

Abstract
Objective:
We aimed to evaluate the relationship of serum activin A levels with risk factors, clinical presentation, biochemical marker levels, extent, and severity of atherosclerotic coronary artery disease (CAD).
Methods:
In total, 310 CAD patients [92 with ST-segment elevation myocardial infarction (STEMI), 111 with non-STEMI (NSTEMI), and 107 with unstable angina (UA)] and 207 healthy subjects (controls) were enrolled. Activin A levels in all participants were measured using ELISA. Angiographic measurements were performed in patients and not in the healthy subjects.
Results:
Activin A levels were higher in all patient groups than in controls (patients vs. controls, p=0.041; NSTEMI vs. UA, p=0.744; STEMI vs. UA, p=0.172; NSTEMI vs. STEMI, p=0.104). According to the cut-off value of activin A level, patients with high and low activin A levels had a similar distribution of clinical and biochemical variables but the prevalence of severe stenosis was observed in groups with high activin A levels. Our results revealed that activin A levels did not decrease as thrombolysis in myocardial infarction (risk score increased (p=0.590). The area under the ROC curve for activin A levels in patients was 0.590±0.047 (95% CI: 0.439–0.591, p=0.193). In multiple analysis of the overall population, male gender (β=–0.260; 95% CI: –617.39 to –110.04; p=0.005) was an independent predictor of activin A levels.
Conclusion:
This study indicated that activin A can not be a predictive marker in CAD and is not associated with extensive and severe CAD. In contrast, the increase in activin A levels in patients, especially in patients with different clinical groups of acute coronary syndromes, suggested its involvement in atherosclerosis.
Keywords: Activin A, coronary artery disease, risk factors, extent, severity
Introduction
Atherosclerosis, an inflammatory condition, is associated with multiple risk factors and is characterized by a complex pathophysiology. Vessel bifurcations or branched regions are susceptible to atherosclerotic events (1), and the development of interventional cardiology over the last few years has led to an improved access to invasive diagnostics of ischemic heart disease. Previously, the diagnosis of the severity of coronary artery disease (CAD) relied on the assessment of the extent of advanced atherosclerotic stages and concomitant worsening of circulation in major coronary arteries (2). Convincing evidence has suggested that CAD is an inflammatory process, and a multitude of pro-inflammatory markers associated with atherogenesis have been identified (3). It remains unknown whether the association of these markers with CAD is direct or requires interaction with pro-atherogenic factors. Furthermore, it remains unknown whether systemic biomarkers associated with different stages of atherosclerosis are more closely associated with the extent of atherosclerosis or with the composition of coronary atherosclerotic plaque (4).
In the vascular system, members of the TGF-β superfamily play a vital role in the regulation of vasculogenesis, angiogenesis, and vascular remodeling (5); activin A, which belongs to this family, was recently recognized as a multifunctional cytokine expressed by several tissues and cells. Activin A effects include the regulation of wound repair, cell differentiation, apoptosis, and embryogenesis (6); its role in atherogenesis has been hypothesized based on the findings that activin A modulates the proliferation and differentiation cells involved in atherogenesis, notably endothelial cells (ECs), macrophages, and smooth muscle cells (SMCs) (7). Activin A inhibits the propagation of human ECs and enhances the differentiation of monocytes into macrophages (7).
The present study aimed to examine serum activin A levels under different coronary clinical presentations [ST-segment elevation myocardial infarction (STEMI), non-STEMI (NSTEMI), and unstable angina (UA)] and to explore whether serum activin A is associated with thrombolysis in myocardial infarction (TIMI) risk score, angiographic CAD prevalence or severity, traditional cardiovascular risk factors, and inflammatory markers. In addition, the ideal cut-off value of activin A level for predicting CAD occurrence was also assessed.
Methods
Study subjects
In this case-control study, 310 CAD patients who were admitted to the Department of Cardiology, University Hospital (Monastir, Tunisia), and 207 healthy subjects were included. The latter were mostly blood donors and were recruited from the Blood Bank Department of the University Hospital (Mahdia, Tunisia). As their physical examination was unremarkable, along with the absence of personal or family history of CAD and reasons to suspect CAD, they were classified as healthy. CAD diagnosis was based on the presence (by angiography) of luminal diameter stenosis of ≥50% in at least one major coronary artery [left anterior descending (LAD), left circumflex (LCx) or right coronary artery (RCA), or major branches]. STEMI was defined as the presence of chest pain symptoms (>20 min) associated with electrocardiographic ST-segment elevation of ≥0.1 mV in two or more limb leads or ≥0.2 mV in two or more precordial leads and an increase in cardiac-specific troponin I levels. NSTEMI included NSTEMI and UA. NSTEMI was defined as ischemic symptoms with elevated cardiac-specific troponin I levels without new ST elevation on electrocardiogram. UA was defined as having newly developed and accelerated chest symptoms on exertion or rest angina without a significant increase in cardiac-specific troponin I levels. Multivessel CAD was confirmed when at least two of the major coronary arteries had significant atherosclerosis. Angiographic measurements were performed in patients but not in the healthy subjects.
