

Abstract
The widespread use of next generation sequencing for clinical testing is detecting an escalating number of variants in noncoding regions of the genome. The clinical significance of the majority of these variants is currently unknown, which presents a significant clinical challenge. We have screened over 6,000 early‐onset and/or familial breast cancer (BC) cases collected by the ENIGMA consortium for sequence variants in the 5′ noncoding regions of BC susceptibility genes BRCA1 and BRCA2, and identified 141 rare variants with global minor allele frequency < 0.01, 76 of which have not been reported previously. Bioinformatic analysis identified a set of 21 variants most likely to impact transcriptional regulation, and luciferase reporter assays detected altered promoter activity for four of these variants. Electrophoretic mobility shift assays demonstrated that three of these altered the binding of proteins to the respective BRCA1 or BRCA2 promoter regions, including NFYA binding to BRCA1:c.‐287C>T and PAX5 binding to BRCA2:c.‐296C>T. Clinical classification of variants affecting promoter activity, using existing prediction models, found no evidence to suggest that these variants confer a high risk of disease. Further studies are required to determine if such variation may be associated with a moderate or low risk of BC.
Keywords: breast cancer, BRCA1, BRCA2, promoter, transcription, variants of unknown clinical significance (VUS)
1. INTRODUCTION
Genetic susceptibility to breast cancer (BC) is complex. Multiple germline variants have been identified over the past 25 years that are broadly categorized as high, moderate, and low risk. High‐risk variants are generally rare, have a major deleterious effect on gene function, are sufficient to confer a high risk of disease, and are highly penetrant within a family. Nonsense, splicing, large deletions, and some missense changes in BRCA1 and BRCA2 fall into this category (reviewed in Walsh et al., 2006). There is also evidence that some alleles confer a moderate risk of cancer. These can include hypomorphic variants in known “high‐risk” cancer syndrome genes (Shimelis et al., 2017; Spurdle et al., 2012), or clear loss‐of‐function alleles in other genes such as CHEK2, PALB2, and ATM (Couch et al., 2017). Low‐risk variants, largely identified by genome‐wide association studies, are usually common and cause subtle functional effects, such as small but significant changes in gene expression due to altered activity of proximal and distal regulatory elements (reviewed in Bogdanova, Helbig, & Dork, 2013; Ghoussaini, Pharoah, & Easton, 2013; Skol, Sasaki, & Onel, 2016). Evidence suggests that combinations of low, moderate, and high‐risk variants could confer a clinically significant risk of disease (Ding et al., 2012; Kuchenbaecker et al., 2017; Sawyer et al., 2012). Identification and evaluation of all such variants is therefore crucial for accurately predicting BC risk.
Use of next generation sequence analysis for germline clinical testing of cancer cases is identifying an increasing number of variants in noncoding regions of cancer susceptibility genes, including promoters, untranslated regions (UTRs), and introns. There are currently no firm recommendations for assessing the relevance of noncoding region variants to clinical testing of Mendelian disease genes, and so the vast majority of such variants are deemed of uncertain clinical significance. This adds to the clinical challenge presented by variants of uncertain significance, namely that they complicate test reporting and genetic counseling, limit patient eligibility for intensive surveillance and gene‐targeted therapies, and prevent gene testing and guided management of relatives (reviewed in Amendola et al., 2015; Eccles et al., 2013; Plon et al., 2011). It is therefore essential that the functional and clinical significance of variants mapping to noncoding regions of the genome is determined.
Gene expression is controlled at many levels with key regulatory elements being housed in noncoding regions of the genome, such as gene promoters, introns, long‐range elements, and 5′ and 3′ UTRs. The BRCA1 gene is regulated at the transcriptional and posttranscriptional levels, with functional proximal and distal regulatory elements being described in the promoter, introns, and UTRs, by us and others (Brewster et al., 2012; Brown et al., 2002; Santana dos Santos et al., 2017; Saunus et al., 2008; Tan‐Wong, French, Proudfoot, & Brown, 2008; Wardrop, Brown, & kConFab, 2005; Wiedemeyer, Beach, & Karlan, 2014). Although less studied, the BRCA2 promoter has also been mapped and characterized (reviewed in Wiedemeyer et al., 2014).
Common and rare variations in regulatory elements upstream of genes have been shown to alter gene expression and be associated with disease risk (reviewed in Betts, French, Brown, & Edwards, 2013; Diederichs et al., 2016; Millot et al., 2012). We and others have described germline cancer‐associated variants in the regulatory regions, including large deletions in the BRCA1 promoter (Brown et al., 2002), and single nucleotide variants in the promoter and/or 5′ UTR of BRCA1 and BRCA2 (Evans et al., 2018; Santana dos Santos et al., 2017), MLH1 promoter (Hitchins et al., 2011), POLG promoter (Popanda et al., 2013), PTEN promoter (Heikkinen et al., 2011), TERT promoter (Horn et al., 2013), KLHDC7A and PIDD1 promoters (Michailidou et al., 2017), BRCA1 3′ UTR (Brewster et al., 2012), and BC‐associated Single Nucleotide Polymorphisms (SNPs) in long‐range enhancers of CCND1 (French et al., 2013).
Cancer risk‐associated variants within regulatory regions are anticipated to mediate an effect on trans‐acting regulatory factors (e.g., transcription factors [TFs] and miRNAs), by disrupting binding of regulatory factors and interactions between regulatory elements, such as promoter–enhancer interactions. For example, a variant in a Cyclin D1 transcriptional enhancer has been associated with altered binding of the ELK4 TF (French et al., 2013) and a variant within the BRCA1 3′UTR has been shown to introduce a functional mir‐103 binding site (Brewster et al., 2012). In addition, a dominantly inherited 5′ UTR BRCA1 variant was recently shown to be associated with BRCA1 promoter hypermethylation, which is known to impact TF binding, and associated allelic loss of BRCA1 expression in two families affected by breast and ovarian cancers (Evans et al., 2018).
In this paper, we describe 141 germline variants in the BRCA1 and BRCA2 promoter, identified by members of the ENIGMA consortium in early onset or familial BC patients with no known pathogenic variants in the coding region of these genes. Using a combination of bioinformatic and experimental analyses, we have prioritized and analyzed a subset of variants that are most likely to affect the regulation of BRCA1 and BRCA2 and thus have the most potential to contribute to BC risk. TF binding site affinity changes resulting from these variants were subsequently analyzed by information theory (IT)‐based analyses. In parallel, we have assessed if these variants exhibited the features expected for a high‐risk pathogenic BRCA1 or BRCA2 variant, on the basis of available clinical and population data.
2. MATERIALS AND METHODS
2.1. Study design
An overview of the study design is shown in Figure 1. Collection of variants at all sites enabled an initial catalogue of variants from which variants were prioritized for functional analysis. Additional screening was carried out at three sites, Maastricht (M), Santiago (S), and Prague (Pr), that included additional patients (M, S, and Pr) and controls (Pr) that expanded the list of variants (Pr), the number of patients (M, S, and Pr), and included control subjects (Pr).
Figure 1.
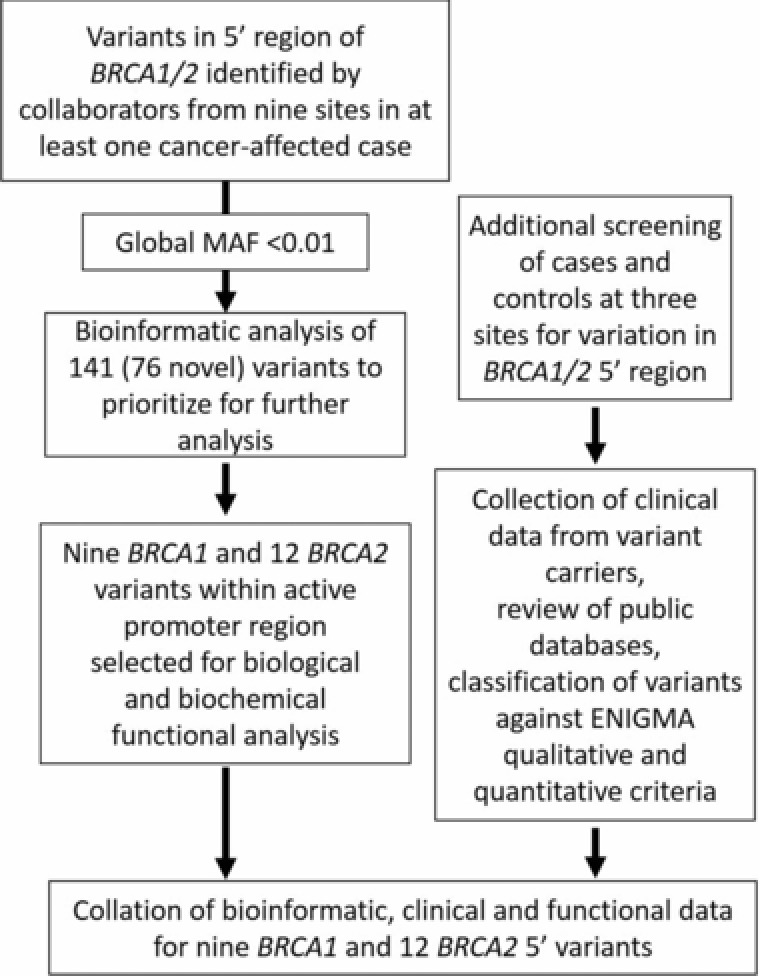
Overview of study design. Outline of the workflow of variant collection, prioritization and analysis
2.2. Clinical and control samples
Clinical and genetic data were collected and analyzed in accordance with local human ethics guidelines of the institutions contributing to this study. All participating individuals provided informed consent for their data to be used for research purposes. An overview of the samples analyzed is shown in Table 1. Clinical samples were collected from nine European sites and were originally selected for BRCA1 and BRCA2 testing using ascertainment criteria that included family history and young age of BC diagnosis. Female patients who did not carry a pathogenic variant in BRCA1 or BRCA2 coding regions or splice junctions were selected for testing of variation in the BRCA1 and BRCA2 5′ regions. The controls were as follows: 661 healthy female individuals recruited through the Immunohematology and Transfusion Medicine Service of INT and Associazione Volontari Italiani Sangue (AVIS) of Milan; 312 healthy females above 60 years of age and with no malignancy in the first filial generation recruited through First Faculty of Medicine, Charles University in Prague (Lhota et al., 2016; Soukupova, Zemankova, Kleiblova, Janatova, & Kleibl, 2016); and 130 healthy females without cancer diagnosis recruited in Santiago de Compostela.
