

Abstract
Mycosis fungoides (MF) is the most common cutaneous T‐cell lymphoma (CTCL). Causative genetic alterations in MF are unknown. The low recurrence of pathogenic small‐scale mutations (ie, nucleotide substitutions, indels) in the disease, calls for the study of additional aspects of MF genetics. Here, we investigated structural genomic alterations in tumor‐stage MF by integrating whole‐genome sequencing and RNA‐sequencing. Multiple genes with roles in cell physiology (n = 113) and metabolism (n = 92) were found to be impacted by genomic rearrangements, including 47 genes currently implicated in cancer. Fusion transcripts involving genes of interest such as DOT1L, KDM6A, LIFR, TP53, and TP63 were also observed. Additionally, we identified recurrent deletions of genes involved in cell cycle control, chromatin regulation, the JAK‐STAT pathway, and the PI‐3‐K pathway. Remarkably, many of these deletions result from genomic rearrangements. Deletion of tumor suppressors HNRNPK and SOCS1 were the most frequent genetic alterations in MF after deletion of CDKN2A. Notably, SOCS1 deletion could be detected in early‐stage MF. In agreement with the observed genomic alterations, transcriptome analysis revealed up‐regulation of the cell cycle, JAK‐STAT, PI‐3‐K and developmental pathways. Our results position inactivation of HNRNPK and SOCS1 as potential driver events in MF development.
Keywords: cutaneous T‐cell lymphoma, genomic rearrangements, JAK‐STAT signaling, mycosis fungoides, RNA sequencing, whole‐genome sequencing
1. INTRODUCTION
Mycosis fungoides (MF) is the most common type of cutaneous T‐cell lymphoma (CTCL), a heterogeneous group of neoplasms derived from malignant skin‐homing T cells. MF typically evolves from erythematous cutaneous patches and/or plaques to tumors. Patients with tumor‐stage MF have a 10‐year survival of 42%, which shows the need of a better understanding of the disease and more effective treatments.1
Inactivation of tumor suppressors CDKN2A and CDKN2B are established genetic alterations in MF, whereas mutations in FAS have been reported in subsets of patients.2, 3, 4 In recent years, the copy number alteration (CNA), micro‐RNA (miRNA), and mutational profiles of MF have been investigated using genome‐wide array technologies and next generation sequencing (NGS).
Common CNAs include losses within chromosomes 1, 5, 9, and 13, and gains within chromosomes 7 and 17.5 Highlights of miRNA expression are up‐regulation of oncomirs miR‐93 and miR‐155.6 Gain‐of‐function single nucleotide variants (SNVs) found in solitary or few cases include JAK3 (p.A573V), MAPK1 (p.E322K), STAT3 (p.Y640F), PLCG1 (p.S345F, p.S520F), and TNFRSF1B (p.T377I).7, 8, 9, 10, 11
Even though this body of information has shed some light on the pathogenetics of MF, driver genetic alterations remain unknown. Particularly, the low recurrence of pathogenic small‐scale mutations (ie, SNVs, indels) manifests the need of research on additional facets of MF genetics.
To date, no study has provided insight into the landscape of genomic rearrangements underlying MF. Consequently, we performed an integrated whole‐genome sequencing (WGS)/RNA‐sequencing (RNA‐seq) analysis of tumor‐stage MF to investigate structural aberrations at base‐level resolution.
Our results reveal numerous rearrangements associated with CNAs, and affecting genes involved in signal transduction and transcriptional regulation. Moreover, we identify two recurrently deleted tumor suppressors, HNRNPK and SOCS1, that are novel to MF genetics. These findings broaden our understanding of MF and provide new potential targets for treatment.
2. MATERIALS AND METHODS
2.1. Patient selection
Frozen skin biopsies from nine patients with tumor‐stage MF (IIB‐IVA2, Supporting Information Table 1) were selected for this study. Diagnosis was based on the criteria of the WHO‐EORTC classification for primary cutaneous lymphomas1 and confirmed by an expert panel of dermatologist and pathologists. We subjected eight tumor biopsies to WGS and RNA‐seq, and one biopsy to WGS only. Eighteen additional frozen tumor biopsies (IIB‐IVA2, Supporting Information Table 1) were used as a validation cohort. Whenever available, formalin‐fixed paraffin‐embedded (FFPE) tumor biopsies were used for validation experiments (sequenced and extension cohorts) by fluorescence in situ hybridization (FISH). Frozen and FFPE tumor biopsies contained ≥70% malignant T cells. Patient material was approved by the Leiden University Medical Center institutional review board and informed consent was obtained in accordance with the declaration of Helsinki.
2.2. DNA and RNA isolation
Genomic DNA was isolated using Genomic‐tip 20/G kit (Qiagen) following the manufacturer's protocol. DNA purity (A260/280 and A260/230 ratios) was evaluated using a Nanodrop 1000 system (Nanodrop Technologies, Wilmington, CA). DNA integrity was verified by gel electrophoresis (0.7% agarose, ethidium bromide). Total RNA was isolated using RNeasy mini kit (Qiagen). RNA integrity was verified with an Agilent 2100 Bioanalyzer.
2.3. Sequencing
DNA and RNA were sequenced by the Beijing Genomics Institute (BGI). For whole‐genome sequencing, DNA libraries were prepared using TruSeq Nano DNA HT sample prep kit (Illumina), which involves DNA fragmentation by Covaris sonication, end‐repair, A‐tailing, adapter ligation, and fragment enrichment. Purified DNA fragments were subjected to paired‐end sequencing (2 × 150 bp) on the Illumina HiSeq X‐Ten platform. For RNA sequencing, total RNA was depleted from rRNA using Ribo‐Zero Magnetic kit (Epicentre Biotechnologies, Madison, WI), fragmented, and followed by first‐strand cDNA synthesis, second‐strand cDNA synthesis (with dUTP instead of dTTP), end repair, A‐tailing, adapter ligation, uracil‐N‐glycosylase treatment, and cDNA library enrichment. Purified cDNA libraries were subjected to paired‐end sequencing (2 × 100 bp) on the Illumina HiSeq 4000 platform. All NGS data have been deposited in the European Genome‐Phenome Archive (EGA) under study number EGAS00001002860.
2.4. Sequence data processing
For whole‐genome sequencing, raw reads were processed using FastQC (v0.11.2), SeqTK (v1.0‐r63), Cutadapt, and Sickle (v1.33). Clean reads were aligned to the human reference genome Hg19 using BWA‐mem (v0.7.10) (Supporting Information Table 2). For RNA sequencing, raw reads were processed with FastQC (v0.10.1), Cutadapt (v1.5), and Sickle (v1.33). Clean reads were aligned to human reference genome Hg19 using GSNAP (release December 23, 2014). SAM alignments were compressed and indexed with Picard (v1.120), and fragment counts were obtained with HTseq (v0.6.1p1) using UCSC RefSeq annotations (downloaded July 1, 2015).
2.5. Discovery of DNA rearrangements and fusion transcripts
Genome structural variation (SV) analysis was performed using a set of tools that included Pindel (v0.2.5b8), CleverSV (v2.0rc3), Breakdancer‐max (v1.4.4), and Delly (v0.6.7). Postprocessing of the SV calls included sorting and merging of the calls using a local script and pySVTools (v0.1.3). Each of the structural variant callers were used with default settings and following best practices. SV calls were manually verified and curated using the Integrative Genomic Viewer12 (IGV, v2.3.78). Select events were validated by PCR or FISH. Star Fusion13 (v0.8.0) and FusionCatcher14 (v0.99.6a) were used to detect fusion transcripts in RNA‐seq data. Fusion transcript calls were contrasted with DNA SV data and visually verified on DNA level using IGV. Rearranged genes implicated in cancer were identified using the Network of Cancer Genes 5.0 (NCG 5.0) and literature search.
2.6. Detection of CNAs
Copy number alterations (CNAs) were identified by Control‐FREEC15 using a window size of 50 kb. The output was then subjected to a Wilcoxon rank test and a Kolmogorov‐Smirnov test to generate a list of genomic regions with statistically supported CNAs. CNA regions were visually verified using IGV and select CNA events were validated by digital droplet PCR (ddPCR). Subclonal CNA events were visually detected (coverage changes + supporting reads) using IGV. Select subclonal events were validated by Sanger sequencing.
2.7. Discovery of pathogenic SNVs
SNVs were detected using GATK (v3.5). SNVs present in the dbSNP database were filtered out. We searched for pathogenic SNVs in 1461 genes involved in signaling pathways and cellular processes previously reported as affected in CTCL.7, 8, 9, 16, 17, 18, 19, 20, 21, 22 Gene lists were retrieved from the PathCards database (http://pathcards.genecards.org/). Only SNVs predicted to produce highly deleterious amino acid substitutions by both SIFT and PolyPhen‐2 were further investigated on ClinVar, COSMIC, and literature.