Exclusion criteria included the presence of severe respiratory disease, liver disease, kidney disease, concomitant inflammatory diseases such as infections and autoimmune disorders, or malignancy. This study was approved by the National Committee for Medical and Research Ethics of Farhat Hached University Hospital (Sousse, Tunisia), which complied with the ethical principles of the WMA Declaration of Helsinki. Informed consent was obtained from all participants.
Laboratory evaluation
Serum samples were collected from study participants and stored in aliquots at –80°C until use. Triglyceride, total cholesterol, high-density lipoprotein cholesterol, high-sensitivity C-reactive protein (hs-CRP), glucose, urea, and creatinine levels in all patients were measured using a Cobas Integra 600 analyzer (Roche Diagnostic, Germany) on admission. Low-density lipoprotein cholesterol levels were estimated using the Friedewald equation. Serum interleukin-6 (IL-6) levels were measured by electrochemiluminescence immunoassay using a Cobas E601 analyzer (Roche Diagnostics, Germany).
Activin A measurement
Activin A levels were quantified using a commercially available ELISA kit (AbFrontier, Young In Frontier Co., Ltd., Korea). All samples were assayed once according to the manufacturer’s instructions. The optical density of contents in each well was determined using a microplate reader at 450 nm.
Coronary angiography and assessment of CAD severity
All patients underwent coronary angiography after admission, and all angiograms were reviewed by trained cardiologists who were blinded to procedural and clinical data. In case of disagreement, the final decision was reached by consensus. CAD was defined as >50% luminal narrowing of at least one major epicardial vessel. CAD severity was ascertained by assessing the multivessel disease extent [one-, two-, or three-vessel disease (>50% stenosis)]. Accordingly, patients were categorized as having no disease or having one-, two- or three-vessel disease. The degree of coronary stenosis was classified as moderate (50–70% stenosis) and severe (>70% stenosis) according to previously published guidelines (8). Acute coronary syndrome (ACS) patients were divided into three subgroups according to TIMI risk score: group 1 [low TIMI risk score (1-2)], group 2 [intermediate TIMI risk score (3-4)], and group 3 [high TIMI risk score (≥5)]. Echocardiographic characteristics of the study groups were obtained from hospital records.
Statistical analysis
Statistical Package for Social Sciences version 23 was used for data analysis. Categorical data were expressed as percentages, and comparisons between patients were performed using chi-square test. Normally distributed data were expressed as mean±standard deviation (SD), and comparisons between patients were performed using Student’s t test. Non-normally distributed data were expressed as mean±SD and medians, and comparisons between patients were performed using the Mann–Whitney U test. A p value of <0.05 was considered to indicate statistically significant difference. Kruskal–Wallis tests or one-way analysis of variance tests were used to compare the three groups. Univariate and multivariate linear analyses were applied to identify the predictors of activin A levels.
Results
Baseline characteristics and the prevalence of risk factors among patient and control groups are shown in Table 1. In total, 310 patients (74.8% males; age, 60.3±11.0 years) and 207 healthy subjects (29.5% males; age, 33.2±12.2 years) were enrolled in this study. Menopause (90.8%), diabetes (48.4%), and hypertension (47.1%) were the most prevalent identifiable CAD risk factors.
Table 1.
Baseline characteristics of the study population
| Characteristics | Controls N=207 | All patients N=310 | P |
|---|---|---|---|
| Age, years | 33.2±12.2 | 60.3±11.0 | <0.001 |
| Male, n, % | 61(29.5) | 232(74.8) | <0.001 |
| BMI, kg/m2 | 25.1±3.6 | 27.5±4.3 | <0.001 |
| Smoker, % | 8.2 | 41.0 | <0.001 |
| Menopause, % | 10.3 | 90.8 | <0.001 |
| Obesity, % | 8.3 | 22.6 | <0.001 |
| Hypertension, % | 0 | 47.1 | <0.001 |
| Diabetes, % | 0 | 48.4 | <0.001 |
| Dyslipidemia, % | 0 | 23.5 | <0.001 |
| Personal history of CAD, % | 0 | 33.2 | <0.001 |
| Personal history of ACS, % | 0 | 27.4 | <0.001 |
| Peripheral artery diseases, % | 0 | 1.6 | <0.001 |
| History of bypass surgery, % | 0 | 3.2 | <0.001 |
ACS-acute coronary syndrome, BMI-body mass index, CAD-coronary artery diseasea. Data are expressed as mean±SD or n (%). P<0.05
Patients were then divided into low activin A (<429.0 pg/mL) and high activin A (>429.0 pg/mL) groups based on activin A cut-off value in healthy controls. Among patients, the LAD artery was identified as the affected vessel (72.7%), followed by the LCx (50.5%) and RCA (48.6%). Multivessel disease was observed in 56.5% of patients, 67.0% of whom had severe stenosis (>70%). Among patient subgroups, the distribution of clinical and biochemical variables was similar between low and high activin A groups, and no significant differences were noted in these variables (Table 2).
