

Abstract
Objective:
Celastrol, a major active constituent of Tripterygium wilfordii, has antioxidant, anti-inflammatory, and anticancer effects. However, whether celastrol can exert protective effect on myocardial ischemia–reperfusion injury (MIRI) is unknown. The aim of this study was to test the protective effect of celastrol on MIRI and elucidate its underlying mechanism.
Methods:
Cardiomyocytes (H9c2 cells) were subjected to hypoxia for 8 h followed by reoxygenation for 4 h to create hypoxia/reoxygenation (H/R) model, an in vitro MIRI model. Celastrol was added to the medium 60 min before the H/R process. Cell viability was detected using MTT assay. Myocardial injury was evaluated by measuring lactate dehydrogenase (LDH) and creatine kinase MB isoenzyme (CK-MB) activity. Changes in mRNA and protein expression of TNF-α, IL-1β, and nuclear factor-KB (NF-KB) were measured with RT-qPCR assay and western blot analysis.
Results:
Results showed that low-dose celastrol (20 and 50 nM) treatment significantly increased cell viability and decreased LDH and CK-MB activity in the condition of H/R, but high-dose celastrol (200 and 400 nM) resulted in extra injury to cardiomyocytes. Moreover, treatment with 50 nM celastrol significantly downregulated mRNA and protein expression of TNF-α and IL-1β. Meanwhile, NF-KB mRNA and protein in the nucleus were also correspondingly reduced.
Conclusion:
Our study demonstrated that low-dose celastrol could prevent MIRI in cardiomyocytes by inhibiting the activation of NF-KB, and celastrol may be a potential therapeutic agent for preventing MIRI.
Keywords: celastrol, myocardial ischemia–reperfusion injury, nuclear factor-KB, TNF-α, IL-1β
Introduction
Ischemic heart disease is one of the major causes of death worldwide (1). Instant recovery of blood supply (reperfusion) is considered one of the most optimal methods to prevent myocardial necrosis in ischemia. However, reperfusion itself may aggravate the extent of myocardial injury, which is termed as myocardial ischemia–reperfusion injury (MIRI) (2). MIRI is a complicated pathological process that involves in multiple factors including oxidative stress (free radical damage), inflammatory response, calcium overloading, and apoptosis in cardiomyocytes (3). Although the exact mechanism of MIRI remains unknown, it is widely accepted that inhibition of damage with oxidative stress, inflammatory response, calcium overloading, and apoptosis is a strategy for relieving MIRI (4).
Tripterygium wilfordii, a well-known traditional Chinese medicine, has been used for the treatment of inflammation, pain, tumor, and immune regulation for centuries (5). Celastrol, a pentacyclic triterpenoid, is a major active constituent isolated from the root of T. wilfordii (6). Celastrol has been demonstrated to possess multiple pharmacological actions, including antioxidant (7), anti-inflammatory (8), anticancer (9, 10), and obesity-controlling effects (11, 12). Of note, a recent study has demonstrated that celastrol protects the kidneys against ischemia reperfusion-induced injury in rats (13). However, whether celastrol possesses protective effect against MIRI remains unknown.
Therefore, in the present study, we first tested the protective effect of celastrol on H9c2 cells under the condition of the hypoxia/reoxygenation (H/R) model, an in vitro MIRI model. Furthermore, we explored the role of inflammatory factors and nuclear factor-KB (NF-KB) in the cardioprotection of celastrol.
Methods
Cell culture and H/R process
H9c2 cell lines obtained from the Shanghai Institutes for Biological Sciences (Shanghai, China) was routinely cultivated in Dulbecco’s modified eagle medium (Hyclone, Logan, USA) supplemented with 10% fetal bovine serum (Sijiqing Biotechnology Company, Hangzhou, China), 100 units/mL penicillin and 100 µg/mL streptomycin in a humidified atmosphere at 37°C with 5% CO2.
H/R process was performed as previously described (14). Briefly, H9c2 cells were first transferred to the medium without serum and glucose and then placed in a hypoxic chamber saturated with a mixed gas containing 5% CO2 and 95% N2 at 37°C for 8 h. Then, cells were reoxygenated by incubating in a fresh medium with serum and glucose under normoxic conditions for 4 h.
