

Abstract
Steroid refractory inflammation is an unmet medical need in the management of inflammatory diseases. Thus, mechanisms, improving steroid sensitivity and simultaneously decreasing inflammation have potential therapeutic utility. The FK506‐binding protein 51 (FKBP51) is reported to influence steroid sensitivity in mental disorders. Moreover, biochemical data highlight a connection between FKBP51 and the IKK complex. The aim of this study was to elucidate whether FKBP51 inhibition had utility in modulating steroid resistant inflammation by increasing the sensitivity of the glucocorticoid receptor (GR) signalling and simultaneously inhibiting NFκB‐driven inflammation. We have demonstrated that FKBP51 silencing in a bronchial epithelial cell line resulted in a 10‐fold increased potency for dexamethasone towards IL1beta‐induced IL6 and IL8, whilst FKBP51 over‐expression of FKBP51 reduced significantly the prednisolone sensitivity in a murine HDM‐driven pulmonary inflammation model. Immunoprecipitation experiments with anti‐FKBP51 antibodies, confirmed the presence of FKBP51 in a complex comprising Hsp90, GR and members of the IKK family. FKBP51 silencing reduced NFκB (p50/p65) nucleus translocation, resulting in reduced ICAM expression, cytokine and chemokine secretion. In conclusion, we demonstrate that FKBP51 has the potential to control inflammation in steroid insensitive patients in a steroid‐dependent and independent manner and thus may be worthy of further study as a drug target.
Keywords: FKBP51, glucocorticoid receptor, IKK, NFκB, steroid sensitivity
Introduction
Glucocorticoids are the most used anti‐inflammatory drugs in a multitude of inflammatory (autoimmune‐) diseases, such as IBD 1, multiple sclerosis 2, rheumatoid arthritis 3, lupus 4, COPD 5 or asthma 6. Steroids bind to the GRα causing it to shuttle to the nucleus, thereafter, the anti‐inflammatory properties of steroids are mediated either by trans‐activation of genes possessing a glucocorticoid receptor element (GRE), e.g. the anti‐inflammatory protein Gilz (Tsc22d) or by trans‐repression, where ligand bound‐GR interacts directly with nuclear factor‐κ B (NFκB) or activator protein‐1 (AP‐1) transcription factors, repressing their function 7. The pleiotropic mechanisms of glucocorticoids make them outstanding anti‐inflammatory drugs and able to achieve disease control in many patients. However, there are a number of patients who are relatively insensitive to normal doses and resort to the use of high doses of orally administered steroids. In many cases prolonged intake of high dose steroids do not achieve pharmacological disease control but do increase the risk for side effects 8.
Unligated GR is sequestered in the cytoplasm by a protein complex containing HSP90 and other co‐chaperones, such as the immunophilins FKBP51 and FKBP52 7. These FKBPs share approximately 70% sequence homology and have each a cis‐trans peptidyl‐prolyl isomerase domain (FK1 domain) on their N‐terminal site, one central enzyme‐dead FK2 domain and tetratricopeptide repeats (TPR) on their C‐terminus which are responsible for Hsp90‐binding 9. Although these two immunophilins are structurally similar, FKBP51 and FKBP52 have opposite effects on the steroid affinity status of the GR‐Hsp90 complex. Indeed, steroid signaling is controlled by the equilibrium balance between FKBP51 and FKBP52 10. FKBP51 is bound to the GR‐Hsp90 complex in the ligand unbound state, reducing the hormone‐binding affinity. Upon steroid binding, FKBP51 is exchanged by FKBP52, which increases the hormone‐binding affinity and directs hormone‐receptor complex shuttling into the nucleus 11, 12. Consequently, FKBP51 is transcribed as a steroid responsive gene to produce a short negative feedback loop and terminate nuclear localization and signaling 13. Studies showing that steroid efficacy/sensitivity being affected by FKBP51 over‐expression have been reported in new world primates, here, the relative steroid resistance in squirrel monkeys compared to humans was attributed to a 13‐fold higher level of FKBP51 in conjunction with lower levels of FKBP52 leading to reduced steroid binding to the GR 28. Because FKBP51 and FKBP52 are direct competitors for Hsp90 binding, an increase of FKBP51 levels may affects GR‐steroid sensitivity in two ways: (i) a shift in the FKBP51/FKBP52 binding equilibrium in favor of FKBP51 to inhibit shuttling of the GR complex into the nucleus and (ii) maintaining the Hsp90‐GR complex in a low‐affinity state for steroid binding
One of the master regulators of cellular inflammation and apoptosis is the NFκB pathway. The canonical NFκB transcription factor consists primarily of the subunits p65 (RelA) and p50 (NFκB1) 14. In the absence of an inflammatory trigger, NFκB dimers are bound to the inhibitor IκB and the complex remains in the cytoplasm. Upon stimulation, IκB becomes phosphorylated by the IKK complex, followed by ubiquitination and proteasomal degradation 14, 15. The degradation of IκB enables the p65/p50 dimer to translocate into the nucleus and activate the gene expression of many anti‐apoptotic genes, chemotactic factors (e.g. CXCL1 and CXCL2), cytokines and adhesion proteins (e.g. ICAM). The IKK complex itself consists of three IKKs, IKKα, IKKβ and IKKγ (NEMO). Within this complex, NEMO functions as a regulator of IKKα/β, which are the functional kinases, phosphorylating IκB and activating indirectly the NFκB dimer 14, 15.
Recent studies, mainly in the field of mental disorders, showed that different SNPs and epigenetic changes in the FKBP51 locus maybe linked to depression, anxiety or post‐traumatic stress disorders (PTSD) by manipulating endogenous cortisol function in the hypothalamic–pituitary–adrenal (HPA) axis 16, 17, 18. Previously, biochemical studies have highlighted an interaction between FKBP51 and the IKK complex 19, 20 but the functional consequences of this putative interaction are largely unexplored. Using an NFκB‐luciferase activity assay, these authors also demonstrated that RNAi‐mediated inhibition of FKBP51 reduced NFκB activation by ∼80% after stimulation with TNFα 19. This work contrasts to findings from Erljman et al. 21 where the authors observed diminished RelA nuclear translocation after stimulation with PMA or TNFα and an over‐expression of FKBP51, leading also to reduced inflammatory markers in HEK293 cells. Instead they claimed that FKBP52 is responsible for the activation of NFκB and its translocation into the nucleus upon activation. The objective of the present study therefore was to clarify whether the manipulation of FKBP51 expression levels/function is able to influence steroid sensitivity and can also affect NFκB activation in a steroid‐independent manner. By modulation of FKBP51 levels in vitro and in vivo we showed that steroid sensitivity is indeed affected. In addition to GR and HSP90, components of the NFκB pathway were co‐immunoprecipitated with FKBP51, consistent with effects on both pathways. Moreover, we demonstrated that genetic suppression of FKBP51 results in the inhibition of IL1beta‐driven p50/p65 nuclear translocation (i.e. NFκB pathway activation) and a subsequent reduction in the release of pro‐inflammatory cytokines.