2.8. Differential expression analysis
EdgeR (v3.14.0) was used to normalize fragments per gene counts and determine differentially expressed (DE) genes. Each MF sample was compared individually to a control group formed by seven CD4+ T‐cell subsets (Tnaïve, TH1, TH2, TH17, Treg, TEM, TCM; five samples per subset). RNA‐seq data of T‐cell controls were generated by Ranzani et al. and downloaded from EBI (https://www.ebi.ac.uk/).23 Testing was performed using negative binomial generalized log‐linear models. Only genes found to be commonly up‐regulated or down‐regulated (FDR < 0.01) in all MF samples were regarded as DE genes. DE genes implicated in cancer were identified using NCG 5.0 and literature search.
2.9. Functional annotation, GSEA, and pathway analysis
Functional annotation of rearranged genes was performed using Panther24 (v11.1). Gene set enrichment analysis25 (GSEA, v2.2.4) was run as a preranked analysis with 1000 permutations using the hallmarks gene set from the Molecular Signatures Database (MSigDB). Normalized enrichment scores (NES) were calculated to determine expression signatures. FDR q values were obtained. Pathway analysis with DE genes was performed with DAVID26 (v6.8) using default settings.
2.10. FISH
FISH for CLEC16A was performed on FFPE tumor and plaque biopsies using bacterial artificial chromosome (BAC) probes. For all tumor samples except one (MF5), probe mix A (RP11‐727C18, RP11‐916G12, and RP11‐959J23; telomeric/5′) and probe mix B (RP11‐722I5, RP11‐829F21, and RP11‐936M1; centromeric/3′) were employed (Supporting Information Figure 1; Supporting Information Table 3). For sample MF5, probe mix A (telomeric/5′) and RP11‐396B14 (centromeric/3′) were used. For the two plaque samples included in this study, we used a combination of break apart and fusion FISH. Break apart probe mixes A and B were used together with patient‐specific fusion probes (MF3: RP11‐107L10, RP11‐1097K16, and RP11‐421N18; MF4: RP11‐625M5 and RP11‐1083M15) (Supporting Information Figure 1; Supporting Information Table 3). Probes were purchased from BACPAC resources at Children's Hospital Oakland (CHORI) and their identity confirmed by FISH on metaphase controls and Sanger sequencing. DNA was isolated from BAC clones by alkaline extraction, and labeled with haptens (digoxigenin (DIG)‐, biotin (BIO)‐ or dinitrophenyl (DNP)‐coupled dideoxynucleotides) by nick translation. FFPE tissue sections were subjected to deparaffinization in xylene, pre‐treatment in 10 mM citrate buffer, digestion in 0.4% pepsin, co‐denaturation, and hybridization with the probes. Hybridized sections were then incubated with fluorescein isothiocyanate (FITC)‐conjugated mouse anti‐DIG antibodies, Cy3‐conjugated streptavidin, and rabbit anti‐DNP antibodies; and subsequently, incubated with Alexa 488‐conjugated goat antimouse antibodies and Cy5‐conjugated goat antirabbit antibodies. Finally, sections were counterstained with 4,6 diamidino‐2‐phenylindole (DAPI). Analysis was performed by manually scoring 100 tumor cells per section.
2.11. Digital droplet PCR
Select CNAs were validated by using Bio‐Rad QX200 ddPCR system (Supporting Information Figure 2) following the manufacturer's guidelines. In short, 20–40 ng of genomic DNA was mixed with a frequent‐cutting restriction enzyme, ddPCR supermix, FAM‐labeled probes against the gene of interest and HEX‐labeled probes against the reference gene in a 96‐well PCR plate. Each 20 μL reaction was then transferred to a droplet generation cartridge, partitioned into nano droplets, and pipetted back to a fresh 96‐well PCR plate by using Bio‐Rad QX200 automated droplet generator. The plate containing the emulsified samples was sealed with foil and amplified on a Bio‐Rad T100 thermocycler. PCR program was the following: 95°C for 10 min, 39 cycles of 94°C for 30 s, and 60°C for 1 min, and 98°C for 10 min. The plate containing the post‐PCR nano droplets was then placed into Bio‐Rad QX200 droplet reader, which aspirates droplets and measures FAM/HEX fluorescence one droplet at a time. Copy number values were determined with Bio‐Rad Quantasoft software v1.7.4. Reported copy numbers of HNRNPK and SOCS1 in samples from the validation cohort are the average of three independent measurements using different reference genes.
3. RESULTS
3.1. Landscape of genomic rearrangements
The number of rearrangements ranged from 13 to 62 per patient (352 total events; mean/patient ± SD, 39 ± 18) (Figures 1 and 2A; Supporting Information Table 4). Fifty‐two percent of events were interchromosomal (range/patient, 35%–85%) (Figure 2B). Thirty‐one percent of events fused two annotated genes, while the rest joined either a gene with a nongenic region or two nongenic regions, or reshuffled sequences within a single gene. Seven percent of rearrangements resulted in fusion transcripts (mean fusions/patient, 3; range, 1–5 fusions/patient) (Figure 2C; Supporting Information Table 5). We also observed chromothripsis‐like events in three patients (ie, MF1, MF6, MF9) who carry numerous complex rearrangements in chromosomes 1 and X, 6 and 10, and 1 and 5, respectively (Figures 1 and 2D).
Figure 1.
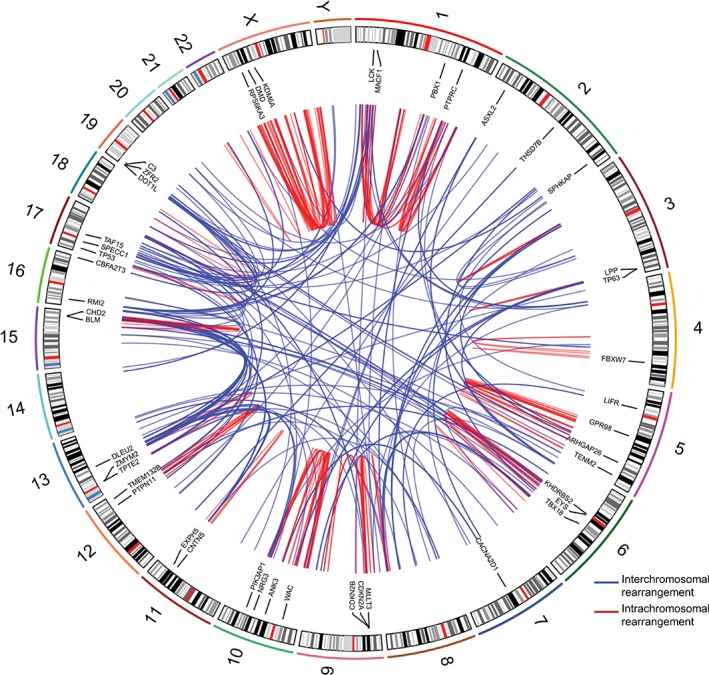
Landscape of genomic rearrangements in mycosis fungoides. Circos plot depicting 352 genomic rearrangements identified in nine MF genomes by WGS. The outer ring consists of chromosome ideograms arranged circularly end to end. The inner area in the plot shows arcs that represent interchromosomal (blue) and intrachromosomal (red) rearrangements. The ring between the chromosome ideograms and the arcs contains labels indicating rearranged genes implicated in cancer [Color figure can be viewed at http://wileyonlinelibrary.com]
Figure 2.
