

Abstract
Aging affects the core processes of almost every organism, and the functional decline at the cellular and tissue levels influences disease development. Recently, it was shown that the methylation of certain CpG dinucleotides correlates with chronological age and that this epigenetic clock can be applied to study aging‐related effects. We investigated these molecular age loci in non‐small cell lung cancer (NSCLC) tissues from patients with adenocarcinomas (AC) and squamous cell carcinomas (SQC) as well as in matched tumor‐free lung tissue. In both NSCLC subtypes, the calculated epigenetic age did not correlate with the chronological age. In particular, SQC exhibited rejuvenation compared to the corresponding normal lung tissue as well as with the chronological age of the donor. Moreover, the younger epigenetic pattern was associated with a trend toward stem cell‐like gene expression patterns. These findings show deep phenotypic differences between the tumor entities AC and SQC, which might be useful for novel therapeutic and diagnostic approaches.
Keywords: non‐small cell lung cancer, aging, epigenetic clock, stem cells, methylome, transcriptome
Short abstract
What's new?
Chronological age is correlated with the methylation status of CpG sites in the genome, enabling the study of aging‐related phenomena. Here, investigation of molecular age loci in cells from patients with non‐small cell lung cancer (NSCLC) reveals remarkable differences in NSCLC cell epigenetic age compared to the host's chronological age. Adenocarcinomas showed a higher epigenetic age than squamous cell carcinomas (SQC). Reduced SQC epigenetic age was accompanied by increased expression of stem cell gene signatures, suggesting an increased abundance of stem cells in SQC. Elevated stem cell levels could have clinical implications, as stems cells often show therapeutic resistance.
Abbreviations used
- NSCLC
Non‐small cell lung cancer
- AC
adenocarcinoma
- SQC
squamous cell carcinoma
- DNAm age
epigenetic age
- CSCs
cancer stem cells
- iPSCs
induced‐pluripotent stem cells
Introduction
Life is limited by the progressive functional decline of the organism at multiple levels that is known as aging and that is, for most organisms, inevitable, and irreversible,1, 2 with a few exceptions were negligible senescence can occur.3 Humans develop a plethora of age‐related pathologies that lead to diseases such as atherosclerosis, osteoporosis, pulmonary insufficiency, and cancer.1 Changes during the process of aging are not only limited to cellular functions, morphological changes or impaired organelles but also extend to the epigenome level and are likely influenced by epigenetic changes.2
The cytosine‐5 methylation status of CpG sites can be used to estimate the epigenetic age (DNAm age) and to define an epigenetic clock, referred to as Horvath's clock, which correlates with the chronological age of the organism in multiple tissues. Interestingly, the calculated DNAm age is reduced in embryonic and induced‐pluripotent stem cells and correlates well with the cell passage number.4 Moreover, different tumor types display DNAm age alterations and differ considerably from the chronological age of the host.4, 5 Additionally, other CpG loci have been identified by correlation analyses to show age‐dependent DNA methylation patterns.6
Here, we analyzed the DNAm age in a cohort of non‐small cell lung cancer (NSCLC) samples and the corresponding tumor‐free tissues to obtain insight into possible DNAm age changes and the underlying transcriptional processes.
Material and Methods
Sample origin
Tissues from 33 patients who underwent surgery with curative intent due to lung cancer, composed of 15 adenocarcinomas (AC) and 18 squamous cell carcinomas, (SQC) were analyzed together with their matched tumor‐free control tissues. Ethical permission was obtained from the University of Lübeck through BioMaterialBank North (Ref. 12–220). Tumor specimens were micro‐dissected to enrich the tumor content for the methylome analysis7 and transcriptome analysis.8 For this, non‐tumor content was scratched off the tissue sections under a microscope to deplete non‐tumor cells and enrich NSCLC cells prior to the extraction of the nucleic acids.
Methylome analysis
After extraction of the DNA, methylome data was generated as previously reported using the HumanMethylation450K BeadChip (Illumina, Inc., San Diego, CA).7 Data were stored in the gene expression omnibus (GEO) (accession number GSE75008). CpG loci located on gonosomes as well as CpG loci with detection P‐values> 0.01 in at least one sample were excluded from further analyses. Omics Explorer 3.2 (Qlucore, Lund, Sweden) was used for cluster analyses, principal component analysis (PCA) and data presentation.
To validate the results obtained from our cohort, DNA methylation data on lung adenocarcinoma (LUAD) and squamous cell carcinoma (LUSC) made publicly available by The Cancer Genome Atlas (TCGA) project was included. The data was accessed from http://gdac.broadinstitute.org/runs/stddata__2014_09_02/data on December 1, 2017.