Table 2.
Association between activin A and clinical and biochemical variables
| Clinical and biochemical characteristics | Patients | P | ||
|---|---|---|---|---|
| All N=310 | Low <429.0 pg/mL | High >429.0 pg/mL | ||
| SBP, cm Hg | 13.1±2.2 | 13.1±1.9 | 12.6±2.3 | 0.190 |
| DBP, cm Hg | 7.5±1.3 | 7.7±1.2 | 7.2±1.4 | 0.021 |
| Heart rate, b/min | 75.5±16.2 | 76.9±17.4 | 72.5±14.0 | 0.111 |
| LVEF, % | 49.5±14.7 | 46.0±16.3 | 51.0±13.0 | 0.298 |
| Vessel involved, % | 6.9 | 8.7 | 7.5 | 0.792 |
| LMA | 50.5 | 62.3 | 40.3 | 0.010 |
| LCx | 72.7 | 78.3 | 67.2 | 0.146 |
| LAD | 48.6 | 44.9 | 47.8 | 0.740 |
| RCA | ||||
| Stenosis, % | ||||
| <50% | 5.8 | 12.2 | 2.9 | 0.047 |
| (50-70) % | 27.2 | 31.7 | 14.7 | |
| >70% | 67.0 | 56.1 | 82.4 | |
| Single vessel, % | 43.5 | 31.9 | 50.7 | 0.025 |
| Multivessel, % | 56.5 | 68.1 | 49.3 | |
| Insulin use, % | 37.5 | 40.0 | 42.1 | 0.364 |
| Statins, % | 62.6 | 68.2 | 79.2 | 0.379 |
| β Blokers, % | 45.5 | 60.0 | 33.3 | 0.077 |
| CEI, % | 69.2 | 71.4 | 79.2 | 0.547 |
| Aspirin, % | 68.1 | 54.5 | 69.2 | 0.295 |
| Furosemid, % | 19.1 | 9.1 | 30.4 | 0.074 |
| Glucose, mmol/L | 9.8±5.6 | 8.4±4.8 | 8.6±4.2 | 0.762 |
| Urea, mmol/L | 6.3±3.3 | 5.8±3.7 | 6.0±2.4 | 0.650 |
| Creatinin, µmol/L | 93.7±28.9 | 88.9±24.3 | 86.7±25.9 | 0.594 |
| Uric acid, µmol/L | 337.4±125.3 | 263.3±111.1 | 296.3±96.2 | 0.206 |
| TC, mmol/L | 4.3±1.4 | 4.2±1.4 | 4.5±1.4 | 0.160 |
| TG, mmol/L | 1.4(0.3-12.1) | 1.3(0.3-4.0) | 1.4(0.5-12.1) | 0.280 |
| HDL-c, mmol/L | 1.0±0.7 | 0.9±0.3 | 0.9±0.4 | 0.844 |
| LDL-c, mmol/L | 1.7±1.3 | 2.6±1.1 | 2.8±1.2 | 0.384 |
| Lp (a), mg/L | 107.0(14.5-1230.0) | 108.0(14.5-780.0) | 103.5(23.0-230.0) | 0.409 |
| ApoA1, g/L | 1.0±0.3 | 0.9±0.2 | 1.0±0.3 | 0.846 |
| ApoB, g/L | 0.8±0.3 | 0.7±0.3 | 0.8±0.3 | 0.711 |
| Hs-CRP, mg/L | 5.0(0.2-1020.0) | 6.1(0.2-1020) | 5.0(0.2-500.0) | 0.770 |
| IL-6, pg/mL | 8.2(1.5-3640.0) | 8.5(2.2-3640.0) | 8.1(1.5-855.4) | 0.241 |
ApoA-1-apolipoprotein A-1, ApoB-apolipoprotein B, CEI-conversion enzyme inhibitor, DBP-diastolic blood pressure, HDL-c-high density lipoprotein cholesterol, hs-CRP-high sensitivity C-reactive protein, IL-6-Interleukin 6, LAD-left anterior descending, LCx-left circumflex, LDL-c-low density lipoprotein cholesterol, LMCA-left main coronary artery, Lp(a)-lipoprotein(a), LVEF-left ventricular ejection fraction, RCA-right coronary artery, SBP-systolic blood pressure, TC-total cholesterol, TG-triglyceride. Values are expressed as the mean±SD. P<0.05
CAD extent and severity
The associations of altered activin A levels with CAD extent and severity are shown in Table 3. Significantly difference was observed in activin A levels according to the degree of stenosis, and high activin A levels were associated with severe stenosis (>70%). In addition, patients with multivessel disease had lower activin A levels, and difference was significant (380.0 vs. 566.0 pg/mL; p=0.038).