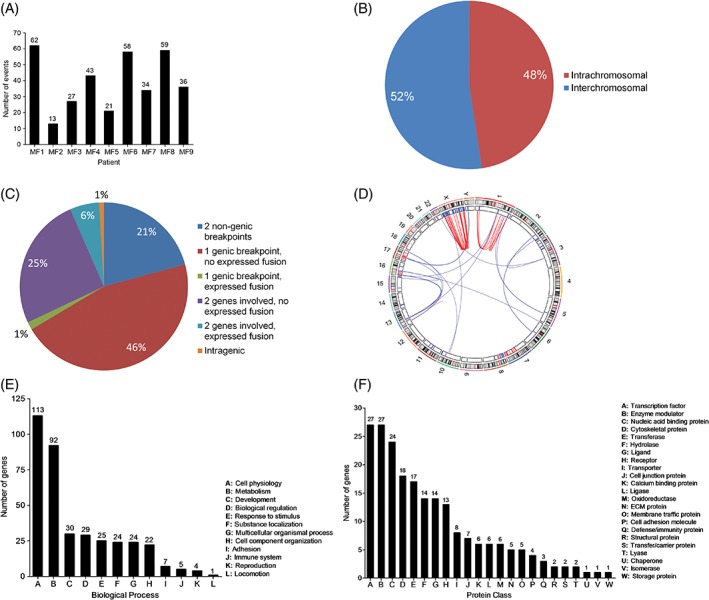
Distribution and functional annotation of genomic rearrangements in mycosis fungoides. A, Number of genomic rearrangements per sample. B, Distribution of inter‐ and intrachromosomal rearrangements. C, Distribution of genomic rearrangements based on the type of DNA sequences involved in the event (genic or nongenic) and the expression of fusion sequences determined through integration of WGS and RNA‐seq data. D, Circos plot illustrating chromothripsis‐like events in chromosomes 1 and X of sample MF1. The plot shows that complex rearrangements are associated with deletion of genomic regions. E, Distribution of rearranged genes according to the biological process their protein products take part in. F, Distribution of rearranged genes according to the protein class they encode (143 of 270 rearranged genes were assigned to a protein class by Panther) [Color figure can be viewed at http://wileyonlinelibrary.com]
A total of 270 genes were found to be rearranged (Supporting Information Table 6), 47 of which are currently implicated in cancer (Supporting Information Table 7). This group includes genes previously associated with MF or Sézary Syndrome (SS) (ie, CDKN2A, CDKN2B, DLEU2, KDM6A, TP53, TP63, and VAV1)5, 19 and genes implicated in other hematological malignancies (eg, ARHGAP26, CBFA2T3, CHD2, DOT1L, LCK, LPP, PBX1, PTPN11, MLLT3, TAF15, SPECC1, ZMYM2) (Figure 1).27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38
Functional annotation of rearranged genes reveals a diverse set of biological processes (Figure 2E; Supporting Information Table 8), being cell physiology (n genes = 113) and metabolism (n genes = 92) the highest ranking categories. Breakdown of these two categories shows that cell communication (n genes = 34) and cell cycle (n genes = 16) are the most affected physiological processes while nucleic acid metabolism (n genes = 48, esp. transcription) leads the group of impacted biochemical processes (Supporting Information Table 8).
In support of the postulated deleterious effects of genomic rearrangements on normal cellular functions, at least 100 rearranged genes (127/270 genes could not be assigned to a protein class by Panther) encode proteins with relevant roles in signal transduction (eg, ligands, receptors, enzyme modulators) and transcriptional regulation (eg, transcription factors, chromatin regulators) (Figure 2F; Supporting Information Table 9). Nonetheless, rearranged genes do not group into a single or a few signaling pathways, but take part in numerous different pathways/processes.
We found nine recurrently rearranged genes, ARHGAP26 (two of nine patients), ATXN1 (two of nine), CLEC16A (four of nine), ELF1 (two of nine), EYS (two of nine), RBPJ (two of nine), RPS6KA3 (two of nine), SLC24A2 (two of nine), and SSH2 (two of nine) in our sequenced cohort. However, in all cases fusion partners differ between patients and the resulting chimeric sequences are expressed only in single patients (five of nine rearranged genes) or not expressed (four of nine). Interestingly, 50% of recurrent CNAs containing cancer genes are associated with inter‐ or intrachromosomal rearrangements. For instance, ARID1A, CDKN2A/B, PTPRC, SOCS1, and STK11 are deleted as a consequence of chromosomal rearrangements in two or more patients. This fact, together with the small percentage of expressed gene fusions, suggests that in MF, rearrangements more often mediate inactivation of tumor suppressors, rather than generate oncogenic fusions.
3.2. Aneuploidies and CNAs
Large‐scale numerical abnormalities (>3 Mb) included trisomy 4 (one of nine patients), 7 (two of nine) and 18 (two of nine), as well as deletions within 9q (four of nine patients), 10q (two of nine) and 16q (three of nine), and gains within 3q (three of nine patients), 5p (three of nine), 7q (two of nine), 8q (two of nine) and 17q (four of nine) (Figures 3 and 4).
Figure 3.

Overview of CNAs in mycosis fungoides. Human chromosome ideograms showing regions of gain and loss identified by WGS in nine MF genomes. Red bars to the left of each chromosome represent regions of gain while blue bars to the right of each chromosome represent regions of loss [Color figure can be viewed at http://wileyonlinelibrary.com]
Figure 4.

Distribution of recurrent CNAs and pathogenic SNVs in mycosis fungoides. Upper panel, broad CNAs (>3 Mb); middle panel, focal MCRs (≤3 Mb) shared by CNAs. Bona fide cancer genes contained within each focal MCR are indicated; bottom panel, pathogenic SNVs. Only SNVs for which functional validation has been reported in literature are shown [Color figure can be viewed at http://wileyonlinelibrary.com]
We identified 18 focal (≤3 Mb) minimal common regions (MCRs) shared by CNAs, with 15/18 MCRs containing cancer genes (Figures 3 and 4; Supporting Information Table 10). These focal MCRs affect genes primarily involved in cell cycle control, chromatin regulation, the JAK‐STAT pathway, and the PI‐3‐K pathway.
The most frequent focal MCR was 9p21.3 deletion, found in seven of nine patients. This region encloses exon 2 and 3 of CDKN2A. We found deletions at 16p13.13 and 9q21.32 in five of nine patients, which include JAK‐STAT pathway regulator SOCS1 and TP53‐dependent p21 co‐activator HNRNPK, respectively.
Deletions at 1q31.3 and 13q14.3 were observed in four of nine patients. The former involves JAK‐STAT inhibitor PTPRC while the latter contains the DLEU2/Mir‐15a/16‐1 locus, which is frequently deleted in chronic lymphocytic leukemia (CLL).39
Additionally, three of nine patients had deletions at 1p36.11, 9q21.31, 10q23.31, 13q14.11, 19p13.3, and 20q13.13, which include tumor suppressors ARID1A, TLE4, PTEN, FOXO1, STK11 (alongside TCF3) and PTPN1, respectively. Lastly, two of nine patients presented deletions at 5q15‐21.1, 6p22.3, and 16q24.3 which involve (putative) tumor suppressors CHD1, JARID2, and CBFA2T3, respectively.
In contrast, focal MCRs within gain areas were rare (n = 3), with gain at 8q24.21 (involving MYC) found in two of nine patients, being the only event containing a cancer gene.
3.3. Pathogenic SNVs
Prior studies showed that recurrent pathogenic SNVs in MF are rare.7, 8, 9, 19 Nevertheless, we looked for pathogenic SNVs in exonic sequences of genes involved in the JAK–STAT pathway, the MAPK pathway, the NF‐κB pathway, the PI‐3‐K pathway, the T‐cell receptor (TCR) pathway, cell cycle control, chromatin organization, and genes that are presumed drivers19 in CTCL (Supporting Information Table 11).
Recurrent SNVs were found in two genes, FGFR4 (p.G388R,40 three of nine patients; expressed: 1/3) and JAK3 (p.A573V,41 two of nine patients), both of which are known gain‐of‐function mutations. The remaining SNVs predicted as pathogenic occur in single patients only. These include gain‐of‐function SNVs in BRAF (p.G466E42), JAK3 (p.V722I43), KRAS (p.G13C44), MYD88 (p.L273P45), and STAT3 (p.Y640F46), which have been reported either in CTCL or other malignancies and functionally validated elsewhere (Figure 4; Supporting Information Figure 3); also, SNVs in CHEK2 (p.I200T47) and MITF (p.E419K48), which are germline risk factors for breast cancer and melanoma, respectively (Figure 4); and 47 other patient‐specific SNVs (predicted as highly deleterious by PolyPhen‐2 and SIFT) (Supporting Information Table 12) located in genes with relevant roles in the aforesaid pathways. Importantly, pathogenic SNVs in genes from the JAK‐STAT and MAPK pathway are not mutually exclusive.
