

Abstract
The design, synthesis and formulation of non‐viral gene delivery vectors is an area of renewed research interest. Amongst the most efficient non‐viral gene delivery systems are lipopolyplexes, in which cationic peptides are co‐formulated with plasmid DNA and lipids. One advantage of lipopolyplex vectors is that they have the potential to be targeted to specific cell types by attaching peptide targeting ligands on the surface, thus increasing both the transfection efficiency and selectivity for disease targets such as cancer cells. In this paper, we have investigated two different modes of displaying cell‐specific peptide targeting ligands at the surface of lipopolyplexes. Lipopolyplexes formulated with bimodal peptides, with both receptor binding and DNA condensing sequences, were compared with lipopolyplexes with the peptide targeting ligand directly conjugated to one of the lipids. Three EGFR targeting peptide sequences were studied, together with a range of lipid formulations and maleimide lipid structures. The biophysical properties of the lipopolyplexes and their transfection efficiencies in a basal‐like breast cancer cell line were investigated using plasmid DNA bearing genes for the expression of firefly luciferase and green fluorescent protein. Fluorescence quenching experiments were also used to probe the macromolecular organisation of the peptide and pDNA components of the lipopolyplexes. We demonstrated that both approaches to lipopolyplex targeting give reasonable transfection efficiencies, and the transfection efficiency of each lipopolyplex formulation is highly dependent on the sequence of the targeting peptide. To achieve maximum therapeutic efficiency, different peptide targeting sequences and lipopolyplex architectures should be investigated for each target cell type.
Keywords: gene delivery, lipopolyplex, liposome, targeting peptide
1. INTRODUCTION
The delivery of oligonucleotide or genetic material to specific cells has been a long‐term goal for treatment of intractable diseases such as cancer, cystic fibrosis, retinal disorders, and cardiovascular disease. A range of potential gene delivery systems, both viral and non‐viral, have been used for the delivery of pDNA, siRNA, mRNA, and miRNA.1, 2 Viral gene delivery systems have high efficiencies and generally show good transfection properties in vitro and in vivo, with several in clinical trials, and one (Glybera) recently approved in the EU.3 However, in recent years, concerns about the potential safety of such approaches have led to a renewed interest in non‐viral gene delivery vectors, as these have lower immunogenicity, have the potential to deliver large payloads, and can be functionalised to target specific cell types. A range of nanoparticle‐based systems have been developed for gene delivery,2 with the most common vectors being those based on cationic lipids (lipoplexes), cationic polymers (polyplexes), or a combination of cationic lipids and cationic polymers (lipopolyplexes).1
Despite the advantages of non‐viral gene delivery vectors, in the past they have been slow to progress to clinical use due to their generally lower efficiency of gene delivery. Recent understanding of the barriers to efficient non‐viral vector delivery, such as nanoparticle instability in vivo, poor targeting to specific cells, and inefficient transport through biological barriers such as the cell membrane, has led to an increased number of candidate vectors currently in clinical trials.2 However, further improvements in these areas are still needed to realise the potential of gene‐based therapies, in particular in the treatment of cancers, where approaches such as suicide gene therapy,4 regulation of gene expression by delivery of miRNA,5 p53 replacement gene therapy,6 and redirection of T‐cell specificity towards cancer cells7 have recently shown promise. Targeting of nanoparticles to tumors can be passive or active. Nanoparticles of 100 to 200 nm in diameter tend to accumulate in tumours, through a combination of leaky tumor endothelium and ineffective lymphatic drainage, a phenomenon known as the enhanced permeability and retention effect (EPR).8 Whilst this passive targeting to tumors is undoubtedly important, inter‐ and intra‐tumoral heterogeneity of the tumor microenvironment means that the EPR effect may be more pronounced in some tumors than others.9 Moreover, it is also clear that accumulation in tumors is necessary, but accumulation on its own is not sufficient for cellular uptake.10 This frequently needs to be enhanced by the presence of cell‐specific targeting ligands on the surface