Table 3.
Activin A levels according to angiographic characteristics of CAD
| Characteristics | Activin A, pg/mL | P | ||
|---|---|---|---|---|
| Mean±SD | Median | V Min; V Max | ||
| Stenosis | ||||
| <50% | 272.2±130.4 | 290.0 | 85.0; 457.0 | |
| (50-70)% | 413.8±255.0 | 316.0 | 1.8; 203.3 | 0.031 |
| >70% | 669.9±507.0 | 504.0 | 85.0; 2126.0 | |
| Number of stenotic coronary arteries | ||||
| Single vessel disease | 750.5±652.5 | 566.0 | 85.0; 4188.0 | 0.038 |
| Multi-vessel disease | 589.5±490.9 | 380.0 | 85.0; 2660.0 | |
Max-maximum value, Min-minimum value. Values are expressed as the mean±SD. P<0.05
Activin A levels in different clinical groups
Activin A levels were significantly higher in all CAD patients (660.1±557.7 pg/mL) than in controls (373.7±95.7 pg/mL; p=0.041) (Table 4). Activin A levels were comparable (p=0.104) between patients who presented with STEMI (647.5±714.0 pg/mL; median: 380.0 pg/mL) and those who presented with NSTEMI (663.8±478.7 pg/mL; median: 461.0 pg/mL) (Table 4).
Table 4.
Levels of Activin A in different clinical groups
| Marker | Controls N=207 | Patients N=310 | P | UA N=107 | NSTEMI N=111 | STEMI N=92 |
|---|---|---|---|---|---|---|
| Activin A, pg/mL | 373.7±95.7 | 660.1±557.7 | 669.6±443.7 | 666.7±458.8 | 647.5±714.0 | |
| Median | 373.7 | 435.0 | 0.041 | 581.0 | 538.0 | 380.0 |
| V Min; V Max | 162.0; 512.0 | 85.0; 4188.0 | 85.0; 2126.0 | 85.0; 126.0 | 111.0; 4188.0 |
NSTEMI-non ST elevation myocardial infarction, STEMI-ST elevation myocardial infarction, UA-unstable angina, V max-maximum value, V Min- minimum value, P (NSTEMI vs. UA)=0.744; P (STEMI vs. UA)=0.172; P (NSTEMI vs. STEMI)=0.104. P values <0.05 statistical signifiance
Activin A levels and scoring system (TIMI risk score)
Serum activin A levels were not significantly decreased with the increase in TIMI risk score in ACS patients. As illustrated in Figure 1, activin A levels were not significantly different (p=0.590) among group 1 (629.8±446.8 pg/mL), group 2 (665.5±653.9 pg/mL), and group 3 (680.4±504.0 pg/mL).
Figure 1.
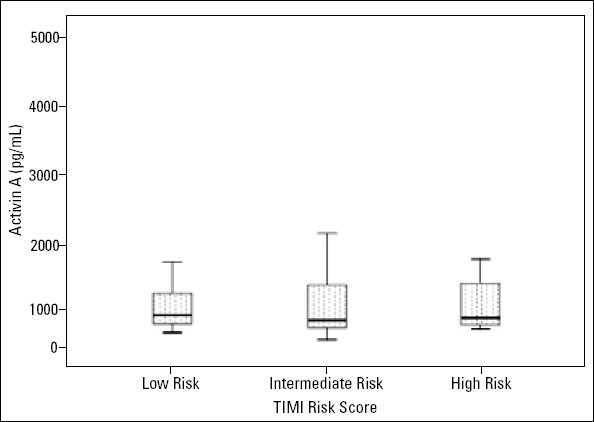
Distribution of serum activin A levels in low-, intermediate-, and high-risk groups. ACS patients were divided into three subgroups according to TIMI risk score: group 1 [low TIMI risk score (1-2)], group 2 [intermediate TIMI risk score (3-4)], group 3 [high TIMI risk score (≥5)]. Activin A levels were 629.8±446.8 pg/mL in the low-risk group, 665.5±653.9 pg/mL in the intermediate-risk group, and 680.4±504.0 pg/mL in the high-risk group (P=0.590)
Linear regression analysis
Univariate linear regression analysis was performed for assessing the contribution of potential confounders on altered activin A levels in CAD. These confounders included age, gender, body mass index, diabetes, hypertension, hyperlipidemia, smoking, and diagnosis at admission. Results from Table 5 show that male gender (β=–0.286; 95% CI: –623.6 to –176.4; p=0.001) and smoking (β=–0.177; 95% CI: –380.59 to –14.49; p=0.035) were independent predictors of activin A levels. Multivariate analysis demonstrated that male gender (β=–0.260; 95% CI: –617.39 to –110.04; p=0.005) was the only independent predictor of activin A levels (Table 5).