Celastrol treatment
Celastrol powder (Meilun Biotechnology Company, Dalian, China) was dissolved in dimethyl sulfoxide (DMSO) (Sigma, St. Louis, USA) to prepare celastrol reserve solution (1 mM). Different doses of celastrol (10, 20, 50, 100, 200, and 400 nM) were added to the medium 60 min before the H/R process. The optimal dose of celastrol for treatment was determined based on the ratio of cell viability and was used in the follow-up study.
Measurement of cell viability
MTT assay was used to determine cell viability after the H/R process and celastrol treatment. Briefly, H9c2 cells were seeded in a 96-well plate and treated with various concentrations of PA as described above. After 12 h, cell medium was removed and replaced with 200 µL of 5 mg/mL MTT solution (Sigma) per well. After incubation at 37°C for 4 h, MTT solution was removed and each well was washed with PBS three times. The precipitated formazan in each well was solubilized by DMSO and optical density was measured using an automated microplate reader (Thermo, USA).
Measurement of cell injury
Lactate dehydrogenase (LDH) and creatine kinase MB isoenzyme (CK-MB) activity of cell medium was used to determine the injury to H9c2 cells using a commercial LDH assay kit (Jiancheng Bioengineering Institute, Nanjing, China) and CK-MB assay kit (Jiancheng Bioengineering Institute), according to the manufacturer’s instructions.
RT-qPCR assay
mRNA expression was examined by RT-qPCR assay as previously described (15). In brief, total RNA was first extracted from cells using Trizol reagent (Takara, Dalian, China) and was then reverse transcribed onto cDNA using PrimeScriptTM RT reagent Kit with gDNA Eraser (Takara) according to the manufacturer’s instructions. qPCR was performed using the ABI 7500 Real Time PCR system (Applied Biosystems, Foster City, USA) with SYBR® Premix Ex Taq™ II (Takara) and specific primers (Table 1) according to the amplifying condition (95°C for 30 min, 40 cycles of 95°C for 5 s and 60°C for 34 s). Human GAPDH was used for mRNA normalization.
Table 1.
Primer information
| Primer name | Sequence (5’to 3’) |
|---|---|
| TNF-α(F) | GAAGAGAACCTGGGAGTAGATAAGG |
| TNF-α(R) | GTCGTAGCAAACCACCAAGC |
| IL-1b(F) | TCGTTGCTTGTCTCTCCTTG |
| IL-1b(R) | AAAAATGCCTCGTGCTGTCT |
| NF-kB(F) | GACCTGGAGCAAGCCATTAG |
| NF-Kb(R) | CACTGTCACCTGGAAGCAGA |
| GAPDH(F) | ATCATCAGCAATGCCTCC |
| GAPDH(R) | CATCACGCCACAGTTTCC |
F-forward; R-reverse
Western blot analysis
Cells were harvested after the H/R process and celastrol treatment, membrane and cytosol proteins were extracted from cells after centrifugation using Membrane and Cytosol Protein Extraction Kit (Beyotime, Shanghai, China), and nuclear proteins were also extracted from cells after centrifugation using Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime). Protein concentration was measured using Enhanced BCA Protein Assay Kit (Beyotime). For western blot analysis, 50 µg protein denatured by heating was subjected to 10% SDS polyacrylamide gel for separation and then transferred to PVDF membranes. The membrane was blocked using 5% skim milk for 1 h and then incubated overnight at 4°C with specific primary antibodies including monoclonal anti-TNF-α (1:500) (Wanleibio, Shenyang, China), anti- IL-1β (1:500) (Wanleibio), anti-NF-KB (1:500) (Wanleibio), and anti-β-actin (1:5000) (Golden Bridge Biotechnology, Beijing, China). After this, membranes were incubated with horseradish peroxidase-conjugated goat anti-rabbit or rabbit anti-mouse immunoglobulin G (1:10000) (Golden Bridge Biotechnology) at 37°C for 2 h. Detection of protein band was performed using an enhanced chemiluminescence for Western Blotting Kit (Santa Cruz, California, USA) according to the manufacturer’s instructions. Levels of phosphorylated proteins were normalized to their corresponding total protein levels. Relative densitometry was calculated using Image J2x analysis software (NIH, USA).