of the nanoparticles. Such targeting ligands both enhance the selectivity of nanoparticles for cancer cells and may also trigger internalisation via mechanisms such as receptor‐mediated endocytosis. For example, exploiting the fact that epidermal growth factor receptor (EGFR) is over‐expressed on the surface of many cancer cell types, such as basal‐like breast cancer cells, has been very effective in targeting nanoparticles to such tumors.11 A range of preclinical studies have now demonstrated that active targeting improves the efficacy of nanoparticle‐based therapies for several cancer types,12, 13 and several targeted nanoparticle therapies are now in clinical trials.13 In addition, liposome‐based delivery systems and other nanoparticles that are shielded from the reticuloendothelial system with a surface coating of poly (ethyleneglycol) (PEG) or n‐ethylene glycol (n‐EG) have a significantly longer half‐life in vivo, allowing more of the nanoparticles to localise to the tumor.14
Lipopolyplex gene delivery systems combine the desirable features of lipoplexes and polyplexes with high in vivo transfection efficiencies and a nanoscale size (100‐200 nm). They are self‐assembling nanoparticles which can be formulated from a wide range of components, enabling them to be tailored to many different applications and have multiple functionalities (reviewed in Rezaee et al15). For example: formulation of LPD nanoparticles using cationic lipids and peptide sequences derived from protamine or histone resulted in enhancement of cell transfection in vitro16; in early work, RGD‐targeted LPD revealed a 30‐fold increase in cell transfection compared with the use of naked DNA17; lipopolyplexes incorporating a fusion protein consisting of the carboxy‐terminal domain of histone H1 and a nuclear localization signal gave transfection efficiencies up to 20‐fold higher than lipofectin/DNA complexes.18 We have previously developed targeted, environmentally responsive lipid:peptide:DNA (LPD) lipopolyplexes for gene delivery. These lipopolyplex formulations contain a bimodal peptide with a cationic sequence to bind and condense pDNA,19 a linker sequence (RVRR) which can be cleaved by enzymes within the endosome, and a targeting sequence.20 The formulation of the lipopolyplexes also includes cationic lipids such as DOTMA, and the helper lipid 1,2‐dioleoyl‐sn‐glycero‐3‐phosphoethanolamine (DOPE). The latter is believed to mediate release of the nanoparticle components from the endosome by fusion to the endosomal membrane and perturbing the structure to a non‐lamellar HII phase.21 In the original paper describing the LPD vector22 and in more recent work23 the order of mixing of the lipid, peptide, and plasmid DNA were studied in detail, and it was shown that this order of mixing was crucial to ensure high transfection efficiencies. We have previously24 used a combination of FCS, freeze‐fracture electron microscopy, and fluorescence quenching experiments to prove the stoichiometry of the complex and demonstrate that the DNA is tightly condensed to the peptide in an inner core, which is surrounded by a disordered lipid layer, from which the integrin‐targeting sequence of the peptide partially protrudes, mediating internalization through receptor‐mediated endocytosis. Indeed, it has been shown by several groups (most recently Munye et al25) that cationic peptides more efficiently condense and package DNA than cationic liposomes, a phenomenon attributed to the higher charge density of the peptide molecules.26 We have recently developed lipopolyplexes which are sterically shielded by a shallow but even coverage of n‐EG conferred by incorporating novel cationic lipids with short n‐EG at the headgroup (n = 2‐6)20 and have studied the cellular uptake of the lipopolyplexes and the intracellular distribution of the components by confocal microscopy. We have also shown that liposomes formulated including these n‐EG lipids form nanoparticles that are shielded with a shallow, homogeneous n‐EG layer, and that these have much better cellular uptake than liposomes formulated with 1,2‐distearoyl‐sn‐glycero‐3‐phosphoethanolamine‐N‐[carboxy (polyethyleneglycol)2000] (DSPE‐PEG2000).27 We have recently used this approach to formulate lipopolyplexes that selectively transfected tumor cells with pDNA coding for a FRET biosensor and used this to monitor EGFR inhibition by tyrosine kinase inhibitors in vivo using quantitative FRET‐FLIM imaging.28 In this work, the bifunctional peptide incorporated peptide sequences targeting EGFR, conferring tumor selectivity and active targeting on these lipopolyplexes.