Calculation of DNA methylation age
Horvath's epigenetic clock is a method of predicting a specimen's age based on the DNA methylation values of the 353 CpG loci present on the HumanMethylation450K BeadChip. The epigenetic age (DNAm age) was calculated as described before4 using the DNAm age Calculator (accessible at https://dnamage.genetics.ucla.edu/).
Detection of aging‐associated methylation regions
The DNA methylation profiles of 40 control lung specimens (chronological age 41–82 years; mean: 66 years, median: 66 years) were determined as described above. Using the R software package (version 3.4.1; http://www.r-project.org), Pearson's correlation coefficients of the donors’ chronological ages and DNA methylation values (average beta values) were calculated for every CpG locus. CpG loci with absolute Pearson's correlation coefficients >0.6 were included in further analyses.
Transcriptome analysis
Gene expression data for 18 NSCLC tumors and matched tumor‐free lungs from the GEO dataset GSE74706, for which methylome data (GSE75008) was also available, were selected for the transcriptome analysis and annotated with the difference in DNAm ages between the sample and the matched tumor‐free lung (DNAm age [lung]—DNAm age [tumor]): 9495–10 (DNAm age diff: −37.6), 12097–07 (DNAm age diff: −25.5), 13230–12 (DNAm age diff: −23.9), 12928–12 (DNAm age diff: −23.7), 12799–12 (DNAm age diff: −10.8), 1888–12 (DNAm age diff: −9.3), 2508–11 (DNAm age diff: −5.3), 6495–08 DNAm age diff: −1.1), 15982–07 DNAm age diff: 15.5), 17962–08 (DNAm age diff: 27.6), 1799–12 (DNAm age diff: 31), 22024–11 (DNAm age diff: 34.2), 12893–12 (DNAm age diff: 37.4), 21918–11 (DNAm age diff: 38.3), 21577–08 (DNAm age diff: 44.3), 11560–12 (DNAm age diff: 51.5), 3302–12 (DNAm age diff: 62.1), and 5622–12 (DNAm age diff: 79.5). Samples were analyzed using GeneSpring software version 13.0 (Agilent Technologies, Böblingen, Germany) as described elsewhere.8 One‐way ANOVA (1 W‐ANOVA) was used for the analysis of >2 classes with a Benjamini‐Hochberg multiple testing correction cut‐off of P ≤ 0.05 considered significant. Tukey post hoc testing was used for the selection of genes specific for each experimental condition. For unsupervised hierarchical clustering of normalized intensity values on genes and samples, the Pearson centered similarity measure was used together with Ward's linkage rule. Clusters were selected for further analysis by their distance metric, and gene symbols from each cluster were used to query the Molecular Signature Database (MSigDB) version 6 (http://software.broadinstitute.org/gsea/msigdb) for the enrichment of significant GO terms from biological processes, with an FDR q‐value cut‐off of P ≤ 0.05 considered significant.
Gene set enrichment analysis (GSEA)
Gene set enrichment analysis (GSEA) software was downloaded from the BROAD Institute at https://software.broadinstitute.org/gsea/index.jsp. Next, 20 gene sets that contain stem cell gene expression signatures or genes that are upregulated in stem cells,9 regulate tumor stemness or are involved in embryonic stem cells were retrieved from the Molecular Signatures Database (MSigDB) and uploaded into the GSEA software. Normalized transcriptome data that contain the gene expression signatures of tumor samples with younger or older DNAm ages than the respective tumor‐free lung samples were loaded into GSEA software, and the analysis was run with the default parameters. Normalized enrichment scores (NES) were used to retrieve genes with an enrichment of > 1 for detailed analysis and correlation with the DNAm age. Hierarchical clustering of selected genes from the GSEA was conducted using the Multi Experiment Viewer version 4 (http://mev.tm4.org) with the Pearson centered algorithm. Selected genes and their normalized expression from the enrichment results were plotted against the DNAm age difference of each patient. A linear regression line was fitted, and the 95% confidence intervals were displayed. Pearson's correlation was conducted on plotted values using GraphPad Prism version 7.
Results and Discussion
Epigenetic analysis of NSCLC tissues revealed reduced DNAm age of SQC and accelerated DNAm age of AC
Given the accuracy of the Horvath clock's ability to reflect the host's age based on the methylation level, we investigated the DNAm age of NSCLC tissues and matched tumor‐free lung tissues in comparison to their chronological ages. Overall, cancer tissues exhibited a different DNAm age than the tumor‐free tissues. Furthermore, AC behaved differently than squamous cell carcinomas (SQC) (Fig. 1 a) and seemed to have a higher DNAm age than the tumor‐free lungs and an even more pronounced increased age compared to that of SQC. This was also true for the differences between the DNAm age of the tumors and the matched tumor‐free tissues (Fig. 1b). Overall, SQC tissue exhibited a significantly decreased DNAm age compared to that of AC tissue (p‐value<1.32x10−5, Fig. 1 c) and their respective tumor‐free tissues. Since the investigated dataset was comprised of a limited number of samples, we further investigated publicly available datasets from TCGA. In line with our data, SQC exhibited a significantly (P‐value<2.2x10−16) lower delta age (DNAm age—chronological age) than that of AC specimens (Figs. 1 d–1f). However, in contrast to our data set age acceleration in AC was detected only in some of the TCGA samples.