Statistical analysis
Data were expressed as mean±SD values. Statistical analysis was performed using SPSS 17.0 version (SPSS, Inc., Chicago, USA). Differences between groups were first evaluated using one-way analysis of variance, and if the differences were significant, multiple comparison analysis was further performed using Fisher’s least significant difference test. All p-values of <0.05 were considered statistically significant.
Results
Effect of celastrol on cell viability
To explore the optimal dose of celastrol for the treatment of H/R, different doses of celastrol were used. As shown in Figure 1, cell viability was significantly increased by 20 and 50 nM celastrol treatment compared with the H/R group (20 nM celastrol group vs. H/R group: 75.26%±2.16% vs. 70.50%±2.26%, p=0.027; 50 nM celastrol group vs. H/R group: 81.93%±2.15% vs. 70.50%±2.26%, p<0.001). In addition, cell viability was higher in the 50 nM celastrol group than in the 20 nM celastrol group (50 nM celastrol group vs. 20 nM celastrol group: 81.93%±2.15% vs. 75.26%±2.16%, p=0.003). However, cell viability was not significantly increased in the 100 nM celastrol group (100 nM celastrol group vs. H/R group: 69.86%±1. 64% vs. 70.50%±2.26%, p=0.774) and even significantly reduced in the 200 and 400 nM celastrol group (200 nM celastrol group vs. H/R group: 44.35%±4.01% vs. 70.50%±2.26%, p<0.001; 400 nM celastrol group vs. H/R group: 28.26%±2. 90% vs. 70.50%±2.26%, p<0.001). Taken together, 50 nM celastrol was considered the optimal dose for the treatment of H/R-induced myocardial injury and was further used in the follow-up study.
Figure 1.
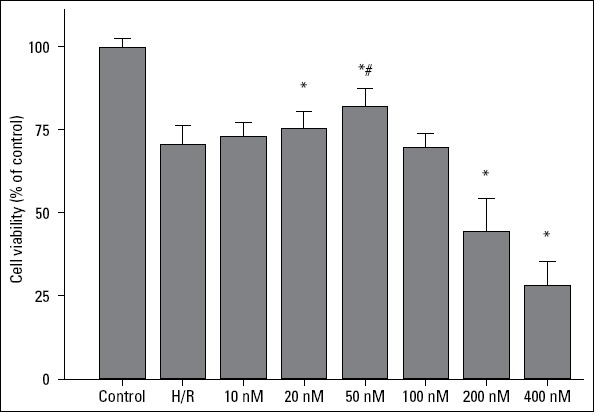
The effect of celastrol on cell viability. Control: H9c2 cells are cultured under normoxic condition for 12 h; H/R: H9c2 cells are subjected to hypoxia for 8 h followed by reoxygenation for 4 h; *represents P<0.05 vs. H/R group; # represents P<0.05 vs. 20 nM celastrol group
Effect of celastrol on the release of LDH and CK-MB
As shown in Figure 2, H/R promoted the release of a large amount of LDH and CK-MB from cardiomyocytes into the culture medium (H/R group vs. control group: for LDH, 313.97 IU/L±16.04 IU/L vs.109.86 IU/L±14.41 IU/L, p<0.001; for CK-MB, 201.00 IU/L±56.04 IU/L vs. 56.67 IU/L±23.03 IU/L, p<0.001), but the releases of LDH and CK-MB were remarkably decreased by 50 nM celastrol treatment (H/R+ celastrol group vs. H/R group: for LDH, 208.96 IU/L±18.89 IU/L vs. 313.97 IU/L±16.04 IU/L, p<0.001; for CK-MB, 136.00 IU/L±10.54 IU/L vs. 201.00 IU/L±56.04 IU/L, p<0.001), which suggested that 50 nM celastrol could prevent H/R-induced myocardial injury.
Figure 2.

