serfs*25) | c.DNA position | Uttarilli et al. (2015) | |
| c.31091insCCTGAAG_delATACT | — | c.750_754delinsCCTGAAG | p.(Glu250Aspfs*4) | Reference sequence | Jurecka et al. (2011b) | The reference sequence used by Jurecka likely is NC_000005.9 |
| c.356358 del | p.(Y86del) | c.356_358del | p.(Pro119_Ser120delinsArg) | Amino acid position | Karageorgos et al. (2004) | |
| c.375_376insT | p.(Glu126*) | c.375dup | p.(Glu126*) | c.DNA position | Jurecka et al. (2012a) | |
| c.436G>T | p.(Trp146Leu) | c.437G>T | p.(Trp146Leu) | c.DNA position | Simonaro and Schuchman (1995) | |
| c.496delT | p.(Phe166Leufs*18) | c.498del | p.(Phe166Leufs*18) | c.DNA position | Uttarilli et al. (2015) | |
| c.592C>T | p.(R197*) | c.589C>T | p.(Arg197*) | c.DNA position | Petry et al. (2005) | Also reported by Karageorgos et al. (2007b) |
| c.629_635del | p.(Tyr210*) | c.630_636del | p.(Tyr210*) | c.DNA position | Karageorgos et al. (2007b) | |
| c.659_660del | p.(I220fs*5) | c.659_660del | p.(Ile220Serfs*5) | Annotation error | Uttarilli et al. (2015) | |
| c.785insA | p.(Asn262Lysfs*14) | c.785dup | p.(Asn262Lysfs*14) | c.DNA position | Chistiakov et al. (2014) | |
| c.883_884dupTT | p.(F295Ffs*42) | c.883_884dup | p.(Ile296Serfs*43) | Amino acid position, annotation | Ferla et al. (2015) | |
| c.1036delG | p.(E346Sfs*11) | c.1036del | p.(Glu346Serfs*13) | Amino acid position, annotation | Ferla et al. (2015) | |
| c.1285_1286insT | p.(His430Profs*5) | c.1286dup | p.(His430Profs*5) | c.DNA position | Isbrandt et al. (1994) | |
| c.1325C>T | p.(T442R) | c.1325C>T | p.(Thr442Met) | Annotation error | Karageorgos et al. (2007b) | |
| c.1348G>C | p.(Trp450Cys) | c.1350G>C | p.(Trp450Cys) | c.DNA position | Mathew et al. (2015) | |
| c.1457A>G | p.(D486V) | c.1457A>T | p.(Asp486Val) | Annotation error | Nouri et al. (2012) | |
| c.1531_1553del | p.(V512Pfs*3) | c.1534_1556del | p.(Val512Profs*3) | c.DNA position | Petry et al. (2005) | Also reported by Garcia et al. (2010) |
For the variants collected, all the evidence available was exploited for variants analysis and clinical classification, according to the approach recommended by ACMG (Richards et al., 2015). Finally, all variants were submitted to ClinVar database with associated evidence and literature references (ClinVar accession numbers from SCV000802937 to SCV000803138).
2.1. ARSB gene structure
The ARSB gene is located on chromosome 5q13‐14 and spans a region of 206 kb. ARSB contains eight exons which encode an mRNA transcript of 2228 base pairs and a protein of 533 amino acids, including a signal peptide at the N‐terminal (Brands et al., 2013a; Karageorgos et al., 2007b; Litjens et al., 1989; Litjens & Hopwood, 2001; Peters et al., 1990). ARSB contains an alkaline phosphatase‐like domain and two conserved sulfatase regions near the N‐terminal (Figure 2; Bond et al., 1997; Peters et al., 1990).
Figure 2.
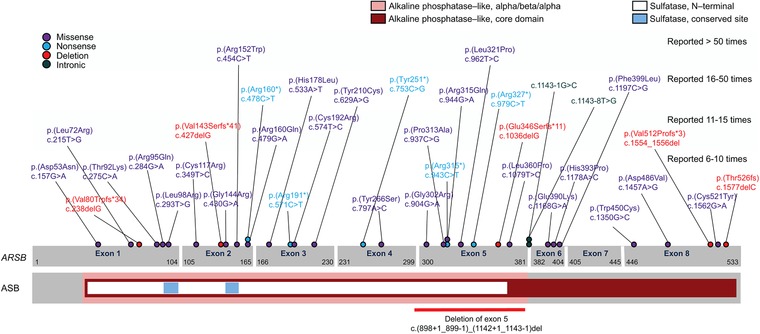
ARSB gene structure including coding variants reported >5 times
Location of selected MPS VI variants in the human ARSB gene and ASB peptide. Exons are represented by grey boxes; cDNA coordinates are indicated within the exons. ASB protein domains are color coded as shown by legend. Individual variants are color coded by mutation type
3. VARIANTS
3.1. Overview and summary statistics
A uniform summary of all ARSB variants identified in individuals with MPS VI was created based on the literature and database searches. The PICO search identified 208 records, the targeted case report search identified 198 records, and 5 additional records were identified from other sources. Removal of duplicates led to a total of 380 unique records. Manual screening yielded 74 publications containing 615 individuals with ARSB variant information. Duplicate entries were eliminated based on direct references (“patient reported previously”) and if the entries contained no conflicting attributes along with identical genotypes, ethnicities, gender, and age of diagnosis/start of ERT. This ultimately resulted in the aggregation of 908 alleles from 478 individuals diagnosed with MPS VI (Supp. Mat. S2). No additional unpublished pathogenic variants were identified through the public database search. The 908 alleles encompassed 198 unique non‐polymorphic variants (Supp. Mat. S3). However, Supp. Mat. S3 also reports three benign variants, highlighted in blue, which previously had been erroneously reported as disease‐causing variants. A shorter version of Supp. Mat. S3, containing only variants described in more than one independent family, is represented by Table 2.
Table 2.
ARSB sequence variants identified in more than one independent family
| Exon/Intron | Nucleotide Change | Predicted Amino Acid Change | Number of Reports | Clinical Significance | Variant Identifier (ClinVar ID and/or dbSNP Number, if Available) |
|---|---|---|---|---|---|
| Exon 1 | c.157G>A | p.(Asp53Asn) | 6 | Likely pathogenic | |
| Exon 1 | c.160G>A | p.(Asp54Asn) | 5 | Likely pathogenic | |
| Exon 1 | c.171G>C | p.(Trp57Cys) | 2 | Uncertain significance | |
| Exon 1 | c.175G>A | p.(Asp59Asn) | 2 | Uncertain significance | |
| Exon 1 | c.215T>G | p.(Leu72Arg) | 13 | Likely pathogenic | |
| Exon 1 | c.238del | p.(Val80Cysfs*34) | 6 | Pathogenic | ClinVar Allele ID: 15920; rs750845916 |
| Exon 1 | c.245del | p.(Leu82Argfs*32) | 2 | Likely pathogenic | |
| Exon 1 | c.245T>G | p.(Leu82Arg) | 3 | Likely pathogenic | rs750845916 |
| Exon 1 | c.262C>T | p.(Gln88*) | 4 | Pathogenic | |
| Exon 1 | c.275C>A | p.(Thr92Lys) | 9 | Uncertain significance | |
| Exon 1 | c.275C>T | p.(Thr92Met) | 2 | Uncertain significance | |
| Exon 1 | c.284G>A | p.(Arg95Gln) | 6 | Likely pathogenic | ClinVar Allele ID: 15923 |
| Exon 1 | c.289C>T | p.(Gln97*) | 4 | Likely pathogenic | |
| Exon 1 | c.293T>A | p.(Leu98Gln) | 4 | Likely pathogenic | |
| Exon 1 | c.293T>C | p.(Leu98Pro) | 2 | Uncertain significance | |
| Exon 1 | c.293T>G | p.(Leu98Arg) | 8 | Likely pathogenic | |
| Exon 1 | c.305G>A | p.(Arg102His) | 2 | Uncertain significance | |
| Exon 1 | c.312G>C | p.(Gln104His) | 4 | Uncertain significance | rs150087888 |
| Exon 2 | c.332A>C | p.(His111Pro) | 5 | Uncertain significance | rs775780931 |
| Exon 2 | c.349T>C | p.(Cys117Arg) | 6 | Likely pathogenic | ClinVar Allele ID: 15917 |
| Exon 2 | c.384_386del | p.(Leu129del) | 3 | Uncertain significance | ClinVar Allele ID: 106583 |
| Exon 2 | c.395T>C | p.(Leu132Pro) | 3 | Likely pathogenic | |
| Exon 2 | c.427del | p.(Val143Serfs*41) | 12 | Pathogenic | rs746206847 |
| Exon 2 | c.430G>A | p.(Gly144Arg) | 8 | Likely pathogenic | |
| Exon 2 | c.437G>C | p.(Trp146Ser) | 2 | Uncertain significance | |
| Exon 2 | c.438G>A | p.(Trp146*) | 4 | Likely pathogenic | |
| Exon 2 | c.440A>C | p.(His147Pro) | 2 | Uncertain significance | |
| Exon 2 | c.454C>T | p.(Arg152Trp) | 62 | Pathogenic | |
| Exon 2 | c.464G>A | p.(Cys155Tyr) | 3 | Uncertain significance | |
| Exon 2 | c.478C>T | p.(Arg160*) | 16 | Pathogenic | |
| Exon 2 | c.479G>A | p.(Arg160Gln) | 13 | Likely pathogenic | |
| Exon 2 | c.499G>A | p.(Gly167Arg) | 5 | Uncertain significance | |