Figure 1.
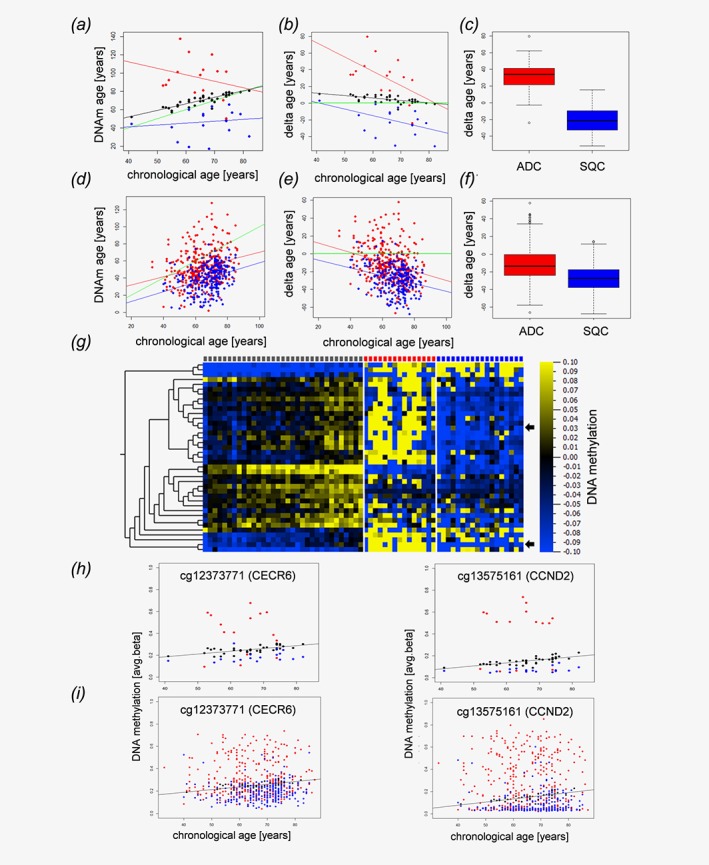
NSCLC tissues display epigenetic age distortions. Dot plot (a) and Bland–Altman‐Plot (b) showing correlations between the chronological age of the host and the epigenetic DNAm age of matched tumor tissues from normal control specimens (black dots), adenocarcinomas (red) and squamous cell carcinomas (blue). (c) Boxplot of the deviations of DNAm age from the chronological age in adenocarcinomas (ADC, red) and squamous cell carcinomas (SQC, blue). The delta age of cancer entities differs significantly (p‐value<1.32x10−5, Wilcoxon rank sum test). Validation cohort (public available TCGA data set): Dot plot (d) and Bland–Altman‐Plot (e) showing correlations between the chronological age and the epigenetic DNAm age of adenocarcinomas (red) and squamous cell carcinomas (blue) in an independent validation cohort. (f) The delta age of adenocarcinomas (ADC, red) and squamous cell carcinomas (SQC, blue) differs significantly (P‐value<2.2 x 10−16, Wilcoxon rank sum test). The delta age is calculated as DNAm age (years)—chronological age (years). (a, b, c, d) Black, red and blue lines represent the regression lines for control samples, adenocarcinomas, and squamous cell carcinomas, respectively. The green line indicates a perfect match of chronological age and DNAm age (chronological age = DNAm age). DNA methylation changes of distinct CpG loci associated with aging and alterations of these patterns in lung cancer (g–i). Heatmap (g) of CpG loci with changing DNA methylation during aging. Pearson correlation analysis of avg. beta values of all CpG loci included in our study in normal lung tissue (controls) with the chronological age of the sample donors identified 39 loci with │Pearson correlation coefficients│ > 0.6 (Table 1). The DNA methylation values of these loci are presented separately for controls (gray squares, top lane), adenocarcinoma (red squares) and squamous cell carcinoma (blue squares). Samples are ordered according to donors’ chronological ages. Heatmap: blue, low DNA methylation values; yellow, high DNA methylation values, for visualization purposes, the DNA methylation values were normalized to zero for each locus, and the bar below the heatmap indicates the range and color code. Arrows indicate randomly selected loci detailed in H and I. (h) Detailed presentation of data from two randomly selected loci. DNA methylation of cg12373771 (CECR6) and cg13575161 (CCND2) increases during aging in control samples (black spheres). Compared to the controls, squamous cell carcinoma (blue spheres) are characterized by lower DNA methylation values, while DNA methylation is increased in adenocarcinoma (red spheres), putatively reflecting decelerated and accelerated aging, respectively. (i) A similar pattern is seen also in an independent dataset.