Results
FKBP51 over‐expression leads to steroid insensitivity in an HDM‐mediated asthma model
Motivated by the clinical observations of increased FKBP51 expression being correlated by steroid insensitivity 16, 17, 18, we wanted to test whether an over‐expression of FKBP51 could result in decreased steroid sensitivity in an in vivo HDM asthma model. For this we generated FKBP51 over expressing AAVs (serotype 6.2), which were capable of selectively transducing airway epithelial cells after intra‐tracheal administration 22. FKBP51‐AAVs were instilled intra‐tracheally to HDM sensitized mice on day 4 (see Fig. 1A). On the last 2 days before sacrificing the mice to get the bronchial alveolar lavage fluid, we challenged the mice daily with 25μg HDM intratracheally to trigger an inflammatory lung response. Animals of the steroid groups received prednisolone b.i.d. on the same days. A codon‐optimized murine FKBP51 gene sequence was used for AAV‐mediated transfection allowing us to discriminate between endogenous and exogenous expressed FKBP51 RNA; no virally induced FKBP51 RNA could be detected in the sham or Stuffer‐AAV control groups comparable expression levels were achieved in experimental groups receiving the FKBP51‐AAV (Fig. 1B).
Figure 1.
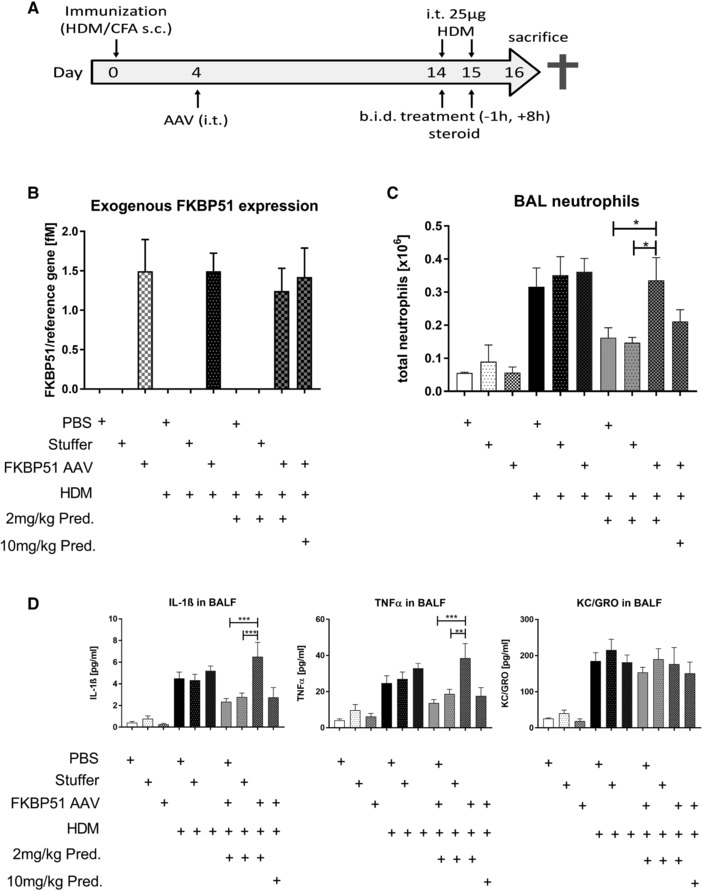
AAV‐mediated over‐expression of FKBP51 in a murine HDM asthma model. (A). Condensed outline of the conducted HDM asthma model. Detailed information is provided in the Material and Methods. Lungs were lavaged at day 16 for neutrophil and cytokine quantification. The experiment was done once with n = 4 animals per control group and n = 8 animals per HDM‐treated group (B) Gene expression levels of AAV delivered FKBP51. Total amount of exogenous FKBP51 RNA is normalized by the reference gene RNA Pol II. Units are given in femto molar (fM). (C) Total neutrophil count in BALF. Neutrophils were quantified in BALF of all treated mice on day 16. Neutrophil counting was performed automated using a Sysmex device. (D) Detection of cytokines in BALF on day 16. Cytokines were detected with exactly the same samples using a MSD Multiplex assay. (B–D) Data/statistics was calculated with n = 4 individuals for the control group and n = 8 individuals per HDM‐treated group of one animal experiment. Statistics: All data are given as means ±SEM. One‐way Annova, Dunnet post‐test, *p < 0.05, **p < 0.01, ***p < 0.001.
Because AAV (serotype 6.2) transduction was restricted to epithelial cells 22, we focused our steroid‐responsive in vivo readouts on epithelial derived or epithelial dependent markers in order to test the influence of FKBP51 over‐expression. One such readout was neutrophil cell count in bronchoalveolar fluid (BALF). In HDM untreated animals, BALF neutrophil counts were negligible. Treatment with HDM significantly increased the neutrophil cell count in all three groups (PBS, AAV‐Stuffer AAV and AAV‐FKBP51) up to 350 000 cells/BAL. AAV treatment itself did not influence neutrophil cell count versus the PBS group. Treatment with prednisolone (2 mg/kg p.o.) significantly reduced neutrophil cell count to ∼50% in the mice that were treated with PBS or the AAV‐Stuffer; however, the mice that over‐expressed FKBP51 were refractory to 2 mg/kg prednisolone and required a 5‐fold higher dose (10 mg/kg) to achieve a 37% reduction in neutrophil cell count.
Analyses of IL‐1β and TNF‐α levels in BALF were performed in the same experiment. HDM treated mice had higher levels of both cytokines. A 2 mg/kg prednisolone treatment reduced these cytokines in PBS or AAV‐Stuffer treated groups, but not in the FKBP51 over‐expressing mice (Fig. 1D). In line with our observations on neutrophil counts, upon FKBP51 over‐expression, a five‐fold higher prednisolone dose (10 mg/kg) was required to reduce IL‐1β and TNF‐α to the same extent as a 2 mg/kg dose in the PBS and AAV‐Stuffer groups. Interestingly KC, the murine equivalent to human IL‐8, was not affected by prednisolone at all, neither in the PBS or AAV‐Stuffer group nor in the FKBP51‐AAV group (Fig. 1D).
FKBP51 is highly regulated by corticosteroids in human cells
To understand the molecular mechanism of the induced steroid sensitivity that we observed in vivo we performed further experiments in human cells. First, we wanted to test whether the negative feedback loop of FKBP51 expression that is reported in neuronal cells upon treatment with corticosteroids also holds true for myeloid and epithelial cells. We isolated PBMCs from four different donors and treated them with increasing doses of dexamethasone in order to quantify FKBP51 and FKBP52 RNA levels. For FKBP51 there was a linear positive correlation between dexamethasone concentration and FKBP51 expression, whereas FKBP52 remained unchanged at baseline levels (Fig. 2A). This upregulation of FKBP51 upon dexamethasone treatment was confirmed representatively in two donors by detecting increased protein level at 24 h by western blotting (Fig 2B). Significant increases in FKBP51 expression were observed at pharmacologically relevant concentrations of dexamethasone (1–5 nM).