| Exon 3 | c.511G>A | p.(Gly171Ser) | 3 | Likely pathogenic | |
| Exon 3 | c.533A>T | p.(His178Leu) | 29 | Likely pathogenic | |
| Exon 3 | c.571C>T | p.(Arg191*) | 7 | Pathogenic | |
| Exon 3 | c.574T>C | p.(Cys192Arg) | 15 | Likely pathogenic | |
| Exon 3 | c.589C>T | p.(Arg197*) | 5 | Pathogenic | rs118203943 |
| Exon 3 | c.629A>G | p.(Tyr210Cys) | 46 | Pathogenic | ClinVar Allele ID: 15924 |
| Intron 3 | c.691‐1G>A | — | 3 | Likely pathogenic | rs6870443 |
| Exon 4 | c.707T>C | p.(Leu236Pro) | 2 | Uncertain significance | ClinVar Allele ID: 15918 |
| Exon 4 | c.710C>A | p.(Ala237Asp) | 4 | Likely benign | |
| Exon 4 | c.716A>G | p.(Gln239Arg) | 3 | Uncertain significance | |
| Exon 4 | c.753C>G | p.(Tyr251*) | 43 | Pathogenic | |
| Exon 4 | c.765T>A | p.(Tyr255*) | 3 | Likely pathogenic | rs749015246 |
| Exon 4 | c.797A>C | p.(Tyr266Ser) | 7 | Uncertain significance | |
| Exon 5a | c.(898+1_899‐1)_(1142+1_1143‐1)del | — | 6 | Pathogenic | |
| Exon 5 | c.899‐1341_1142+1051del | — | 1 | Pathogenic | |
| Exon 5 | c.903C>G | p.(Asn301Lys) | 4 | Likely pathogenic | rs779378413 |
| Exon 5 | c.904G>A | p.(Gly302Arg) | 10 | Uncertain significance | |
| Exon 5 | c.908G>A | p.(Gly303Glu) | 5 | Likely pathogenic | |
| Exon 5 | c.937C>G | p.(Pro313Ala) | 11 | Likely pathogenic | |
| Exon 5 | c.943C>T | p.(Arg315*) | 10 | Likely pathogenic | rs727503809 |
| Exon 5 | c.944G>A | p.(Arg315Gln) | 22 | Likely pathogenic | ClinVar Allele ID: 177363 |
| Exon 5 | c.960C>G | p.(Ser320Arg) | 3 | Uncertain significance | |
| Exon 5 | c.962T>C | p.(Leu321Pro) | 60 | Pathogenic | |
| Exon 5 | c.966G>A | p.(Trp322*) | 2 | Pathogenic | rs398123125 |
| Exon 5 | c.971G>T | p.(Gly324Val) | 5 | Likely pathogenic | ClinVar Allele ID: 98267; rs773492223 |
| Exon 5 | c.979C>T | p.(Arg327*) | 16 | Pathogenic | rs201168448 |
| Exon 5 | c.1001G>T | p.(Ser334Ile) | 3 | Uncertain significance | |
| Exon 5 | c.1036del | p.(Glu346Serfs*11) | 13 | Pathogenic | |
| Exon 5 | c.1057T>A | p.(Trp353Arg) | 2 | Uncertain significance | |
| Exon 5 | c.1079T>C | p.(Leu360Pro) | 6 | Likely pathogenic | |
| Exon 5 | c.1127T>A | p.(Val376Glu) | 4 | Likely pathogenic | |
| Exon 5 | c.1142+2T>A | — | 2 | Likely pathogenic | |
| Exon 5 | c.1142+2T>C | — | 4 | Pathogenic | |
| Intron 5 | c.1143‐1G>C | — | 16 | Pathogenic | ClinVar Allele ID: 15926 |
| Intron 5 | c.1143‐8T>G | — | 24 | Likely pathogenic | ClinVar Allele ID: 15927; rs431905496 |
| Exon 6 | c.1151G>A | p.(Ser384Asn) | 9 | Benign | ClinVar Allele ID: 98263; rs25414 |
| Exon 6 | c.1168G>A | p.(Glu390Lys) | 6 | Likely pathogenic | |
| Exon 6 | c.1178A>C | p.(His393Pro) | 10 | Uncertain significance | ClinVar Allele ID: 15925 |
| Exon 6 | c.1178A>G | p.(His393Arg) | 3 | Uncertain significance | |
| Exon 6 | c.1197C>G | p.(Phe399Leu) | 17 | Likely pathogenic | rs200793396 |
| Intron 6 | c.1213+5G>A | — | 2 | Uncertain significance | |
| Intron 6 | c.1213+6T>C | — | 3 | Pathogenic | |
| Exon 7 | c.1214G>A | p.(Cys405Tyr) | 3 | Uncertain significance | ClinVar Allele ID: 15919; rs118203941 |
| Exon 7 | c.1289A>G | p.(His430Arg) | 3 | Likely pathogenic | |
| Exon 7 | c.1325C>T | p.(Thr442Met) | 3 | Uncertain significance | ClinVar Allele ID: 368534 |
| Exon 7 | c.1336G>A | p.(Gly446Ser) | 3 | Uncertain significance | |
| Intron 7 | c.1336+2T>G | — | 7 | Pathogenic | rs768012515 |
| Exon 8 | c.1340G>T | p.(Cys447Phe) | 4 | Uncertain significance | |
| Exon 8 | c.1350G>C | p.(Trp450Cys) | 11 | Likely pathogenic | rs555785323 |
| Exon 8 | c.1366C>T | p.(Gln456*) | 4 | Pathogenic | rs200188234 |
| Exon 8 | c.1415T>C | p. (Leu472Pro) | 2 | Likely pathogenic | |
| Exon 8 | c.1450A>G | p.(Arg484Gly) | 2 | Uncertain significance | ClinVar Allele ID: 187113 |
| Exon 8 | c.1457A>T | p.(Asp486Val) | 6 | Uncertain significance | |
| Exon 8 | c.1534_1556del | p.(Val512Profs*3) | 13 | Uncertain significance | |
| Exon 8 | c.1562G>A | p.(Cys521Tyr) | 9 | Uncertain significance | |
| Exon 8 | c.1577del | p.(Thr526Metfs*48) | 8 | Pathogenic | |
| Exon 8 | c.1582_1596del | p.(Val528_Trp532del) | 2 | Uncertain significance |
Variants classified per Richards et al. (2015). ClinVar accessed 06/05/2017.
Deletion of exon 5 without defined boundaries.
Individual characteristics of the patients were highly variable, highlighting the clinical heterogeneity of MPS VI. Patients displayed a wide range of clinical manifestations, ages at diagnosis, and national/geographic and ethnic backgrounds. Of the identified individuals, 54.8% (262/478) were homozygous for pathogenic ARSB variants, 35.6% (170/478) were heterozygous, 9.2% (44/478) had only one allele reported, and two individuals (0.4%) had both alleles unidentified. For alleles not characterized, we considered also those alleles carrying a polymorphism erroneously considered a disease‐causing variant. Patient gender was reported in 40% (190/478) of individuals and was split roughly evenly between genders (55% male; 45% female). Age at diagnosis (reported in 172 individuals) ranged from <1 year to 45 years of age, with a mean of 7.8 years and median of 5.0 years. Two patients, both with an older affected sibling, were diagnosed at birth (Furujo, Kubo, Kosuga, & Okuyama, 2011; Mendelsohn et al., 2013). Approximately one‐third (31.7%) of unique variants appeared only once in the population assessed, with an additional 28.5% appearing twice.
In agreement with previous reports (Karageorgos et al., 2007b), most unique variants were missense (59.5%); followed by small deletions (13.5%), nonsense (12.0%), splice site or intronic variants (5.0%), small duplications (3.0%), and large deletions (3.0%) (Figure 1). Stop‐loss mutations were identified in 1.0% of variants.
Figure 1.
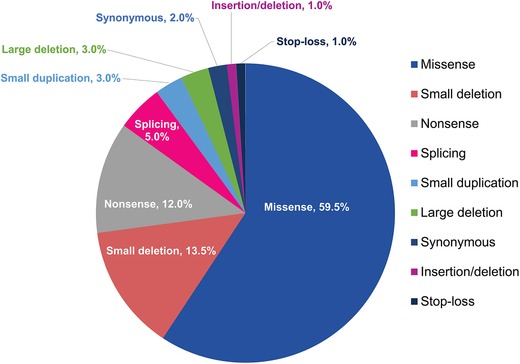
Unique non‐polymorphic ARSB variants classified by mutation type, most common being missense and small deletion (N = 198 variants)
Commonly reported variants occur throughout the length of the ARSB gene, and include missense, nonsense, deletion, and intronic alleles (Figure 2). Only two alleles were reported more than 50 times in our sample taken globally: c.962T>C [p.(Leu321Pro)] and c.454C>T [p.(Arg152Trp)] (Table 2). These alleles are located in exons 5 and 2, respectively.
The most commonly described nonsense allele, c.979C>T [p.(Arg327*)], was reported 16 times (1.8%) in our sample, and is expected to result in premature truncation of ARSB midway through exon 5. c.979C>T is identified in various populations, including individuals from North and South America, Europe, and Asia, and was detected in a homozygous state in five individuals (5 of 11, 45.5%).
The missense mutation c.284G>A [p.(Arg95Gln)] is present in six individuals in our sample, but never in the homozygous state. Of note, Arg95 is the final residue making up the N‐terminal sulfatase domain (CTPSR sequence), which is conserved in several sulfatases including human arylsulfatase A, human steroid sulfatase, and sea urchin arylsulfatase (Peters et al., 1990). Individuals with this variant vary in phenotypic severity, presumably based on the allele present in the other ARSB parental allele.