The DNAm age deviations from the chronological ages of the tumor‐free lung tissues might be explained by the possible presence of COPD or smoking habits.10 Among these, patients showing an age acceleration of more than 8 years (7 cases) smoked 63.1 pack‐years, while patients showing a deceleration of the DNAm age in the tumor‐free lungs (3 cases) smoked 23.3 pack‐years. There was no correlation between the pack‐years, GOLD‐grade, or emphysema and the DNAm age of the tumor‐free lung tissues; however, it has been shown that DNAm age acceleration measured in the blood are predictive of various diseases including cancer.11 The strongly decreased DNAm age of SQC (mean: 25.1 years) and the accelerated DNAm age of AC (mean + 25.9 years) raises questions about the underlying biological mechanisms.
Age‐related DNA methylation patterns are disturbed in lung carcinoma
In addition to Horvath's epigenetic clock, which relies on a comprehensive mathematical approach,4 additional age‐related changes in the DNA methylation values of multiple genes have been described.6 Given the alterations in the Horvath loci, we wondered whether DNA methylation patterns directly relate to age, indicating aberrant epigenome aging in NSCLC. Therefore, we determined the correlation coefficients between donors’ chronological ages and the DNA methylation values for every CpG locus in 40 matched controls. In so doing, we identified 40 CpG loci with absolute Pearson's correlation coefficients > 0.6 (Fig. 1 g). Of these, one locus (cg22901919; CLGN) was removed from further analysis due to insufficient data from the malignant specimens. The remaining 39 loci are allocated to 26 genes (Table 1). Many of these loci have been previously described as being either expressed or methylated in an age‐related or disease‐related manner in other tissues12, 13 or as being affected in lung cancer.14 The 39 loci were significantly enriched for DNase I hypersensitive sites (DHS, OR = 5.541, RR = 5.540, p < 0.0001; chi‐square test) and known differentially methylated regions (DMR, OR = 8.873, RR = 8.869, P < 0.0001) according to array annotation, suggesting a possible role in gene regulation. Twenty‐four (61.5%) of these loci were differentially methylated between SQC and their corresponding controls and the DNA methylation of 10 loci (25.6%) differentiated between AC tissues and their corresponding controls, while 9 loci (23.1%) were differentially methylated between AC and SQC of the lung (Student's t‐test, two‐sided). Interestingly, 22 of the loci that were aberrantly methylated in SQC showed DNA hypomethylation compared to the controls. Only two loci showed hypermethylation (cg12206199, cg15936446). In contrast, 6/10 aberrantly methylated loci in adenocarcinoma showed DNA hypermethylation compared to the corresponding controls. All loci differentially methylated between AC and SQC had lower methylation levels in SQC. As low DNA methylation at these loci is indicative of lower age (Figs 1 g–1i), these findings support our conclusion that AC are “epigenetically older” than squamous cell carcinomas.
Table 1.
DNA methylation changes in distinct CpG loci associated with aging in normal lung tissue
| TargetID | Chromosome | MAPINFO | UCSC refgene name | Pearson correlation coefficient |
|---|---|---|---|---|
| cg23040782 | 1 | 6762215 | DNAJC11 | 0.6081 |
| cg23813012 | 1 | 14026482 | PRDM2 | 0.7119 |
| cg08169949 | 1 | 28241532 | RPA2 | 0.6041 |
| cg12100751 | 1 | 109203672 | C1orf59 | 0.6230 |
| cg03105929 | 1 | 161591659 | 0.6254 | |
| cg02375320 | 2 | 38978881 | SFRS7 | 0.6013 |
| cg12206199 | 2 | 39187543 | LOC375196 | 0.6168 |
| cg23606718 | 2 | 131513927 | FAM123C | 0.6033 |
| cg03545227 | 2 | 220173100 | PTPRN | 0.7443 |