For these lipopolyplexes, the tumor selectivity and transfection efficiency both depend on how well the targeting moiety is displayed at the surface of the nanoparticle. A range of approaches for mounting the targeting moiety at the surface are possible29: as well as the bimodal peptide approach that we have adopted in previous work, other groups have successfully conjugated targeting peptides directly to the surface of liposomes,30 lipoplexes,31 and polyplexes.32 However, it is imperative to understand how subtle changes in the structures of the toolbox components can affect both the macromolecular architecture of the nanoparticles and also the selectivity, stability, and effective transfection in vivo of the resulting imaging probe.
In this paper, we have for the first time directly compared two different modes of attaching the targeting moiety to the lipopolyplex, via the bimodal peptide approach versus direct conjugation to the lipid. We have determined the effect that these two approaches have on the macromolecular structure of the lipopolyplex and its transfection efficiency. To develop this technology, we have used pDNA that has optical readouts through the expression of either firefly luciferase33, 34 or green fluorescent protein (GFP)35 with the aim that the lipopolyplex formulations could then be applied to other targeted gene‐based therapy approaches.
2. MATERIALS AND METHODS
2.1. General methods, reagents, and chemical synthesis
General methods for chemical synthesis are included in the Supporting Information. All chemicals were of commercial quality and have been used without additional purification. Unless otherwise stated chemicals were bought from Sigma Aldrich Co. Ltd. 1,2‐Dioleoyl‐sn‐glycero‐3‐phosphoethanolamine (DOPE) was purchased from Avanti Polar Lipids Inc. 1,2‐Di‐O‐octadecenyl‐3‐trimethylammonium propane (DOTMA) was synthesized according to literature procedures.36 2,3‐Di‐((9Z)‐octadecenyloxy)propyl‐N‐(2‐{2‐[2‐(2‐hydroxyethoxy)ethoxy]ethoxy}ethyl)‐N,N‐dimethylammonium bromide (DODEG‐4) was synthesised as previously described.37 Experimental procedures for the synthesis, purification, and characterisation of the novel maleimide lipids DiOleyl‐Dimethyl‐Spacer‐Maleimide (DODSM) 1 and DiOleyl‐Spacer‐EthyleneGlycol3‐Maleimide (DOSEG3M) 2 are described in the Supporting Information. All peptides (Table S1) were synthesized via solid‐phase peptide synthesis using Fmoc chemistry. The synthetic procedures, purification methods, and compound characterisations are reported in the Supporting Information.
2.2. Plasmid DNA
The lentiviral transfer vector plasmid pSEW38 was engineered for the transient expression of firefly luciferase (5x FLuc)34 for bioluminescence, along with the enhanced GFP (eGFP) as a marker for fluorescence‐activated cell sorting (FACS) analysis.35 Plasmid DNA was amplified in bacteria (One Shot Top10 competent cells, Invitrogen) grown overnight in LB Broth with 100 μg/mL ampicillin, following heat‐shock transfection with the plasmid DNA. DNA was extracted and purified using a Qiagen Plasmid Maxi kit, according to the manufacturer's instructions, and eluted in de‐ionised water. DNA concentration and purity were measured using a spectrophotometer (NanoDrop 2000, Thermofisher), and the DNA stock was stored at −20°C until use.