Three large deletions within the alkaline phosphatase‐like domain have also been reported. One large deletion that includes exons 2 and 3 was identified in homozygosity in a male Chinese individual with a classical, rapidly progressing MPS VI phenotype (Zheng et al., 2014); also a 13.8 kb deletion encompassing exons 2 and 3, whose breakpoints were defined, was reported in homozygosity in a boy with disease onset at 3 years (Ittiwut et al., 2017). A deletion of exon 4 [c.(690+1_691‐1)_(898+1_899‐1)del; breakpoints unknown] in its entirety was reported in homozygosity in one female Taiwanese individual. This deletion is expected to result in frameshift, premature truncation, and loss of significant portions of the catalytic domain (Lin, Ke, Chou, Wang, & Tsai, 2015). Finally, a genomic deletion of exon 5 [c.(898+1_899‐1)_(1142+1_1143‐1)del] was reported eight times, once in homozygosity in a male Italian individual, who displayed a classical, severe MPS VI phenotype (Ferla et al., 2015). In only one case, the boundaries were defined (Villani et al., 2010).
3.2. Most common alleles
Of the 12 most commonly reported alleles, 7 of these are missense mutations, 3 are nonsense mutations, and 2 are intronic/splice site mutations. The seven most commonly reported alleles (c.454C>T, c.962T>C, c.629A>G, c.753C>G, c.533A>T, c.1143‐8T>G, c.944G>A) make up 31.5% of all alleles reported. Together, the three most frequently reported alleles make up 18.5% of all alleles reported (Table 3). Importantly, the allele frequency reported here may not represent the real‐world distribution of alleles, as well‐characterized alleles are less likely to be published and therefore may be underrepresented in the literature.
Table 3.
Most common ARSB pathogenic and likely pathogenic variants reported in the assessed populationa
| Nucleotide Change | Amino Acid Change | Times Reported, n (%) N = 908 alleles |
|---|---|---|
| c.454C>T | p.(Arg152Trp) | 62 (6.8) |
| c.962T>C | p.(Leu321Pro) | 60 (6.6) |
| c.629A>G | p.(Tyr210Cys) | 46 (5.1) |
| c.753C>G | p.(Tyr251*) | 43 (4.7) |
| c.533A>T | p.(His178Leu) | 29 (3.2) |
| c.1143‐8T>G | — | 24 (2.6) |
| c.944G>A | p.(Arg315Gln) | 22 (2.4) |
| c.1197C>G | p.(Phe399Leu) | 17 (1.9) |
| c.1143‐1G>C | — | 16 (1.8) |
| c.478C>T | p.(Arg160*) | 16 (1.8) |
| c.979C>T | p.(Arg327*) | 16 (1.8) |
| c.574T>C | p.(Cys192Arg) | 15 (1.7) |
Nucleotide changes are per NM_0000446.4; protein changes are per NP_000037.2.
As it is uncommon to publish findings of well‐characterized alleles, these alleles may be underrepresented in the literature.
3.3. Geographic distribution
Information regarding ethnic or national origin was available for 406 individuals (85% of sample), and individuals from Africa, Asia, Australia, Europe, North America, and South America were included, although most of them came from the northern hemisphere. Allelic heterogeneity varied among individual populations, although rare or private mutations were prominent in all populations studied. Individuals from Italy, the United States, and Colombia were the most genetically heterogeneous, with the largest proportion of rare or private mutations (≈84% of individuals) in the populations studied (Figure 3).
Figure 3.
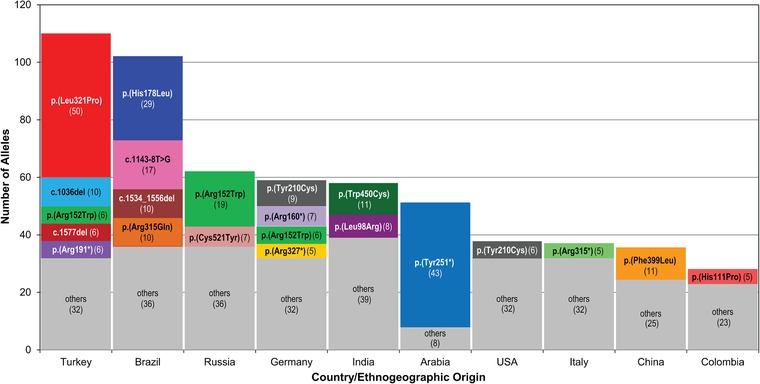
Most common alleles for the 10 most frequent nationalities/ethnic backgrounds reported. The number of times an allele was encountered in a population is shown in parentheses. In grey other variants, reported less or equal to 5 times
Turkey was the most heavily represented national origin, with information reported for 55 Turkish individuals (13.5% of total reported). Of the major populations studied, the Turkish population was the most genetically homogenous, and 54 of 55 Turkish individuals were homozygous. Variant p.(Leu321Pro) accounted for nearly half of all alleles reported in Turkish individuals (50/110). c.1036del was heavily represented in this population. Interestingly, p.(Leu321Pro) did not appear to be associated with a phenotypic signature in this population, possibly due to the relatively high rates of consanguineous marriages in Turkey, leading to a higher proportion of homozygous alleles across the genome and variable clinical phenotypes (Koc, 2008; Zanetti et al., 2014).
Mutations in Trp450 [p.(Trp450Cys) and p.(Trp450Leu)] were found exclusively in individuals from India, and resulted in a range of clinical phenotypes. Variant p.(Phe399Leu) (c.1197C>G) was commonly reported in individuals from both China (59% of individuals) and Taiwan (38% of individuals), and was associated with a classical phenotype in these patients.
3.4. ASB structure and variants of special interest
Arylsulfatase B includes two domains; a larger N‐terminal domain and a smaller C‐terminal domain; and reported to function as a monomer (Mathew et al., 2015; McGovern, Vine, Haskins, & Desnick, 1982). ASB contains two conserved sulfatase regions near the N‐terminal (Bond et al., 1997; Peters et al., 1990). The active site pocket is located near the surface of the folded enzyme, and consists of Asp53, Asp54, Cys91, Pro93, Ser94, Arg95, Lys145, His147, His242, Asp300, and Lys318. A cluster of conserved cysteine residues is also thought to be required for enzymatic activity (Bond et al., 1997; Brooks et al., 1995).
Pathogenic variants in ARSB do not appear to be concentrated in any particular region or domain of the ASB protein. Although pathogenic variants in or near the active site pocket were identified, many are located remotely from the active site (Figure 4). Importantly, genotype–phenotype correlations in MPS VI have remained elusive, due (at least in part) to the large proportion of rare or private ARSB mutations, limited phenotypic reporting, and overall heterogeneity of the disease (Vairo et al., 2015; Valayannopoulos et al., 2010). This is especially true for missense mutations, whose effect on ASB enzyme function may not be immediately apparent (Valayannopoulos et al., 2010). In addition, residual ASB enzyme activity does not clearly correlate with the severity of clinical phenotype in patients with MPS VI. While detailed structural analysis or genotype–phenotype correlation are beyond the scope of this publication, our observations mentioned previously are generally consistent with previous reports (e.g., Karageorgos et al., 2007a,b; Lin et al., 2008; Saito, Ohno, Sugawara, & Sakuraba, 2008).
Figure 4.
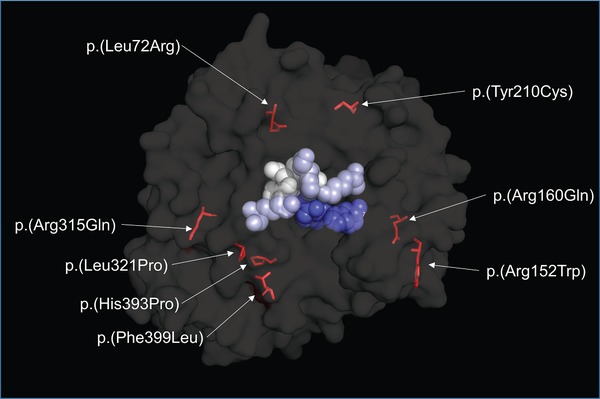
ASB structure and variants of special interest
Three‐dimensional structure of mature human ASB protein overlaid with variants of interest. The enzyme active site pocket is indicated by colored space‐filling models
c.215T>G [p.(Leu72Arg)] is associated with early onset and classical, rapidly progressing disease, consistent with a lack of detectable ASB protein and enzymatic activity when expressed in cultured fibroblasts (Karageorgos et al., 2004, 2007a,b; Petry et al., 2005). Leu72 resides in the 3′ coil of ASB, and a leucine‐to‐arginine change is predicted to disrupt formation of this coil and result in severe disturbance of ASB structure (Karageorgos et al., 2007a). Of note, Leu72 is conserved in certain other sulfatases, including galactose‐3‐sulfatase (ASA) and N‐acetylgalactosamine‐6‐sulfatase (GALNS) (Bond et al., 1997).