The effect of celastrol on the release of cardiac enzymes. (a) The release of LDH in culture medium. (b) The release of CK-MB in culture medium. Control: H9c2 cells are cultured under normoxic condition for 12 h; celastrol: H9c2 cells was treated with 50 nM celastrol for 1 h followed by culture under normoxic condition for 12 h. H/R: H9c2 cells are subjected to hypoxia for 8 h followed by reoxygenation for 4 h; H/R+ celastrol: H9c2 cells were treated with 50 nM celastrol for 1 h followed by hypoxia for 8 h and reoxygenation for 4 h. *represents P<0.05 vs. control group; # represents P<0.05 vs. H/R group
Effect of celastrol on the expression of inflammatory factor
As shown in Figure 3, TNF-α was significantly upregulated in the condition of H/R, as evidenced by the increase in its mRNA and protein expression (H/R group vs. control group: 2.91-fold increase in mRNA expression; 1.44-fold increase in protein expression). However, 50 nM celastrol treatment could significantly reduce mRNA and protein expression of TNF-α (H/R+celastrol group vs. H/R group: 2.43 0.09 vs. 2.91±0.12, p<0.001 for mRNA; 1.26±0.07 vs. 1.44±0.11, p=0.035 for protein). Similarly, 50 nM celastrol treatment also decreased mRNA and protein expression of IL-1β in the condition of H/R (H/R+celastrol group vs. H/R group: 2.53±0.08 vs. 2.90±0.22, p=0.006 for mRNA; 1.26±0.05 vs. 1.47±0.13, p=0.026 for protein) (Fig. 4). These results demonstrated that celastrol could downregulate the expression of TNF-α and IL-1β.
Figure 3.

The effect of celastrol on the expression of TNF-α. (a) Relative TNF-α mRNA expression. (b) Relative TNF-α protein expression. Control: H9c2 cells are cultured under normoxic condition for 12 h; celastrol: H9c2 cells was treated with 50 nM celastrol for 1 h followed by culture under normoxic condition for 12 h. H/R: H9c2 cells are subjected to hypoxia for 8 h followed by reoxygenation for 4 h, H/R+ celastrol: H9c2 cells were treated with 50 nM celastrol for 1 h followed by hypoxia for 8 h and reoxygenation for 4 h. *represents P<0.05 vs. control group, # represents P<0.05 vs. H/R group
Figure 4.

The effect of celastrol on the expression of IL-1β. (a) Relative IL-1β mRNA expression. (b) Relative IL-1β protein expression. Control: H9c2 cells are cultured under normoxic condition for 12 h; celastrol: H9c2 cells were treated with 50 nM celastrol for 1 h followed by culture under normoxic condition for 12 h. H/R: H9c2 cells are subjected to hypoxia for 8 h followed by reoxygenation for 4 h; H/R+ celastrol: H9c2 cells were treated with 50 nM celastrol for 1 h followed by hypoxia for 8 h and reoxygenation for 4 h. *represents P<0.05 vs. control group, # represents P<0.05 vs. H/R group
Effect of celastrol on the expression of NF-KB
To explore whether celastrol can modulate the activation of NF-KB, we further evaluated the change in NF-KB mRNA and protein expression. Results showed that 50 nM celastrol treatment significantly reduced NF-KB mRNA expression in the condition of H/R (H/R+ celastrol group vs. H/R group: 1.28 0.03 vs. 1.57±0.07, p<0.001) (Fig. 5a). In addition, 50 nM celastrol treatment significantly increased NF-KB protein expression in the cytoplasm (H/R+ celastrol group vs. H/R group: 0.750 0.051 vs. 0.504±0.175, p=0.035), whereas it decreased the protein expression in the nucleus (H/R+ celastrol group vs. H/R group: 1.206±0.055 vs. 1.427±0.082, p=0.047) (Fig. 5b), suggesting that celastrol attenuates the activation of NF-KB in the nucleus.
Figure 5.