Table 1.
Samples used in this study
| Location | Institution | Samples | Gene region |
|---|---|---|---|
| Paris | Institut Curie, Saint Cloud | 686 cases | BRCA1 5′region, BRCA2 5′region |
| Milan | IFOM, Fondazione Instituto FIRC di Oncologia Molecolare |
|
BRCA1 5′region |
| Pisa | Department of Translational Research and New Technologies in Medicine, University of Pisa | 80 cases | BRCA1 5′region, BRCA2 5′region |
| Santiago de Compostela | Fundación Pública Galega de Medicina Xenómica‐SERGAS, Grupo de Medicina Xenómica‐USC, CIBERER, IDIS |
|
BRCA1 5′region, BRCA2 5′region |
| Copenhagen | Center for Genomic Medicine | 1157 cases | BRCA1 5′region, BRCA2 5′region |
| Ghent | Center for Medical Genetics, Ghent University Hospital | 357 cases | BRCA1 5′region, BRCA2 5′region |
| Barcelona | Vall d'Hebron Institute of Oncology | 192 cases | BRCA1 5′region, BRCA2 5′region |
| Prague | CZECANCA – CZEch CAncer panel for Clinical Aplication, Institute of Biochemistry and Experimental Oncology |
|
BRCA1 5′region, BRCA2 5′region |
| Maastricht | Department of Clinical Genetics, Maastricht University Medical Centre | 900 cases | BRCA2 5′region |
2.3. Identification of variants
Regions containing the BRCA1 and BRCA2 promoter and 5′ UTR were sequenced using a range of standard DNA sequencing technologies, and bioinformatic filtering pipelines. Variants mapping to the 2,400 bp region (hg19; chr17:41,278,514 – 41,276,114) of BRCA1 and the 2,000 bp region (hg19; chr13: 32,888,597‐32,890,597) of BRCA2 were considered for further analysis. The identified variants in BRCA1 and BRCA2 5′ noncoding regions are numbered whereby the first translated nucleotide of the translation initiation codon is +1 (https://varnomen.hgvs.org/) using the Mutalyzer website (https://mutalyzer.nl/). BRCA1 is described using NC_000017.10 (hg19 genomic sequence) and NM_007294.3 (transcript). BRCA2 is described using NC_000013.10 (hg19 genomic sequence) and NM_000059.3 (transcript).
2.4. Bioinformatic analysis of variants
As an initial screen, each variant submitted for study was assessed for population frequency using intersection of the variants with dbSNP (version 138 or 150, as the study progressed) within the UCSC Genome browser and Variant Effect Predictor at ENSEMBL (https://www.ensembl.org/info/docs/tools/vep/index.html). Variants with a global minor allele frequency (MAF) of < 0.01 were included in subsequent bioinformatic analyses. Further details of bioinformatics analyses to map active regulatory elements and prioritize variants for functional assays are contained in Supporting Information Methods. Variants were considered to be high priority for experimental analysis if they contained all of the following features: (1) resided in DNaseI or formaldehyde‐assisted isolation of regulatory elements (FAIRE) peaks, (2) coincided with high scores for DNaseI (Base Overlap Signal > 40) or FAIRE (Base Overlap Signal > 10) in a breast cell line, (3) resided in a region of breast cell specific TF binding, (4) overlapped with a TF consensus motif, and (5) were within an evolutionarily conserved element with a high Phastcons score (>0.75). Medium priority variants lacked one or two of these features, whereas low priority variants had only one or none of these features.
2.4.1. In silico TF binding analysis
All rare variants were analyzed in silico using an IT‐based method (Caminsky et al., 2016; Mucaki et al., 2016) and a modified version of the Shannon pipeline utilizing TF information models built from ENCODE ChIP‐seq datasets (Lu, Mucaki, & Rogan, 2017) to assess potential effects of variants on TF binding. Details of analyses are contained in Supporting Information Methods.
2.5. Experimental analysis of variants
2.5.1. Promoter reporter assays
The 499 bp BRCA1 (chr17:41,277,787‐41,277,289) and 750 bp BRCA2 (chr13:32,889,230‐32,889,979) promoter regions were cloned into pCR‐Blunt vector (Thermo Fisher, Waltham, MA). Site‐directed mutagenesis was used to introduce variants using the primers listed in Supporting Information Table S1. Plasmids were purified using the QIAprep miniprep kit (Qiagen, Hilden, Germany) as per the manufacturer's instructions. Plasmid preparations were validated using restriction digest and DNA sequencing and inserts were shuttled into pGL3‐Basic luciferase reporter vector (Promega, Madison, WI). All plasmids for transfection were analyzed for DNA conformation on a 1% w/v agarose gel and only plasmids possessing a supercoiled conformation were used for transfections. Transfection details are described in Supporting Information Methods.
The luciferase‐based reporter assay was performed as described previously (Brewster et al., 2012). Positive controls were B1‐Ets, BRCA1:c.‐330_‐329delinsTT, that decreases BRCA1 promoter activity in MCF7 cells (Atlas, Stramwasser, Whiskin, & Mueller, 2000) and B2‐Ets (E2Fmut1: BRCA2:c.‐282_‐281delinsAA), that has been shown to decrease BRCA2 promoter activity in MCF7 cells (Davis, Miron, Andersen, Iglehart, & Marks, 1999). Statistical analyses were performed in GraphPad Prism using one‐way analysis of variance followed by Tukey's post hoc test and values P < 0.05 were deemed statistically significant.
2.5.2. Electrophoretic mobility shift assays
Nuclear proteins were extracted as described in Supporting Information Methods and electrophoretic mobility shift assays (EMSAs) were carried out using a Pierce LightShift Chemiluminescent EMSA Kit (Thermo Fisher, Waltham, MA) with modifications described in Supporting Information Methods. For competition and supershift studies, nuclear extracts were initially incubated with unlabeled double‐stranded (ds) competitor probes or antibodies in binding buffer before addition of the biotinylated probe and incubation at room temperature. Positive controls for BRCA1 and BRCA2 DNA binding were sequences surrounding the B1‐Ets and B2‐Ets mutations described above.
2.6. Qualitative and quantitative classification of variants
Variants were classified according to the ENIGMA classification criteria for variation in BRCA1 and BRCA2 (https://enigmaconsortium.org/) to determine whether any of the prioritized variants were associated with a high risk of disease. See Supporting Information Methods for further details.
3. RESULTS
3.1. Identification and prioritization of sequence variants in BRCA1 and BRCA2 5´ noncoding regions
The 5′ noncoding regions of BRCA1 and BRCA2 in early onset or familial BC patients with no known BRCA1 or BRCA2 germline pathogenic variant were sequenced at nine different sites as part of an approved ENIGMA (https://enigmaconsortium.org/) project. For the BRCA1 5′ region, 6,475 patients were sequenced at eight different sites along with 1,103 controls. For the BRCA2 5′ region, 6,603 patients were sequenced at eight different sites as well as 442 controls.
After excluding variants with global MAF > 0.01 at time of variant identification, a total of 141 unique single nucleotide variants and short insertions/deletions were identified, 81 in BRCA1 and 60 in BRCA2 (Supporting Information Tables S2 and S3). Theses variants have been submitted to the LOVD databases, http://www.lovd.nl/BRCA1 and http://www.lovd.nl/BRCA2. To evaluate the potential of these rare variants to impact gene regulation, we initially undertook a comprehensive bioinformatic analysis. Promoter regions of BRCA1 and BRCA2 were defined by bioinformatic predictors including chromatin marks (Figure 2). These regions show the characteristic histone H3 epigenetic marks, including H3K4me3, H3K27ac, and H3K9ac, as well as occupancy by multiple TFs. Of the variants identified in cases only, 22 BRCA1 and 23 BRCA2 variants resided within the minimal promoter regions.
Figure 2.
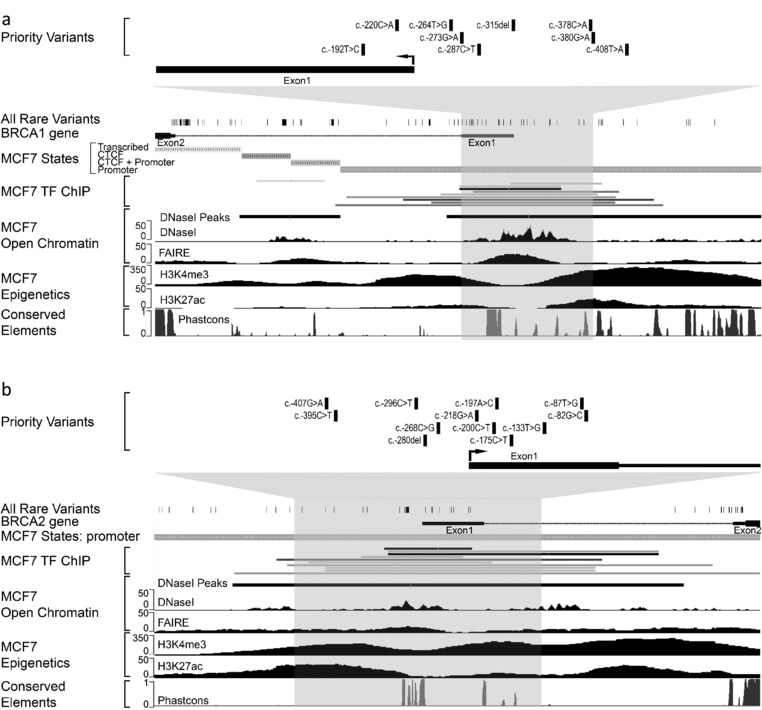
Variants identified in the 5′ regions of BRCA1 and BRCA2 map to predicted regulatory elements. Snapshots of the UCSC genome browser showing regions of BRCA1 (a) and BRCA2 (b) analyzed by targeted sequencing with available ENCODE regulatory marks derived from MCF7 cells. Chromatin segregation states from regulatory region annotation are shown (MCF7 states). The BRCA1 and BRCA2 genomic regions used for functional analyses are highlighted in grey. Prioritized variants within these regions are indicated
To predict the potential impact of variants on promoter activity, we prioritized variants using breast cell specific data for chromatin accessibility and TF occupancy along with evolutionary conservation. Due to the limited breast cell specific TF ChIP‐seq data, we also included ENCODE TF ChIP‐seq and TF consensus motif data from all cell lines. A total of nine BRCA1 and 12 BRCA2 variants were selected for further functional analysis (Figure 2; Tables 2 and 3).