c.454C>T [p.(Arg152Trp)] was the most frequently reported allele in our sample (62/916; 6.8%). p.(Arg152Trp) was detected in several distinct ethnic populations, including individuals from Turkey, Russia, Northern Europe, Western Europe, and Eastern Europe, where specifically it is thought to be a founder mutation (Chistiakov et al., 2014; Jurecka et al., 2012a). p.(Arg152Trp) has been identified in both heterozygous and homozygous states, and is associated with non‐classical, slowly progressing phenotypes in homozygotes, with some patients living into their thirties (Brands et al., 2013a; Chistiakov et al., 2014; Ferla et al., 2015; Jurecka et al., 2011b, 2012a; Karageorgos et al., 2007b; Thümler et al., 2012). Interestingly, p.(Arg152Trp) has been associated with a primarily cardiac and musculoskeletal phenotype in certain patients, but whether this indicates a genuine genotype–phenotype correlation remains unclear (Brands et al., 2013a; Jurecka et al., 2011a). Trp152 is located near the conserved hexapeptide sequence 144‐Gly‐Lys‐Trp‐His‐Leu‐Gly‐149, the largest connecting stretch of identity between arylsulfatases (Peters et al., 1990; Voskoboeva, Isbrandt, von Figura, Krasnopolskaya, & Peters, 1994).
c.479G>A [p.(Arg160Gln)] results from a transition in a CpG dinucleotide (Litjens & Hopwood, 2001). p.(Arg160Gln) has been observed in both heterozygous and homozygous states (Kantaputra et al., 2014; Mathew et al., 2015; Villani et al., 2010; Voskoboeva et al., 1994). The mechanism by which the arginine‐to‐glutamine alteration contributes to disease is not well understood, as its effect on enzyme structure is predicted to be minimal (Mathew et al., 2015). However, Arg160 immediately precedes the dipeptide sequence 161‐Gly‐Phe‐162, which is conserved in arylsulfatases (Peters et al., 1990; Voskoboeva et al., 1994). Interestingly, wild‐type arylsulfatase A (ASA) also contains a glutamine in the position immediately preceding the conserved dipeptide sequence (Peters et al., 1990).
Of note, a nonsense mutation [p.(Arg160*)] has also been identified at Arg160, and thus Arg160 has been suggested to be a mutational hot spot in the ARSB gene (Kantaputra et al., 2014; Voskoboeva et al., 1994). Further supporting this idea is the observation that Arg160 mutations have been found in diverse populations and ethnic groups, including patients of Belarusian, German, Indian, Italian, Russian, Spanish, and Taiwanese descent (Garrido et al., 2007; Jurecka et al., 2012a; Kantaputra et al., 2014; Thümler et al., 2012; Voskoboeva et al., 1994).
c.629A>G [p.(Tyr210Cys)], previously reported as the most common pathogenic variant in patients with MPS VI (Karageorgos et al., 2007b), was the third most frequently reported allele (49/916, 5.3%) in this analysis, found in 11.1% of individuals collected. Individuals with the p.(Tyr210Cys) mutation spanned several ethnic populations, including individuals from Australia and Western Europe. Previous work from Bradford, Litjens, Parkinson, Hopwood, and Brooks (2002) has demonstrated that the substitution of Tyr210 with cysteine causes impaired protein processing, trafficking, and instability of the mutant ASB protein, ultimately leading to decreased levels of mature, functional protein in lysosomes. Tyr210 is located between the sixth beta‐strand and fifth alpha‐helix of ASB (Saito et al., 2008). The tyrosine‐to‐cysteine change is predicted to result in a relatively stable enzyme which should retain some enzymatic activity (Saito et al., 2008). As such, patients with the p.(Tyr210Cys) mutation generally exhibit a non‐classical, slowly progressing form of MPS VI, and the p.(Tyr210Cys) mutation may reduce the severity of other ARSB pathogenic variants when present in a compound heterozygous state (Bradford et al., 2002; Brands et al., 2013a; Litjens, Brooks, Peters, Gibson, & Hopwood, 1996; Saito et al., 2008). Interestingly, this allele has only been reported in a homozygous state twice (Gottwald et al., 2011; Mendelsohn et al., 2013), despite having a relatively high allele frequency reported in the Genome Aggregation Database (gnomAD; gnomad.broadinstitute.org, accessed October 26, 2017), a large population database. Both individuals had a non‐classical form of the disease (Gottwald et al., 2011; Mendelsohn et al., 2013).
c.944G>A [p.(Arg315Gln)] has been reported in both heterozygous and homozygous states (Petry et al., 2005; Villani et al., 1999). Patients with two p.(Arg315Gln) alleles demonstrate a range of clinical phenotypes, from intermediate to classically severe (Di Natale et al., 2008; Karageorgos et al., 2007b; Petry et al., 2005; Villani et al., 1999). Arg315 is predicted to form a salt bridge with Asp466, as well as hydrogen bonds with Gly448 and Glu483 (Garrido et al., 2007; Mathew et al., 2015). Thus, the arginine‐to‐glutamine alteration is predicted to hinder both substrate binding and metal ion coordination (Villani et al., 1999). Two additional mutations at Arg315 have also been reported—the missense mutation p.(Arg315Pro) and the nonsense mutation p.(Arg315*), which produces a polypeptide roughly one‐third the length of wild‐type and is predicted to lack enzymatic activity (Di Natale et al., 2008; Ferla et al., 2015; Lin et al., 2008; Mathew et al., 2015; Villani et al., 2010). Both p.(Arg315Gln) and p.(Arg315Pro) result from an alteration in the CpG dinucleotide c.944G, suggesting this nucleotide may be a mutational hot spot in ARSB (Litjens & Hopwood, 2001; Mathew et al., 2015).
c.962T>C [p.(Leu321Pro)] was the second most common allele identified in the literature search (60/916; 6.6%). p.(Leu321Pro) was found primarily (but not exclusively) in patients of Turkish origin, where it is believed to be a founder mutation (Kantaputra et al., 2014). Interestingly, the c.962T>C mutation is not found in gnomAD (gnomad.broadinstitute.org, accessed October 26, 2017), further supporting the hypothesis that this is primarily a population‐specific variant. Individuals homozygous for p.(Leu321Pro) demonstrated a range of clinical phenotypes, from a classic, rapidly progressing phenotype to a non‐classical, slowly progressing phenotype (Isbrandt et al., 1994; Kantaputra et al., 2014; Zanetti et al., 2014). Of note, Leu321 is not conserved between the arylsulfatases, and the effect of the p.(Leu321Pro) mutation on the secondary structure of ASB is not known (Isbrandt et al., 1994).
c.1197C>G [p.(Phe399Leu)] was observed solely in individuals from China and Taiwan, and was heavily represented in both populations. p.(Phe399Leu) was associated with a severe phenotype in homozygous individuals (Lin et al., 2008; Yang, Wu, Lin, & Tsai, 2001; Zheng et al., 2014). One report suggests that the p.(Phe399Leu) allele may be associated with the polymorphisms c.1072G>A and c.1191G>A, and that these alleles may share a common origin (Lin et al., 2008).
3.5. Homozygosity
Homozygote subjects are particularly represented in the analyzed population of MPS VI patients, 55% of all reported cases. Analysis of homozygotes may be useful for the in vivo genotype–phenotype correlation and also for the comprehension of the geographical distribution of the pathogenic variants. This analysis has identified 55 variants which were present in a homozygote condition only once; several other variants were present in the examined population 2–5 times. Only eight variants were present six or more times in homozygosis. Interestingly, these variants represent a particular geographical/ethnographical distribution, each being mostly present in only one ethnic group (Supp. Mat. S4). Of the 30 homozygote subjects with the variant p.(Leu321Pro), 25 are of Turkish background. All 21 homozygote subjects with the p.(Tyr251*) variant are Arabic, and 11 of the 16 homozygote subjects with the p.(Arg152Trp) variant are from Russia or from Baltic countries. Lastly, all 14 homozygote subjects with the variant p.(His178Leu) are Brazilian, and belong to a cluster of MPS VI patients living in Northeast Brazil. These subjects all show the same haplotype (Costa‐Motta et al., 2014), which is likely due to a founder effect, with the contemporary presence of parental consanguinity and endogamy.
Our analysis of the genotype–phenotype correlation based on homozygotes was poorly informative for most variants, except p.(Tyr251*), p.(Arg152Trp), and p.(Cys192Arg). Variant p.(Tyr251*) was associated with a severe phenotype in most subjects (17/21), p.(Arg152Trp) was associated with an attenuated phenotype in almost all subjects (14/15), and p.(Cys192Arg) was associated with an attenuated phenotype in half of the subjects reported (3/6).
3.6. Conflicting issues
Two polymorphisms, or benign variants erroneously reported as disease‐causing variants, were collected in this review, despite only collecting variants reported as linked to disease. c.1151G>A [p.(Ser384Asn)] and c.98C>T [p.(Ala33Val)] are now known to be benign. These variants are highlighted in blue in Supp. Mat. S3, and also reported in Supp. Mat. S2. Moreover, a list of variants in ARSB with frequency>1% in gnomAD (gnomad.broadinstiture.org, accessed October 26, 2017) is reported in Supp. Mat. S5. As for variant c.1151G>A [p.(Ser384Asn)], it has been determined to not be a disease‐causing variant a few years ago (Zanetti et al., 2009), but it had been reported as such in nine alleles in data collected from previously published literature.