| cg07720856 | 2 | 232572668 | PTMA | 0.6784 |
| cg12141030 | 3 | 44803447 | KIAA1143 | 0.6760 |
| cg07303143 | 3 | 44803452 | KIAA1143 | 0.7257 |
| cg10806820 | 3 | 48699090 | CELSR3 | 0.6388 |
| cg19614811 | 3 | 98250723 | GPR15 | 0.6763 |
| cg09401099 | 3 | 156534380 | 0.6094 | |
| cg05991454 | 4 | 147558435 | 0.6354 | |
| cg15936446 | 5 | 42952369 | 0.6168 | |
| cg04268670 | 5 | 76926341 | OTP | 0.6019 |
| cg26921969 | 5 | 92948217 | 0.6318 | |
| cg03873281 | 5 | 131608955 | PDLIM4 | −0.6160 |
| cg22736354 | 6 | 18122719 | NHLRC1 | 0.6632 |
| cg20899581 | 6 | 27841230 | HIST1H4L | 0.6873 |
| cg20692569 | 7 | 72848481 | FZD9 | 0.6563 |
| cg21186299 | 7 | 100808810 | VGF | 0.6358 |
| cg26830108 | 7 | 100813299 | 0.6216 | |
| cg05418199 | 8 | 6756832 | 0.6723 | |
| cg18245316 | 8 | 144962981 | 0.6002 | |
| cg14138050 | 9 | 14693504 | ZDHHC21 | 0.6029 |
| cg14506657 | 9 | 86755443 | 0.6481 | |
| cg08715791 | 11 | 66189297 | NPAS4 | 0.6451 |
| cg13575161 | 12 | 4381792 | CCND2 | 0.7322 |
| cg03036557 | 13 | 92050720 | GPC5 | 0.6086 |
| cg12678562 | 13 | 92050726 | GPC5 | 0.6196 |
| cg11082362 | 14 | 36003181 | INSM2 | 0.6182 |
| cg07850604 | 14 | 36003443 | INSM2 | 0.6071 |
| cg08151705 | 17 | 33775917 | SLFN13 | 0.6324 |
| cg04179438 | 17 | 75082479 | −0.6289 | |
| cg07873590 | 19 | 17858298 | FCHO1 | 0.6030 |
| cg12373771 | 22 | 17601381 | CECR6 | 0.6639 |
RNA expression levels of SQC/younger DNAm age patients and AC/older DNAm age patients reveal the underlying biological processes
To further decipher the processes that differentiate tumors with an accelerated DNAm age from tumors with a lower DNAm age, we investigated the transcriptome data of those samples for which DNAm age and gene expression data were available. Microarray data were annotated for the calculated DNAm age difference of each patient, and significantly differentially expressed genes were identified, separating AC from SQC and tumor‐free lungs (Fig. 2 a). Specific gene signatures attributed to either AC or SQC were extracted from the 1 W‐ANOVA results (Fig. 2 b). To identify the possible underlying processes, GO analysis was conducted on twofold upregulated genes, resulting in strikingly different outcomes. SQC/younger DNAm age samples exhibited various enriched terms for developmental processes such as “tissue development”, “regulation of multicellular organismal development” or “embryo development” (Fig. 2 b), suggesting cell fate regulating processes. In contrast, AC/older DNAm age samples displayed enriched terms for metabolic activity such as “ion transport” or “homeostatic process”.
Figure 2.
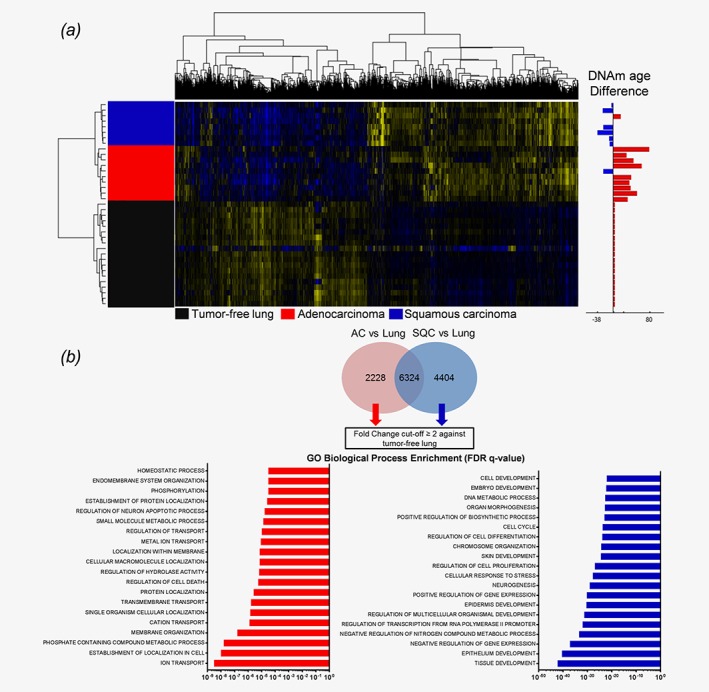
NSCLC tumors display a differential gene expression pattern associated with differentiation and developmental processes in SQC. (a) NSCLC tumors and matched tumor‐free lungs were analyzed by 1 W‐ANOVA for the significantly different expression of genes. The difference in DNAm age for each tumor compared to the chronological age of the host is displayed as an adjacent annotation track. Hierarchical clustering clearly separated all groups. (b) Specific gene expression signatures of AC and SQC tumors were derived from genes found to be significantly differentially expressed between adenocarcinomas and tumor‐free lungs and between squamous cell carcinomas and tumor‐free lungs, as indicated by the Tukey HSD post hoc test. From both gene lists, shared genes were excluded, and specific signatures were extracted. Only those genes with a fold‐change of ≥ 2 compared to the tumor‐free lung tissues were subjected to GO enrichment analysis. Gene Ontology analysis displays the top 20 most significantly enriched GO terms, as determined by the FDR q‐values.