Table 2.
BRCA1 prioritized variants
| Gene | hg19 position (chr17) | Variant namea | rsID | Global MAF in dbSNP | TF motif (ENCODE)b | Bioinformatic priority |
|---|---|---|---|---|---|---|
| BRCA1 | g.41277676A>T | c.‐408T>A | Novel | CEBPB | High/medium | |
| BRCA1 | g.41277648C>T | c.‐380G>A | Novel | RXRA | High/medium | |
| BRCA1 | g.41277646G>T | c.‐378C>A | rs186775935 | 0.00040 | RXRA | High/medium |
| BRCA1 | g.41277583del | c.‐315del | rs901029407 | 0.00003 | ATF1,2,3, CREB1c | Medium |
| BRCA1 | g.41277555G>A | c.‐287C>T | Novel | NFYA, NFYB | High/medium | |
| BRCA1 | g.41277541C>T | c.‐273G>A | rs112960339 | 0.00499 | Medium | |
| BRCA1 | g.41277532A>C | c.‐264T>G | rs904148166 | 0.00003 | Medium | |
| BRCA1 | g.41277488G>T | c.‐220C>A | Novel | Medium | ||
| BRCA1 | g.41277460A>G | c.‐192T>C | rs113323025 | 0.00519 | Medium |
TF, transcription factors.
Based on NM_007294.3.
Overlap with TF motif in ENCODE TF‐ChIP datasets from all cells.
Variant overlaps this motif, but the deletion does not alter the motif sequence.
Table 3.
BRCA2 prioritized variants
| Gene | hg19 Position (Chr13) | Variant namea | rsID | Global MAF in dbSNP | TF motif (ENCODE)b | Bioinformatic priority |
|---|---|---|---|---|---|---|
| BRCA2 | g.32889437G>A | c.‐407G>A | rs36221751 | 0.0018 | Medium | |
| BRCA2 | g.32889449C>T | c.‐395C>T | Novel | Medium | ||
| BRCA2 | g.32889548C>T | c.‐296C>T | rs563971900 | 0.0004 | PAX5 | High/medium |
| BRCA2 | g.32889564delG | c.‐280del | Novel | ELF1, GABPA, ELK1,4 | High | |
| BRCA2 | g.32889576C>G | c.‐268C>G | Novel | High/medium | ||
| BRCA2 | g.32889626G>A | c.‐218G>A | Novel | Medium | ||
| BRCA2 | g.32889644C>T | c.‐200C>T | Novel | MAZ | Medium | |
| BRCA2 | g.32889647A>C | c.‐197A>C | rs370721506 | NA | MAZ | Medium |
| BRCA2 | g.32889669C>T | c.‐175C>T | rs55880202 | 0.0058 | Medium | |
| BRCA2 | g.32889711T>G | c.‐133T>G | Novel | Medium | ||
| BRCA2 | g.32889757T>G | c.‐87T>G | Novel | Medium/low | ||
| BRCA2 | g.32889762G>C | c.‐82G>C | Novel | Medium/low |
NA, no data available, TF, transcription factors.
Based on NM_000059.3.
Overlap with TF motif in ENCODE TF‐ChIP datasets from all cells.
3.2. BRCA1 and BRCA2 promoter activity is altered by 5′ noncoding sequence variants
To examine the potential effect of the 21 prioritized BRCA1 and BRCA2 5′ noncoding variants on regulatory activity, promoter activity was measured using luciferase assays in MCF7 and MDA‐MB‐468 BC cell lines. Two of the nine prioritized BRCA1 variants decreased BRCA1 promoter activity relative to the wild‐type (WT) construct (Figure 3a and 3b). BRCA1:c.‐315del significantly decreased the BRCA1 promoter luciferase activity in both cell lines, whereas BRCA1:c.‐192C decreased luciferase activity in the MCF7 cell line. Furthermore, one variant, BRCA1:c.‐287T, displayed increased activity relative to the WT construct in the MCF7 cell line. For BRCA2, one of the 12 variants, BRCA2:c.‐296T, decreased BRCA2 promoter activity relative to the WT construct in the MCF7 cell line (Figure 3c and 3d).
Figure 3.
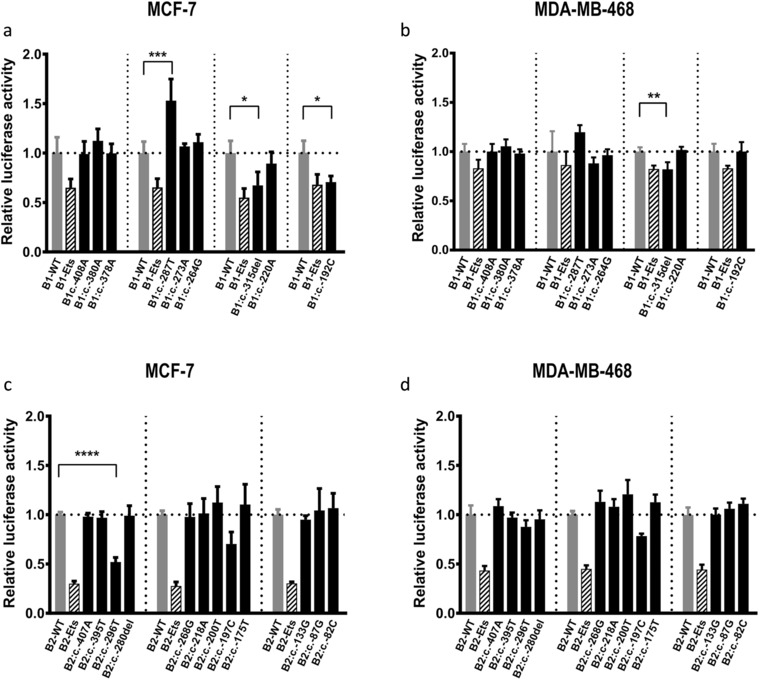
Variants mapping to the 5′ regions of BRCA1 and BRCA2 alter promoter activity in MCF7 and MDA‐MB‐468 breast cancer cells. MCF7 (a and c) and MDA‐MB‐468 cells (b and d) were transfected with pGL3 vectors where luciferase expression is controlled by a portion of the BRCA1 (B1) (a and b) or BRCA2 (B2) (c and d) promoter. Cells were transfected with plasmids containing the wild‐type (WT) promoter sequence (grey bars), positive control (B1‐Ets or B2‐Ets; striped bars) or the indicated variants (black bars). Luciferase expression was normalized to a cotransfected pRL‐TK plasmid. Data represent the average of three independent biological replicates ± standard deviation (SD). The horizontal dotted line represents WT promoter activity set at 1.0‐fold. The vertical dotted lines demarcate individual experiments that include WT, positive control, and variant containing plasmids. (* P ˂ 0.05; ** P ˂ 0.01, *** P ˂ 0.005, **** P ˂ 0.0001)
3.3. In silico analyses of BRCA1 and BRCA2 5′ variants predict alterations in TF binding
BRCA1 and BRCA2 promoters are regulated by a complex array of DNA‐binding proteins and transcriptional coactivators and corepressors (reviewed in McCoy, Mueller, & Roskelley, 2003; Mueller & Roskelley, 2003; Wiedemeyer et al., 2014). In silico analysis was carried out to examine whether the BRCA1 and BRCA2 promoter variants shown to alter luciferase activity (see above) are likely to affect binding of trans‐acting protein factors in breast cells.
Interrogation of ENCODE ChIP‐seq datasets derived from breast cell lines show that, although the number of datasets is limited, TFs bind to regions encompassing the prioritized variants (Figure 2 and Supporting Information Figure S1). ENCODE ChIP‐seq data from other cell lines indicate that some variants are located within consensus motifs for specific TFs associated with these regions (Tables 2 and 3; Supporting Information Figure S1). BRCA1:c.‐287C>T overlaps with the consensus binding motif for CCAAT Box binding factors and BRCA2:c.‐296C>T is located within the consensus motif for PAX5.
IT analysis of the prioritized variants showed that the binding strengths of several TFs are predicted to be altered by the BRCA1 and BRCA2 variants (Table 4 and Supporting Information Table S4). All of the variants that altered promoter activity were predicted to have consequences on TF binding. BRCA1:c.‐287C>T and BRCA2:c.‐296C>T are predicted to disrupt binding of CCAAT Box binding factors and PAX5, respectively. BRCA1:c.‐315del is predicted to disrupt the binding of TCF7L2 but creates a POU2F2 (also known as Oct‐2) binding site. BRCA1:c.‐192T>C is predicted to strengthen a RFX5 site and creates an ETS1 site.
Table 4.