Figure 2.
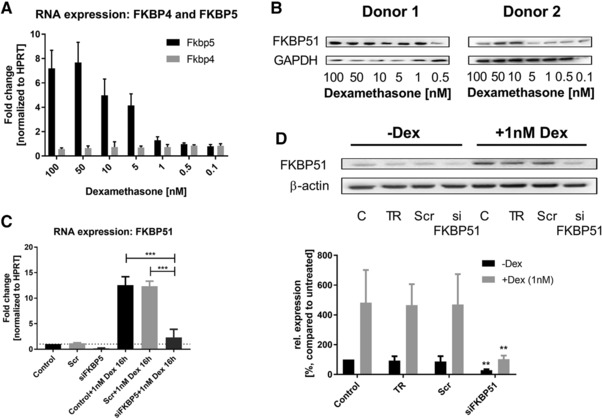
Steroid responsiveness of FKBP51 and its repression by RNAi knock down. (A) Gene expression of FKBP5 and FKBP4 in isolated PBMCs of 4 different donors (n = 4) 24 h after dexamethasone treatment. Gene expression was normalized to HPRT and referred to untreated samples. (B) FKBP51 protein expression in PBMCs of donor 1 and donor 2 (n = 2), 24h after dexamethasone treatment. Dexamethasone doses decreased half logarithmic (100, 50, 10, 5, 1, 0.5 and 0.1nM). (C) Fkbp5 RNA expression in A549 bronchial epithelial cells normalized to HPRT. Cells were transfected with a scrambled (Scr) siRNA (negative control) or a siRNA, knocking down FKBP51 (siFKBP51). Cells were treated with Dexamethasone and were then tested for Fkbp5 RNA expression. (D) FKBP51 protein expression in A549 cells. A549 cells were stimulated 16h with dexamethasone. Transfection with siRNAs was performed as described above. Samples indicated with TR, only received equal amounts of transfection reagent. Statistics: All values are represented in means ±SD of 3–4 independent experiments. (C) One‐way Annova, Tukey post‐test and (D) Two way ANOVA, Tukey post‐test; **p < 0.01, ***p < 0.001.
Similarly, A549 cells also showed a significant up‐regulation of FKBP51 mRNA (Fig. 2C) and protein (Fig. 2D) in response to 16h treatment with 1 nM dexamethasone. These cells were used for RNAi knockdown studies to further elucidate the actions of FKPB51. Treatment of A549 cells with transfection reagent (TR) alone or transfection with a scrambled siRNA (Scr) did not influence the expression of FKBP51; however, transfection with a specific pool of FKBP51‐siRNAs resulted in a FKBP51 knockdown of ∼85% under dexamethasone‐independent conditions and prevented the FKBP51 up‐regulation when treated with dexamethasone (Fig. 2C and 2D).
FKBP51 knockdown improves steroid sensitivity
Gilz (gene name: Tsc22d), a robust steroid‐responsive gene [23], was used as a marker of GR pathway activation. A 16h stimulation of A549 cells with dexamethasone resulted in a 14‐ and 11‐fold induction in the TR and Scr control groups, respectively. Upon FKBP51 knockdown, we observed a significant enhancement of the Gilz induction when compared to the TR or Scr control groups (25 and 40%, respectively; Fig. 3A). To verify that this effect translated into disease‐relevant readouts, we quantified the concentration‐response effect of dexamethasone on IL1beta‐induced levels of IL8 and IL6, with and without FKBP51 knockdown. No differences were observed between the TR and Scr groups (Fig. 3B). For both, IL8 and IL6 readouts, there was an approximately 10‐fold shift in dexamethasone sensitivity upon FKBP51 knockdown.
Figure 3.
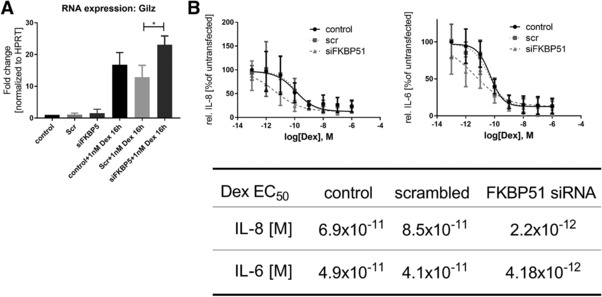
FKBP51 knock down increases steroid sensitivity. (A) RNA expression of Gilz (Tsc22d) 16 h after dexamethasone treatment in A549 cells. Cells were transfected with a scrambled (Scr) siRNA (negative control) or a siRNA, knocking down FKBP51 (siFKBP51) Gilz expression was referred to the control and normalized with HPRT. A549 cells were transfected like it is described in the Materials and methods. Data are shown as means ± SD of three independent experiments. One‐way ANOVA, Tukey post‐test; *p < 0.05. (B) Dexamethasone dose response curves. A549 cells were siRNA transfected and stimulated with decreasing amounts of dexamethasone and IL‐1β. Supernatants were used to determine IL‐8 and IL‐6 levels by ELISA. The table indicates the EC50 concentrations of dexamethasone for IL‐8 and IL‐6 reduction in the control‐, the scrambled‐ and the siFKBP51 treated group. Statistics: Fitted curves are means ± SD of three independent experiments. Extra sum‐of‐square F test to test the hypothesis for same EC50 values; EC50s are different between the siFKBP51 group and the controls for IL‐8 and IL‐6 with p < 0.0009.
FKBP51 binds to Hsp90 and to the IKK complex
One mechanistic hypothesis for how FKBP51 could cause decreased steroid sensitivity is via its binding to the glucocorticoid receptor chaperone, Hsp90. To elaborate this hypothesis, we transfected A549 cells with SBP‐tagged FKBP51 and immune‐precipitated FKBP51 and identified putative interaction partners using tryptic mass‐spectrometry. MS/MS analysis using Mascot validated Hsp90 as a FKBP51 binding partner. Peptide fragments derived from IKKα and IKKβ were also extracted from the same Coomassie‐band (Fig. 4A and Supporting Information Table 1). To further validate these MS‐based findings, we repeated the IPs with SBP‐tagged FKBP51 and Hsp90 several times using western blot readouts on the flow through (FT) and the immune‐precipitated (IP) fractions (Fig. 4B). Immunoblots for FKBP51 showed a clear signal in the FKBP51 IP‐fraction, proving the efficiency of the immune precipitation. Detection of Hsp90 in the same fraction validated the results from the MS analysis. In the MS‐analysis we were able to identify peptides for IKKα and IKKβ, but not IKKγ. Nevertheless, we used also an IKKγ detection‐antibody to test whether this is a real observation or just a false negative result due to sensitivity limitations of the mass‐spectrometry method. In fact, using an antibody, we were able to detect IKKγ together with IKKβ in the FKBP51 IP fraction.