2.3. Lipopolyplex formulation
Bimodal peptide targeted lipopolyplexes: For lipopolyplexes bearing their EGFR targeting peptide on the oligo‐lysine peptide, a mixture of 40 mol% DODEG‐4, 20 mol% DOTMA, and 40 mol% DOPE (formulation F7, Table 1) was prepared at a concentration of 1 mM in chloroform. The lipid mixture was slowly evaporated under reduced pressure to form a lipid thin film and further dried under high vacuum for at least 2 hours to ensure the complete removal of organic solvents. The thin film was then hydrated with deionised water to give a liposome solution with a final concentration of 1 mM lipids in water and sonicated in a VWR ultrasonic bath (45 kHz, effective power 80 W) to give an average size of around 200 nm. Subsequently, liposomes were diluted to a concentration of 100 μM and a solution of the corresponding bimodal peptide P1 (K16‐RVRR‐YHWYGYTPQNVI), P2 (K16‐RVRR‐LARLLT), or P3 (K16‐RVRR‐AEYLR) was added to a concentration of 5 μM, mixed and left standing for 5 minutes. Plasmid DNA was added to give a concentration of 1 μg/100 μL DNA per liposome‐peptide mixture to give bimodal peptide targeted lipopolyplexes.
Table 1.
Molecular composition of lipids used for liposomal formulations F1 to F9 for the preparation of surface targeted and bimodal lipopolyplexes. Liposomes were made up to a total concentration of 1 mM lipid and subsequently diluted with de‐ionised water to give the appropriate concentrations for the preparation of lipopolyplexes (100 μM unless otherwise noted)
| Formulation | DODEG3 (Mol%) | DOPE (Mol%) | DOTMA (Mol%) | CHOL (Mol%) | DODSM (1) (Mol%) | DOSEG3M (2) (Mol%) | |
|---|---|---|---|---|---|---|---|
| Surface targeted | F1 | 40 | 40 | 10 | 0 | 10 | 0 |
| Surface targeted | F2 | 40 | 35 | 10 | 0 | 15 | 0 |
| Surface targeted | F3 | 40 | 30 | 10 | 0 | 20 | 0 |
| Surface targeted | F4 | 40 | 30 | 20 | 0 | 10 | 0 |
| Surface targeted | F5 | 40 | 15 | 10 | 15 | 10 | 0 |
| Surface targeted | F6 | 30 | 25 | 10 | 20 | 15 | 0 |
| Bimodal | F7 | 40 | 40 | 20 | 0 | 0 | 0 |
| Bimodal | F8 | 40 | 20 | 20 | 20 | 0 | 0 |
| Surface targeted | F9 | 40 | 40 | 10 | 0 | 0 | 10 |
Surface‐targeted lipopolyplexes: For liposomes bearing the EGFR targeting peptide sequence on the lipid component, mixtures of DODEG‐4, DOTMA, DODSM 1 or DODEG3SM 2 and DOPE were prepared in the ratios shown in Table 1, at a concentration of 1 mM in chloroform. The lipid mixture was slowly evaporated under reduced pressure to form a lipid thin film and further dried under high vacuum for at least 2 hours to ensure complete removal of organic solvents. The thin film was then hydrated with deionised water to give liposome solutions F1 to F6 and F9 (Table 1) with a final concentration of 1 mM lipids in water. These liposome solutions were then sonicated in a VWR ultrasonic bath (45 kHz, effective power 80 W) to give liposomes with an average size of approximately 200 nm. Peptide targeting sequences P4 (CYHWYGYTPQNVI), P5 (CLARLLT), and P6 (CAEYLR), all with Cys residues at the N‐terminus, were then added to a final concentration of 1 mM and incubated for 2 hours. The liposomes were diluted to a lipid concentration of 500 μM and dialysed against deionised water (BioDesignDialysis Tubing (D001), 14000 MWCO) over 16 hours during which the water was changed 3 times. Subsequently, the peptide‐covered liposomes were diluted to a concentration of 100 μM, and a solution of oligo‐lysine (K16, P7) was added to a concentration of 5 μM, mixed and left standing for 5 minutes. Plasmid DNA was added to a concentration of 1 μg/100 μL DNA per liposome peptide mixture to give lipid bound targeted lipopolyplexes.
The final composition of all lipid formulations used for the preparation of both surface targeted and bimodal liposomes is summarised in Table 1.