Information theory analysis of prioritized BRCA1/2 variants
| Variant name | TF motif (ENCODE) | Consequences |
|---|---|---|
| BRCA1:c.‐408T>A | CEBPB | CEBPB site weakened (did not meet stringent filtering thresholds) |
| BRCA1:c.‐380G>A | RXRA | Weak RXRA and IRF3 sites weakened, HNF4G site weakened. |
| BRCA1:c.‐378C>A | RXRA | RXR unchanged, HSF1 site lost and GR site created |
| BRCA1:c.‐315del | ATF1,2,3, CREB1a | TCF7L2 site lost and POU2F2 created |
| BRCA1:c.‐287C>T | NFYA, NFYB | NFYA and NFYB sites lost, weak PBX3 site created |
| BRCA1:c.‐273G>A | Altered TF strength did not fulfill stringent filtering thresholdsb | |
| BRCA1:c.‐264T>G | BHLHE32 and MYC sites created. | |
| BRCA1:c.‐220C>A | Altered TF strength did not fulfill stringent filtering thresholdsb | |
| BRCA1:c.‐192T>C | ETS1 site created, weak RFX5 site strengthened. | |
| BRCA2:c.‐407G>A | Weak MEF2A site strengthened, GATA2 site lost. | |
| BRCA2:c.‐395C>T | TEAD4 site lost. | |
| BRCA2:c.‐296C>T | PAX5 | PAX5 site weakened. |
| BRCA2:c.‐280del | ELF1, GABPA, ELK1,4 | GABPA site unchanged, MXI1 andTCF3 sites lost. |
| BRCA2:c.‐268C>G | Altered TF strength did not meet filtering thresholdsb | |
| BRCA2:c.‐218G>A | Altered TF strength did not meet filtering thresholdsb | |
| BRCA2:c.‐200C>T | MAZc | KLF1 site abolished. |
| BRCA2:c.‐197A>C | MAZc | SP4 weakened, GR site weakened, TCF3 site created |
| BRCA2:c.‐175C>T | Altered TF strength did not fulfill stringent filtering thresholdsb | |
| BRCA2:c.‐133T>G | Altered TF strength did not fulfill stringent filtering thresholdsb | |
| BRCA2:c.‐87T>G | Altered TF strength did not fulfill stringent filtering thresholdsb | |
| BRCA2:c.‐82G>C | Altered TF strength did not fulfill stringent filtering thresholdsb |
Variant overlaps this motif, but the deletion does not alter the motif sequence.
Change in information did not fulfill stringent filtering criteria, where [A] site R i < R sequence–1 standard deviation of TF model, or [B] where ΔR i < 4 bits.
No MAZ binding model available.
3.4. 5′ variants in BRCA1 and BRCA2 alter protein–DNA interactions in EMSA analyses
To examine potential alterations in the binding of nuclear proteins from breast cells by the BRCA1 and BRCA2 promoter variants that altered luciferase activity, we carried out EMSA analysis. For BRCA1, two of three analyzed variants, c.‐315del and c.‐287C>T, displayed allele‐specific protein binding (Figure 4). For probes containing the region surrounding the BRCA1:c.‐315del variant, changing the WT sequence to the variant sequence resulted in the enhanced binding of a slower migrating band (Figure 4a and 4b). For probes containing the region surrounding the BRCA1:c.‐287C˃T variant, introduction of the variant sequence resulted in almost complete loss of protein binding to the probe (Figure 4a).
Figure 4.
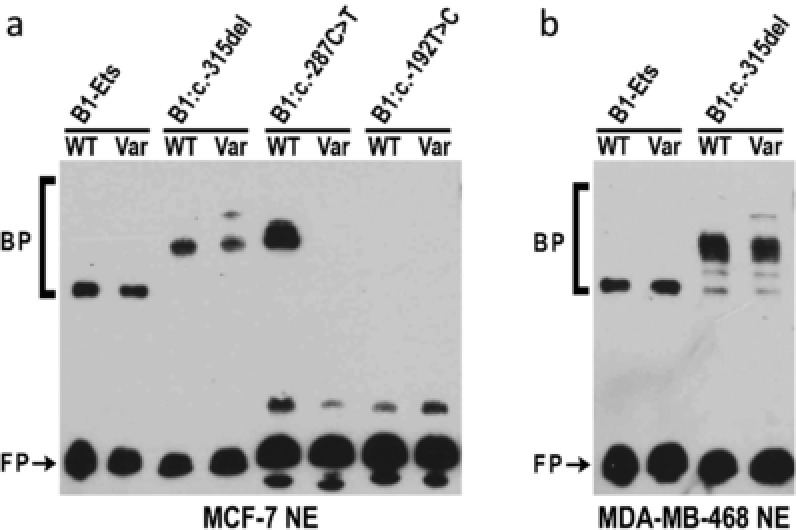
Variants in the 5′ regions of BRCA1 alter DNA:protein complex formation. Electrophoretic mobility shift assay (EMSA) reactions were performed with 3′ biotinylated double‐stranded DNA probes from the BRCA1 5′ region and nuclear extracts (NE) from (a) MCF7 or (b) MDA‐MB‐468 cells. DNA probes contained either wild‐type (WT) or variant (Var) sequences. Free unbound probe (FP) and probe bound by nuclear proteins (BP) are indicated
To determine if the DNA‐protein interactions were specific, competition experiments were performed. In the case of BRCA1:c.‐315del, all bands were competed by both the WT and the variant containing probes in two cell lines (Figure 5a and 5b). For BRCA1:c.‐287C>T, only the WT probe was able to compete for binding (Figure 5c). The nonspecific probe from an unrelated region of the BRCA1 promoter did not compete any bands showing that the bands seen in the EMSA were specific.
Figure 5.
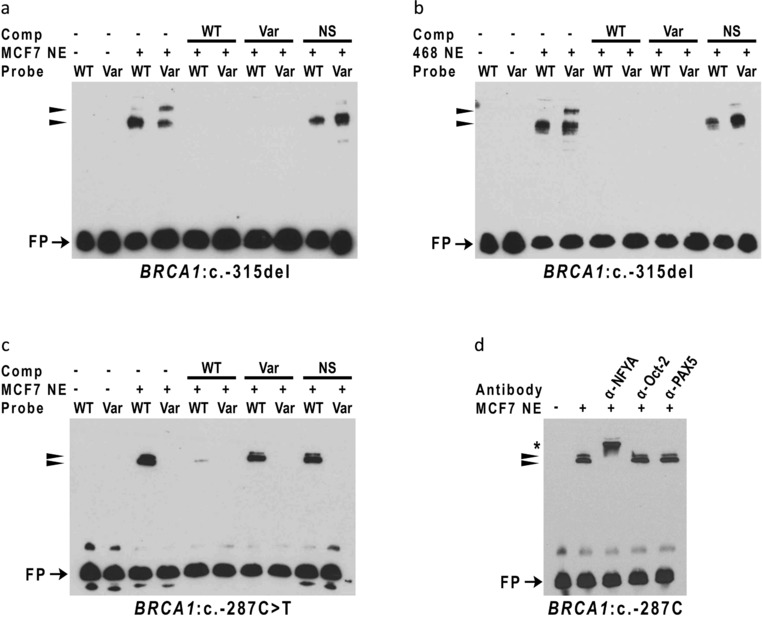
Variant sequences in the BRCA1 5′ region alter specific DNA:protein complex formation. Competition electrophoretic mobility shift assay (EMSAs) were performed using 3′ biotinylated double‐stranded DNA probes containing sequences from the BRCA1 5′ region surrounding the B1:c.‐315del (a and b) and B1:c.‐287C>T (c) variants. DNA probes containing the wild‐type (WT) or variant (Var) sequence were incubated with nuclear extracts from MCF7 cells (MCF7 NE) or MDA‐MB 468 cells (468 NE) in the presence (+) or absence (–) of unlabeled WT, Var, or nonspecific (NS) competitor (Comp) DNA. Free unbound probe (FP) and specific DNA:protein complexes (arrowheads) are indicated. Supershift experiments (d) were performed with the BRCA1:c.‐287C (WT) probe and antibodies to NFYA, Oct‐2 (POU2F2) and PAX5. The supershifted NFYA complex is indicated by asterisk (*)
Analysis of the regions of the BRCA2 promoter using EMSA revealed that region containing the BRCA2:c.‐296C>T variant bound nuclear proteins from MCF7 nuclear extracts and that this interaction was dramatically reduced by introduction of the variant sequence (Figure 6a). Competition experiments showed that these interactions were specific and not competed by a nonspecific probe from an unrelated region of the BRCA1 promoter (Figure 6a).
Figure 6.
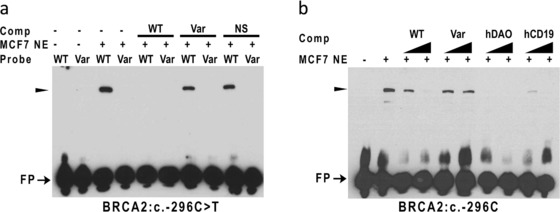
Variants in the 5′ region of BRCA2 alter specific DNA:protein complex formation. Competition electrophoretic mobility shift assay (EMSAs; a) were performed using 3′ biotinylated double‐stranded (ds) DNA probes containing sequences from the BRCA2 5′ region surrounding the BRCA2:c.‐296C>T variant. DNA probes containing the wild‐type (WT) or variant (Var) sequence were incubated with nuclear extracts from MCF7 cells (MCF7 NE) in the presence (+) or absence (–) of unlabeled WT, Var, or nonspecific (NS) competitor (Comp) DNA. Cross‐competition EMSAs (b) contained BRCA2 WT sequences and increasing concentrations of ds competitor DNA containing unlabeled WT, Var, or PAX5 binding sites from the hCD19 gene and D‐amino acid oxidase gene (hDAO). Free unbound probe (FP) and specific DNA:protein complexes (arrowheads) are indicated
To determine the effect of these variants on the binding of specific TFs, competition and supershift analyses were performed. BRCA1:c.‐287C>T overlaps with the consensus binding motif for CCAAT Box binding factors, NFYA and NFYB (Table 2 and Supporting Information Figure S1a), and IT analysis predicts that the variant disrupts binding of these TFs (Table 4). Consistent with these predictions, supershift experiments show that BRCA1:c.‐287C>T disrupts binding of NFYA to this region (Figure. 5d). In addition, we analyzed BRCA2:c.‐296C>T, which maps within the consensus binding motif for PAX5 (Table 2 and Supporting Information Figure S1b), and is predicted by IT analysis to disrupt binding of PAX5 (Table 4), by cross‐competition experiments using known PAX5 binding sites from hCD19 (Kozmik, Wang, Dorfler, Adams, & Busslinger, 1992) and hDAO (Tran et al., 2015) genes. These experiments show that known PAX5 binding sites compete efficiently for binding of nuclear proteins to the BRCA2 promoter region, indicating that PAX5 binding is reduced as a consequence of the nucleotide sequence change (Figure. 6b). In contrast, supershift experiments for POU2F2 (Oct‐2) showed no evidence for BRCA1:c.‐315del causing a change in binding of POU2F2 in the cell line used (data not shown).