Figure 4.
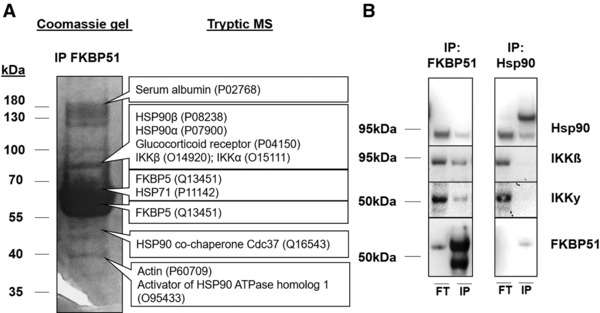
Co‐Immunoprecipitation of FKBP51 and Hsp90. (A) Mass spectrometry (MS)‐based analysis of FKBP51 co‐immunoprecipitates. A549 cells were transfected with SBP‐tagged FKBP51. IP‐fraction was used for a Coomassie gel and cropped single bands were used for tryptic based MS analysis (MS analysis was done from one IP (n = 1) and verified by four independent IPs with western blot detection). Listed are a couple of relevant proteins. A detailed table of analyzed peptides and MASCOT Scores is depicted in the Supporting Section. (B) Co‐ IP with FKBP51 and Hsp90. Depicted are representative Western Blots of four independent experiments. “FT” stands for flow through fraction, “IP” for the eluted, precipitated fraction. FT‐ and IP fractions do not contain equal amounts of loaded protein.
By performing orthogonal IPs with SBP‐tagged Hsp90 we could detect two bands for Hsp90 in the IP‐fraction, one for the tagged Hsp90 and one for the endogenous Hsp90. The detection of both the tagged and untagged HSP90 variants in the IP fraction suggests that they build homo‐dimers. We were also able to detect a band for FKBP51 in the Hsp90 IP fraction consistent with an interaction between these two proteins. IKKγ and IKKβ were not detectable in the Hsp90 IP fraction in all four repeats of these experiments.
FKBP51 regulates NFκB signaling
Nuclear translocation assays were performed for p65 and p50, following IL‐1β stimulation with and without RNAi‐mediated knockdown of FKBP51, to test whether the binding of FKBP51 to IKKs also has a direct impact on NFκB activation (Fig. 5A and B). Separation of lamin a/c as a nuclear marker and β‐tubulin as a cytoplasmic marker was clearly visible, indicating a pure separation of the two compartments was achieved. Treatment with 0.5 ng/mL IL1β increased p50 and p65 in the nucleus. Transfection with the Scr negative RNAi control did not affect p50 and p65 nucleus translocation. In contrast, inhibition of FKBP51 expression significantly reduced p50/p65 expression in the nucleus after IL‐1β stimulation (Fig 5 A and B).
Figure 5.
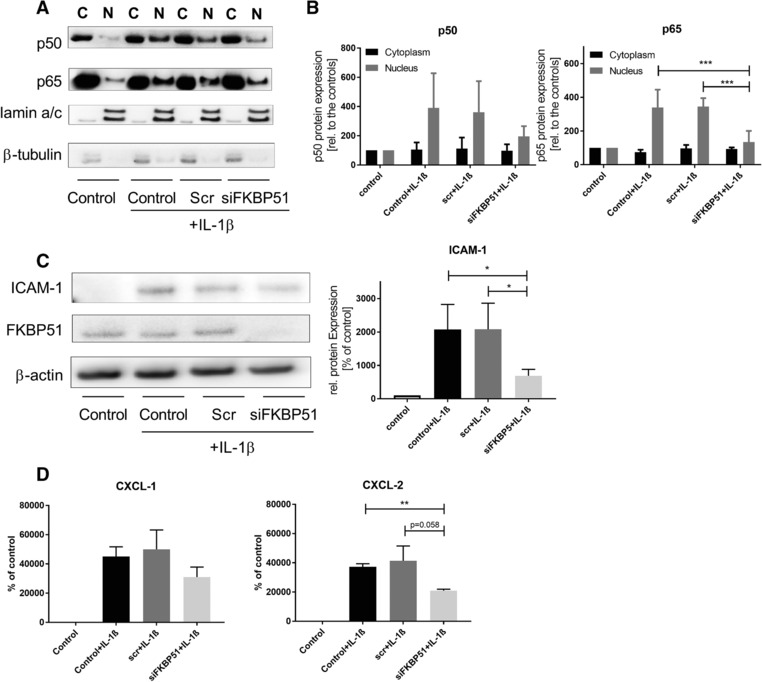
Modulation of NFκB signaling by FKBP51 knock down. (A) Nucleus translocation of p50/p65 after IL‐1ß stimulation. A549 cells were transfected with siRNAs and treated with IL‐1β. Cytoplasmic (C) and nucleic (N) fractions were separated as it is described in the material and method part. One representative western blot of three independent experiments is shown. The order of the western blot samples is the same like in the western blot quantitation of B. (B) Quantitation of p50/p65 cytoplasm and nucleus fractions. Western blots were quantified using densitometry of the single bands, normalized with the appropriate housekeeper (lamin for nucleic fractions, beta tubulin for cytoplasmic fractions). Cytoplasmic samples were relatively compared to the cytoplasmic control, nucleic samples were relatively compared to the nucleic control. Bar graphs are means ±SD of three independent experiments (n = 3). One way ANOVA, Dunnett's multiple comparisons test. *p < 0.05 C) Protein expression of ICAM‐1 in A549 cells. One representative western blot of four independent experiments is shown (n = 4). A549 cells were RNAi transfected and treated with 0.5 ng/mL IL‐1ß. Samples were relatively compared to the control. Bar graph shows mean values ± SD of all four independent experiments (n = 4). One way ANOVA, Dunnett's multiple comparisons test. *p < 0.05 D) Quantitation of NFκB mediated release of CXCL‐1/2. A549 cells were treated like described above. Supernatants were collected 16 h after stimulation and samples are compared to the control and given as relative values. Statistics: Both graphs in (D) show means ±SEM of 4 independent experiments (CXCL‐1) or 3 independent experiments (CXCL‐2). One‐way ANOVA, Tukey or Dunnet post‐test **p<0.01.
ICAM‐1, a well‐established target gene for NFκB signaling 22, was dependent on FKBP51. ICAM‐1 was not detectable in A549 cells under basal conditions; the dramatic upregulation upon IL1beta treatment was suppressed by ∼50% by the knockdown of FKBP51 (Fig. 5C). To strengthen the evidence for FKBP51s involvement in NFκB‐mediated inflammation, we expanded the analysis to additional molecules whose secretion from epithelial cells is dependent on NFκB pathway activation, CXCL‐1(GRO‐1) and CXCL‐2 (MIP‐2a). For both chemokines, there was no expression in unstimulated A549 cells and there was a significant up‐regulation on IL1beta treatment. Similar to ICAM‐1, this induction of both CXCL‐1 and CXCL‐2 was approximately halved by the knockdown of FKBP51 (Fig. 5D). These data are consistent with FKBP51 inhibition having potential for a broad anti‐inflammatory profile.