3.4. Differentially expressed genes and fusion transcripts
We identified differentially expressed (DE) genes by comparing expression in our MF cohort with expression in normal CD4+ T cells. Since the cell of origin of MF remains unidentified, transcriptome analysis was performed using a control group formed by several CD4+ T‐cell subsets (see Materials and Methods) with the aim of detecting aberrant expression patterns that are absent in a range of normal CD4+ phenotypes. A total of 733 genes (450 up‐regulated, 283 down‐regulated, FDR <0.01) were found to be differentially expressed (Figure 5A, Supporting Information Table 13). We next used NCG 5.0 to pinpoint DE genes implicated in cancer. Eighty‐one cancer genes (51 up‐regulated, 30 down‐regulated) were identified (Supporting Information Table 14). Up‐regulated genes include oncogenes MALAT1, MECOM, PBX1, TTK, and WWTR1, whereas down‐regulated genes include tumor suppressors BRD7, CDKN1B, CYLD, HNRNPK, TSC1, and XPA. The expression profile also comprises up‐regulation of developmental genes GLI3, JAG1, and NOTCH3, and down‐regulation of transcriptional repressor FOXP1 and cell proliferation inhibitors GPS2 and RHOH (Figure 5B–D).
Figure 5.
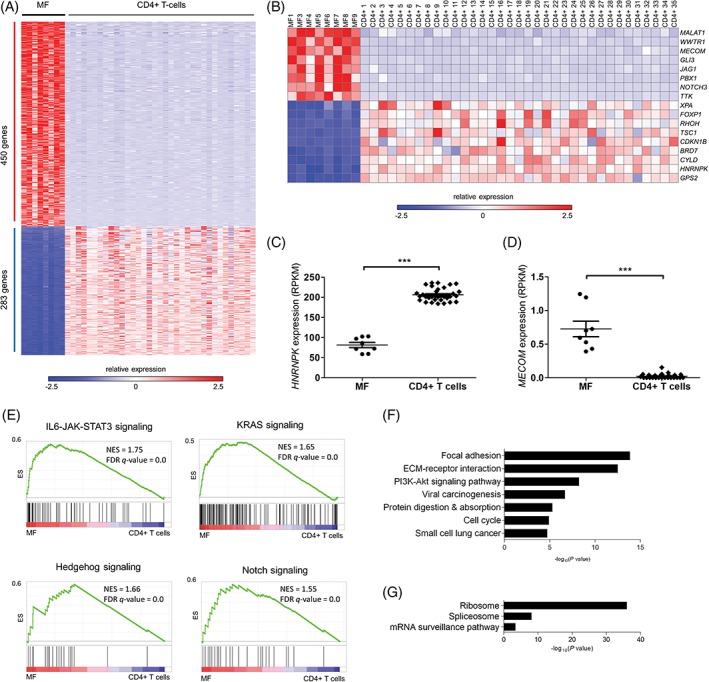
RNA‐seq identifies differentially expressed cancer genes and deregulated signaling pathways in mycosis fungoides. A, Heat map of differentially expressed (DE) genes (FDR < 0.01) in MF when compared to CD4+ T cells. Of 733 DE genes, 450 were commonly up‐regulated and 283 were commonly down‐regulated. B, Oncogenes and tumor suppressors with roles in cell cycle control and development are among the group of DE genes. C and D, HNRNPK and MECOM, whose deregulation (down‐ and up‐, respectively) are reported to enhance the JAK‐STAT pathway, are differentially expressed in MF (HNRNPK: −2.5‐fold average, ***P < 1 × 10−4; MECOM: 31‐fold average, ***P < 1 × 10−4, Mann‐Whitney test). E, Gene set enrichment analysis. Select GSEA plots showing up‐regulation of STAT3 signaling (upper left), KRAS signaling (upper right), Hedgehog signaling (lower left) and Notch signaling (lower right) in MF compared to CD4+ T cells (see Supporting Information Table 15 for a complete list of GSEA signatures). NES, normalized enrichment score; FDR q‐value, false discovery rate q‐value. F and G, Pathway analysis by DAVID reveals up‐regulation of the PI‐3‐K/Akt pathway, the cell cycle and cancer signatures, and down‐regulation of ribosome, spliceosome and mRNA surveillance (see Supporting Information Table 16 for a complete list of enriched terms/processes) [Color figure can be viewed at http://wileyonlinelibrary.com]
We detected 24 patient‐specific fusion transcripts (Table 1), including 6 (ie, ATXN1‐TP63, CCR7‐DOT1L, KDM6A‐IL1RAPL1, LMF1‐TAF15, TP53‐GPR3, and YTHDF3‐LIFR) that contain genes implicated in cancer. To our knowledge, with the exception of ATXN1‐TP63, 49 all these chimeric transcripts are reported for the first time.
Table 1.
Fusion transcripts detected by RNA‐seq in tumor‐stage MF
| Sample | Fusion transcript | Breakpoints (DNA) | Breakpoint type | Event class | WGS confirmed |
|---|---|---|---|---|---|
| MF1 | KDM6A–IL1RAPL1 | chrX:44746566–chrX:29451290 | Genic–genic | ITX | Yes |
| MF1 | CHIC1–RP2 | chrX:72844450–chrX:46680435 | Genic–nongenic | ITX | Yes |
| MF3 | ANKRD13A–CUL9 | chr12:110448655–chr6:43160142 | Genic–genic | CTX | Yes |
| MF3 | CLEC16A–SCARB1 | chr16:11067010–chr12:125350896 | Genic–nongenic | CTX | Yes |
| MF3 | SSH2–GRAP2 | chr17:28059210–chr22:40314573 | Genic–genic | CTX | Yes |
| MF3 | LMF1–TAF15 | chr16:986148–chr17:34145925 | Genic–genic | CTX | Yes |
| MF3 | ATXN1–TP63 | chr6:16307814–chr3:189470345 | Genic–genic | CTX | Yes |
| MF4 | CCR7–DOT1L | chr17:38718403–chr19:2181252 | Genic–genic | CTX | Yes |
| MF5 | PHACTR4–EPB41 | chr1:28755797–chr1:29246304 | Genic–genic | iDel | Yes |
| MF5 | ADAM12–MMRN2 | chr10:127935628–chr10:88698606 | Genic–genic | iDel | Yes |
| MF5 | TRAPPC10–TRPM2 | chr21:45487448–chr21:45795335 | Genic–genic | iDel | Yes |
| MF5 | ARHGAP26–TENM2 | chr5:142272242–chr5:167448836 | Genic–genic | ITX | Yes |
| MF6 | ANK3–RNLS | chr10:62168031–chr10:90101461 | Genic–genic | ITX | Yes |
| MF6 | ELF1–SATB2 | chr13:41540637–chr2:200369768 | Genic–nongenic | CTX | Yes |
| MF7 | TP53–GPR3 | chr17:7579754–chr1:27718138 | Genic–nongenic | CTX | Yes |
| MF7 | CLPP–NR3C1 | chr19:6365544–chr5:142800539 | Genic–genic | CTX | Yes |
| MF7 | SARNP–WRAP53 | chr12:56161974–chr17:7593927 | Genic–genic | CTX | Yes |
| MF8 | SETD5–RNF19A | chr3:9497374–chr8:101391443 | Genic–nongenic | CTX | Yes |
| MF8 | SUDS3–TMEM132B | chr12:118847216–chr12:125987360 | Genic–genic | ITX | Yes |
| MF8 | AACS–STAB2 | chr12:125625503–chr12:104094623 | Genic–genic | ITX | Yes |
| MF8 | RPUSD3–RNF19A | chr3:9882804–chr8:101303862 | Genic–genic | CTX | Yes |
| MF8 | YTHDF3–LIFR | chr8:64081882–chr5:38586949 | Genic–genic | CTX | Yes |
| MF9 | DPM1–UBE2V1 | chr20:49574368–chr20:48703893 | Genic–genic | iDel | Yes |
| MF9 | KCNAB2–ESPN | chr1:6071941–chr1:6493075 | Genic–genic | iDel | Yes |
Abbreviations: CTX, interchromosomal translocation. ITX, intrachromosomal translocation. iDel, interstitial deletion.
3.5. Deregulated signaling pathways
To look for evidence of deregulated pathways in MF, we performed GSEA using annotated gene sets from MSigDB to look for expression signatures. The analysis revealed up‐regulation of IL6‐JAK‐STAT3 signaling (NES = 1.75, FDR q‐value = 2.62 × 10−4), KRAS signaling (NES = 1.65, FDR q‐value = 1.8 × 10−3), Hedgehog signaling (NES = 1.66, FDR q‐value = 1.8 × 10−3), and Notch signaling (NES = 1.55, FDR q‐value = 0.01) (Figure 4E; Supporting Information Table 15).
In addition, we performed DAVID pathway analysis with up‐ and down‐regulated genes separately. Cellular processes associated with integrin‐mediated signaling (ie, ECM‐receptor interaction, focal adhesion), PI‐3‐K/Akt signaling (P = 4.6 × 10−10), cancer signatures (ie, viral carcinogenesis, small cell lung cancer) and cell cycle (P = 1.1 × 10−5) are prominent up‐regulated profiles in MF (Figure 5F; Supporting Information Table 16). Down‐regulated profiles include ribosome (P = 1.0 × 10−36) and spliceosome (P = 8.7 × 10−9) activity, and mRNA surveillance (P = 4.1 × 10−4) (Figure 5G; Supporting Information Table 16).