Table 5.
Univariate and multiple analysis for Activin A
| Factors | Activin A | |||
|---|---|---|---|---|
| Univariate | Multiple | |||
| β (95% CI) | P | β (95% CI) | P | |
| Age | 0.074 [–4.95; 12.41] | 0.397 | – | – |
| Men | –0.286 [–623.61; –176.41] | 0.001* | –0.260 [-617.39; –110.04] | 0.005 |
| BMI | –0.023 [–24.92; 20.04] | 0.829 | – | – |
| Smoking | –0.177 [–380.59; –14.49] | 0.035* | –0.055 [–263.94; 140.43] | 0.547 |
| Diabetes | 0.043 [–138.77; 235.72] | 0.610 | – | – |
| Dyslipidemia | –0.016 [–244.25; 201.78] | 0.851 | – | – |
| Hypertension | 0.044 [–137.64; 235.92] | 0.604 | – | – |
| History of CAD | –0.031 [–236.46; 161.88] | 0.712 | – | – |
| Menopause | 0.089 [–34.23; 106.74] | 0.311 | – | – |
| Ejection fraction | 0.064 [–9.6; 14.92] | 0.697 | – | – |
| Heart rate | –0.031 [–7.58; 5.30] | 0.729 | – | – |
| SBP | –0.28 [–38.43; 53.21] | 0.750 | – | – |
| DBP | –0.006 [–77.46; 71.89] | 0.941 | – | – |
| Creatinin | –0.131 [–4.95; 0.719] | 0.142 | – | – |
| Glucose | –0.123 [–40.40; 9.03] | 0.211 | – | – |
| CT | 0.089 [–34.23; 106.74] | 0.311 | – | – |
| Lp(a) | 0.130 [–129.73; 852.66] | 0.148 | – | – |
| TG | 0.082 [–41.34; 115.64] | 0.351 | – | – |
| LDL-c | 0.040 [–60.24; 92.98] | 0.673 | – | – |
| HDL-c | –0.066 [–365.937; 174.81] | 0.485 | – | – |
| ApoA1 | –0.150 [–756.65; 65.10] | 0.098 | – | – |
| ApoB | –0.070 [–514.53; 227.84] | 0.446 | – | – |
*: P<0.05
ROC Curve for activin A in predicting CAD
The area under the ROC curve for activin A levels in CAD patients was 0.513±0.041 (95% CI: 0.443–0.594, p=0.749). In contrast to IL-6 and hs-CRP levels, activin A level was not a significant predictor of CAD severity. The area under the ROC curve for IL-6 levels was higher than that for the serum hs-CRP level [IL-6: 0.860±0.038, (95% CI: 0.785–0.935), p<0.001 vs. hs-CRP: 0.816±0.038, (95% CI: 0.742–0.889), p<0.001] (Fig. 2).
Figure 2.