A re‐analysis of molecular data of the entire gene in those patients with only one allele identified, or with one allele carrying a known polymorphism, should be strongly encouraged, searching for potential missed variants such as large deletions, intronic or promotorial variants. On the contrary, all the synonymous variants encountered in our analysis [c.246G>A, p.(Leu82Leu); p.(Ile114Ile); p.(Leu124Leu); c.1515C>T, p.(Tyr505Tyr)] were considered polymorphisms. Actually these variants should be considered potentially pathogenetic, given their absence from gnomAD database, with the exception of p.(Leu82Leu) which has a frequency of 0.003858%. In fact, despite their name, synonymous or silent variants are no longer considered “neutral” variants with no functional consequences on protein, but are considered variants that may affect the function of translated protein through different cellular mechanisms (Hunt, Simhadri, Iandoli, Sauna, & Kimchi‐Sarfaty, 2014). However, not enough information is available from the literature for the classification of these ARSB synonymous variants according to ACMG criteria, therefore in this study they are reported as with “not enough evidence.” Moreover, an underestimation of this kind of nucleotide substitution in our study has to be taken into account and, in light of this fact, a re‐evaluation of those patients missing the identification of one (or both) variant(s) or with one allele (or both alleles) carrying only a polymorphism should be performed, as mentioned above.
3.7. ACMG classification of ARSB variants
A classification of ARSB sequence variants was performed according to ACMG criteria. This analysis evidenced that, for most variants (about 45%), there is not enough evidence in the literature for their classification, and thus they are variants of “uncertain significance.” The remaining variants were classified in “likely pathogenic” (37%) and “pathogenic” (16%). One variant (c.113_121del), despite having a frequency < 1% (0.1719%) resulted “benign” using the ACMG approach. Hence, this in‐frame deletion seems to be a polymorphism, if we consider only the South Asian population in which it has a frequency of 1.37%.
These results provide evidence for the need of a deeper analysis of the variants diagnosed in MPS VI patients: this should include both in vitro and in silico analysis. Moreover, specific attention has to be paid to the annotation of the variants: the use of the most updated gene variant nomenclature suggested by HGVS (v15.11) is recommended so as to use shared rules that could unequivocally describe a specific variant with no risk of misreporting.
4. CLINICAL RELEVANCE
4.1. Diagnostic strategies and complications
Mucopolysaccharidoses, including MPS VI, are generally diagnosed by a combination of clinical findings and laboratory tests. Often, the first biochemical test performed upon clinical suspicion of MPS is the analysis of urinary glycosaminoglycans (uGAGs). While commonly used, a positive uGAG test is not sufficient for an MPS diagnosis, and a negative uGAG test is not sufficient to rule out a diagnosis of MPS (Cobos, Steglich, Santer, Lukacs, & Gal, 2015; Vairo et al., 2015). Additionally, individuals with non‐classical MPS VI and/or older individuals with MPS VI may not show strong uGAG elevation (Wood et al., 2012). While tandem mass spectrometry‐based (MS/MS) testing of uGAGs appears to have increased sensitivity in these individuals, the MS/MS format of the uGAG test is not widely available. Due to the lack of MS/MS availability and the potential to misdiagnose MPS, a lysosomal enzyme panel is often preferable for MPS screening (Wood et al., 2012).
When clinical suspicion for MPS is high (or after a finding of elevated uGAGs), individuals undergo testing for lysosomal enzyme activity via fibroblasts, leukocytes, or dried blood spots (DBS), which are commonly used due to their low cost, ease of collection, and simple storage requirements (Cobos et al., 2015; Vairo et al., 2015; Wood et al., 2012). Although enzyme activity varies among individuals (e.g., due to the specific ARSB variant, assay method, etc.), most individuals with MPS VI demonstrate ASB activity <10% of the lower limit of normal level across the disease spectrum (Valayannopoulos et al., 2010).
A confirmation of MPS VI diagnosis does require the specific pathogenic variants in ARSB to be determined. Moreover, variant identification is beneficial to individuals with MPS VI and their relatives as it can render genetic counseling possible and potentially inform treatment decisions. Although most genotype–phenotype correlations remain uncertain in MPS VI, some variants (e.g., large deletions, nonsense mutations, frameshift, and certain missense mutations) do appear to be correlated with a classic, rapidly progressing phenotype (Karageorgos et al., 2007b).
Importantly, delays in diagnosis of MPS are quite common, especially in patients with non‐classical patterns of disease. These patients may present later in life without the typical physical features of MPS, resulting in a low‐clinical suspicion of disease and a delay to diagnosis (Fernández‐Marmiesse et al., 2014; Vairo et al., 2015). In addition, clinical features of many MPS overlap, making a specific diagnosis difficult based on clinical presentation alone. Finally, some attenuated patients may display a normal result on a urine GAG screen despite having disease‐causing variants and clinical symptoms of MPS (Fernández‐Marmiesse et al., 2014). This diagnostic delay is particularly troubling in MPS disorders where treatment options are available (as for MPS VI), as certain clinical manifestations may not be reversible once they arise.
4.2. Treatment options
The primary goal of therapy in individuals with LSDs is to preserve or restore function by removing excess material accumulated in the lysosome and/or to prevent future buildup of substrate. While treatment options for certain LSDs are still limited, therapeutic options for MPS have advanced significantly over the past decade. Enzyme replacement therapy (ERT) is now available for four MPS disorders (MPS I, MPS II, MPS IVA, and MPS VI), and hematopoietic stem cell transplantation has demonstrated effectiveness in some patients (Biffi, 2017; Vairo et al., 2015; Valayannopoulos, 2013). In addition, novel therapeutic approaches are currently being studied for LSDs including gene therapy, substrate reduction therapy, and chemical chaperone therapy (Macauley, 2016; Rastall & Amalfitano, 2015; Valayannopoulos, 2013).
Since initial U.S. approval in 2005, ERT is available for patients with MPS VI in many countries (Naglazyme, BioMarin Pharmaceutical Inc., 2013). Galsulfase, a purified human enzyme, has been shown to improve walking capacity, stair‐climbing capacity, pulmonary function, hepatosplenomegaly, cardiac function, and survival in individuals with MPS VI (Giugliani et al., 2014; Harmatz et al., 2006, 2008, 2010, 2014; Naglazyme, BioMarin Pharmaceutical Inc., 2013; Quartel et al., 2018). As multiple studies have demonstrated that early intervention with ERT (before irreversible complications arise) is beneficial (Furujo et al., 2011; McGill et al., 2010; Meikle & Hopwood, 2003; Muenzer, 2014; Wang et al., 2011), it is vital that diagnosis be made swiftly and accurately in order to maximize the therapeutic potential of ERT.
5. FUTURE PROSPECTS
One potential remedy to delayed diagnosis is the wider adaptation of newborn screening, particularly in regions where MPS disorders are common, or prenatal screening when a family history of MPS is known (Wang et al., 2011). In fact, pilot programs of this nature are already underway in many regions of the world, including parts of Austria, Brazil, Mexico, Spain, Taiwan, and the United States, several of which have proven successful at identifying lysosomal storage disorders (including MPS) in newborns prior to the onset of clinical symptoms (Acosta, Abe‐Sandes, Giugliani, & Bittles, 2013; Colón et al., 2017; Hopkins et al., 2015; Lin et al., 2013; Mechtler et al., 2012; Navarrete‐Martínez et al., 2017). While these types of screening programs can be somewhat costly and time‐consuming, the systematic reporting and classification of alleles has the potential to reduce ethical complications in cases where novel variants with unknown pathogenicity are identified (Wang et al., 2011).
Another appealing option for diagnosis of MPS is next‐generation sequencing (NGS), a platform capable of performing the sequencing of multiple genes in a single reaction. NGS is currently the method of choice for gene panels, whole exome, and whole genome analyses, and is particularly powerful for pathogenic variant identification in disorders such as MPS VI which are rare, clinically heterogeneous, and demonstrate a large proportion of private mutations (Lohmann & Klein, 2014). For example, whole exome sequencing (WES) was recently demonstrated to successfully diagnose a symptomatic patient with MPS IIIB, revealing compound heterozygous mutations, one of which was novel (Zeng et al., 2017). Although NGS is more expensive than other commonly used diagnostic techniques, the price has steadily decreased since the advent of NGS and will likely continue to do so, potentially allowing for more widespread use of NGS in practice (Lohmann & Klein 2014; Vairo et al., 2015). In addition, a genetic diagnosis allows for genetic counseling and rapid treatment initiation, both of which are vital for patients with MPS and their families (Fernández‐Marmiesse et al., 2014). Of note, while NGS is promising, this technique could fail to detect certain pathogenic variants, including complete gene deletions and genetic inversions or rearrangements, potentially leading to false negatives in some cases (Vairo et al., 2015).
Finally, increased reporting of pathogenic variants in ARSB, specifically in publicly available databases, coupled with a clinical description and other patient information may help to shorten the time for diagnosis, prevent misdiagnosis, and assist in understanding genotype–phenotype interactions where present. At present, variants can be submitted to, and reviewed at, the clinically‐focused, public database ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/intro/). Additionally, variants associated with MPS VI can be submitted to the locus‐specific database (http://mps6-database.org) by email to Hitoshi Sakuraba (sakuraba@my-pharm.ac.jp). These considerations are especially important in MPS VI due to the severity of the disease, broad clinical spectrum, and high level of genetic diversity displayed in individuals with MPS VI, as well as the availability of ERT for these patients.
Supporting information
Supporting information
Supporting information
Supporting information
ACKNOWLEDGMENTS
Medical writing support was provided by Stacie Dilks, PhD, of Health Interactions, Inc. and funded by BioMarin Pharmaceutical Inc. Additional editorial support was provided by Sara Hawley, MS, and Christine Ly, PharmD, of BioMarin Pharmaceutical Inc. The University of Padova (Italy) has provided support to R. Tomanin and A. Zanetti within the Strategic Project “BIOINFOGEN” (Bioinformatics for Personal Genomics).