3.6. SOCS1 and HNRNPK are recurrently deleted
From all structural alterations revealed by our analysis, deletion of HNRNPK and SOCS1 stand out because of their novelty and recurrence. Notably, apart from being deleted in five of nine sequenced patients, HNRNPK is down‐regulated in eight of eight transcriptomes (2.5‐fold average, P < 1 × 10−4) (Figures 5C and 7A) whereas SOCS1 deletions are invariably focal (≤3 Mb) in five of five affected patients (MCR: 305 kb, three genes) (Figures 6 and 7B; Supporting Information Figure 4). Consequently, we evaluated copy number of HNRNPK and SOCS1 in 18 additional tumor biopsies by ddPCR. In this extension cohort we found HNRNPK deletion in five patients and SOCS1 deletion in four patients (Figure 7C,D). Taking together the sequenced and extension cohorts, HNRNPK was deleted in 10 of 27 (37%) patients and SOCS1 in 9 of 27 (33%) patients.
Figure 7.
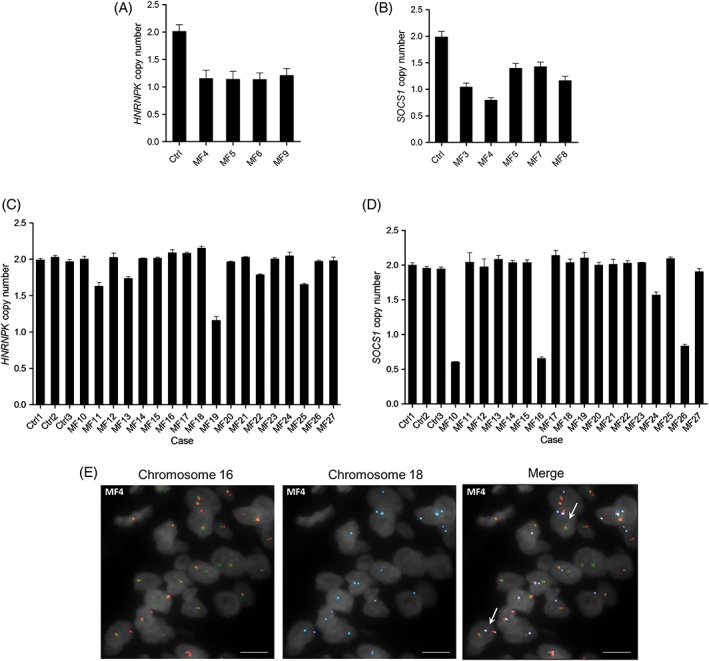
HNRNPK and SOCS1 are recurrently deleted in mycosis fungoides. Deletion of (A) HNRNPK and (B) SOCS1 in sequenced tumor samples was confirmed by ddPCR. Deletion of (C) HNRNPK and (D) SOCS1 was also identified in samples from the extension cohort by ddPCR. E, The translocation responsible for SOCS1 deletion in sample MF4 was found in (early‐stage) plaque tissue by FISH. Ctrl, CD4+ T cells. Scale bar, 10 μm [Color figure can be viewed at http://wileyonlinelibrary.com]
Figure 6.
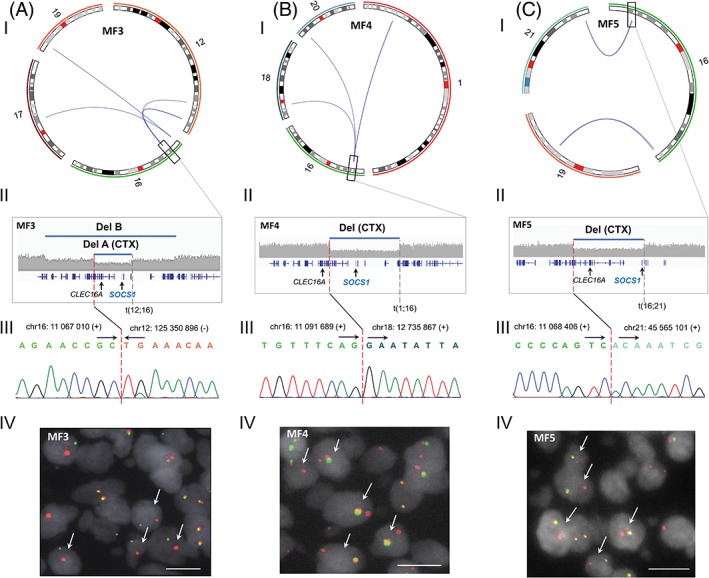
Genomic rearrangements at 16q13.13 are associated with focal SOCS1 deletions in mycosis fungoides. (I) Circos plots displaying genomic rearrangements at 16q13.13. (II) Magnified views of deletions at 16q13.13 resulting from structural alterations. Genomic rearrangements at 16q13.13 validated by (III) Sanger sequencing and (IV) break apart FISH in (A) MF3, (B) MF4, and (C) MF5. Del, deletion. CTX, interchromosomal translocation. Scale bar, 10 μm [Color figure can be viewed at http://wileyonlinelibrary.com]
3.7. Deletion of SOCS1 can be found at early stage
We seized upon the fact that SOCS1 deletions mostly result from translocations in our sequenced cohort to investigate their occurrence at early stage. We used a combination of break‐apart and fusion FISH to search for SOCS1‐deleting translocations in available plaque‐stage tissue from two sequenced patients (MF3 and MF4) with confirmed SOCS1‐deleting translocations in tumor‐stage tissue (Figure 6). Plaque biopsies from patients MF3 and MF4 were procured, respectively, 3 years and 8 months prior to tumor development. We found that patient MF4 bears the translocation at plaque‐stage too (Figure 7E), suggesting that SOCS1 deletion is an early event in this individual.
4. DISCUSSION
This study represents the first integrated analysis (DNA/RNA) of genomic rearrangements in MF. The analysis reveals that MF displays a complex and heterogeneous landscape of inter‐ and intrachromosomal rearrangements. We observed, among others, translocations leading to deletion of ARID1A, CDKN2A/B, PTPRC, SOCS1, and STK11. This suggests that rearrangements mediate the deletion of tumor suppressors involved in pathways that are commonly deregulated in MF patients. We detected 270 rearranged genes, of which at least 100 play diverse roles in signal transduction and transcriptional regulation, and 47 are currently implicated in neoplasms. Our analysis identified 24 fusion transcripts, including 6 containing bona fide cancer genes, which though not recurrent, may contribute to MF development in individual cases.
All potentially deleterious SNVs we observed are patient‐specific, with the exception of FGFR4 (p.G388R) and JAK3 (p.A573V). Still, pathogenic SNVs may play relevant roles in signaling deregulation in some individuals. Importantly, we observed numerous SNVs in two genes reported as putative drivers in CTCL (ie, KMT2C, NCOR1), which albeit predicted as highly deleterious, were not found to be expressed in the RNA‐seq data. This highlights the importance of integrating DNA and RNA analyses to evaluate mutational data.
Although phenotypic resemblance between MF cells and several CD4+ T‐cell subsets (ie, TH2,50 TH17,51 TRM 52) has been documented in previous years, there is no definitive proof of any of these potential origins. Despite the cell of origin of MF remains undetermined, aberrant expression detected in our MF cohort using a “pan”‐ CD4+ control group matches earlier observations made by other groups. For instance, overactivation of JAK‐STAT53, 54 and NOTCH55 signaling, and mutations that enhance RAS‐mediated signaling56 have been previously described in MF. Yet, our transcriptome data should be interpreted with caution as further confirmation is required once the exact CD4+ T‐cell subset giving rise to MF is identified.
Interestingly, transcriptome analysis reveals a subset of DE cancer genes that play roles in cell cycle regulation and development. Tumor suppressors BRD7, CDKN1B, GPS2, and HNRNPK, which are down‐regulated in MF, are known to prompt cell cycle arrest at G1/S.57, 58, 59 TSC1, down‐regulated too, sustains quiescence in naïve T cells and its abrogation results in rapid cycling behavior.60 PBX1, a direct transcriptional repressor of CDKN2B, is consistently up‐regulated in MF.61 Additionally, up‐regulation of mitotic checkpoint kinase TTK might contribute to genomic instability in MF, since its expression has been shown to prevent aneuploidy‐induced cell death.62
On the other hand, up‐regulation of developmental genes GLI3, JAG1, and NOTCH3 might contribute to boost cell proliferation. Notably, NOTCH3 overactivation has been shown to induce an auto‐sustaining JAG1 expression loop in T‐cell acute lymphoblastic leukemia (T‐ALL), which in turn, enhances expression of Notch target genes responsible for the progression of the disease.63
Moreover, transcriptome analysis also shows that processes related to transcription (ie, spliceosome activity, mRNA surveillance) are flawed in MF, which might be linked to the considerable number of transcription‐related genes affected by genomic rearrangements. Taken together, the structural and expression analyses show that the cell cycle, the JAK‐STAT pathway, the PI‐3‐K pathway, and developmental pathways are deregulated in MF.