Stem cell signatures show a trend towards enrichment in tumor tissues classified younger in DNAm age
We next hypothesized that this increased activity of cell fate decisions or differentiation processes might be caused by stem cell activity. Therefore, gene sets describing embryonic‐like stem cell genes9 and others were investigated for enrichment between younger and older DNAm age patients. Both groups exhibited an enrichment of gene sets, with 18/20 found to be upregulated in epigenetically younger tumors compared to 2/20 upregulated in epigenetically older tumors. However, only four gene sets from the epigenetically younger tumors passed the generic GSEA FDR cut‐off (< 0.25). These sets indicated a trend towards enrichment of stem cell signatures. The results from the GSEA are shown in Figure 3, with Figure 3 a summarizing the enrichment results in epigenetically younger tumors. The significantly enriched gene sets and their enrichment plots are shown in Figure 3 b. Here, the gene set “Benporath_Es_Core_Nine” contains embryonic stem cell transcription regulators that are also overexpressed in high‐grade, ER‐negative breast cancers, while “Benporath_Proliferation” contains crucial genes for proliferation. The gene sets “Benporath_ES_1 and ES_2” are the results of meta‐analysis of genes found to be overexpressed in human embryonic stem cells from 5 out of 20 profiling studies. The enrichment plots in all gene sets show a positive correlation of matched hits with the samples of a younger DNAm age, as the hits aggregate on the left side of each curve (Fig. 3 b). Next, those genes that displayed an NES of > 1 were selected for more detailed analyses. Hierarchical clustering of the expression patterns of these genes among the investigated tumors revealed a clear pattern. The enriched genes from the stem cell gene sets were found to be overexpressed and clustered with tumors of a younger DNAm age (Fig. 3 c). The top six genes from this list of enriched genes were used to plot the normalized gene expression against each patient's DNAm age difference. Linear regression was computed to insert a best fit line, and the data were correlated. All six genes showed significant and strong negative correlations with the DNAm age differences (Fig. 3 d). The higher the expression levels of these genes were, the lower the calculated DNAm age difference between the tumor and the tumor‐free lung. One of the enriched genes from the gene sets with a strong negative correlation with DNAm age was SOX2. SOX2 is involved in the pluripotency of human embryonic stem cells15, 16 and is indicative of squamous histology17 as well as being an antagonist of the tumor‐suppressive Hippo pathways, leading to maintenance of cancer stem cells18 (CSCs). This observation might reflect the nature of lung SQC, exhibiting an increased stem cell activity accompanied by decreased DNAm age, as possible CSC populations have been identified in NSCLC cell lines and tissues,19 with SQC exhibiting a higher fraction of stem cells leading to speculation about the respective cells of origin.20 Furthermore, embryonic or induced‐pluripotent stem cells (iPSCs) have DNAm ages close to zero,4 which is, at least in part, mirrored by the observed reduced DNAm age in SQC, thereby further strengthening the observation. Whether increased stem cell activity or the processes leading to reduced DNAm age are causative during carcinogenesis or vice versa constitutes a question that might lead to future therapies, as active interference with the epigenome has gained widespread attention.21 Even more, these data might be reflected in the clinic by observations of SQC patients who exhibit lower 1‐year survival22 or overall survival23 rates compared to those of AC patients. CSCs display increased chemotherapy resistance,24 hence contributing to the reduced effectiveness of chemotherapy for SQC.
Figure 3.