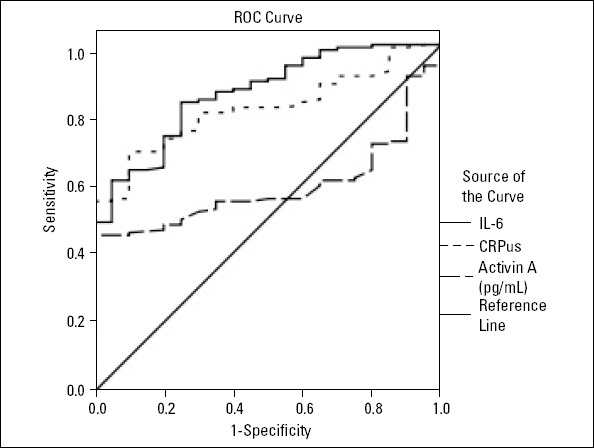
ROC analysis for activin A, hs-CRP, and IL-6 levels in predicting CAD. Area under the ROC curve for activin A levels was 0.590±0.047 (95% CI: 0.439–0.591, P=0.193); for hs-CRP levels was 0.816±0.038 (95% CI: 0.742–0.889, P<0.001); and for IL-6 levels was 0.860±0.038 (95% CI: 0.785–0.935, P<0.001)
Discussion
In this study, we demonstrated increased activin A levels in CAD patients compared with those in control subjects. When stratified according to activin A cut-off values, patients with high and low activin A levels had similar clinical and biochemical profile, except for severe stenosis, which was significantly higher in the high activin A group than in the low activin A group. High activin A levels were observed in patients with severe stenosis, which appears to correlate with the degree of severity. On the other hand, activin A levels did not affect TIMI risk score, and serum activin A levels were not predictive of CAD. Although not completely addressed, this may result from the contribution of other risk factors, such as obesity and diabetes, to the difference in time to admission, to the use of drugs, and also to the relatively low number of participants in this study. Significant differences in baseline characteristics, such as age, hypertension, and diabetes, observed between the control and patient groups might be due to the multifactorial property of CAD and selected profile of controls participant.
Based on the increased activin A levels in different clinical groups vs. controls, we hypothesized that activin A is involved in atherosclerosis and is associated with the degree of stenosis. The association of high activin A levels in patients with multivessel disease can be explained by the fact that one-vessel disease may be multi-stenotic and can thus present with another atherosclerosis progression step, such as plaque and lesion formation.
Previous studies have shown that activin A is a multifunctional cytokine, with roles in cell growth and differentiation, regulation of inflammatory responses, and tissue repair and regeneration (6). This implicates altered activin A levels in the initiation and progression of atherosclerotic lesions in the vessel wall (9). Increased evidence has shown that activin A levels are increased in several inflammatory diseases, such as inflammatory bowel disease, septicemia, meningitis, and arthropathies (6, 10). Increased activin A levels in CAD patients reported here confirms a role of activin A in atherosclerosis, which is consistent with the findings of earlier studies showing that serum activin A levels are increased in patients with stable angina pectoris and UA pectoris (10); this is correlated with the clinical severity of heart failure (6).
On the other hand, two independent studies have shown that activin A levels are markedly higher in STEMI and do not correlate with the number of diseased vessels (11, 10). This was in apparent disagreement with our results, which showed that activin A levels were associated with the degree of stenosis, but not with the number of diseased vessels. These differences could be explained by the fact that activin A levels might reflect plaque vulnerability, but not the plaque volume of coronary arteries (10). It has been postulated that high activin A levels during clinical and experimental heart failure indicate an involvement of activin A in the pathogenesis of heart failure (12). It is noteworthy that activin A markedly attenuates leukemia inhibitory factor-induced increase in cardiomyocyte length and perimeter and sarcomeric organization (13).
Reportedly, activin A plays an inflammatory role in vascular tissues at different stages of atherosclerosis (9) and exerts anti-inflammatory properties in atherosclerosis (6). It has been suggested that activin A attenuates vascular inflammation and oxidative stress in inflammatory disorders, such as atherosclerosis and type 2 diabetes (14), and inhibits the inflammatory response of cultured macrophages and uptake of modified lipoproteins by phagocytic cells (9). In addition, activin A is involved in plaque stabilization, in part by inhibiting foam cell formation and by the induction of the differentiation of contractile, non-proliferative phenotype in cultured smooth muscle cells (6). Reportedly, activin A plays a protective role against cardiac endoplasmic reticulum stress in diabetes, attenuates the release of IL-8 from ECs, enhances the expression of the antioxidative enzyme metallothionein. This suggests that activin A protects the heart and kidneys against diabetes-induced pathophysiology (14). Activin A has been shown to be beneficial in preventing the formation of SMC-rich lesions in mice, which are similar to those formed in in-stent restenosis in humans, in contrast to TGF-β, which aggravates vascular lesions due to excessive extracellular matrix synthesis (9). Activin A decreased the release of IL-6, IL-8, and macrophage inflammatory protein-1-alpha from mononuclear cells in patients with angina, suggesting anti-inflammatory potential of activin A (11).