The effect of celastrol on the expression of NF-KB. (a) Relative NF-KB mRNA expression. (b) Relative NF-KB protein expression in the cytoplasm and nucleus. Control: H9c2 cells are cultured under normoxic condition for 12 h; celastrol: H9c2 cells were treated with 50 nM celastrol for 1 h followed by culture under normoxic condition for 12 h. H/R: H9c2 cells were subjected to hypoxia for 8 h followed by reoxygenation for 4 h; H/R+ celastrol: H9c2 cells were treated with 50 nM celastrol for 1 h followed by hypoxia for 8 h and reoxygenation for 4 h. *represents P<0.05 vs. control group; # represents P<0.05 vs. H/R group
Discussion
Several recent studies have provided evidence that celastrol has a beneficial effect on cardiovascular diseases. For instance, Yu et al. (16) suggested that celastrol attenuated hypertension-induced inflammation and oxidative stress in vascular smooth muscle cells via heme oxygenase-1 induction. Gu et al. (17) found that celastrol prevented atherosclerosis by inhibiting LOX-1 and oxidative stress. More importantly, celastrol showed protective effect on acute myocardial infarction and doxorubicin-induced cardiomyopathy (18, 19). Although Der Sarkissian et al. (18) suggested that celastrol is suitable for human applicability in the clinical setting of an acute myocardial infarction without reperfusion; of note, the effect of celastrol on MIRI is still unknown at present. In this present study, we first found that low-dose celastrol could attenuate MIRI as evidenced by the increase in cell viability and decline in the release of LDH and CK-MB. Moreover, we also cannot ignore the fact that high-dose celastrol showed no beneficial effect on MIRI, and instead led to a decline in cell viability. The reason we speculated is that high-dose celastrol may induce cell apoptosis (20, 21).
Inflammatory response has been shown to play an important role in the occurrence of MIRI (22). During MIRI, numerous leukocytes (in particular neutrophils) were promptly activated, resulting in capillary blockage and microcirculation obstacle (23), which is a major cause of no-reflow, a serious clinical manifestation of MIRI. Simultaneously, some inflammatory mediators, such as TNF-α and IL-1, were substantially released from activated neutrophils and resulted in inflammatory damage to myocardial cells (24). In the present study, our results displayed that celastrol treatment significantly decreased mRNA and protein expression of TNF-α and IL-1β in myocardial cells in the condition of H/R. These results are consistent with results of other studies founding that celastrol reduced TNF-β in lung infiltration model and inhibited IL-1β-induced inflammation in orbital fibroblasts (25, 26). Therefore, we suggested that celastrol attenuated MIRI by relieving TNF-α- and IL-1β-mediated inflammatory damage to cardiomyocytes.
NF-KB is the key transcription factor that regulates cellular responses to inflammation and is responsible for the regulation of a wide variety of proinflammatory genes. Some proinflammatory cytokines, such as TNF-α and IL-1β, are substantially increased after NF-KB is activated (27). In the present study, we found that NF-KB was activated in the nucleus of cardiomyocytes during the H/R process. More importantly, we found that celastrol treatment could attenuate the activation of NF-KB in the nucleus, which is consistent with the results of other studies (28, 29). Therefore, we speculated that celastrol prevents cardiomyocytes from MIRI by inhibiting the activation of NF-KB.
Study limitations
We must acknowledge the two limitations that existed in this study. First, we only tested the protective effect of celastrol in in vitro model, and its cardioprotection still needs to be examined in an animal model. Second, we only examined the change in the activation of NF-KB in cardiomyocytes after celastrol treatment; however, the NF-KB-specific activator should be further used to confirm the role of NF-KB in the cardioprotection of celastrol.
Conclusion
In conclusion, our study demonstrated that low-dose celastrol could prevent MIRI in cardiomyocytes by inhibiting the activation of NF-KB, and celastrol may be considered as a potential therapeutic agent for preventing MIRI.
Footnotes
Disclosure statement: The authors declare that they have no competing interests.
Peer-review: Externally peer-reviewed.
Authorship contributions: Concept – X.L., D.J.; Design – D.J.; Supervision – X.L., N.W., L.Z.; Fundings – D.J.; Materials – X.L., N.W., L.Z.; Data collection &/or processing – X.L., N.W., L.Z.; Analysis &/or interpretation – X.L., N.W.; Literature search – X.L., N.W.; Writing – X.L., N.W.; Critical review – D.J.

From Prof. Dr. İstemi Nalbantgil’s collections.