3.5. Clinical classification of BRCA1 and BRCA2 5′ noncoding sequence variants
Variants were classified according to the ENIGMA guidelines, which are calibrated for classification of variants as high risk, using available population frequency and/or clinical data (Supporting Information Tables S5 and S6). In this context, the term pathogenicity refers to a variant that confers a high risk of disease. Importantly, these classification guidelines do not identify those variants that confer a moderate or low risk of disease.
Of those variants identified in cases only, 26/70 (37%) of BRCA1 variants had been reported in dbSNP at study initiation (maximum global frequency = 0.006; Supporting Information Table S2), and 22/54 (41%) of BRCA2 variants observed in cases only were identified in dbSNP (maximum global frequency = 0.006; Supporting Information Table S3). Review of variant frequency in public reference groups identified 21 variants that were classifiable, as Not Pathogenic, based on frequency in control groups (Supporting Information Table S5): six BRCA1 and five BRCA2 variants were observed at >1% frequency in population subgroups (stand‐alone evidence against pathogenicity, when detected in a nonfounder outbred population group); six BRCA1 and four BRCA2 variants occurred at frequency 0.001–0.01 (range 0.0014–0.0076) in at least five individuals in the reference set, which combined with a low assumed prior is considered sufficient as evidence against pathogenicity (Supporting Information Table S5). Frequency data from controls screened for this study also supported the frequency‐based classifications for eight of these 21 variants (Supporting Information Table S5).
Segregation analysis for seven informative families aided classification for six variants, whereas histopathology likelihood ratios (LRs) derived for 24 tumors altered classification for 10 variants (Supporting Information Table S6). Combining findings from qualitative and quantitative methods, most variants (113/141; 80%) remained Class 3 Uncertain, largely due to a lack of data.
A total of 27/141 (19%) variants were classified as Not Pathogenic or Likely Not Pathogenic. Of the 21 variants prioritized for functional analysis, eight variants (38%) were classified as Not Pathogenic or Likely Not Pathogenic based on frequency information and/or multifactorial analysis (Table 5), including two variants (BRCA1:c.‐192T>C and BRCA2:c.‐296 C > T) that were shown to decrease promoter activity and in the case of BRCA2:c.‐296 C>T also resulted in perturbed TF binding. Taken together this analysis indicates that none of the variants shown to affect function in this study are associated with a high risk of disease. This analysis is silent, however, on whether these variants may confer a moderate or low risk of disease.
Table 5.
Classification of prioritized variants
| Gene | Genomic location (hg19) | HGVS c. nomenclature | Luciferase result | Combined interpretation of frequency data & multifactorial analysis | Highest MAF (population, database) | Prior probability of pathogenicity | Segregation Bayes score (# families) | Tumor histopathology likelihood ratio (# tumors) | Combined odds for causality | Posterior probability of pathogenicityc |
|---|---|---|---|---|---|---|---|---|---|---|
| BRCA1 | g.41277676A>T | c.‐408T>A | No effect | Uncertain | 0.02 | |||||
| BRCA1 | g.41277648C>T | c.‐380G>A | No effect | Uncertain | 0.02 | 1.67 (1) | 1.67 | NA | ||
| BRCA1 | g.41277646G>T | c.‐378C>A | No effect | Uncertain | 0.0015 (African, 1,000 Genomes) | 0.02 | ||||
| BRCA1 | g.41277583del | c.‐315del | Decrease | Uncertain | 0.02 | |||||
| BRCA1 | g.41277555G>A | c.‐287C>T | Increase | Uncertain | 0.02 | 0.64 (1) | 0.64 | NA | ||
| BRCA1 | g.41277541C>T | c.‐273G>A | No effect | Not pathogenica | 0.0159 (African, 1,000 Genomes) | 0.02 | ||||
| BRCA1 | g.41277532A>C | c.‐264T>G | No effect | Uncertain | 0.02 | 0.51 (1) | 0.51 | NA | ||
| BRCA1 | g.41277488G>T | c.‐220C>A | No effect | Uncertain | 0.02 | |||||
| BRCA1 | g.41277460A>G | c.‐192T>C | Decrease | Not pathogenica | 0.0159 (African, 1,000 Genomes) | 0.02 | ||||
| BRCA2 | g.32889437G>A | c.‐407G>A | No effect | Not pathogenicb | 0.0080 (Prague, this study) | 0.02 | 0.55 (6) | 0.55 | NA | |
| BRCA2 | g.32889449C>T | c.‐395C>T | No effect | Uncertain | 0.02 | |||||
| BRCA2 | g.32889548C>T | c.‐296C>T | Decrease | Not pathogenicb | 0.0080 (Prague, this study) | 0.02 | 3.07 (1) | 1.91 (8) | 5.87 | 0.1069 |
| BRCA2 | g.32889564delG | c.‐280del | No effect | Uncertain | 0.02 | 0.69 (1) | 0.69 | NA | ||
| BRCA2 | g.32889576C>G | c.‐268C>G | No effect | Uncertain | 0.02 | |||||
| BRCA2 | g.32889626G>A | c.‐218G>A | No effect | Likely not pathogenic | 0.02 | 0.52 (1) | 0.72 (1) | 0.38 | 0.0076 | |
| BRCA2 | g.32889644C>T | c.‐200C>T | No effect | Likely not pathogenic | 0.02 | 0.37 (1) | 0.37 | 0.0075 | ||
| BRCA2 | g.32889647A>C | c.‐197A>C | No effect | Not pathogenicb | 0.0014 (African, FLOSSIES) | 0.02 | 1.08 (1) | 1.08 | NA | |
| BRCA2 | g.32889669C>T | c.‐175C>T | No effect | Not pathogenica | 0.0197 (African, FLOSSIES) | 0.02 | ||||
| BRCA2 | g.32889711T>G | c.‐133T>G | No effect | Uncertain | 0.02 | |||||
| BRCA2 | g.32889757T>G | c.‐87T>G | No effect | Uncertain | 0.02 | |||||
| BRCA2 | g.32889762G>C | c.‐82G>C | No effect | Uncertain | 0.02 |
NA, not applicable: multifactorial classification not assigned as the combined odds of causality were insufficient (≥0.5 and ≤2) to derive a posterior probability of pathogenicity (Vallee et al., 2016).
Not pathogenic based on frequency > 1% in an outbred sampleset.
Variant allele assigned a low prior probability of pathogenicity of 0.02 assuming conservatively that 2/100 of such variants might be associated with a high risk of cancer and allele frequency ≥0.001 and < 0.01 in outbred sample set.
Posterior probabilities used to assign IARC 5‐tier class as described in Plon et al., 2008.
4. DISCUSSION
Next generation sequencing and gene panel testing enable rapid analysis of gene regions that have previously not been included in standard screening procedures, including promoters, UTRs, introns, and extragenic regions. It is hypothesized that variants in these regions have potential to modulate gene expression (Stranger et al., 2005; Stranger et al., 2007) and impact on relative disease risk, possibly in collaboration with multiple other low‐, moderate‐, and high‐risk variants (Manolio et al., 2009). This extends and validates our previous study (Santana dos Santos et al., 2017) by using a larger number patients analyzed over nine geographical locations, identifying additional BC‐associated variants, and showing that a subset of these variants modulate binding of specific TFs. Further, we have compared results from our bioinformatics and functional analysis to variant classifications based on ENIGMA BRCA1/2 guidelines for high‐risk variation in these genes.
Through targeted sequencing of over 6,000 early onset/familial BC patients, we identified 141 single nucleotide variants and small indels mapping to the 5′ noncoding regions of BRCA1 and BRCA2. Of these, four (BRCA1:c.‐315del, BRCA1:c.‐287C>T, BRCA1:c.‐192T>C, and BRCA2:c.‐296C>T) caused a significant change in promoter activity. The observed alterations in BRCA1 and BRCA2 promoter activity are of a similar magnitude to that seen with other germline variants associated with BC risk (Michailidou et al., 2017), including a variant in the TERT promoter, which creates a new binding site for Ets factors and results in a 1.2–1.5‐fold increase in luciferase activity in a promoter reporter assay (Horn et al., 2013), and variants in the promoters of KLHDC7A and PIDD1 (Michailidou et al., 2017). Although this supports the hypothesis that moderate change in promoter activity can be associated with disease risk, further work is needed to confirm this.
One of the four variants significantly altered luciferase activity in both tested cell lines, whereas the remaining three variants only affected luciferase activity in MCF7 cells. This may reflect the differential availability of crucial TFs in MDA‐MB‐468 cells (Kao et al., 2009) and highlights the importance of undertaking that assays for functional activity of variants in more than one cell line. Three variants, BRCA1:c.‐380G>A, BRCA2:c.‐296C>T, and BRCA2:c.‐218G>A, were also analyzed in our earlier paper (Santana dos Santos et al., 2017). Although the cell lines used in the two studies were different (MDA‐MB‐231 in Santana dos Santos et al., 2017 and MCF7 and MDA‐MB‐468 here), the trends are the same in five out of six analyses. The difference for BRCA2:c.‐296C>T, which causes a significant decrease in MDA‐MB‐231 and MCF7 cells, but not MDA‐MB‐468 cells, may again be indicative of differential gene expression in BC cell lines (Kao et al., 2009). Overall, however, the consistency of results performed in two separate laboratories underscores the robustness of the assay system.
Some variants were associated with a decrease in promoter activity, whereas others were associated with an increase. As TFs can function as activators or repressors, a variant‐associated change in TF binding can result in either a decrease or an increase in promoter (or other regulatory element) activity. Differences in the quanta and direction of promoter activity have been reported previously (e.g., Fraile‐Bethencourt et al., 2018; Santana dos Santos et al., 2017) and have also been shown to differ between cell lines potentially reflecting the availability of TFs or cofactors (e.g., Zn).
Three of the variants, BRCA1:c.‐315del, BRCA1: c.‐287C>T, and BRCA2:c.‐296C>T, altered protein binding. ENCODE ChIP‐seq data from BC cell lines indicate candidate proteins that are bound to the genomic regions containing these variants (Figure 2 and Supporting Information Figure S1). These include E2F1, CEBPB, GATA3, Max, ELF1, GABP, and FOXA1 for BRCA1 and E2F1, MYC, ELF1, GABP, Max, and PML for BRCA2. Interestingly, a number of these factors have previously been implicated in BC.