The novelty of our study is in analyzing the relationship between serum activin A levels and TIMI risk score for ACS, a validated clinical risk score that is a short- and long-term predictor of death and cardiovascular complications (15). Activin A levels did not increase as the TIMI risk score increased, suggesting that activin A does not predict CAD complication. Of the cardiovascular risk factors studied here, only gender was associated with altered activin A levels. An autopsy examination has suggested that both genders present a different change in signaling pathway apoptosis in infarct zone, indicating that the activation of the apoptotic cascade induces protection against ischemia in females (16). The differences between males and females in early death and cardiac rupture after myocardial infarction may be due to the difference in necrosis repair mechanisms, extracellular matrix alteration, myocyte slippage, and myocardial tissue remodeling (17). It should be noted that gender with other clinical research needs to be addressed when examining the association with activin A. Despite its production in response to inflammatory mediators, especially IL-6 (18), hs-CRP did not show correlation with activin A. It has been reported that hs-CRP levels are correlated with other inflammatory and necrosis markers in different cardiovascular diseases, such as acute myocardial infarction and heart failure (18). The lack association between activin A and the inflammatory markers IL-6 and hs-CRP can be explained by the fact that activin A does not have a direct effect on inflammatory cytokines, although it may act as a mediator in acute stress, such as inflammation and oxidative stress (10).
Study limitations
First, the small number of participants may have contributed to the lack of statistically significant differences. Second, because few variables showed p values of <0.05 in univariate analyses, it is possible that lipid-lowering drugs, for example, affect the involvement of lipid variables in circulating activin A stability. Third, in some participants, high hs-CRP, IL-6, and activin A level might have been affected by unknown infections. Forth, the mean age and other baseline characteristics of the control group were significantly different from those of the patient groups, and this can be explained by the importance of male gender and age as cardiovascular risk factors and the young age of Tunisian donors. Finally, in this study, we did not consider medications before admission that might have affected the prognosis of patients.
Conclusion
The absence of significant association between activin A and angiographic characteristics and the area under the ROC curve for activin A levels following the ROC analysis indicated that this cytokine is not a predictive marker for CAD severity and cannot be a valuable tool in the atherogenic risk assessment of CAD patients. Serum activin A levels were not associated with TIMI risk score, risk factors, and biochemical markers. In contrast, activin A levels were significantly elevated in patients compared with those in healthy subjects, suggesting a potential role of activin A in atherosclerosis.
Acknowledgments:
The authors are grateful to the patients and volunteers for their collaboration; to Pr. Wassim Youssef Almawi (Biochemistry Department, Arabian Gulf University, Kingdom of Bahrain) for his excellent technical assistance and advice; to Pr. Habib Gamra (Cardiology A Department, Fattouma Bourguiba University Hospital, Monastir, Tunisia), Pr. Faouzi Maatouk (Cardiology B Department, Fattouma Bourguiba University Hospital, Monastir, Tunisia), and the entire team for collecting blood samples from patients; and the staff members of the Biochemistry Laboratory (Farhat Hached University Hospital, Sousse, Tunisia) for their technical support.
Footnotes
Disclosure statement: The authors declare that they have no competing interests.
Conflict of interest: The authors have no conflicts of interest to declare.
Peer-review: Externally peer-reviewed.
Authorship contributions: Concept – N.B.; Design – A.M.; Supervision – F.B.; Fundings – A.M.; Materials – S.F.; Data collection &/or processing – N.B.; Analysis &/or interpretation – F.B.; Literature search – F.M.; Writing – H.G.; Critical review – S.F. – Other- A.M.

Ayla Bağ, from EFSAD’s collections.
References
- 1.Park CS, Hong OK, Kim MK, Chung WB, Choi YS, Baek KH, et al. Serum Bone Morphogenic Protein-4 contributes to discriminating coronary artery disease severity. Medicine. 2015;94:e1530. doi: 10.1097/MD.0000000000001530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krecki R, Drozdz J, Szczesniak P, Orszulak-Michalak D, Krzeminska-Pakula M. Novel atherogenesis markers for identification of patients with a multivessel coronary artery disease. Kardiol Pol. 2008;66:1173–80. [PubMed] [Google Scholar]