We report for the first time recurrent deletion of HNRNPK and SOCS1 not only in MF, but any CTCL. Furthermore, we found evidence that SOCS1 deletion is an early event in one of two patients with available plaque‐stage material by FISH. Importantly, while the incidence of deletion of both genes in the extension cohort was lower compared to the sequenced cohort, this difference was not statistically significant (P > .05, Fisher's exact test). Moreover, we can rule out the existence of clinical differences between the tumors from the two cohorts.
hnRNP‐K is a nuclear ribonucleoprotein implicated in leukemogenesis of acute myeloid leukemia (AML).59 Interestingly, studies have shown that haploinsufficiency of HNRNPK not only down‐regulates p21, but also up‐regulates STAT3 signaling and give rise to B‐ and T‐cell lymphomas in a mouse model.59 On the other hand, SOCS1, which is silenced in several cancers including multiple myeloma (MM),64 inhibits JAK‐STAT signaling by suppressing the tyrosine kinase activity of JAK proteins.65
A noteworthy fact is that miR‐155, which is often up‐regulated in MF,6 has been found to target SOCS1 in breast cancer and laryngeal carcinoma, leading to constitutive STAT3 activation in both cancers.66, 67 We observed that two of three patients without SOCS1 deletions express much higher levels (sevenfold average) of miR‐155 precursor MIR155HG than patients with SOCS1 deletions (Supporting Information Figure 5), which suggests that miR‐155 levels rise to inhibit SOCS1 in patients with functional copies of the gene. Moreover, SOCS1 might be suppressed in MF in one additional way. MECOM, which is consistently up‐regulated in our sequenced cohort, has been found to inhibit the expression of several regulators of the JAK‐STAT pathway in AML, particularly SOCS1.68 This evidence suggests that deregulation of STAT3 signaling via inactivation of HNRNPK and SOCS1 might be important in the pathogenesis of MF. Future studies with cells and animal models will be essential to functionally confirm the association between these tumor suppressors, their regulators, and STAT3 signaling in MF. In this scenario, targeting miR‐155 and/or MECOM to treat patients with functional SOCS1 alleles constitute potential novel therapeutic strategies.
Overall, the findings in this study reveal that genomic rearrangements and CNAs play relevant roles in the pathogenetics of MF and position HRNRPK and SOCS1 as putative drivers of MF development.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
Supporting information
Supplementary Figure 1 Genomic location of BAC probes used in FISH experiments. (A) Break‐apart BAC probes for the validation of DNA breaks at 16q13.13 in tumor‐stage MF. (B) Fusion BAC probes for the detection of t(12;16) in plaque‐stage tissue of sample MF3. (C) Fusion BAC probes for the detection of t(16;18) in plaque‐stage tissue of sample MF4. See Supplementary Table 3 for exact genomic positions of BAC probes used in this study.
Supplementary Figure 2. Validation of select CNAs by ddPCR. (A) Deletion of tumor suppressors (I) PTEN and (II) TLE4 in tumor‐stage MF were confirmed by ddPCR. (B) Gain of oncogenes (I) MYC and (II) TERT in tumor‐stage MF were confirmed by ddPCR. Ctrl, CD4+ T cells.
Supplementary Figure 3. Sanger sequencing validation of select SNVs. (A) Sanger chromatograms of tumor‐stage MF DNA showing select point mutations with known pathogenic effects (ie, gain‐of‐function, susceptibility to disease). (B) Sanger chromatograms of tumor‐stage MF DNA showing select point mutations with predicted deleterious effects on protein function. Genomic coordinates according to reference genome Hg19.
Supplementary Figure 4. Validation of genomic alterations at 16q13.13 associated with focal SOCS1 deletions in mycosis fungoides. (A) Deletion event at 16q13.13 in MF7 confirmed by Sanger sequencing. (B) (I) Circos plot displaying a genomic rearrangement at 16q13.13 in MF8. (II) Magnified view of deletions (Del A and Del B) at 16q13.13 resulting from structural alterations in MF8. (III) Genomic events (Del A and Del B) at 16q13.13 validated by Sanger sequencing in MF8. Del, deletion. CTX, interchromosomal translocation.
Supplementary Figure 5. MIR155HG expression in tumor‐stage MF. Two out of three MF samples with intact copies of SOCS1 (SOCS1 +) express higher levels (7‐fold average) of transcript MIR155HG compared to MF samples with SOCS1 deletions (SOCS1 ‐). MIR155HG hosts miR‐155, a known inhibitor of SOCS1.
Supplementary Table 1 Clinical information of samples used in the study
Supplementary Table 2 Mapping and Coverage metrics of samples subjected to WGS
Supplementary Table 3 BAC probes used in FISH experiments
Supplementary Table 4 Genomic rearragements detected by WGS
Supplementary Table 5 Fusion transcripts detected by RNA‐seq
Supplementary Table 6 Genes affected by genomic rearrangements
Supplementary Table 7 Genes affected by genomic rearrangements and implicated in cancer
Supplementary Table 8 Functional annotation (Biological process) of genes affected by genomic rearrangements
Supplementary Table 9 Functional annotation (Protein Class) of genes affected by genomic rearrangements
Supplementary Table 10 Minimal common regions (MCRs) between copy number alterations
Supplementary Table 11 Genes screened for deleterious SNVs
Supplementary Table 12 SNVs predicted as deleterious by Polyphen‐2 & SIFT
Supplementary Table 13 Differentially expressed genes in MF when compared to CD4+ T cells
Supplementary Table 14 Differentially expressed genes implicated in cancer
Supplementary Table 15 GSEA: Expression signatures enriched in MF when compared to CD4+ T cells
Supplementary Table 16 Up‐regulated and down‐regulated processes/pathways in MF
ACKNOWLEDGMENTS
The authors thank D. de Jong, from the Department of Cell and Chemical Biology, Leiden University Medical Center, Leiden, The Netherlands, for providing assistance in performing FISH.
AUTHOR CONTRIBUTIONS
A.N.B.T. and C.P.T. designed the project and wrote the manuscript. A.N.B.T. and D.C. performed the bioinformatic analyses and analyzed the data. A.N.B.T. performed all the experiments. A.N.B.T., D.C., H.M., K.S., M.H.V., and C.P.T. reviewed the data and the manuscript.