DISCLOSURE STATEMENT
R. Tomanin, L. Karageorgos, A. Zanetti, and J.J. Hopwood have no conflicts of interest to declare. H. Sakuraba reports grants and personal fees from Sumitomo Dainippon, Co., Ltd. outside of the submitted work and grants from Shire Japan, Co. outside of the submitted work. M. AlSayed has received honorarium and travel reimbursement from BioMarin Pharmaceutical Inc., Shire, and Sanofi Genzyme. M. Bailey and N. Miller are employees and stockholders of BioMarin Pharmaceutical Inc.
Tomanin R, Karageorgos L, Zanetti A, et al. Mucopolysaccharidosis type VI (MPS VI) and molecular analysis: Review and classification of published variants in the ARSB gene. Human Mutation. 2018;39:1788–1802. 10.1002/humu.23613
Communicated by Johannes Zschocke
REFERENCES
- Acosta, A. X. , Abe‐Sandes, K. , Giugliani, R. , & Bittles, A. H. (2013). Delivering genetic education and genetic counseling for rare diseases in rural Brazil. Journal of Genetic Counseling, 22(6), 830–834. [DOI] [PubMed] [Google Scholar]
- Baehner, F. , Schmiedeskamp, C. , Krummenauer, F. , Miebach, E. , Bajbouj, M. , Whybra, C. , … Beck, M. (2005). Cumulative incidence rates of the mucopolysaccharidoses in Germany. Journal of Inherited Metabolic Disease, 28(6), 1011–1017. [DOI] [PubMed] [Google Scholar]
- Biffi, A. (2017). Hematopoietic stem cell gene therapy for storage disease: Current and new indications. Molecular Therapy, 25(5), 1155–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond, C. S. , Clements, P. R. , Ashby, S. J. , Collyer, C. A. , Harrop, S. J. , Hopwood, J. J. , & Guss, J. M. (1997). Structure of a human lysosomal sulfatase. Structure, 5(2), 277–389. [DOI] [PubMed] [Google Scholar]
- Bradford, T. M. , Litjens, T. , Parkinson, E. J. , Hopwood, J. J. , & Brooks, D. A. (2002). Mucopolysaccharidosis type VI (Maroteaux–Lamy syndrome): A Y210C mutation causes either altered protein handling or altered protein function of N‐acetylgalatosamine 4‐sulfatase at multiple points in the vacuolar network. Biochemistry, 41(15), 4962–4971. [DOI] [PubMed] [Google Scholar]
- Brands, M. M. , Hoogeveen‐Westerveld, M. , Kroos, M. A. , Nobel, W. , Ruijter, G. J. , Özkan, L. , … Reuser, A. J. (2013a). Mucopolysaccharidosis type VI phenotypes‐genotypes and antibody response to galsulfase. Orphanet Journal of Rare Diseases, 8(1), 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks, D. A. , Robertson, D. A. , Bindloss, C. , Litjens, T. , Anson, D. S. , Peters, C. , … Hopwood, J. J. (1995). Two site‐directed mutations abrogate enzyme activity but have different effects on the conformation and cellular content of the N‐acetylgalactosamine‐ 4‐sulphatase protein. Biochemical Journal, 307, 457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chistiakov, D. A. , Savost'anov, K. V. , Kuzenkova, L. M. , Gevorkyan, A. K. , Pushkov, A. A. , Nikitin, A. G. , … Baranov, A. A. (2014). Molecular characteristics of patients with glycosaminoglycan storage disorders in Russia. Clinica Chimica Acta, 436, 112–120. [DOI] [PubMed] [Google Scholar]
- Cobos, P. N. , Steglich, C. , Santer, R. , Lukacs, Z. , & Gal, A. (2015). Dried blood spots allow targeted screening to diagnose mucopolysaccharidosis and mucolipidosis. JIMD Reports, 15, 123–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colón, C. , Alvarez, J. V. , Castaño, C. , Gutierrez‐Solana, L. G. , Marquez, A. M. , O'Callaghan, M. , … Couce, M. L. (2017). A selective screening program for the early detection of mucopolysaccharidosis: Results of the FIND project – a 2‐year follow‐up study. Medicine, 96(19), e6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa‐Motta, F. M. , Acosta, A. X. , Abé‐Sandes, K. , Bender, F. , Schwartz, I. V. , Giugliani, R. , & Leistner‐Segal, S. (2011). Genetic studies in a cluster of mucopolysaccharidosis type VI patients in Northeast Brazil. Molecular Genetics and Metabolism, 104(4), 603–607. [DOI] [PubMed] [Google Scholar]
- Costa‐Motta, F. M. , Bender, F. , Acosta, A. , Abé‐Sandes, K. , Machado, T. , Bomfim, T. , … Leistner‐Segal, S. (2014). A community‐based study of mucopolysaccharidosis type VI in Brazil: The influence of founder effect, endogamy, and consanguinity. Human Heredity, 77(1–4), 189–196. [DOI] [PubMed] [Google Scholar]
- Di Natale, P. , Villani, G. R. D. , Parini, R. , Scarpa, M. , Parenti, G. , Pontarelli, G. , … Fiumara, A. (2008). Molecular markers for the follow‐up of enzyme‐replacement therapy in mucopolysaccharidosis type VI disease. Biotechnology and Applied Biochemistry, 49, 219–223. [DOI] [PubMed] [Google Scholar]
- Ferla, R. , Claudiani, P. , Savarese, M. , Kozarsky, K. , Parini, R. , Scarpa, M. , … Auricchio, A. (2015). Prevalence of anti‐adeno‐associated virus serotype 8 neutralizing antibodies and arylsulfatase B cross‐reactive immunologic material in mucopolysaccharidosis VI patient candidates for a gene therapy trial. Human Gene Therapy, 26(3), 145–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández‐Marmiesse, A. , Morey, M. , Pineda, M. , Eiris, J. , Couce, M. L. , Castro‐Gago, M. , … Cocho, J. A. (2014). Assessment of a targeted resequencing support tool in the diagnosis of lysosomal storage disorders. Orphanet Journal of Rare Diseases, 9(1), 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furujo, M. , Kubo, T. , Kosuga, M. , & Okuyama, T. (2011). Enzyme replacement therapy attenuates disease progression in two Japanese siblings with mucopolysaccharidosis type VI. Molecular Genetics and Metabolism, 104(4), 597–602. [DOI] [PubMed] [Google Scholar]
- Garcia, P. , Sousa, S. B. , Ling, T. P. , Conceição, M. , Seabra, J. , White, K. K. , & Diogo, L. (2010). Skeletal complications in mucopolysaccharidosis VI patients: Case reports. Journal of Pediatric Rehabilitation Medicine, 3(1), 63–69. [DOI] [PubMed] [Google Scholar]
- Garrido, E. , Chabás, A. , Coll, M. J. , Blanco, M. , Domínguez, C. , Grinberg, D. , … Cormand, B. (2007). Identification of molecular defects in Spanish and Argentinian mucopolysaccharidosis VI (Maroteaux–Lamy syndrome) patients, including 9 novel mutations. Molecular Genetics and Metabolism, 92(1‐2), 122–130. [DOI] [PubMed] [Google Scholar]
- Golda, A. , Jurecka, A. , & Tylki‐Szymanska, A. (2012). Cardiovascular manifestations of mucopolysaccharidosis type VI (Maroteaux–Lamy syndrome). International Journal of Cardiology, 158(1), 6–11. [DOI] [PubMed] [Google Scholar]
- Gottwald, I. , Hughes, J. , Stewart, F. , Tylee, K. , Church, H. , & Jones, S. A. (2011). Attenuated mucopolysaccharidosis type VI (Maroteau‐Lamy syndrome) due to homozygosity for the p.Tyr201Cys mutation in the ARSB gene. Molecular Genetics and Metabolism, 103(3), 300–302. [DOI] [PubMed] [Google Scholar]
- Giugliani, R. , Lampe, C. , Guffon, N. , Ketteridge, D. , Leão‐Teles, E. , Wraith, J. E. , … Harmatz, P. (2014). Natural history and galsulfase treatment in mucopolysaccharidosis VI (MPS VI, Maroteaux–Lamy syndrome) – 10‐year follow‐up of patients who previously participated in an MPS VI Survey Study. American Journal of Medical Genetics Part A, 164A(8), 1953–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmatz, P. , Giugliani, R. , Schwartz, I. , Guffon, N. , Teles, E. L. , Miranda, M. C. , … Swiedler, S. J. (2006). Enzyme replacement therapy for mucopolysaccharidosis VI: A phase 3, randomized, double‐blind, placebo‐controlled, multinational study of recombinant human N‐acetylgalactosamine 4‐sulfatase (recombinant human arylsulfatase B or rhASB) and follow‐on, open‐label extension study. Journal of Pediatrics, 148(4), 533–539. [DOI] [PubMed] [Google Scholar]
- Harmatz, P. , Giugliani, R. , Schwartz, I. V. , Guffon, N. , Teles, E. L. , Miranda, M. C. , … Decker, C. (2008). Long‐term follow‐up of endurance and safety outcomes during enzyme replacement therapy for mucopolysaccharidosis VI: Final results of three clinical studies of recombinant human N‐acetylgalactosamine 4‐sulfatase. Molecular Genetics and Metabolism, 94(4), 469–475. [DOI] [PubMed] [Google Scholar]
- Harmatz, P. , Yu, Z. F. , Giugliani, R. , Schwartz, I. V. D. , Guffon, N. , Teles, E. L. , … Decker, C. (2010). Enzyme replacement therapy for mucopolysaccharidosis VI: Evaluation of long‐term pulmonary function in patients treated with recombinant human N‐acetylgalactosamine 4‐sulfatase. Journal of Inherited Metabolic Disease, 33(1), 51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmatz, P. , Garcia, P. , Guffon, N. , Randolph, L. M. , Shediac, R. , Braunlin, E. , … Decker, C. (2014). Galsulfase (Naglazyme®) therapy in infants with mucopolysaccharidosis VI. Journal of Inherited Metabolic Disease, 37(2), 277–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart, R. , Blyth, B. , Chiu, T. , Fusaro, V. , Garcia, J. , Hare, E. , … Westbrook, J. (2014). CLINVITAE: An open database of clinically observed variants, and other open source tools from Invitae. Poster Presented at Pacific Symposium on Biocomputing. Hawaii, USA.