- 3.Rothenbacher D, Müller-Scholze S, Herder C, Koenig W, Kolb H. Differential expression of chemokines, risk of stable coronary heart disease, and correlation with established cardiovascular risk markers. Arterioscler Thromb Vasc Biol. 2006;26:194–9. doi: 10.1161/01.ATV.0000191633.52585.14. [DOI] [PubMed] [Google Scholar]
- 4.Caselli C, De Graaf MA, Lorenzoni V, Rovai D, Marinelli M, Del Ry S, et al. HDL cholesterol, leptin interleukine-6 predtict higher risk coronary anatomy assessed by CT angiography in patients with stable chest pain. Atherosclerosis. 2015;241:55–61. doi: 10.1016/j.atherosclerosis.2015.04.811. [DOI] [PubMed] [Google Scholar]
- 5.Hayashi Y, Maeshima K, Goto F, Kojima I. Activin A as a critical mediator of capillary formation: Interaction with the fibroblast growth factor action. Endocr J. 2007;54:311–8. doi: 10.1507/endocrj.k06-222. [DOI] [PubMed] [Google Scholar]
- 6.Smith C, Yndestad A, Halvorsen B, Ueland T, Waehre T, Otterdal K, et al. Potential anti-inflammatory role of activin A in acute coronary syndromes. J Am Coll Cardiol. 2004;44:369–75. doi: 10.1016/j.jacc.2004.03.069. [DOI] [PubMed] [Google Scholar]
- 7.Engelse MA, Neele JM, van Achterberg TA, van Aken BE, van Schaik RH, Pannekoek H, et al. Human activin-A is expressed in the atherosclerotic lesion and promotes the contractile phenotype of smooth muscle cells. Circ Res. 1999;85:931–9. doi: 10.1161/01.res.85.10.931. [DOI] [PubMed] [Google Scholar]
- 8.Mogensen UM, Jensen T, Køber L, Kelbæk H, Mathiesen AS, Dixen U, et al. Cardiovascular autonomic neuropathy and subclinical cardiovascular disease in normoalbuminuric Type 1 diabetic patients. Diabetes. 2012;61:1822–30. doi: 10.2337/db11-1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Groenendijk BC, Benus GF, Klous A, Pacheco YM, Volger OL, Fledderus JO, et al. Activin A induces a non-fibrotic phenotype in smooth muscle cells in contrast to TGF- β. Exp Cell Res. 2011;317:131–42. doi: 10.1016/j.yexcr.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 10.Miyoshi T, Hirohata S, Uesugi T, Hirota M, Ohnishi H, Nogami K, et al. Relationship between activin A level and infarct size in patients with acute myocardial infarction undergoing successful primary coronary intervention. Clin Chim Acta. 2009;401:3–7. doi: 10.1016/j.cca.2008.10.027. [DOI] [PubMed] [Google Scholar]
- 11.Lin JF, Hsu SY, Teng MS, Wu S, Hsieh CA, Jang SJ, et al. Activin A Predicts Left Ventricular Remodeling and Mortality in Patients with ST-Elevation Myocardial Infarction. Acta Cardiol Sin. 2016;32:420–7. doi: 10.6515/ACS20150415A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yndestad A, Ueland T, Øie E, Florholmen G, Halvorsen B, Attramadal H, et al. Elevated levels of activin A in heart failure: potential role in myocardial remodeling. Circulation. 2004;109:1379–85. doi: 10.1161/01.CIR.0000120704.97934.41. [DOI] [PubMed] [Google Scholar]
- 13.Florholmen G, Halvorsen B, Beraki K, Lyberg T, Sagen EL, Aukrust P, et al. Activin A inhibits organization of sarcomeric proteins in cardiomyocytes induced by leukemia inhibitory factor. J Mol Cell Cardiol. 2006;41:689–97. doi: 10.1016/j.yjmcc.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 14.Andersen GØ, Ueland T, Knudsen EC, Scholz H, Yndestad A, Sahraoui A, et al. Activin A levels are associated with abnormal glucose regulation in patients with myocardial infarction: potential counteracting effects of activin A on inflammation. Diabetes. 2011;60:1544–51. doi: 10.2337/db10-1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang CH, Chang CC, Kuo CL, Huang CS, Chiu TW, Lin CS, et al. Serum Iron Concentration, but Not Hemoglobin, Correlates with TIMI Risk Score and 6-Month Left Ventricular Performance after Primary Angioplasty for Acute Myocardial Infarction. PLoS One. 2014;9:e104495. doi: 10.1371/journal.pone.0104495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mallat Z, Fornes P, Costagliola R, Esposito B, Belmin J, Lecomte D, et al. Age and gender effects on cardiomyocyte apoptosis in the normal human heart. J Gerontol A Biol Sci Med Sci. 2001;56:M719–23. doi: 10.1093/gerona/56.11.m719. [DOI] [PubMed] [Google Scholar]
- 17.Piro M, Della Bona R, Abbate A, Biasucci LM, Crea F. Sex-Related Differences in Myocardial Remodeling. J Am Coll Cardiol. 2010;55:1057–65. doi: 10.1016/j.jacc.2009.09.065. [DOI] [PubMed] [Google Scholar]
- 18.Altay S, Çakmak HA, Öz TK, Karadeniz FÖ, Türer A, Erer HB, et al. Long-term prognostic significance of pentraxin-3 in patients with acute myocardial infarction:5-year prospective cohort study. Anatol J Cardiol. 2017;17:202–9. doi: 10.14744/AnatolJCardiol.2016.7307. [DOI] [PMC free article] [PubMed] [Google Scholar]