Discussion
In this work we showed that FKBP51 gene expression is induced by dexamethasone in PBMCs and the A549 bronchial epithelial cell line. This steroid‐dependent induction of FKBP51 across several cell types has previously been exploited in experiments and the clinic as a potential biomarker of steroid treatment 23, 24, 25, 26. For instance, Kelly et al. demonstrated the steroid dependent expression of FKBP51 and GILZ in bronchial biopsies of mild atopic asthmatics 23. In accordance with Kelly et al., Chun et al. showed that FKBP51 levels were increased dramatically after dexamethasone treatment in PBMCs and lung tissue from asthmatics 26. Interestingly severe asthmatics showed much higher FKBP51 levels than mild‐to moderate asthmatics 26, 27. In line with these observations, there are several studies which demonstrated an inverse correlation of FKBP51 levels with lung function improvement upon fluticasone treatment, reflecting an impaired steroid response associated with higher FKBP51 levels 24, 26, 28. This hypothesis is supported by our in vivo results, where AAV‐mediated FKBP51 over‐expression reduced the efficacy of prednisolone (Fig. 1).
Whether up‐regulation of FKBP51 is solely a physiological response to high doses of steroids or, alternatively, there is an aberrant gene regulation of FKBP51 in inflammatory diseases is unclear. There is, however, compelling evidence that (epi)genetic modifications and SNPs in the FKBP5 gene are associated with several mental disorders such as depression, bipolar disorders, PTSD or suicidal behavior 16, 17, 18. In these diseases, enhanced expression of FKBP51 seems to interfere with the regulation of the HPA axis, impairing the function of endogenous produced cortisol. If these genetic modifications also play a role in other disease populations, e.g. steroid refractory pulmonary diseases, is currently unknown and needs further evaluation. In the present studies, we attempted to address the question whether FKBP51 levels are informative as only a marker of steroid action or if FKBP51 is functionally important as a regulator of steroid action, with potential utility as a therapeutic target.
Since FKBP51 is an important regulator of GR sensitivity and function, it is a candidate target for improving steroid sensitivity in patients with impaired steroid responses; however, there are very few studies showing a direct effect on inflammation or steroid responsive genes via reducing FKBP51 levels, which for the design of new therapeutics is more informative. Here we could show that inhibition of FKBP51 protein expression leads to a higher induction of GILZ upon steroid treatment as well as improved dexamethasone‐dependent suppression of IL‐6 and IL‐8. This corroborates previous reports that FKBP51 knockout in MEFs led to a significant increase of two prominent steroid responsive genes, Sgk and Gilz, after dexamethasone treatment compared to the WT cells 10. In contrast and in agreement with our hypothesis, the knockout of FKBP52 led to the opposite effect, decreasing steroid sensitivity. Furthermore, FKBP51 knockout led to increased phosphorylation of the murine GR at Ser220 and 234, which resulted in increased GR activity. Conversely, the FKBP52 knockout increased the phosphorylation of Ser212, which in turn diminished the activity of the GR 10. Subsequently, it was shown that phosphorylation of GR at Ser 220 and 234 in the FKBP51 knockout cells is mediated by the Akt‐p38 axis, which was significantly activated in the absence of FKBP51 indicating that indirectly at least, FKBP51 can modify GR responses 29. Thus, the dissociated phosphorylation of the GR by FKBP51/FKBP52 demonstrates an additional mechanism how both immunophilins could influence steroid sensitivity. In our study we did not measure GR phosphorylation, but we could verify in pull‐down experiments with FKBP51 that both Hsp90 and the GR receptor are associated to FKBP51.
In addition to these steroid‐related binding partners, we were able to detect the three members of the IKK complex (IKKα, β, γ) in the immunoprecipitated fraction of FKBP51 which builds on the work of Bouwmeester et al. 19 and this affords a plausible explanation for the reported and potentially therapeutically attractive modulation of the NFκB pathway by FKBP51. However, to translate these FKBP51 research observations into findings that support a drug design program, it is important to understand the nature of the functional complexes responsible for the GR‐dependent and NFκB‐dependent biology and whether the IKK‐FKBP51association is Hsp90 dependent or independent fashion. In our studies, IKKs were not detected in pull‐down experiments using Hsp90 as the bait and therefore we conclude that IKKs bind directly to FKBP51 and not indirectly via the association between FKBP51 and Hsp90 and which supports inferences from earlier studies 20. We were unable to detect IKKs in the Hsp90 pull‐down experiment which is supported by the findings of Romano et al. that FKBP51 can bind to the IKK complex in an Hsp90‐independent manner. Their results suggest that FKBP51 employs its FK1 and TPR domains to stabilize the IKK complex as a scaffolding protein that acts as connector to TRAF2 upon pro‐inflammatory signals like TNFα stimulation 20. In detail, mutation of the TPR domain prevented TRAF2 and IKKα/β/γ binding and mutation of the PPIase (FK1) domain reduced IKKγ binding to FKBP51 as well. The authors concluded that FKBP51 acts as a scaffolding protein to support assembly and stabilization of the IKK complex and, in turn, promoting the phosphorylation of IκB. This previously published observation is supported by our work showing that the depletion of the FKBP51 protein via siRNA reduced p50/p65 translocation into the nucleus and reduced consequently the expression of a broad range of NFκB responsive genes/proteins like ICAM‐1, CXCL‐1 or CXCL‐2.
In this study, we characterized FKBP51 as a potential drug target and were able to show the steroid‐sensitizing and anti‐inflammatory effects of FKBP51 inhibition in a wide range of cellular systems (in mice, epithelial cells and myeloid cells). This leads us to hypothesize that FKBP51 mediated NFκB regulation is a general mechanism, ubiquitous distributed in a wide range of cells, tissues and species. We predict that antagonism of FKBP51 would simultaneously improve steroid sensitivity and reduce NFκB‐mediated inflammation (Fig. 6), which could be of help for many patients with chronic inflammation suffering from steroid insensitivity. To find advanced potent and selective inhibitors will be the challenge of the near future.
Figure 6.