In addition, ENCODE ChIP‐seq data from cell lines derived from tissues other than breast indicate that the variants that affect protein binding are located within consensus motifs for specific TFs associated with these regions (Tables 2 and 3; Supporting Information Figure S1). BRCA1:c.‐287C>T overlaps with the consensus binding motif for CCAAT Box binding factors, BRCA1:c.‐315del is located in a consensus motif for CREB/ATF proteins, although the deletion does not modify this motif, and BRCA2:c.‐296C>T is located within the consensus motif for PAX5. IT analysis also predicts that all these variants alter TF binding (Table 4 and Supporting Information Table S4). We show that BRCA1:c.‐287C>T disrupts the binding of NFYA to the BRCA1 promoter region. Furthermore, we present evidence that BRCA2:c.‐296C>T disrupts the binding of PAX5. BRCA1:c.‐315del lies in the so‐called positive regulator region that has been shown to bind GABPα, CREB, and AP‐1 proteins (Atlas et al., 2000; Atlas, Stramwasser, & Mueller, 2001; Graves, Zhou, MacDonald, Mueller, & Roskelley, 2007; Suen & Goss, 1999; Thakur & Croce, 1999). Although these proteins are generally considered activators of transcription, repression of promoter activity by BRCA1:c.‐315del suggests the recruitment of an additional transcriptional repressor or corepressor to this region. IT analysis predicts creation of a binding site for POU2F2, a known repressor; however, we found no evidence to suggest that this variant increased POU2F2 binding in the cell line used, although it is possible that changes may be observable in other cell lines. Biochemical studies, including mass spectrometry, will be required to validate and discover other alterations in TF binding.
One variant, BRCA1:c.‐287C>T, increased promoter activity and decreased protein:DNA interactions. This increase in promoter activity was unanticipated because this variant is within a consensus motif for the CCAAT box binding proteins, NFYA and NFYB, and mutation of this CCAAT box has previously been shown to reduce BRCA1 promoter activity in MCF7 cells (Bindra et al., 2005; Xu, Chambers, & Solomon, 1997). This variant also decreases promoter activity in MDA‐MB‐231 cells (Santana dos Santos et al., 2017). Here, we show that the BRCA1:c.‐287C>T variant reduces NFYA binding. Importantly, NFY proteins can function as transcriptional activators or repressors depending on recruitment of corepressors or coactivators (Peng & Jahroudi, 2002; Peng et al., 2007) and recruitment of TFs to neighboring sequences (Zhu et al., 2012) indicating possible mechanisms for divergent activities of NFY proteins at this site.
BRCA1:c.‐192T>C, which lies in the 5′UTR, decreased reporter activity but did not bind any proteins from MCF7 nuclear extracts in EMSA analysis. Possibly, EMSA binding conditions are not optimal for binding of factors to this sequence or alternatively, this reduction in promoter activity could be by posttranscriptional mechanisms as seen for BRCA2:c.‐26G>A (Gochhait et al., 2007).
Using existing prediction models developed for high risk variants, population frequency and clinical information classified 27 variants as "Not Pathogenic" or "likely Not Pathogenic." This included two BRCA1 and six BRCA2 variants with functional assay data available, six with no statistically significant effect on promoter activity, and two that decreased promoter activity in vitro. These two variants, BRCA1:c.‐192T>C and BRCA2:c.‐296C>T, were observed in population subgroup controls; notably BRCA1:c.‐192T>C was observed at a frequency of >1%, which is considered stand‐alone evidence against pathogenicity (defined as high risk of cancer) for BRCA1/2 variation. This suggests that promoter region variants, irrespective of bioinformatic prediction or functional assay results, are unlikely to be associated with a high risk of cancer. This is consistent with current evidence from ENIGMA studies (de la Hoya et al., 2016), which suggest that an allele resulting in only ∼20–30% expression of BRCA1 transcript/s encoding functional transcripts is not associated with high risk of BC. The low impact of these variants on risk is likely to reflect the complex interplay of TFs and DNA elements, and possible redundancy in the system. For example, a variant in one TF binding site within a cluster may be buffered by other binding sites and thus insufficient on its own to reduce gene expression markedly (Lu & Rogan, 2018).
Given that moderate‐ and low‐risk variants often occur in >1% of the population, and that the remaining 13 variants had insufficient evidence available to assess clinical significance, we cannot exclude the possibility that BRCA1/2 promoter region variants, in particular those with proven functional effect, may be associated with a moderate or low risk of cancer. This indicates an urgent need to further develop prediction models to accommodate criteria for moderate‐ or low‐risk variants by extending the BRCA1/2‐specific criteria developed by ENIGMA (https://www.enigmaconsortium.org/), or even the generic variant classification criteria developed by the American College of Medical Genetics for Mendelian disorders (Richards et al., 2015).
This study has evaluated the significance of single nucleotide variants and small indels mapping to the 5′ noncoding region of BRCA1 and BRCA2 using bioinformatic, biological, and biochemical analyses in combination with consideration of clinical data that inform qualitative and quantitative variant classification. We present data to suggest that a subset of these variants have functional effects on gene regulation. We also present evidence that variants mapping to and affecting the function of BRCA promoters are not likely to be associated with a high risk of cancer. We propose that studies of differing design, such as very large‐scale case‐control sequencing studies able to detect rare variation, will be required to address if a low to moderate risk of cancer may be associated with BRCA1/2 regulatory region variation that has not been captured to date by genome‐wide association genotyping platforms. We believe that the bioinformatic and functional analysis presented will be important to define the design and interpretation of such future sequencing studies. We also believe that this study highlights the challenges associated with classifying variants with respect to low or moderate disease risk, and the need to be cautious in the clinical use of information on individual variants that is likely to be one of many factors contributing to disease risk.
Supporting information
Supplementary Figure S1. Variants in BRCA1 and BRCA2 overlap with potential TF binding sites. Snapshots of the UCSC genome browser showing BRCA1 (A) and BRCA2 (B) prioritized variants and ENCODE ChIP‐seq data from multiple cell lines and available breast cell specific TF ChIPseq data. TF consensus motifs within the ENCODE ChIP‐seq dataset tracks are displayed in green. Genomic position of variants that alter luciferase activity are indicated by vertical lines.
Supplementary Table S1. Oligonucleotides used in this study
Supplementary Table S2. Overview of rare variants in the BRCA1 5'upstream region in patients and controls.*
Supplementary Table S3. Overview of rare variants in the BRCA2 5'upstream region in patients and controls.*
Supplementary Table S4. Information Theory Analysis of Prioritized Variants
Supplementary Table S5. Clinical classification of BRCA1 and BRCA2 5' noncoding variants
Supplementary Table S6. Tumour histopathology status for BRCA1 and BRCA2 5' noncoding variants
Supplementary methods
ACKNOWLEDGEMENTS
The authors would like to acknowledge all the patients that were involved in this study. This work was supported by grants from the National Health and Medical Research Council (ID1104808) and Cancer Council Queensland (ID1044008 and ID1026095) to M.A.B. A.B.S. is supported by an NHMRC Senior Research Fellowship (ID1061779). S.L.E. is supported by an NHMRC Senior Research Fellowship (ID1135932). This work was supported by the grants from the National Cancer Institute (INCa: INCA‐DGOS_8706 to S.M.C.), the Ministry of Health of the Czech Republic (AZV 16–33444A: J.Sevcik, J.S., M.J., P.Z., K.L., L.S., and M.B), and Fondazione Pisa (G. G. and M. C. #2016), the Spanish Instituto de Salud Carlos III (ISCIII) funding (to O.D and S.O.G), an initiative of the Spanish Ministry of Economy and Innovation and partially supported by European Regional Development FEDER Funds: FIS PI12/02585 and PI15/00355 (to O.D.) and PI13/01711 and PI16/01218 (to S.G‐E.). S.G‐E. is supported by the Miguel Servet Progam (CP10/00617). Partial funding also came from a CIBERER grant (ER17P1AC7112/2017) and Fundación Mutua Madrileña to A.V. and a Ghent University Special Research Fund (BOF15/GOA/011) to K.B.M.C. P.K.R. is supported by the Canadian Breast Cancer Foundation, Canadian Foundation for Innovation, Canada Research Chairs Secretariat, and the Natural Sciences and Engineering Research Council of Canada (NSERC Discovery Grant RGPIN‐2015‐06290). T.vO.H and M.R. were supported by The Research Council of The Capital Region of Denmark (Grant E‐22283‐02). P.P. and P.R. were supported by Investigator Grants (#4017 to P.P. and #15547 to P.R.) from the Italian Association for Cancer Research (AIRC).
DISCLOSURE STATEMENT
B.C.S is an employee of and P.K.R is co‐founder of CytoGnomix, which has developed algorithms and software for interpretation of variants within transcription factor binding sites.