Bastidas Torres AN, Cats D, Mei H, et al. Genomic analysis reveals recurrent deletion of JAK‐STAT signaling inhibitors HNRNPK and SOCS1 in mycosis fungoides. Genes Chromosomes Cancer. 2018;57:653–664. 10.1002/gcc.22679
Funding information KWF Kankerbestrijding, Grant/Award Number: UL2013‐6104
REFERENCES
- 1. Willemze R, Jaffe ES, Burg G, et al. WHO‐EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768‐3785. [DOI] [PubMed] [Google Scholar]
- 2. Nicolae‐Cristea AR, Benner MF, Zoutman WH, et al. Diagnostic and prognostic significance of CDKN2A/CDKN2B deletions in patients with transformed mycosis fungoides and primary cutaneous CD30‐positive lymphoproliferative disease. Br J Dermatol. 2015;172:784‐788. [DOI] [PubMed] [Google Scholar]
- 3. Laharanne E, Chevret E, Idrissi Y, et al. CDKN2A‐CDKN2B deletion defines an aggressive subset of cutaneous T‐cell lymphoma. Mod Pathol. 2010;23:547‐558. [DOI] [PubMed] [Google Scholar]
- 4. van Doorn R, Dijkman R, Vermeer MH, Starink TM, Willemze R, Tensen CP. A novel splice variant of the Fas gene in patients with cutaneous T‐cell lymphoma. Cancer Res. 2002;62:5389‐5392. [PubMed] [Google Scholar]
- 5. van Doorn R, van Kester MS, Dijkman R, et al. Oncogenomic analysis of mycosis fungoides reveals major differences with Sezary syndrome. Blood. 2009;113:127‐136. [DOI] [PubMed] [Google Scholar]
- 6. van Kester MS, Ballabio E, Benner MF, et al. miRNA expression profiling of mycosis fungoides. Mol Oncol. 2011;5:273‐280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ungewickell A, Bhaduri A, Rios E, et al. Genomic analysis of mycosis fungoides and Sezary syndrome identifies recurrent alterations in TNFR2. Nat Genet. 2015;47:1056‐1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McGirt LY, Jia P, Baerenwald DA, et al. Whole‐genome sequencing reveals oncogenic mutations in mycosis fungoides. Blood. 2015;126:508‐519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. da Silva Almeida AC, Abate F, Khiabanian H, et al. The mutational landscape of cutaneous T cell lymphoma and Sezary syndrome. Nat Genet. 2015;47:1465‐1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vaque JP, Gomez‐Lopez G, Monsalvez V, et al. PLCG1 mutations in cutaneous T‐cell lymphomas. Blood. 2014;123:2034‐2043. [DOI] [PubMed] [Google Scholar]
- 11. Caumont C, Gros A, Boucher C, et al. PLCG1 gene mutations are uncommon in cutaneous T‐cell lymphomas. J Invest Dermatol. 2015;135:2334‐2337. [DOI] [PubMed] [Google Scholar]
- 12. Robinson JT, Thorvaldsdottir H, Winckler W, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Haas B, Dobin A, Stransky N, et al. STAR‐fusion: fast and accurate fusion transcript detection from RNA‐Seq. bioRxiv. 2017. [Google Scholar]
- 14. Nicorici D, Satalan M, Edgren H, et al. FusionCatcher ‐ a tool for finding somatic fusion genes in paired‐end RNA‐sequencing data. bioRxiv. 2014. [Google Scholar]
- 15. Boeva V, Popova T, Bleakley K, et al. Control‐FREEC: a tool for assessing copy number and allelic content using next‐generation sequencing data. Bioinformatics. 2012;28:423‐425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Choi J, Goh G, Walradt T, et al. Genomic landscape of cutaneous T cell lymphoma. Nat Genet. 2015;47:1011‐1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kiel MJ, Sahasrabuddhe AA, Rolland DC, et al. Genomic analyses reveal recurrent mutations in epigenetic modifiers and the JAK‐STAT pathway in Sezary syndrome. Nat Commun. 2015;6:8470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee CS, Ungewickell A, Bhaduri A, et al. Transcriptome sequencing in Sezary syndrome identifies Sezary cell and mycosis fungoides‐associated lncRNAs and novel transcripts. Blood. 2012;120:3288‐3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Park J, Yang J, Wenzel AT, et al. Genomic analysis of 220 CTCLs identifies a novel recurrent gain‐of‐function alteration in RLTPR (p.Q575E). Blood. 2017;130:1430‐1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Prasad A, Rabionet R, Espinet B, et al. Identification of gene mutations and fusion genes in patients with Sezary syndrome. J Invest Dermatol. 2016;136:1490‐1499. [DOI] [PubMed] [Google Scholar]
- 21. Wang L, Ni X, Covington KR, et al. Genomic profiling of Sezary syndrome identifies alterations of key T cell signaling and differentiation genes. Nat Genet. 2015;47:1426‐1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Woollard WJ, Pullabhatla V, Lorenc A, et al. Candidate driver genes involved in genome maintenance and DNA repair in Sezary syndrome. Blood. 2016;127:3387‐3397. [DOI] [PubMed] [Google Scholar]
- 23. Ranzani V, Rossetti G, Panzeri I, et al. The long intergenic noncoding RNA landscape of human lymphocytes highlights the regulation of T cell differentiation by linc‐MAF‐4. Nat Immunol. 2015;16:318‐325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mi H, Muruganujan A, Thomas PD. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013;41:D377‐D386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545‐15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Huang d W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44‐57. [DOI] [PubMed] [Google Scholar]
- 27. Gruber TA, Larson Gedman A, Zhang J, et al. An Inv(16)(p13.3q24.3)‐encoded CBFA2T3‐GLIS2 fusion protein defines an aggressive subtype of pediatric acute megakaryoblastic leukemia. Cancer Cell. 2012;22:683‐697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Borkhardt A, Bojesen S, Haas OA, et al. The human GRAF gene is fused to MLL in a unique t(5;11)(q31;q23) and both alleles are disrupted in three cases of myelodysplastic syndrome/acute myeloid leukemia with a deletion 5q. Proc Natl Acad Sci USA. 2000;97:9168‐9173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vieira L, Sousa AC, Matos P, et al. Three‐way translocation involves MLL, MLLT3, and a novel cell cycle control gene, FLJ10374, in the pathogenesis of acute myeloid leukemia with t(9;11;19)(p22;q23;p13.3). Genes Chromosomes Cancer. 2006;45:455‐469. [DOI] [PubMed] [Google Scholar]
- 30. Tartaglia M, Martinelli S, Cazzaniga G, et al. Genetic evidence for lineage‐related and differentiation stage‐related contribution of somatic PTPN11 mutations to leukemogenesis in childhood acute leukemia. Blood. 2004;104:307‐313. [DOI] [PubMed] [Google Scholar]
- 31. Ren M, Cowell JK. Constitutive notch pathway activation in murine ZMYM2‐FGFR1‐induced T‐cell lymphomas associated with atypical myeloproliferative disease. Blood. 2011;117:6837‐6847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Morerio C, Acquila M, Rosanda C, et al. HCMOGT‐1 is a novel fusion partner to PDGFRB in juvenile myelomonocytic leukemia with t(5;17)(q33;p11.2). Cancer Res. 2004;64:2649‐2651. [DOI] [PubMed] [Google Scholar]
- 33. Martini A, La Starza R, Janssen H, et al. Recurrent rearrangement of the Ewing's sarcoma gene, EWSR1, or its homologue, TAF15, with the transcription factor CIZ/NMP4 in acute leukemia. Cancer Res. 2002;62:5408‐5412. [PubMed] [Google Scholar]
- 34. Diakos C, Xiao Y, Zheng S, Kager L, Dworzak M, Wiemels JL. Direct and indirect targets of the E2A‐PBX1 leukemia‐specific fusion protein. PLoS One. 2014;9:e87602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Daheron L, Veinstein A, Brizard F, et al. Human LPP gene is fused to MLL in a secondary acute leukemia with a t(3;11) (q28;q23). Genes Chromosomes Cancer. 2001;31:382‐389. [DOI] [PubMed] [Google Scholar]
- 36. Bernt KM, Zhu N, Sinha AU, et al. MLL‐rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011;20:66‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Majolini MB, D'Elios MM, Galieni P, et al. Expression of the T‐cell‐specific tyrosine kinase Lck in normal B‐1 cells and in chronic lymphocytic leukemia B cells. Blood. 1998;91:3390‐3396. [PubMed] [Google Scholar]
- 38. Rodriguez D, Bretones G, Quesada V, et al. Mutations in CHD2 cause defective association with active chromatin in chronic lymphocytic leukemia. Blood. 2015;126:195‐202. [DOI] [PubMed] [Google Scholar]
- 39. Klein U, Lia M, Crespo M, et al. The DLEU2/miR‐15a/16‐1 cluster controls B cell proliferation and its deletion leads to chronic lymphocytic leukemia. Cancer Cell. 2010;17:28‐40. [DOI] [PubMed] [Google Scholar]
- 40. Ulaganathan VK, Sperl B, Rapp UR, Ullrich A. Germline variant FGFR4 p.G388R exposes a membrane‐proximal STAT3 binding site. Nature. 2015;528:570‐574. [DOI] [PubMed] [Google Scholar]
- 41. Koo GC, Tan SY, Tang T, et al. Janus kinase 3‐activating mutations identified in natural killer/T‐cell lymphoma. Cancer Discov. 2012;2:591‐597. [DOI] [PubMed] [Google Scholar]
- 42. Kamata T, Hussain J, Giblett S, Hayward R, Marais R, Pritchard C. BRAF inactivation drives aneuploidy by deregulating CRAF. Cancer Res. 2010;70:8475‐8486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Walters DK, Mercher T, Gu TL, et al. Activating alleles of JAK3 in acute megakaryoblastic leukemia. Cancer Cell. 2006;10:65‐75. [DOI] [PubMed] [Google Scholar]