References
- 1.Moran AE, Forouzanfar MH, Roth GA, Mensah GA, Ezzati M, Murray CJ, et al. Temporal trends in ischemic heart disease mortality in 21 world regions, 1980 to 2010: the Global Burden of Disease 2010 study. Circulation. 2014;129:1483–92. doi: 10.1161/CIRCULATIONAHA.113.004042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bainey KR, Armstrong PW. Clinical perspectives on reperfusion injury in acute myocardial infarction. Am Heart J. 2014;167:637–45. doi: 10.1016/j.ahj.2014.01.015. [DOI] [PubMed] [Google Scholar]
- 3.Ferdinandy P, Hausenloy DJ, Heusch G, Baxter GF, Schulz R. Interaction of risk factors, comorbidities, and comedications with ischemia/reperfusion injury and cardioprotection by preconditioning, postconditioning, and remote conditioning. Pharmacol Rev. 2014;66:1142–74. doi: 10.1124/pr.113.008300. [DOI] [PubMed] [Google Scholar]
- 4.Hausenloy DJ, Yellon DM. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest. 2013;123:92–100. doi: 10.1172/JCI62874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang T, Shen F, Su S, Bai Y, Guo S, Yan H, et al. Comparative analysis of four terpenoids in root and cortex of Tripterygium wilfordii Radix by different drying methods. BMC Complement Altern Med. 2016;16:476. doi: 10.1186/s12906-016-1453-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Salminen A, Lehtonen M, Paimela T, Kaarniranta K. Celastrol: Molecular targets of Thunder God Vine. Biochem Biophys Res Commun. 2010;394:439–42. doi: 10.1016/j.bbrc.2010.03.050. [DOI] [PubMed] [Google Scholar]
- 7.Allison AC, Cacabelos R, Lombardi VR, Alvarez XA, Vigo C, et al. Celastrol, a potent antioxidant and anti-inflammatory drug, as a possible treatment for Alzheimer's disease. Prog Neuropsychopharmacol Biol Psychiatry. 2001;25:1341–57. doi: 10.1016/s0278-5846(01)00192-0. [DOI] [PubMed] [Google Scholar]
- 8.Kim DH, Shin EK, Kim YH, Lee BW, Jun JG, Park JH, et al. Suppression of inflammatory responses by celastrol, a quinone methide triterpenoid isolated from Celastrus regelii. Eur J Clin Invest. 2009;39:819–27. doi: 10.1111/j.1365-2362.2009.02186.x. [DOI] [PubMed] [Google Scholar]
- 9.Raja SM, Clubb RJ, Ortega-Cava C, Williams SH, Bailey TA, Duan L, et al. Anticancer activity of Celastrol in combination with ErbB2-targeted therapeutics for treatment of ErbB2-overexpressing breast cancers. Cancer Biol Ther. 2011;11:263–76. doi: 10.4161/cbt.11.2.13959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wolfram J, Suri K, Huang Y, Molinaro R, Borsoi C, Scott B, et al. Evaluation of anticancer activity of celastrol liposomes in prostate cancer cells. J Microencapsul. 2014;31:501–7. doi: 10.3109/02652048.2013.879932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu J, Lee J, Salazar Hernandez MA, Mazitschek R, Ozcan U. Treatment of obesity with celastrol. Cell. 2015;161:999–1011. doi: 10.1016/j.cell.2015.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma X, Xu L, Alberobello AT, Gavrilova O, Bagattin A, Skarulis M, et al. Celastrol Protects against Obesity and Metabolic Dysfunction through Activation of a HSF1-PGC1βTranscriptional Axis. Cell Metab. 2015;22:695–708. doi: 10.1016/j.cmet.2015.08.005. [DOI] [PubMed] [Google Scholar]
- 13.Chu C, He W, Kuang Y, Ren K, Gou X. Celastrol protects kidney against ischemia-reperfusion-induced injury in rats. J Surg Res. 2014;186:398–407. doi: 10.1016/j.jss.2013.07.048. [DOI] [PubMed] [Google Scholar]
- 14.Gong G, Li Y, Yang X, Geng H, Lu X, Wang L, et al. Protective effects of IL28RA siRNA on cardiomyocytes in hypoxia/reoxygenation injury. Anatol J Cardiol. 2017;18:168–74. doi: 10.14744/AnatolJCardiol.2017.7763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ran B, Li M, Li Y, Lin Y, Liu W, Luo Q, et al. Everolimus (RAD001) inhibits the proliferation of rat vascular smooth muscle cells by up-regulating the activity of the p27/kip1 gene promoter. Anatol J Cardiol. 2016;16:385–91. doi: 10.14744/AnatolJCardiol.2015.6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu X, Tao W, Jiang F, Li C, Lin J, Liu C. Celastrol attenuates hypertension-induced inflammation and oxidative stress in vascular smooth muscle cells via induction of heme oxygenase-1. Am J Hypertens. 2010;23:895–903. doi: 10.1038/ajh.2010.75. [DOI] [PubMed] [Google Scholar]