Burke LJ, Sevcik J, Gambino G, et al. BRCA1 and BRCA2 5′ noncoding region variants identified in breast cancer patients alter promoter activity and protein binding. Human Mutation. 2018;39:2025–2039. 10.1002/humu.23652
Communicated by Peter J. Oefner
REFERENCES
- Amendola, L. M. , Dorschner, M. O. , Robertson, P. D. , Salama, J. S. , Hart, R. , Shirts, B. H. , … Jarvik, G. P. (2015). Actionable exomic incidental findings in 6503 participants: Challenges of variant classification. Genome Research, 25(3), 305–315. 10.1101/gr.183483.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atlas, E. , Stramwasser, M. , & Mueller, C. R. (2001). A CREB site in the BRCA1 proximal promoter acts as a constitutive transcriptional element. Oncogene, 20(48), 7110–7114. 10.1038/sj.onc.1204890 [DOI] [PubMed] [Google Scholar]
- Atlas, E. , Stramwasser, M. , Whiskin, K. , & Mueller, C. R. (2000). GA‐binding protein alpha/beta is a critical regulator of the BRCA1 promoter. Oncogene, 19(15), 1933–1940. 10.1038/sj.onc.1203516 [DOI] [PubMed] [Google Scholar]
- Betts, J. A. , French, J. D. , Brown, M. A. , & Edwards, S. L. (2013). Long‐range transcriptional regulation of breast cancer genes. Genes, Chromosomes and Cancer, 52(2), 113–125. 10.1002/gcc.22020 [DOI] [PubMed] [Google Scholar]
- Bindra, R. S. , Gibson, S. L. , Meng, A. , Westermark, U. , Jasin, M. , Pierce, A. J. , … Glazer, P. M. (2005). Hypoxia‐induced down‐regulation of BRCA1 expression by E2Fs. Cancer Research, 65(24), 11597–11604. 10.1158/0008-5472.CAN-05-2119 [DOI] [PubMed] [Google Scholar]
- Bogdanova, N. , Helbig, S. , & Dork, T. (2013). Hereditary breast cancer: Ever more pieces to the polygenic puzzle. Hereditary Cancer in Clinical Practice, 11(1), 12 10.1186/1897-4287-11-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewster, B. L. , Rossiello, F. , French, J. D. , Edwards, S. L. , Wong, M. , Wronski, A. , … Peterlongo, P. (2012). Identification of fifteen novel germline variants in the BRCA1 3'UTR reveals a variant in a breast cancer case that introduces a functional miR‐103 target site. Human Mutation, 33(12), 1665–1675. 10.1002/humu.22159 [DOI] [PubMed] [Google Scholar]
- Brown, M. A. , Lo, L. J. , Catteau, A. , Xu, C. F. , Lindeman, G. J. , Hodgson, S. , & Solomon, E. (2002). Germline BRCA1 promoter deletions in UK and Australian familial breast cancer patients: Identification of a novel deletion consistent with BRCA1:pSiBRCA1 recombination. Human Mutation, 19(4), 435–442. 10.1002/humu.10055 [DOI] [PubMed] [Google Scholar]
- Caminsky, N. G. , Mucaki, E. J. , Perri, A. M. , Lu, R. , Knoll, J. H. , & Rogan, P. K. (2016). Prioritizing variants in complete hereditary breast and ovarian cancer genes in patients lacking known BRCA mutations. Human Mutation, 37(7), 640–652. 10.1002/humu.22972 [DOI] [PubMed] [Google Scholar]
- Couch, F. J. , Shimelis, H. , Hu, C. , Hart, S. N. , Polley, E. C. , Na, J. , … Dolinsky, J. S. (2017). Associations between cancer predisposition testing panel genes and breast cancer. JAMA Oncology, 3(9), 1190–1196. 10.1001/jamaoncol.2017.0424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, P. L. , Miron, A. , Andersen, L. M. , Iglehart, J. D. , & Marks, J. R. (1999). Isolation and initial characterization of the BRCA2 promoter. Oncogene, 18(44), 6000–6012. 10.1038/sj.onc.1202990 [DOI] [PubMed] [Google Scholar]
- de la Hoya, M. , Soukarieh, O. , Lopez‐Perolio, I. , Vega, A. , Walker, L. C. , van Ierland, Y. , … Spurdle, A. B. (2016). Combined genetic and splicing analysis of BRCA1 c.[594‐2A>C; 641A>G] highlights the relevance of naturally occurring in‐frame transcripts for developing disease gene variant classification algorithms. Human Molecular Genetics, 25(11), 2256–2268. 10.1093/hmg/ddw094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diederichs, S. , Bartsch, L. , Berkmann, J. C. , Frose, K. , Heitmann, J. , Hoppe, C. , … Wullenkord, R. (2016). The dark matter of the cancer genome: Aberrations in regulatory elements, untranslated regions, splice sites, non‐coding RNA and synonymous mutations. EMBO Molecular Medicine, 8(5), 442–457. 10.15252/emmm.201506055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding, Y. C. , McGuffog, L. , Healey, S. , Friedman, E. , Laitman, Y. , & Paluch‐Shimon, S. , … Consortium of Investigators of Modifiers of, B . (2012). A nonsynonymous polymorphism in IRS1 modifies risk of developing breast and ovarian cancers in BRCA1 and ovarian cancer in BRCA2 mutation carriers. Cancer Epidemiology, Biomarkers & Prevention, 21(8), 1362–1370. 10.1158/1055-9965.EPI-12-0229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles, S. A. , Aboagye, E. O. , Ali, S. , Anderson, A. S. , Armes, J. , Berditchevski, F. , … Thompson, A. M. (2013). Critical research gaps and translational priorities for the successful prevention and treatment of breast cancer. Breast Cancer Research, 15(5), R92 10.1186/bcr3493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans, D. G. R. , van Veen, E. M. , Byers, H. J. , Wallace, A. J. , Ellingford, J. M. , Beaman, G. , … Newman, W. G. (2018). A dominantly inherited 5' UTR variant causing methylation‐associated silencing of BRCA1 as a cause of breast and ovarian cancer. American Journal of Human Genetics, 103(2), 213–220. 10.1016/j.ajhg.2018.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraile‐Bethencourt, E. , Valenzuela‐Palomo, A. , Diez‐Gomez, B. , Infante, M. , Duran, M. , Marcos, G. , … Velasco, E. A. (2018). Genetic dissection of the BRCA2 promoter and transcriptional impact of DNA variants. Breast Cancer Research and Treatment. 10.1007/s10549-018-4826-7 [DOI] [PubMed] [Google Scholar]
- French, J. D. , Ghoussaini, M. , Edwards, S. L. , Meyer, K. B. , Michailidou, K. , Ahmed, S. , … Dunning, A. M. (2013). Functional variants at the 11q13 risk locus for breast cancer regulate cyclin D1 expression through long‐range enhancers. American Journal of Human Genetics, 92(4), 489–503. 10.1016/j.ajhg.2013.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoussaini, M. , Pharoah, P. D. P. , & Easton, D. F. (2013). Inherited genetic susceptibility to breast cancer: The beginning of the end or the end of the beginning? The American Journal of Pathology, 183(4), 1038–1051. 10.1016/j.ajpath.2013.07.003 [DOI] [PubMed] [Google Scholar]
- Gochhait, S. , Bukhari, S. I. , Bairwa, N. , Vadhera, S. , Darvishi, K. , Raish, M. , … Bamezai, R. N. (2007). Implication of BRCA2 ‐26G>A 5' untranslated region polymorphism in susceptibility to sporadic breast cancer and its modulation by p53 codon 72 Arg>Pro polymorphism. Breast Cancer Research, 9(5), R71 10.1186/bcr1780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves, M. L. , Zhou, L. , MacDonald, G. , Mueller, C. R. , & Roskelley, C. D. (2007). Regulation of the BRCA1 promoter in ovarian surface epithelial cells and ovarian carcinoma cells. FEBS Letters, 581(9), 1825–1833. 10.1016/j.febslet.2007.03.072 [DOI] [PubMed] [Google Scholar]
- Heikkinen, T. , Greco, D. , Pelttari, L. M. , Tommiska, J. , Vahteristo, P. , Heikkila, P. , … Nevanlinna, H. (2011). Variants on the promoter region of PTEN affect breast cancer progression and patient survival. Breast Cancer Research, 13(6), R130 10.1186/bcr3076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitchins, M. P. , Rapkins, R. W. , Kwok, C. T. , Srivastava, S. , Wong, J. J. , Khachigian, L. M. , … Ward, R. L. (2011). Dominantly inherited constitutional epigenetic silencing of MLH1 in a cancer‐affected family is linked to a single nucleotide variant within the 5'UTR. Cancer Cell, 20(2), 200–213. 10.1016/j.ccr.2011.07.003 [DOI] [PubMed] [Google Scholar]
- Horn, S. , Figl, A. , Rachakonda, P. S. , Fischer, C. , Sucker, A. , Gast, A. , … Kumar, R. (2013). TERT promoter mutations in familial and sporadic melanoma. Science, 339(6122), 959–961. 10.1126/science.1230062 [DOI] [PubMed] [Google Scholar]
- Kao, J. , Salari, K. , Bocanegra, M. , Choi, Y. L. , Girard, L. , Gandhi, J. , … Pollack, J. R. (2009). Molecular profiling of breast cancer cell lines defines relevant tumor models and provides a resource for cancer gene discovery. PLoS One, 4(7), e6146 10.1371/journal.pone.0006146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozmik, Z. , Wang, S. , Dorfler, P. , Adams, B. , & Busslinger, M. (1992). The promoter of the CD19 gene is a target for the B‐cell‐specific transcription factor BSAP. Molecular and Cellular Biology, 12(6), 2662–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchenbaecker, K. B. , McGuffog, L. , Barrowdale, D. , Lee, A. , Soucy, P. , Dennis, J. , … Antoniou, A. C. (2017). Evaluation of polygenic risk scores for breast and ovarian cancer risk prediction in BRCA1 and BRCA2 mutation carriers. Journal of the National Cancer Institute, 109(7). 10.1093/jnci/djw302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lhota, F. , Zemankova, P. , Kleiblova, P. , Soukupova, J. , Vocka, M. , Stranecky, V. , … Kleibl, Z. (2016). Hereditary truncating mutations of DNA repair and other genes in BRCA1/BRCA2/PALB2‐negatively tested breast cancer patients. Clinical Genetics, 90(4), 324–333. 10.1111/cge.12748 [DOI] [PubMed] [Google Scholar]