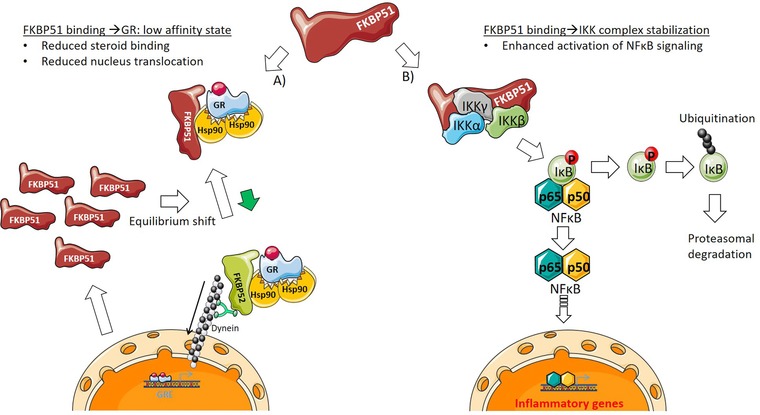
Schematic overview of FKBP51 interaction with steroid‐ and NFκB‐signaling. FKBP51 is able to interact with the Hsp90‐GRα complex as well as the IKK (α, β, γ) complex. (A) Binding to the Hsp90‐GRα complex leads to low affinity state of GR in regard to bind glucocorticoids. Consequently, this results in reduced steroid binding and a shift in the equilibrium between FKBP51/FKBP52 binding to the Hsp90‐GRα complex, which reduces GRα nucleus translocation. If a steroid still is able to bind to GRα, FKBP51 is exchanged by FKBP52, mediating the shuttling of “activated” GRα to the nucleus. As a steroid responsive gene, FKBP51 is massively generated upon GRE activation, shifting the GRα equilibrium further to the low affinity GRα state. (B) In the context of NFκB signaling, FKBP51 could function as a scaffold, stabilizing the IKK complex. Upon an inflammatory stimulus this facilitates phosphorylation of IκB and its degradation by the proteasome. Consequently, NFκB (p50/p65) is released and the associated translocation into the nucleus leads to the activation of NFκB signaling.
Materials and methods
Cell culture and treatment
A549 cells were cultured in F12 Nut Mix+GlutMax (Gibco) containing 10% FBS (Thermo Fisher Scientific). Cells were stimulated with Dexamethasone (Sigma‐Aldrich) and/or IL‐1β (Peprotech) for 16h. To knock down FKBP51, A549 cells were transfected with a pool of several FKBP51 siRNAs (siTOOLs BIOTECH, Germany) using Lipofectamine RNAiMax (Thermo Fisher Scientific). Cells were transfected reversely, according to the guidelines of siTOOLs. Readouts were usually performed 72h after transfection. PBMC were isolated from whole blood using Ficoll‐Paque (GE Healthcare) under standard conditions. Isolated PBMCs were cultured with RPMI 1640 + Glutamax containing 10% FCS. PBMCs (isolation and usage according to local ethical guidelines) cells were stimulated for 24h with decreasing concentrations of Dexamethasone to visualize FKBP51/FKBP52 response.
AAV and molecular cloning of rAAV constructs
First, the pAAV‐MCS plasmid (Agilent Technologies, catalog no. 240071) was modified to generate a self‐complementary AAV (scAAV). Therefore a synthetic piece of DNA harboring the left ITR from the pAAV‐MCS followed by MluI, NotI, NsiI, SalI and SpeI restriction sites, a CMV promoter followed by KpnI, EcoRI, PacI, BamHI and AscI restriction sites, a shortened poly adenylation signal from the simian virus 40 (short SV40 pA) followed by XbaI and BglII restricition sites and an AAV2 ITR without terminal resolution site (ITR del trs) was synthesized (Geneart, Regensburg, Germany) and cloned via the PstI restriction sites into pAAV‐MCS. The resulting plasmid was termed pAAVsc_CMV.
The expression cassette consisting of a Kozak sequence followed by the coding sequence for mouse FKBP51 (UniProt Q64378) was synthesized and at the same time codon‐usage optimized (Geneart, Regensburg, Germany) and cloned into the pAAVsc_CMV via the KpnI and BamHI restriction sites. This plasmid, which was used for production of rAAV6.2, was termed pAAVsc_CMV‐mFKBP51.
For production of rAAV6.2 vectors, the AAV6.2 cap gene (accession no. EU368910.1) was synthesized (Geneart, Regensburg, Germany) and used to replace the AAV2 cap sequence in pAAV‐RC (Agilent Technologies, Waldbronn, Germany). The resulting plasmid was termed pAAV‐Cap6.2 and co‐transfected with pAAVsc_CMV‐mFKBP51 and pHELPER (Agilent Technologies) in order to produce rAAV6.2_CMV‐mFKBP51 vectors.
AAV6.2 vectors were produced by calcium phosphate transfection of human embryonic kidney (HEK)‐293 cells using a three‐plasmid based production protocol (AAV helper free system; no. 240071; Agilent Technologies, Waldbronn, Germany) and purified as described previously 22 with minor modifications.
In vivo HDM‐AAV model
Balb/c mice (Charles River) were purchased from Charles River. The animals were housed under conventional conditions in isolated ventilated cages with free access to water and food. All experiments performed were approved by authorities for the care and use of experimental animals (Regierungspräsidium Tübingen, TVV15‐017). Mice were either treated sub cutaneous (s.c.) with CFA (Complete Freund's Adjuvant; Biomol) alone (control groups; per group n = 4) or with additional 100μg HDM (Greer)/ animal at day 0 (HDM treated groups, per group n = 8). At day 4, 1.5 × 1011 virus genomes of stuffer virus (negative control) or FKBP51‐AAVs were administered intra tracheal (i.t.) to the mice. AAV‐untreated mice received PBS i.t. as negative control. On day 14 and 15 HDM sensitized mice were re‐stimulated with 25μg HDM i.t. On the same days steroid‐treated mice got either 5 or 10 mg/kg prednisolone (Sigma‐Aldrich) b.i.d. via oral administration. Mice were sacrificed on day 16 measuring BALF neutrophils with a Sysmex XT‐1800i‐ device and BALF cytokines using the MSD technology (Meso Scale Discovery). BALF were generated by rinsing the lungs two times with PBS containing 1x cOmplete™ Protease Inhibitor Cocktail (Roche).
PCR
RNA was isolated using the mRNeasy Kit from Qiagen. 500ng RNA was transcribed in cDNA using a High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). Fkbp5 (Hs01561006_m1), Fkbp4 (Hs00427038_g1), Tsc22d (Hs00608272_m1) and Hprt1 (Hs02800695_m1) RNA were quantified with commercial TaqMan assays (Thermo Fisher Scientific) on a ViiA7 PCR device (Applied Biosystems).
ELISAs
Diluted supernatants of treated A549 cells were used for IL‐8 and IL‐6 ELISAs (BD Biosciences, OptEIA Reagent Set B). CXCL‐1 and 2 were measured using Human CXCL1/2 (GROα/MIP2) SimpleStep ELISA Kits (abcam). in vivo cytokine evaluation was performed by using a V‐Plex Pro‐inflammatory Panel 1 (Mesoscale Discovery (MSD), USA) All ELISAS were conducted like it was indicated in the manufacturer manual.
Immunoprecipitation (IP)
FKBP51 k.o. cells or wilde‐type A549 cells were transfected using the Lipofectamine 2000 transfection reagent (Thermo Fisher Scientific) and the according procedure of life technology manual. For transfection, cells were cultured in serum free media. 6h after transfection, media was exchanged with growth media containing 10% FCS. All mutated proteins were expressed using pcDNA 3.1 (+)‐A009 vectors from GeneArt (Thermo Fisher Scientific). All expressed proteins contained an N‐terminal SBP‐tag (streptavidin binding protein) to use them for IP experiments.