- 44. Niemela JE, Lu L, Fleisher TA, et al. Somatic KRAS mutations associated with a human nonmalignant syndrome of autoimmunity and abnormal leukocyte homeostasis. Blood. 2011;117:2883‐2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ngo VN, Young RM, Schmitz R, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011;470:115‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Koskela HL, Eldfors S, Ellonen P, et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med. 2012;366:1905‐1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Roeb W, Higgins J, King MC. Response to DNA damage of CHEK2 missense mutations in familial breast cancer. Hum Mol Genet. 2012;21:2738‐2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yokoyama S, Woods SL, Boyle GM, et al. A novel recurrent mutation in MITF predisposes to familial and sporadic melanoma. Nature. 2011;480:99‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Boddicker RL, Razidlo GL, Dasari S, et al. Integrated mate‐pair and RNA sequencing identifies novel, targetable gene fusions in peripheral T‐cell lymphoma. Blood. 2016;128:1234‐1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vowels BR, Cassin M, Vonderheid EC, Rook AH. Aberrant cytokine production by Sezary syndrome patients: cytokine secretion pattern resembles murine Th2 cells. J Invest Dermatol. 1992;99:90‐94. [DOI] [PubMed] [Google Scholar]
- 51. Ciree A, Michel L, Camilleri‐Broet S, et al. Expression and activity of IL‐17 in cutaneous T‐cell lymphomas (mycosis fungoides and Sezary syndrome). Int J Cancer. 2004;112:113‐120. [DOI] [PubMed] [Google Scholar]
- 52. Campbell JJ, Clark RA, Watanabe R, Kupper TS. Sezary syndrome and mycosis fungoides arise from distinct T‐cell subsets: a biologic rationale for their distinct clinical behaviors. Blood. 2010;116:767‐771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sommer VH, Clemmensen OJ, Nielsen O, et al. In vivo activation of STAT3 in cutaneous T‐cell lymphoma. Evidence for an antiapoptotic function of STAT3. Leukemia. 2004;18:1288‐1295. [DOI] [PubMed] [Google Scholar]
- 54. Nielsen M, Kaltoft K, Nordahl M, et al. Constitutive activation of a slowly migrating isoform of Stat3 in mycosis fungoides: tyrphostin AG490 inhibits Stat3 activation and growth of mycosis fungoides tumor cell lines. Proc Natl Acad Sci USA. 1997;94:6764‐6769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gallardo F, Sandoval J, Diaz‐Lagares A, et al. Notch1 pathway activation results from the epigenetic abrogation of notch‐related MicroRNAs in mycosis Fungoides. J Invest Dermatol. 2015;135:3144‐3152. [DOI] [PubMed] [Google Scholar]
- 56. Kiessling MK, Oberholzer PA, Mondal C, et al. High‐throughput mutation profiling of CTCL samples reveals KRAS and NRAS mutations sensitizing tumors toward inhibition of the RAS/RAF/MEK signaling cascade. Blood. 2011;117:2433‐2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhou M, Liu H, Xu X, et al. Identification of nuclear localization signal that governs nuclear import of BRD7 and its essential roles in inhibiting cell cycle progression. J Cell Biochem. 2006;98:920‐930. [DOI] [PubMed] [Google Scholar]
- 58. Peng YC, Kuo F, Breiding DE, Wang YF, Mansur CP, Androphy EJ. AMF1 (GPS2) modulates p53 transactivation. Mol Cell Biol. 2001;21:5913‐5924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gallardo M, Lee HJ, Zhang X, et al. hnRNP K is a haploinsufficient tumor suppressor that regulates proliferation and differentiation programs in hematologic malignancies. Cancer Cell. 2015;28:486‐499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yang K, Neale G, Green DR, He W, Chi H. The tumor suppressor Tsc1 enforces quiescence of naive T cells to promote immune homeostasis and function. Nat Immunol. 2011;12:888‐897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Koss M, Bolze A, Brendolan A, et al. Congenital asplenia in mice and humans with mutations in a Pbx/Nkx2‐5/p15 module. Dev Cell. 2012;22:913‐926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Stratford JK, Yan F, Hill RA, et al. Genetic and pharmacological inhibition of TTK impairs pancreatic cancer cell line growth by inducing lethal chromosomal instability. PLoS One. 2017;12:e0174863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Pelullo M, Quaranta R, Talora C, et al. Notch3/Jagged1 circuitry reinforces notch signaling and sustains T‐ALL. Neoplasia. 2014;16:1007‐1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Galm O, Yoshikawa H, Esteller M, Osieka R, Herman JG. SOCS‐1, a negative regulator of cytokine signaling, is frequently silenced by methylation in multiple myeloma. Blood. 2003;101:2784‐2788. [DOI] [PubMed] [Google Scholar]
- 65. Yoshimura A, Suzuki M, Sakaguchi R, Hanada T, Yasukawa H. SOCS, inflammation, and autoimmunity. Front Immunol. 2012;3:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jiang S, Zhang HW, Lu MH, et al. MicroRNA‐155 functions as an OncomiR in breast cancer by targeting the suppressor of cytokine signaling 1 gene. Cancer Res. 2010;70:3119‐3127. [DOI] [PubMed] [Google Scholar]
- 67. Zhao XD, Zhang W, Liang HJ, Ji WY. Overexpression of miR −155 promotes proliferation and invasion of human laryngeal squamous cell carcinoma via targeting SOCS1 and STAT3. PLoS One. 2013;8:e56395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Glass C, Wuertzer C, Cui X, et al. Global identification of EVI1 target genes in acute myeloid leukemia. PLoS One. 2013;8:e67134. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1 Genomic location of BAC probes used in FISH experiments. (A) Break‐apart BAC probes for the validation of DNA breaks at 16q13.13 in tumor‐stage MF. (B) Fusion BAC probes for the detection of t(12;16) in plaque‐stage tissue of sample MF3. (C) Fusion BAC probes for the detection of t(16;18) in plaque‐stage tissue of sample MF4. See Supplementary Table 3 for exact genomic positions of BAC probes used in this study.
Supplementary Figure 2. Validation of select CNAs by ddPCR. (A) Deletion of tumor suppressors (I) PTEN and (II) TLE4 in tumor‐stage MF were confirmed by ddPCR. (B) Gain of oncogenes (I) MYC and (II) TERT in tumor‐stage MF were confirmed by ddPCR. Ctrl, CD4+ T cells.
Supplementary Figure 3. Sanger sequencing validation of select SNVs. (A) Sanger chromatograms of tumor‐stage MF DNA showing select point mutations with known pathogenic effects (ie, gain‐of‐function, susceptibility to disease). (B) Sanger chromatograms of tumor‐stage MF DNA showing select point mutations with predicted deleterious effects on protein function. Genomic coordinates according to reference genome Hg19.
Supplementary Figure 4. Validation of genomic alterations at 16q13.13 associated with focal SOCS1 deletions in mycosis fungoides. (A) Deletion event at 16q13.13 in MF7 confirmed by Sanger sequencing. (B) (I) Circos plot displaying a genomic rearrangement at 16q13.13 in MF8. (II) Magnified view of deletions (Del A and Del B) at 16q13.13 resulting from structural alterations in MF8. (III) Genomic events (Del A and Del B) at 16q13.13 validated by Sanger sequencing in MF8. Del, deletion. CTX, interchromosomal translocation.
Supplementary Figure 5. MIR155HG expression in tumor‐stage MF. Two out of three MF samples with intact copies of SOCS1 (SOCS1 +) express higher levels (7‐fold average) of transcript MIR155HG compared to MF samples with SOCS1 deletions (SOCS1 ‐). MIR155HG hosts miR‐155, a known inhibitor of SOCS1.
Supplementary Table 1 Clinical information of samples used in the study
Supplementary Table 2 Mapping and Coverage metrics of samples subjected to WGS
Supplementary Table 3 BAC probes used in FISH experiments
Supplementary Table 4 Genomic rearragements detected by WGS
Supplementary Table 5 Fusion transcripts detected by RNA‐seq
Supplementary Table 6 Genes affected by genomic rearrangements
Supplementary Table 7 Genes affected by genomic rearrangements and implicated in cancer
Supplementary Table 8 Functional annotation (Biological process) of genes affected by genomic rearrangements
Supplementary Table 9 Functional annotation (Protein Class) of genes affected by genomic rearrangements
Supplementary Table 10 Minimal common regions (MCRs) between copy number alterations
Supplementary Table 11 Genes screened for deleterious SNVs
Supplementary Table 12 SNVs predicted as deleterious by Polyphen‐2 & SIFT
Supplementary Table 13 Differentially expressed genes in MF when compared to CD4+ T cells
Supplementary Table 14 Differentially expressed genes implicated in cancer
Supplementary Table 15 GSEA: Expression signatures enriched in MF when compared to CD4+ T cells
Supplementary Table 16 Up‐regulated and down‐regulated processes/pathways in MF