- Lu, R. , & Rogan, P. K. (2018). Information‐dense transcription factor binding site clusters identify target genes with similar tissue‐wide expression profiles and serve as a buffer against mutations. bioRxiv. 10.1101/283267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, R. , Mucaki, E. J. , & Rogan, P. K. (2017). Discovery and validation of information theory‐based transcription factor and cofactor binding site motifs. Nucleic Acids Research, 45(5), e27 10.1093/nar/gkw1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio, T. A. , Collins, F. S. , Cox, N. J. , Goldstein, D. B. , Hindorff, L. A. , Hunter, D. J. , … Visscher, P. M. (2009). Finding the missing heritability of complex diseases. Nature, 461(7265), 747–753. 10.1038/nature08494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy, M. L. , Mueller, C. R. , & Roskelley, C. D. (2003). The role of the breast cancer susceptibility gene 1 (BRCA1) in sporadic epithelial ovarian cancer. Reproductive Biology and Endocrinology, 1, 72 10.1186/1477-7827-1-72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michailidou, K. , Lindstrom, S. , Dennis, J. , Beesley, J. , Hui, S. , Kar, S. , … Easton, D. F. (2017). Association analysis identifies 65 new breast cancer risk loci. Nature, 551(7678), 92–94. 10.1038/nature24284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millot, G. A. , Carvalho, M. A. , Caputo, S. M. , Vreeswijk, M. P. , Brown, M. A. , Webb, M. , … Group, E. C. F. A. W. (2012). A guide for functional analysis of BRCA1 variants of uncertain significance. Human Mutation, 33(11), 1526–1537. 10.1002/humu.22150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucaki, E. J. , Caminsky, N. G. , Perri, A. M. , Lu, R. , Laederach, A. , Halvorsen, M. , … Rogan, P. K. (2016). A unified analytic framework for prioritization of non‐coding variants of uncertain significance in heritable breast and ovarian cancer. BMC Medical Genomics, 9, 19 10.1186/s12920-016-0178-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller, C. R. , & Roskelley, C. D. (2003). Regulation of BRCA1 expression and its relationship to sporadic breast cancer. Breast Cancer Research, 5(1), 45–52. 10.1186/bcr557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, Y. , & Jahroudi, N. (2002). The NFY transcription factor functions as a repressor and activator of the von Willebrand factor promoter. Blood, 99(7), 2408–2417. [DOI] [PubMed] [Google Scholar]
- Peng, Y. , Stewart, D. , Li, W. , Hawkins, M. , Kulak, S. , Ballermann, B. , & Jahroudi, N. (2007). Irradiation modulates association of NF‐Y with histone‐modifying cofactors PCAF and HDAC. Oncogene, 26(54), 7576–7583. 10.1038/sj.onc.1210565 [DOI] [PubMed] [Google Scholar]
- Plon, S. E. , Eccles, D. M. , Easton, D. , Foulkes, W. D. , Genuardi, M. , Greenblatt, M. S. , … Group, I. U. G. V. W. (2008). Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat, 29(11), 1282–1291. 10.1002/humu.20880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plon, S. E. , Cooper, H. P. , Parks, B. , Dhar, S. U. , Kelly, P. A. , Weinberg, A. D. , … Hilsenbeck, S. (2011). Genetic testing and cancer risk management recommendations by physicians for at‐risk relatives. Genetics in Medicine, 13(2), 148–154. 10.1097/GIM.0b013e318207f564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popanda, O. , Seibold, P. , Nikolov, I. , Oakes, C. C. , Burwinkel, B. , Hausmann, S. , … Schmezer, P. (2013). Germline variants of base excision repair genes and breast cancer: A polymorphism in DNA polymerase gamma modifies gene expression and breast cancer risk. International Journal of Cancer, 132(1), 55–62. 10.1002/ijc.27665 [DOI] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … ACMG Laboratory Quality Assurance Committee . (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santana dos Santos, E. , Caputo, S. M. , Castera, L. , Gendrot, M. , Briaux, A. , Breault, M. , … Rouleau, E. (2017). Assessment of the functional impact of germline BRCA1/2 variants located in non‐coding regions in families with breast and/or ovarian cancer predisposition. Breast Cancer Research and Treatment, 10.1007/s10549-017-4602-0 [DOI] [PubMed] [Google Scholar]
- Saunus, J. M. , French, J. D. , Edwards, S. L. , Beveridge, D. J. , Hatchell, E. C. , Wagner, S. A. , … Brown, M. A. (2008). Posttranscriptional regulation of the breast cancer susceptibility gene BRCA1 by the RNA binding protein HuR. Cancer Research, 68(22), 9469–9478. 10.1158/0008-5472.CAN-08-1159 [DOI] [PubMed] [Google Scholar]
- Sawyer, S. , Mitchell, G. , McKinley, J. , Chenevix‐Trench, G. , Beesley, J. , Chen, X. Q. , … James, P. A. (2012). A role for common genomic variants in the assessment of familial breast cancer. Journal of Clinical Oncology, 30(35), 4330–4336. 10.1200/JCO.2012.41.7469 [DOI] [PubMed] [Google Scholar]
- Shimelis, H. , Mesman, R. L. S. , Von Nicolai, C. , Ehlen, A. , Guidugli, L. , & Martin, C. , … for, N. C . (2017). BRCA2 hypomorphic missense variants confer moderate risks of breast cancer. Cancer Research, 77(11), 2789–2799. 10.1158/0008-5472.CAN-16-2568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skol, A. D. , Sasaki, M. M. , & Onel, K. (2016). The genetics of breast cancer risk in the post‐genome era: Thoughts on study design to move past BRCA and towards clinical relevance. Breast Cancer Research, 18(1), 99 10.1186/s13058-016-0759-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soukupova, J. , Zemankova, P. , Kleiblova, P. , Janatova, M. , & Kleibl, Z. (2016). [CZECANCA: CZEch CAncer paNel for clinical application— Design and optimization of the targeted sequencing panel for the identification of cancer susceptibility in high‐risk individuals from the Czech Republic]. Klinická Onkologie, 29, (Suppl 1), S46–54. 10.14735/amko2016S46 [DOI] [PubMed] [Google Scholar]
- Spurdle, A. B. , Whiley, P. J. , Thompson, B. , Feng, B. , Healey, S. , Brown, M. A. , … Consortium, E. (2012). BRCA1 R1699Q variant displaying ambiguous functional abrogation confers intermediate breast and ovarian cancer risk. Journal of Medical Genetics, 49(8), 525–532. 10.1136/jmedgenet-2012-101037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger, B. E. , Forrest, M. S. , Clark, A. G. , Minichiello, M. J. , Deutsch, S. , Lyle, R. , … Dermitzakis, E. T. (2005). Genome‐wide associations of gene expression variation in humans. PLoS Genetics, 1(6), e78 10.1371/journal.pgen.0010078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger, B. E. , Nica, A. C. , Forrest, M. S. , Dimas, A. , Bird, C. P. , Beazley, C. , … Dermitzakis, E. T. (2007). Population genomics of human gene expression. Nature Genetics, 39(10), 1217–1224. 10.1038/ng2142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suen, T. C. , & Goss, P. E. (1999). Transcription of BRCA1 is dependent on the formation of a specific protein‐DNA complex on the minimal BRCA1 Bi‐directional promoter. Journal of Biological Chemistry, 274(44), 31297–31304. [DOI] [PubMed] [Google Scholar]
- Tan‐Wong, S. M. , French, J. D. , Proudfoot, N. J. , & Brown, M. A. (2008). Dynamic interactions between the promoter and terminator regions of the mammalian BRCA1 gene. Proceedings of the National Academy of Sciences of the United States of America, 105(13), 5160–5165. 10.1073/pnas.0801048105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur, S. , & Croce, C. M. (1999). Positive regulation of the BRCA1 promoter. Journal of Biological Chemistry, 274(13), 8837–8843. [DOI] [PubMed] [Google Scholar]
- Tran, D. H. , Shishido, Y. , Chung, S. P. , Trinh, H. T. , Yorita, K. , Sakai, T. , & Fukui, K. (2015). Identification of two promoters for human D‐amino acid oxidase gene: Implication for the differential promoter regulation mediated by PAX5/PAX2. Journal of Biochemistry, 157(5), 377–387. 10.1093/jb/mvu084 [DOI] [PubMed] [Google Scholar]
- Vallee, M. P. , Di Sera, T. L. , Nix, D. A. , Paquette, A. M. , Parsons, M. T. , Bell, R. , … Tavtigian, S. V. (2016). Adding In Silico Assessment of Potential Splice Aberration to the Integrated Evaluation of BRCA Gene Unclassified Variants. Hum Mutat, 37(7), 627–639. 10.1002/humu.22973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh, T. , Casadei, S. , Coats, K. H. , Swisher, E. , Stray, S. M. , Higgins, J. , … King, M. C. (2006). Spectrum of mutations in BRCA1, BRCA2, CHEK2, and TP53 in families at high risk of breast cancer. JAMA, 295(12), 1379–1388. 10.1001/jama.295.12.1379 [DOI] [PubMed] [Google Scholar]
- Wardrop, S. L. , Brown, M. A. , & kConFab, I. (2005). Identification of two evolutionarily conserved and functional regulatory elements in intron 2 of the human BRCA1 gene. Genomics, 86(3), 316–328. 10.1016/j.ygeno.2005.05.006 [DOI] [PubMed] [Google Scholar]
- Wiedemeyer, W. R. , Beach, J. A. , & Karlan, B. Y. (2014). Reversing platinum resistance in high‐grade serous ovarian carcinoma: Targeting BRCA and the homologous recombination system. Frontiers in Oncology, 4, 34 10.3389/fonc.2014.00034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, C. F. , Chambers, J. A. , & Solomon, E. (1997). Complex regulation of the BRCA1 gene. Journal of Biological Chemistry, 272(34), 20994–20997. 10.1074/jbc.272.34.20994 [DOI] [PubMed] [Google Scholar]
- Zhu, X. , Wang, Y. , Pi, W. , Liu, H. , Wickrema, A. , & Tuan, D. (2012). NF‐Y recruits both transcription activator and repressor to modulate tissue‐ and developmental stage‐specific expression of human gamma‐globin gene. PLoS One, 7(10), e47175 10.1371/journal.pone.0047175 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1. Variants in BRCA1 and BRCA2 overlap with potential TF binding sites. Snapshots of the UCSC genome browser showing BRCA1 (A) and BRCA2 (B) prioritized variants and ENCODE ChIP‐seq data from multiple cell lines and available breast cell specific TF ChIPseq data. TF consensus motifs within the ENCODE ChIP‐seq dataset tracks are displayed in green. Genomic position of variants that alter luciferase activity are indicated by vertical lines.
Supplementary Table S1. Oligonucleotides used in this study
Supplementary Table S2. Overview of rare variants in the BRCA1 5'upstream region in patients and controls.*
Supplementary Table S3. Overview of rare variants in the BRCA2 5'upstream region in patients and controls.*
Supplementary Table S4. Information Theory Analysis of Prioritized Variants
Supplementary Table S5. Clinical classification of BRCA1 and BRCA2 5' noncoding variants
Supplementary Table S6. Tumour histopathology status for BRCA1 and BRCA2 5' noncoding variants
Supplementary methods