For immune precipitations cells were lysed by using 1% Triton X‐100/PBS containing 1x HALT protease/phosphatase buffer. To precipitate SBP tagged proteins Streptavidin coated Dynabeads (Dynabeads MyOne Streptavidin T1, Thermo Fisher Scientific) were incubated with lysates for 4h at 4°C. Not precipitated proteins were labeled as flow through (FT) fractions. After five washing steps with 0.1%BSA/PBS, Dynabeads were incubated with 4mM Biotin/PBST solution to elute precipitated proteins. The eluate was labeled as IP‐fraction and used for western blotting.
Cytoplasm/nucleus separation
To isolate cytoplasmic and nucleic fraction from treated A549 cells, cells were detached with trypsin and washed with PBS. Cell pellets were separated into cytoplasmic and nucleic fractions by using the NE‐PER Nuclear and Cytoplasmic Extraction Kit (Thermo Fisher Scientific). Separation of the two fractions was conducted like it is indicated in the manufacturer protocol. To yield best purity NER and CER buffer were used in the highest, recommended volume, containing 1x HALT Protease Inhibitor Cocktail (Thermo Fisher Scientific). Nucleus and Cytoplasm fractions were adjusted to equal amount of protein content and used for Western Blotting.
Immunoblotting
Immunoblots were done according to standard methods using novex gels and according buffers from ThermoFisher and electrophoresis devices from BioRad. All primary antibodies were ordered from Cell Signaling Technology.
In‐gel digest
The Coomassie stained bands were excised and washed in 100 mM triethylammonium bicarbonate buffer (TEAB) and acetonitrile. Disulfides were reduced with 10 mM dithiothreitol (DTT, in 100 mM TEAB) and alkylated with 55 mM iodacetamid (IAA, in 100 mM TEAB). Subsequently the gel pieces were dehydrated in acetonitrile and dried at 60°C in a vacuum concentrator plus (Eppendorf). Proteins were digested by addition of 20 ng/μL trypsin (trypsin Gold mass spec grade, Promega) dissolved in 50 mM TEAB at 37°C overnight. The tryptic peptides were extracted in 3% formic acid/acetonitrile (1:1) for 1 h. The extract was concentrated to full dryness in a vacuum concentrator and resuspended in 0.1% formic acid.
Mass spectrometric analysis
The peptides were analyzed with an Orbitrap Elite (Thermo) mass spectrometer equipped with an Agilent 1290 (Agilent) and an Aeris Widepore XB‐C8 3.6 μm, 100 · 2.1 mm (Phenomonex) column. Solvent A consisted of water and solvent B of acetonitrile, both supplemented with 0.1% formic acid, respectively. A gradient elution profile from 10% to 60% solvent B in 20 min was performed. Full MS mass spectra were recorded between 350 and 1600 m/z at 60 000 resolution and the ten most intense ions were analyzed by fragmentation. The spectra were subjected to a mascot MS/MS search (MASCOT) for identification of proteins. The search parameters were limited to human entries, with up to two missed cleavages. Carbamidomethyl was set as fixed modification and methionine oxidation as well as pyro‐glutamin at the N‐terminus as variable modification. Peptide tolerance was set to 10 ppm and MS/MS tolerance to 0.6 Da.
Conflict of interest
All authors are employees of Boehringer Ingelheim Pharma GmbH & Co. KG. This does not alter our adherence to the European Journal of Immunology policies.
Abbreviations
- AAV
adeno associated virus
- BALF
bronchoalveolar lavage fluid
- FKBP
FK506 binding protein
- Gilz
glucocorticoid induced leucine zipper
- GRE
glucocorticoid responsive element
- HDM
house dust mite
- IBD
inflammatory bowel disease
- IKK
inhibitor of κB kinase
- NFκB
nuclear factor kappa‐light‐chain‐enhancer of activated B cells
- PBMC
peripheral blood mononuclear cells
- TRAF
TNF receptor associated factor
Supporting information
Peer review correspondence
Table S1
Acknowledgments
We would like to thank Martina Keck and Kai Zuckschwerdt for their contribution to the AAV‐ HDM in vivo experiment. This work is supported by Boehringer Ingelheim Pharma GmbH & Co. KG.
References
- 1. Ford, A. C. , Bernstein, C. N. , Khan, K. J. , Abreu, M. T. , Marshall, J. K. , Talley, N. J. and Moayyedi, P. , Glucocorticosteroid therapy in inflammatory bowel disease: systematic review and meta‐analysis. Am J Gastroenterol 2011. 106: 590–599; quiz 600. [DOI] [PubMed] [Google Scholar]
- 2. Le Page, E. , Veillard, D. , Laplaud, D. A. , Hamonic, S. , Wardi, R. , Lebrun, C. , Zagnoli, F. et al., Oral versus intravenous high‐dose methylprednisolone for treatment of relapses in patients with multiple sclerosis (COPOUSEP): a randomised, controlled, double‐blind, non‐inferiority trial. Lancet 2015. 386: 974–981. [DOI] [PubMed] [Google Scholar]
- 3. Singh, J. A. , Saag, K. G. , Bridges, S. L., Jr. , Akl, E. A. , Bannuru, R. R. , Sullivan, M. C. , Vaysbrot, E. et al., 2015 American College of Rheumatology Guideline for the Treatment of Rheumatoid Arthritis. Arthritis Rheumatol 2016. 68: 1–26. [DOI] [PubMed] [Google Scholar]
- 4. Ruiz‐Arruza, I. , Barbosa, C. , Ugarte, A. and Ruiz‐Irastorza, G. , Comparison of high versus low‐medium prednisone doses for the treatment of systemic lupus erythematosus patients with high activity at diagnosis. Autoimmun Rev 2015. 14: 875–879. [DOI] [PubMed] [Google Scholar]
- 5. Snoeck‐Stroband, J. B. , Lapperre, T. S. , Sterk, P. J. , Hiemstra, P. S. , Thiadens, H. A. , Boezen, H. M. , Ten Hacken, N. H. et al., Prediction of long‐term benefits of inhaled steroids by phenotypic markers in moderate‐to‐severe COPD: a randomized controlled trial. PLoS One 2015. 10: e0143793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chung, K. F. , Wenzel, S. E. , Brozek, J. L. , Bush, A. , Castro, M. , Sterk, P. J. , Adcock, I. M. et al., International ERS/ATS guidelines on definition, evaluation and treatment of severe asthma. Eur Respir J 2014. 43: 343–373. [DOI] [PubMed] [Google Scholar]
- 7. Boardman, C. , Chachi, L. , Gavrila, A. , Keenan, C. R. , Perry, M. M. , Xia, Y. C. , Meurs, H. et al., Mechanisms of glucocorticoid action and insensitivity in airways disease. Pulm Pharmacol Ther 2014. 29: 129–143. [DOI] [PubMed] [Google Scholar]