- Hopkins, P. V. , Campbell, C. , Klug, T. , Rogers, S. , Raburn‐Miller, J. , & Kiesling, J. (2015). Lysosomal storage disorder screening implementation: Findings from the first six months of full population pilot testing in Missouri. Journal of Pediatrics, 166(1), 172–177. [DOI] [PubMed] [Google Scholar]
- Hunt, R. C. , Simhadri, V. L. , Iandoli, M. , Sauna, Z. E. , & Kimchi‐Sarfaty, C. (2014). Exposing synonymous mutations. Trends in Genetics, 30(7), 308–321. [DOI] [PubMed] [Google Scholar]
- Isbrandt, D. , Arlt, G. , Brooks, D. A. , Hopwood, J. J. , von Figura, K. , & Peters, C. (1994). Mucopolysaccharidosis VI (Maroteaux–Lamy syndrome): Six unique arylsulfatase B gene alleles causing variable disease phenotypes. American Journal of Human Genetics, 54(3), 454–463. [PMC free article] [PubMed] [Google Scholar]
- Ittiwut, C. , Boonbuamas, S. , Srichomthong, C. , Ittiwut, R. , Suphapeetiporn, K. , & Shotelersuk, V. (2017). Novel mutations, including a large deletion in the ARSB gene, causing mucopolysaccharidosis type VI. Genetic Testing and Molecular Biomarkers, 21(1), 58–62. [DOI] [PubMed] [Google Scholar]
- Jurecka, A. , Golda, A. , Opoka‐Winiarska, V. , Piotrowska, E. , & Tylki‐Szymańska, A. (2011a). Mucopolysaccharidosis type VI (Maroteaux–Lamy syndrome) with a predominantly cardiac phenotype. Molecular Genetics and Metabolism, 104(4), 695–699. [DOI] [PubMed] [Google Scholar]
- Jurecka, A. , Rozdzynska, A. , Marucha, J. , Czartoryska, B. , Wegrzyn, G. , & Tylki‐Szymanska, A. (2011b). Natural history of Polish patients with mucopolysaccharidosis type VI. Central European Journal of Medicine, 6(2), 163–171. [Google Scholar]
- Jurecka, A. , Piotrowska, E. , Cimbalistiene, L. , Gusina, N. , Sobczyńska, A. , Czartoryska, B. , … Tylki‐Szymańska, A. (2012a). Molecular analysis of mucopolysaccharidosis type VI in Poland, Belarus, Lithuania and Estonia. Molecular Genetics and Metabolism, 105(2), 237–243. [DOI] [PubMed] [Google Scholar]
- Jurecka, A. , Zakharova, E. , Cimbalistiene, L. , Gusina, N. , Malinova, V. , Różdżyńska‐Świątkowska, A. , … Tylki‐Szymańska, A. (2014b). Mucopolysaccharidosis type VI in Russia, Kazakhstan, and Central and Eastern Europe. Pediatrics International, 56(4), 520–525. [DOI] [PubMed] [Google Scholar]
- Jurecka, A. , Ługowska, A. , Golda, A. , Czartoryska, B. , & Tylki‐Szymańska, A. (2015). Prevalence rates of mucopolysaccharidoses in Poland. Journal of Applied Genetics, 56(2), 205–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantaputra, P. K. , Kayserili, H. , Guven, Y. , Kantaputra, W. , Balci, M. C. , Tanpaiboon, P. , … Dalal, A. (2014). Clinical manifestations of 17 patients affected with mucopolysaccharidosis type VI and eight novel ARSB mutations. American Journal of Medical Genetics Part A, 164A(6), 1443–1453. [DOI] [PubMed] [Google Scholar]
- Karageorgos, L. , Harmatz, P. , Simon, J. , Pollard, A. , Clements, P. R. , Brooks, D. A. , & Hopwood, J. J. (2004). Mutational analysis of mucopolysaccharidosis type VI patients undergoing a trial of enzyme replacement therapy. Human Mutation, 23(3), 229–233. [DOI] [PubMed] [Google Scholar]
- Karageorgos, L. , Brooks, D. A. , Harmatz, P. , Ketteridge, D. , Pollard, A. , Melville, E. L. , … Hopwood, J. J. (2007a). Mutational analysis of mucopolysaccharidosis type VI patients undergoing a phase II trial of enzyme replacement therapy. Molecular Genetics and Metabolism, 90(2), 164–170. [DOI] [PubMed] [Google Scholar]
- Karageorgos, L. , Brooks, D. A. , Pollard, A. , Melville, E. L. , Hein, L. K. , Clements, P. R. , … Hopwood, J. J. (2007b). Mutational analysis of 105 mucopolysaccharidosis type VI patients. Human Mutation, 28(9), 897–903. [DOI] [PubMed] [Google Scholar]
- Koc, I. (2008). Prevalence and sociodemographic correlates of consanguineous marriages in Turkey. Journal of Biosocial Science, 40(1), 137–148. [DOI] [PubMed] [Google Scholar]
- Lin, S. P. , Lin, H. Y. , Wang, T. J. , Chang, C. Y. , Lin, C. H. , Huang, S. F. , … Chuang, C. K. (2013). A pilot newborn screening program for mucopolysacchardiosis type I in Taiwan. Orphanet Journal of Rare Diseases, 8(1), 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, W. D. , Ke, Y. Y. , Chou, I. C. , Wang, C. H. , & Tsai, F. J. (2015). Deletion of exon 4 in the N‐acetylgalactosamine‐4‐sulfatase gene in a Taiwanese patient with mucopolysaccharidosis type VI. Tohoku Journal of Experimental Medicine, 235(4), 267–273. [DOI] [PubMed] [Google Scholar]
- Lin, W. E. , Lin, S. P. , Wang, C. H. , Hwu, W. L. , Chuang, C. K. , Lin, S. J. , … Tsai, F. J. (2008). Genetic analysis of mucopolysaccharidosis type VI in Taiwanese patients. Clinica Chimica Acta, 394(1–2), 89–93. [DOI] [PubMed] [Google Scholar]
- Litjens, T. , Baker, E. G. , Beckmann, K. R. , Morris, C. P. , Hopwood, J. J. , & Callen, D. F. (1989). Chromosomal localization of ARSB, the gene for human N‐acetylfalactosamine‐4‐sulfatase. Human Genetics, 82(1), 67–68. [DOI] [PubMed] [Google Scholar]
- Litjens, T. , Brooks, D. A. , Peters, C. , Gibson, G. J. , & Hopwood, J. J. (1996). Identification, expression, and biochemical characterization of N‐acetylgalactosamine‐4‐sulfatase mutations and relationship with clinical phenotype in MPS‐VI patients. American Journal of Human Genetics, 58(6), 1127–1134. [PMC free article] [PubMed] [Google Scholar]
- Litjens, T. , & Hopwood, J. J. (2001). Mucopolysaccharidosis type VI: Structural and clinical implications of mutations in N‐acetylgalactosamine‐4‐sulfatase. Human Mutation, 18(4), 282–295. [DOI] [PubMed] [Google Scholar]
- Lohmann, K. , & Klein, C. (2014). Next generation sequencing and the future of genetic diagnosis. Neurotherapeutics, 11(4), 699–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macauley, S. L. (2016). Combination therapies for lysosomal storage diseases: A complex answer to a simple problem. Pediatric Endocrinology Reviews, 13(1), 639–648. [PMC free article] [PubMed] [Google Scholar]
- Mathew, J. , Jagadeesh, S. M. , Bhat, M. , Kumar, S. U. , Thiyagarjan, S. , & Srinivasan, S. (2015). Mutations in ARSB in MPS VI patients in India. Molecular Genetics and Metabolism Reports, 4, 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGill, J. J. , Inwood, A. C. , Coman, D. J. , Lipkie, M. L. , de Lore, D. , Sweidler, S. J. , & Hopwood, J. J. (2010). Enzyme replacement therapy for mucopolysaccharidosis VI from 8 weeks of age—A sibling control study. Clinical Genetics, 77(5), 492–498. [DOI] [PubMed] [Google Scholar]
- McGovern, M. M. , Vine, D. T. , Haskins, M. E. , & Desnick, R. J. (1982). Purification and properties of feline and human arylsulfatase B isozymes. Evidence for feline homodimeric and human monomeric structures. Journal of Biological Chemistry, 257(21), 12605–12610. [PubMed] [Google Scholar]
- Mechtler, T. P. , Stary, S. , Metz, T. F. , De Jesus, V. R. , Greber‐Platzer, S. , Pollak, A. , … Kasper, D. C. (2012). Neonatal screening for lysosomal storage disorders: Feasibility and incidence from a nationwide study in Austria. Lancet, 379, 335–341. [DOI] [PubMed] [Google Scholar]
- Meikle, P. J. , & Hopwood, J. J. (2003). Lysosomal storage disorders: Emerging therapeutic options require early diagnosis. European Journal of Pediatrics, 162, S34–S37. [DOI] [PubMed] [Google Scholar]
- Mendelsohn, N. J. , Wood, T. , Olson, R. A. , Temme, R. , Hale, S. , Zhang, H. , … White, K. K. (2013). Spondyloepiphyseal dysplasias and bilateral Legg‐Calvé‐Perthes disease: Diagnostic considerations for mucopolysaccharidoses. JIMD Reports, 11, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moammar, H. , Cheriyan, G. , Mathew, R. , & Al‐Sannaa, N. (2010). Incidence and patterns of inborn errors of metabolism in the Eastern Province of Saudi Arabia, 1983–2008. Annals of Saudi Medicine, 30(4), 271–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muenzer, J. (2014). Early initiation of enzyme replacement therapy for the mucopolysaccharidoses. Molecular Genetics and Metabolism, 111(2), 63–72. [DOI] [PubMed] [Google Scholar]
- Naglazyme (galsulfase) [package insert]. (2013). Novato, CA: BioMarin Pharmaceutical, Inc.