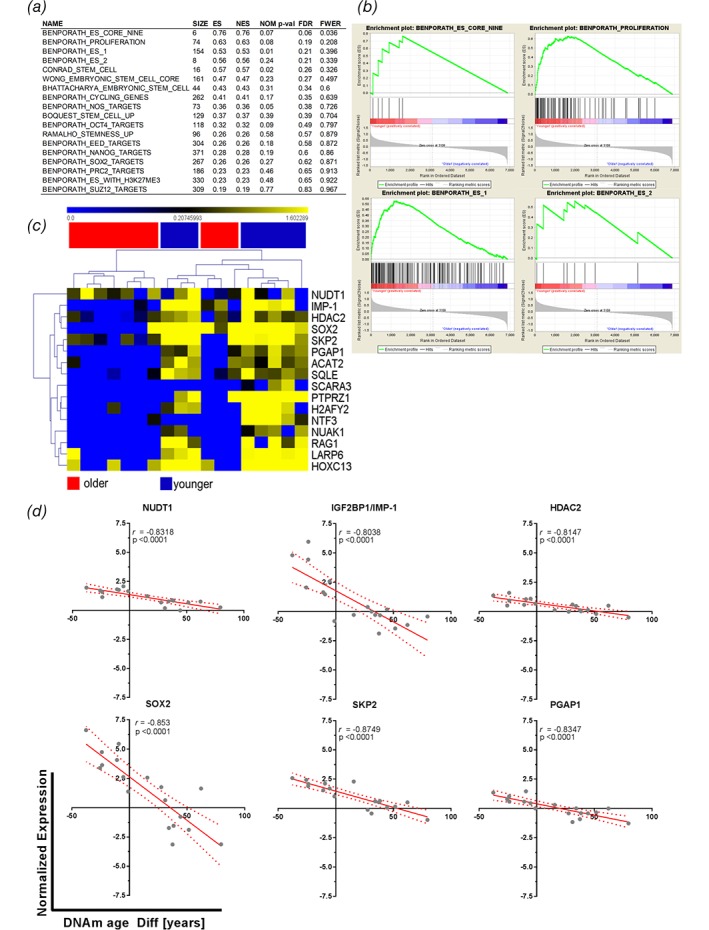
Epigenetically rejuvenated tumors display enriched stem cell gene signatures. (a) Transcriptome data of tumors with an accelerated or “older” and de‐accelerated or “younger” DNAm ages compared to the chronological ages of the same patient's tumor‐free lung tissue were subjected to GSEA analysis. Twenty‐one gene sets from the MSigDB that contained expression profiles of stem cells, stemness‐related gene pattern, or embryonic stem cells were analyzed for possible enrichment within the dataset; 1/21 gene sets were filtered out by the gene set size filter (min = 5, max = 500). Of the remaining 20 gene sets, 18 were found to be upregulated in epigenetically younger tumors, and two were upregulated in epigenetically older tumors. (a) Summary table of GSEA analysis. ES, enrichment score, NES, normalized enrichment score, NOM P‐val, nominal P‐value, FDR, FDR q‐value, FWER, family‐wise error rate. (b) Enrichment plots of gene sets showing a trend to be regulated in younger tumors with an FDR <25%. (c) Hierarchical clustering of genes from the GSEA with a normalized enrichment score > 1 and their normalized gene expression within younger and older samples. (d) The top six genes according to the NES with enrichment of their normalized expression were plotted against the DNAm age difference between the tumor and the tumor‐free lung tissue. The solid red line shows the best fit, and the dotted red lines indicate the 95% confidence intervals. Pearson's correlation coefficients are shown in each quadrant.
Determination of the DNAm age of AC and SQC using Horvath's DNA Methylation Age Calculator revealed a markedly younger DNAm age for SQC tissue compared to matched tumor‐free lung tissue from the same patients. AC tissue seem to show an increased epigenetic age, pointing to the distinctively different biological processes taking place or even having already taken place in the course of carcinogenesis.
Second, we identified loci showing age‐related changes in DNA methylation patterns and subsequently compared those DNA methylation patterns with methylation patterns obtained from corresponding cancer samples. The vast majority of these loci were aberrantly hypomethylated in SQC compared to the controls. Since DNA methylation of these loci increases with age, this suggests either decelerated aging or rejuvenation. Moreover, all the differentially methylated loci between AC and SQC showed higher levels of DNA methylation in AC, which points to accelerated aging in AC.
Although accelerated DNAm age has been already described in various cancer samples4, 5, 11 and has been linked to lung cancer risk,25 our study is the first to report striking differences in DNAm age compared with that of the matched internal control tissues and to attribute reduced DNAm age to increased stem cell gene activity in SQC.
Competing interests
The authors declare that they have no competing interests.
Declarations
Ethics approval and consent to participate
The use of patient material was approved by the local ethics council at the University of Lübeck (AZ‐12‐220).
Funding
This work was funded by the German Center for Lung Research (DZL; 82DZL001A5) and the Deutsche Forschungsgemeinschaft (DR797/3–1). Patient tissues were provided by BioMaterialBank North, which is funded in part by the Airway Research Center North (ARCN, which is a member of the German Center for Lung Research, DZL) and is a member of the popgen 2.0 network (P2N), which is supported by a grant from the German Ministry for Education and Research (01EY1103).
Authors' contributions
SM and LH drafted the manuscript and created the figures. OA conceived of the study. OA and SS prepared and analyzed the methylome data. SM analyzed the transcriptome data. TG, CK, MR, KR and SP edited the manuscript. All authors read and approved the final manuscript.
Acknowledgments
The authors thank Lorena Valles, Jasmin Tiebach, Maria Lammers, and Kristin Wiczkowski for excellent technical assistance.
SM and LH share first authorship.
TG and OA share senior authorship.