- 8. Barnes, P. J. and Adcock, I. M. , Glucocorticoid resistance in inflammatory diseases. Lancet 2009. 373: 1905–1917. [DOI] [PubMed] [Google Scholar]
- 9. Ratajczak, T. , Cluning, C. and Ward, B. K. , Steroid receptor‐associated immunophilins: a gateway to steroid signalling. Clin Biochem Rev 2015. 36: 31–52. [PMC free article] [PubMed] [Google Scholar]
- 10. Hinds, T. D. , Stechschulte, L. A. , Elkhairi, F. and Sanchez, E. R. , Analysis of FK506, timcodar (VX‐853) and FKBP51 and FKBP52 chaperones in control of glucocorticoid receptor activity and phosphorylation. Pharmacol Res Perspect 2014. 2: e00076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mustafi, S. M. , LeMaster, D. M. and Hernandez, G. , Differential conformational dynamics in the closely homologous FK506‐binding domains of FKBP51 and FKBP52. Biochem J 2014. 461: 115–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wochnik, G. M. , Ruegg, J. , Abel, G. A. , Schmidt, U. , Holsboer, F. and Rein, T. , FK506‐binding proteins 51 and 52 differentially regulate dynein interaction and nuclear translocation of the glucocorticoid receptor in mammalian cells. J Biol Chem 2005. 280: 4609–4616. [DOI] [PubMed] [Google Scholar]
- 13. Paakinaho, V. , Makkonen, H. , Jaaskelainen, T. and Palvimo, J. J. , Glucocorticoid receptor activates poised FKBP51 locus through long‐distance interactions. Mol Endocrinol 2010. 24: 511–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Oh, H. and Ghosh, S. , NF‐kappaB: roles and regulation in different CD4(+) T‐cell subsets. Immunol Rev 2013. 252: 41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Clark, K. , Nanda, S. and Cohen, P. , Molecular control of the NEMO family of ubiquitin‐binding proteins. Nat Rev Mol Cell Biol 2013. 14: 673–685. [DOI] [PubMed] [Google Scholar]
- 16. Klengel, T. , Mehta, D. , Anacker, C. , Rex‐Haffner, M. , Pruessner, J. C. , Pariante, C. M. , Pace, T. W. et al., Allele‐specific FKBP5 DNA demethylation mediates gene‐childhood trauma interactions. Nat Neurosci 2013. 16: 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Menke, A. , Klengel, T. , Rubel, J. , Bruckl, T. , Pfister, H. , Lucae, S. , Uhr, M. et al., Genetic variation in FKBP5 associated with the extent of stress hormone dysregulation in major depression. Genes Brain Behav 2013. 12: 289–296. [DOI] [PubMed] [Google Scholar]
- 18. Scheuer, S. , Ising, M. , Uhr, M. , Otto, Y. , von Klitzing, K. and Klein, A. M. , FKBP5 polymorphisms moderate the influence of adverse life events on the risk of anxiety and depressive disorders in preschool children. J Psychiatr Res 2016. 72: 30–36. [DOI] [PubMed] [Google Scholar]
- 19. Bouwmeester, T. , Bauch, A. , Ruffner, H. , Angrand, P. O. , Bergamini, G. , Croughton, K. , Cruciat, C. et al., A physical and functional map of the human TNF‐alpha/NF‐kappa B signal transduction pathway. Nat Cell Biol 2004. 6: 97–105. [DOI] [PubMed] [Google Scholar]
- 20. Romano, S. , Xiao, Y. , Nakaya, M. , D'Angelillo, A. , Chang, M. , Jin, J. , Hausch, F. et al., FKBP51 employs both scaffold and isomerase functions to promote NF‐kappaB activation in melanoma. Nucleic Acids Res 2015. 43: 6983–6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Erlejman, A. G. , De Leo, S. A. , Mazaira, G. I. , Molinari, A. M. , Camisay, M. F. , Fontana, V. , Cox, M. B. et al., NF‐kappaB transcriptional activity is modulated by FK506‐binding proteins FKBP51 and FKBP52: a role for peptidyl‐prolyl isomerase activity. J Biol Chem 2014. 289: 26263–26276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Strobel, B. , Duechs, M. J. , Schmid, R. , Stierstorfer, B. E. , Bucher, H. , Quast, K. , Stiller, D. et al., Modeling pulmonary disease pathways using recombinant adeno‐associated virus 6.2. Am J Respir Cell Mol Biol 2015. 53: 291–302. [DOI] [PubMed] [Google Scholar]
- 23. Kelly, M. M. , King, E. M. , Rider, C. F. , Gwozd, C. , Holden, N. S. , Eddleston, J. , Zuraw, B. et al., Corticosteroid‐induced gene expression in allergen‐challenged asthmatic subjects taking inhaled budesonide. Br J Pharmacol 2012. 165: 1737–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Woodruff, P. G. , Boushey, H. A. , Dolganov, G. M. , Barker, C. S. , Yang, Y. H. , Donnelly, S. , Ellwanger, A. et al., Genome‐wide profiling identifies epithelial cell genes associated with asthma and with treatment response to corticosteroids. Proc Natl Acad Sci U S A 2007. 104: 15858–15863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Menke, A. , Arloth, J. , Putz, B. , Weber, P. , Klengel, T. , Mehta, D. , Gonik, M. et al., Dexamethasone stimulated gene expression in peripheral blood is a sensitive marker for glucocorticoid receptor resistance in depressed patients. Neuropsychopharmacology 2012. 37: 1455–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chun, E. , Lee, H. S. , Bang, B. R. , Kim, T. W. , Lee, S. H. , Kim, J. H. , Cho, S. H. et al., Dexamethasone‐induced FKBP51 expression in peripheral blood mononuclear cells could play a role in predicting the response of asthmatics to treatment with corticosteroids. J Clin Immunol 2011. 31: 122–127. [DOI] [PubMed] [Google Scholar]
- 27. Bigler, J. , Boedigheimer, M. , Schofield, J. P. , Skipp, P. J. , Corfield, J. , Rowe, A. , Sousa, A. R. et al., A severe asthma disease signature from gene expression profiling of peripheral blood from U‐BIOPRED cohorts. Am J Respir Crit Care Med 2016. 195: 1311–1320. [DOI] [PubMed] [Google Scholar]
- 28. Tajiri, T. , Matsumoto, H. , Niimi, A. , Ito, I. , Oguma, T. , Nakaji, H. , Inoue, H. et al., Association of eosinophilic inflammation with FKBP51 expression in sputum cells in asthma. PLoS One 2013. 8: e65284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Stechschulte, L. A. , Hinds, T. D., Jr. , Ghanem, S. S. , Shou, W. , Najjar, S. M. and Sanchez, E. R. , FKBP51 reciprocally regulates GRalpha and PPARgamma activation via the Akt‐p38 pathway. Mol Endocrinol 2014. 28: 1254–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Peer review correspondence
Table S1