- 17.Gu L, Bai W, Li S, Zhang Y, Han Y, Gu Y, et al. Celastrol prevents atherosclerosis via inhibiting LOX-1 and oxidative stress. PLoS One. 2013;8:e65477. doi: 10.1371/journal.pone.0065477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Der Sarkissian S, Cailhier JF, Borie M, Stevens LM, Gaboury L, Mansour S, et al. Celastrol protects ischaemic myocardium through a heat shock response with up-regulation of haeme oxygenase-1. Br J Pharmacol. 2014;171:5265–79. doi: 10.1111/bph.12838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sharma S, Mishra R, Walker BL, Deshmukh S, Zampino M, Patel J, et al. Celastrol, an oral heat shock activator, ameliorates multiple animal disease models of cell death. Cell Stress Chaperones. 2015;20:185–201. doi: 10.1007/s12192-014-0536-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mi C, Shi H, Ma J, Han LZ, Lee JJ, Jin X. Celastrol induces the apoptosis of breast cancer cells and inhibits their invasion via downregulation of MMP-9. Oncol Rep. 2014;32:2527–32. doi: 10.3892/or.2014.3535. [DOI] [PubMed] [Google Scholar]
- 21.Chakravarthy R, Clemens MJ, Pirianov G, Perdios N, Mudan S, Cartwright JE, et al. Role of the eIF4E binding protein 4E-BP1 in regulation of the sensitivity of human pancreatic cancer cells to TRAIL and celastrol-induced apoptosis. Biol Cell. 2013;105:414–29. doi: 10.1111/boc.201300021. [DOI] [PubMed] [Google Scholar]
- 22.Arslan F, de Kleijn DP, Timmers L, Doevendans PA, Pasterkamp G. Bridging innate immunity and myocardial ischemia/reperfusion injury: the search for therapeutic targets. Curr Pharm Des. 2008;14:1205–16. doi: 10.2174/138161208784246090. [DOI] [PubMed] [Google Scholar]
- 23.Vinten-Johansen J. Involvement of neutrophils in the pathogenesis of lethal myocardial reperfusion injury. Cardiovasc Res. 2004;61:481–97. doi: 10.1016/j.cardiores.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 24.Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res. 2002;53:31–47. doi: 10.1016/s0008-6363(01)00434-5. [DOI] [PubMed] [Google Scholar]
- 25.Xu LM, Zheng YJ, Wang Y, Yang Y, Cao FF, Peng B, et al. Celastrol inhibits lung infiltration in differential syndrome animal models by reducing TNF-βand ICAM-1 levels while preserving differentiation in ATRA-induced acute promyelocytic leukemia cells. PLoS One. 2014;9:e105131. doi: 10.1371/journal.pone.0105131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H, Yuan Y, Zhang Y, He Q, Xu R, Ge F, et al. Celastrol inhibits IL-1β-induced inflammation in orbital fibroblasts through the suppression of NF-KB activity. Mol Med Rep. 2016;14:2799–806. doi: 10.3892/mmr.2016.5570. [DOI] [PubMed] [Google Scholar]
- 27.Yamauchi S, Ito H, Miyajima A. IKBn, a nuclear IKB protein, positively regulates the NF-KB–mediated expression of proinflammatory cytokines. Proc Natl Acad Sci U S A. 2010;107:11924–9. doi: 10.1073/pnas.0913179107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee JH, Koo TH, Yoon H, Jung HS, Jin HZ, Lee K, et al. Inhibition of NF-kappa B activation through targeting I kappa B kinase by celastrol, a quinone methide triterpenoid. Biochem Pharmacol. 2006;72:1311–21. doi: 10.1016/j.bcp.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 29.Li Y, He D, Zhang X, Liu Z, Zhang X, Dong L, et al. Protective effect of celastrol in rat cerebral ischemia model: down-regulating p-JNK, p-c-Jun and NF-KB. Brain Res. 2012;1464:8–13. doi: 10.1016/j.brainres.2012.04.054. [DOI] [PubMed] [Google Scholar]