- Navarrete‐Martínez, J. I. , Limón‐Rojas, A. E. , Gaytán‐García, M. J. , Reyna‐Fiqueroa, J. , Wakida‐Kusunoki, G. , Delgado‐Calvillo, M. D. R. , … Cervantes‐Barragán, D. E. (2017). Newborn screening for six lysosomal storage disorders in a cohort of Mexican patients: Three‐year findings from a screening program in a closed Mexican health system. Molecular Genetics and Metabolism, 121(1), 16–21. [DOI] [PubMed] [Google Scholar]
- Nouri, N. , Nouri, N. , Aryani, O. , Kamalidehghan, B. , & Houshmand, M. (2012). Identification of a novel arylsulfatase B gene mutation in three unrelated Iranian mucopolysaccharidosis type‐VI patients with different phenotype severity. Iranian Biomedical Journal, 16(3), 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters, C. , Schmidt, B. , Rommerskirch, W. , Rupp, K. , Zühlsdorf, M. , Vingron, M. , … von Figura, K. (1990). Phylogenetic conservation of arylsulfatases. cDNA cloning and expression of human arylsulfatase B. Journal of Biological Chemistry, 265(6), 3374–3381. [PubMed] [Google Scholar]
- Petry, M. F. G. , Nonemacher, K. , Sebben, J. C. , Schwartz, I. V. D. , Azevedo, A. C. M. , Burin, M. G. , … Leistner‐Segal, S. (2005). Mucopolysaccharidosis type VI: Identification of novel mutations on the arylsulfatase B gene in South American patients. Journal of Inherited Metabolic Disease, 28(6), 1027–1034. [DOI] [PubMed] [Google Scholar]
- Quartel, A. , Harmatz, P. R. , Lampe, C. , Guffon, N. , Ketteridge, D. , Leão‐Teles, E. , … Giugliani, R. (2018). Long‐term galsulfase treatment associated with improved survival of patients with mucopolysaccharidosis VI (Maroteaux–Lamy syndrome): 15‐year follow‐up study from the survey study. Journal of Inborn Errors of Metabolism and Screening, 6, 1–6. [Google Scholar]
- Rastall, D. P. W. , & Amalfitano, A. (2015). Recent advances in gene therapy for lysosomal storage disorders. Application of Clinical Genetics, 8, 157–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito, S. , Ohno, K. , Sugawara, K. , & Sakuraba, H. (2008). Structural and clinical implications of amino acid substitution in N‐acetylgalactosamine‐4‐sulfatase: Insight into mucopolysaccharidosis type VI. Molecular Genetics and Metabolism, 93(4), 419–425. [DOI] [PubMed] [Google Scholar]
- Sandberg, S. , Deanching, M. , Hoganson, G. , Wenger, D. , & Whitley, C. (2008). Pseudo‐deficiency allele of the N‐acetylgalactosamine‐4‐sulfatase gene identified in a family with Maroteaux–Lamy syndrome (mucopolysaccharidosis type VI). Molecular Genetics and Metabolism, 93(2), 34. [Google Scholar]
- Simonaro, C. M. , & Schuchman, E. H. (1995). N‐acetylgalactosamine‐4‐sulfatase: Identification of four new mutations within the conserved sulfatase region causing mucopolysaccharidosis type VI. Biochimica et Biophysica Acta, 1272(3), 129–132. [DOI] [PubMed] [Google Scholar]
- Stenson, P. D. , Ball, E. V. , Mort, M. , Phillips, A. D. , Shiel, J. A. , Thomas, N. S. T. , … Cooper, D. N. (2003). Human Gene Mutation Database (HGMD®): 2003 update. Human Mutation, 21(6), 577–581. [DOI] [PubMed] [Google Scholar]
- Thümler, A. , Miebach, E. , Lampe, C. , Pitz, S. , Kamin, W. , Kampmann, C. , … Mengel, E. (2012). Clinical characteristics of adults with slowly progressing mucopolysaccharidosis VI: A case series. Journal of Inherited Metabolic Disease, 35(6), 1071–1079. [DOI] [PubMed] [Google Scholar]
- Uttarilli, A. , Ranganath, P. , Jain, S. J. M. N. , Prasad, K. , Sinha, A. , Verma, I. C. , … Dalal, A. B. (2015). Novel mutations of the arylsulfatase B (ARSB) gene in Indian patients with mucopolysaccharidosis type VI. Indian Journal of Medical Research, 142, 414–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vairo, F. , Federhen, A. , Baldo, G. , Riegel, M. , Burin, M. , Leistner‐Segal, S. , & Giugliani, R. (2015). Diagnostic and treatment strategies in mucopolysaccharidosis VI. Application of Clinical Genetics, 8, 245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valayannopoulos, V. , Nicely, H. , Harmatz, P. , & Turbeville, S. (2010). Mucopolysaccharidosis VI. Orphanet Journal of Rare Diseases, 5(1), 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valayannopoulos, V. (2013). Enzyme replacement therapy and substrate reduction therapy in lysosomal storage disorders with neurological expression. Handbook of Clinical Neurology, 113, 1851–1857. [DOI] [PubMed] [Google Scholar]
- Villani, G. R. D. , Balzano, N. , Vitale, D. , Saviano, M. , Pavone, V. , & Di Natale, P. (1999). Maroteaux–Lamy syndrome: Five novel mutations and their structural localization. Biochimica et Biophysica Acta, 1453(2), 185–192. [DOI] [PubMed] [Google Scholar]
- Villani, G. R. D. , Grosso, M. , Pontarelli, G. , Chierchia, A. , Sessa, R. , Sibilio, M. , … Di Natale, P. (2010). Large deletion involving exon 5 of the arylsulfatase B gene caused apparent homozygosity in a mucopolysaccharidosis type VI patient. Genetic Testing and Molecular Biomarkers, 14(1), 113–120. [DOI] [PubMed] [Google Scholar]
- Voskoboeva, E. , Isbrandt, D. , von Figura, K. , Krasnopolskaya, X. , & Peters, C. (1994). Four novel mutant alleles of the arylsulfatase B gene in two patients with intermediate form of mucopolysaccharidosis VI (Maroteaux–Lamy syndrome). Human Genetics, 93(3), 259–264. [DOI] [PubMed] [Google Scholar]
- Wang, R. Y. , Bodamer, O. A. , Watson, M. S. , & Wilcox, W. R. (2011). Lysosomal storage diseases: Diagnostic confirmation and management of presymptomatic individuals. Genetics in Medicine, 13(5), 457–484. [DOI] [PubMed] [Google Scholar]
- Wood, T. , Bodamer, O. A. , Burin, M. G. , D'Almeida, V. , Feitz, M. , Giugliani, R. , … Harmatz, P. (2012). Expert recommendations for the laboratory diagnosis of MPS VI. Molecular Genetics and Metabolism, 106(1), 73–82. [DOI] [PubMed] [Google Scholar]
- Yang, C. F. , Wu, J. Y. , Lin, S. P. , & Tsai, F. J. (2001). Mucopolysaccharidosis type VI: Report of two Taiwanese patients and identification of one novel mutation. Journal of the Formosan Medical Association, 100, 820–823. [PubMed] [Google Scholar]
- Zanetti, A. , Ferraresi, E. , Picci, L. , Filocamo, M. , Parini, R. , Rosano, C. , … Scarpa, M. (2009). Segregation analysis in a family at risk for the Maroteaux–Lamy syndrome conclusively reveals c.1151G>A (p.S384N) as to be a polymorphism. European Journal of Human Genetics, 17(9), 1160–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanetti, A. , Önenli‐Mungan, N. , Elcioglu, N. , Özbek, M. N. , Kör, D. , Lenzini, E. , … Tomanin, R. (2014). Molecular analysis of Turkish Maroteaux–Lamy patients and identification of one novel mutation in the arylsulfatase B (ARSB) gene. JIMD Reports, 14, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, Q. , Fan, Y. , Wang, L. , Huang, Z. , Gu, X. , & Yu, Y. (2017). Molecular defects identified by whole exome sequencing in a child with atypical mucopolysaccharidosis IIIB. Journal of Pediatric Endocrinology and Metabolism, 30(4), 463–469. [DOI] [PubMed] [Google Scholar]
- Zheng, J. , Huang, Y. , Zhao, X. , Sheng, H. , Cheng, J. , Zhou, Z. , … Liu, L. (2014). Analysis of clinical features and arylsulfatase B gene mutation in thirteen Chinese children with mucopolysaccharidosis type VI. Chinese Journal of Pediatrics, 52(6), 403–408. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Supporting information
Supporting information