References
- 1. Campisi J. Aging, cellular senescence, and cancer. Annual review of physiology 2013;75:685–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Booth LN, Brunet A. The aging epigenome. Mol Cell 2016;62:728–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kogan V, Molodtsov I, Menshikov LI, et al. Stability analysis of a model gene network links aging, stress resistance, and negligible senescence. Sci Rep 2015;5:13589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Horvath S. DNA methylation age of human tissues and cell types. Genome biology 2013;14:R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lin Q, Wagner W. Epigenetic aging signatures are coherently modified in cancer. PLoS Genet 2015;11:e1005334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hannum G, Guinney J, Zhao L, et al. Genome‐wide methylation profiles reveal quantitative views of human aging rates. Mol Cell 2013;49:359–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Marwitz S, Kolarova J, Reck M, et al. The tissue is the issue: improved methylome analysis from paraffin‐embedded tissues by application of the HOPE technique. Lab Invest 2014;94:927–33. [DOI] [PubMed] [Google Scholar]
- 8. Marwitz S, Depner S, Dvornikov D, et al. Downregulation of the TGFbeta pseudoreceptor BAMBI in non‐small cell lung cancer enhances TGFbeta Signaling and Invasion. Cancer research 2016;76:3785–801. [DOI] [PubMed] [Google Scholar]
- 9. Ben‐Porath I, Thomson MW, Carey VJ, et al. An embryonic stem cell‐like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet 2008;40:499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gao X, Zhang Y, Breitling LP, et al. Relationship of tobacco smoking and smoking‐related DNA methylation with epigenetic age acceleration. Oncotarget 2016;7:46878–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Perna L, Zhang Y, Mons U, et al. Epigenetic age acceleration predicts cancer, cardiovascular, and all‐cause mortality in a German case cohort. Clin Epigenetics 2016;8:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lim SL, Qu ZP, Kortschak RD, et al. HENMT1 and piRNA stability are required for adult male germ cell transposon repression and to define the spermatogenic program in the mouse. PLoS Genet 2015;11:e1005620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bacos K, Gillberg L, Volkov P, et al. Blood‐based biomarkers of age‐associated epigenetic changes in human islets associate with insulin secretion and diabetes. Nat Commun 2016;7:11089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yoon KA, Park S, Hwangbo B, et al. Genetic polymorphisms in the Rb‐binding zinc finger gene RIZ and the risk of lung cancer. Carcinogenesis 2007;28:1971–7. [DOI] [PubMed] [Google Scholar]
- 15. Adachi K, Suemori H, Yasuda SY, et al. Role of SOX2 in maintaining pluripotency of human embryonic stem cells. Genes Cells 2010;15:455–70. [DOI] [PubMed] [Google Scholar]
- 16. Bass AJ, Watanabe H, Mermel CH, et al. SOX2 is an amplified lineage‐survival oncogene in lung and esophageal squamous cell carcinomas. Nat Genet 2009;41:1238–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wilbertz T, Wagner P, Petersen K, et al. SOX2 gene amplification and protein overexpression are associated with better outcome in squamous cell lung cancer. Mod Pathol 2011;24:944–53. [DOI] [PubMed] [Google Scholar]
- 18. Basu‐Roy U, Bayin NS, Rattanakorn K, et al. Sox2 antagonizes the Hippo pathway to maintain stemness in cancer cells. Nat Commun 2015;6:6411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Leung EL, Fiscus RR, Tung JW, et al. Non‐small cell lung cancer cells expressing CD44 are enriched for stem cell‐like properties. PloS one 2010;5:e14062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Giangreco A, Groot KR, Janes SM. Lung cancer and lung stem cells: strange bedfellows? Am J Respir Crit Care Med 2007;175:547–53. [DOI] [PubMed] [Google Scholar]
- 21. Jones PA, Issa JP, Baylin S. Targeting the cancer epigenome for therapy. Nat Rev Genet 2016;17:630–41. [DOI] [PubMed] [Google Scholar]
- 22. Cetin K, Ettinger DS, Hei YJ, et al. Survival by histologic subtype in stage IV nonsmall cell lung cancer based on data from the surveillance, epidemiology and end results program. Clin Epidemiol 2011;3:139–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kogure Y, Ando M, Saka H, et al. Histology and smoking status predict survival of patients with advanced non‐small‐cell lung cancer. Results of West Japan oncology group (WJOG) study 3906L. J Thorac Oncol 2013;8:753–8. [DOI] [PubMed] [Google Scholar]
- 24. Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer 2005;5:275–84. [DOI] [PubMed] [Google Scholar]
- 25. Levine ME, Hosgood HD, Chen B, et al. DNA methylation age of blood predicts future onset of lung cancer in the women's health initiative. Aging (Albany NY) 2015;7:690–700. [DOI] [PMC free article] [PubMed] [Google Scholar]