2.4. Dynamic light scattering and zeta potential
The lipopolyplexes were characterised using dynamic light scattering and zeta potential measurements. Data were obtained using a Malvern Zetasizer Nano‐ZS (Malvern, UK). Aliquots of 10 μL were diluted to 500 μL in deionised water and analysed in triplicates. A representative sample of the prepared liposomal formulations and the resulting lipopolyplexes are reported in the Supporting Information (Tables S2, S3, and S4).
2.5. Cell culture
HCC1954 human breast cancer cells were grown in T175 flasks (Fisher Scientific, UK) in Roswell Park Memorial Institute (RPMI‐1640) medium (Invitrogen, UK), supplemented with 10% heat inactivated fetal calf serum (GIBCO, USA) in a humidified incubator at 37°C with 95% air and 5% CO2. Cells were grown to 80% confluence prior to trypsinisation, counting, and plating for in vitro experiments.
2.6. Liposome‐mediated transfection
HCC1954 cells were plated in six‐well plates (Corning, USA) at a concentration of 5 × 105 cells per well in triplicate and incubated overnight to 80% confluency. Prior to adding liposomes, normal culture medium was aspirated from the wells, and the cells were washed once with phosphate buffered saline (GIBCO, USA), and 2 mL of serum free RPMI‐1640 was added. Then, 100 μL of either liposomes or phosphate buffered saline (control) was added to each well and incubated at 37°C. After 4 hours, 2 mL of normal culture medium was added and the mixture incubated for an additional 20 hours. After 24 hours, the incubation medium was aspirated from the wells and 2 mL of normal RPMI 1640 was added. At 24 and 48 hours after incubation, an in vitro bioluminescence assay (firefly luciferase expression) and FACS (eGFP expression) were performed as a read out of liposome mediated transfection efficiency.
Cell sorting was performed using a BD LSRFortessa (Becton Dickinson, USA) to assess the percentage of eGFP expression in each well. FACS analysis was performed using BD FACSDiva software (version 8.0.1).
The in vitro bioluminescence assay was performed using a IVIS Lumina (PerkinElmer, USA), and images were acquired immediately after adding D‐luciferin (300 μg/mL, beetle luciferin potassium salt, Promega, Madison, WI) using large binning and exposure time of 300 seconds. A region of interest was placed over each well, and the total radiance (photons/s) was quantified using Living Image software (version 4.5.2).
A two‐tailed paired t‐test assuming equal variances was performed to determine significant difference, at the 5% level statistical significance. Errors are given as standard deviation.
2.7. Fluorescence quenching
Fluorescence quenching experiments were carried out on an Agilent Cary Eclipse fluorometer equipped with a single water thermo cell holder. Spectral grade solvents were used for the fluorescence measurements. The preparation of fluorescein‐labelled pDNA and BODIPY‐labelled peptides P8 and P9 is described in the Supporting Information. Lipopolyplexes for fluorescence measurements were prepared at 200‐μM lipid, 0.02 μg/μL plasmid DNA, and 10 mM peptide. The lipopolyplex samples were diluted to a lipid concentration of 30 μM, and a total volume of 1.4 mL before fluorescence spectra were recorded and acrylamide was added.24 Collisional quenching of the fluorescence was plotted using the Stern‐Volmer equation:
where F0 and F are the fluorescence intensities in the absence and presence of quencher Q, respectively, kQ is the bimolecular quenching constant, and τ0 is the lifetime of the fluorophore in the absence of quencher. For species in which a single population of fluorophores is present, all equally accessible to the quenching agent, a plot of F0/F will give a linear graph.39
3. RESULTS AND DISCUSSION
3.1. Design of lipopolyplexes: peptide targeting and lipopolyplex architecture
In order to exploit the overexpression of the EGF receptor at the surface of the basal‐like breast cancer cells used in this study, three peptides were investigated that have recently been reported to target this receptor. The GE11 (YHWYGYTPQNVI),40 D4 (LARLLT),41 and AE (AEYLR)42 peptide sequences were previously identified as suitable ligands, via phage display or in silico peptide library screening. These sequences have been successfully used to target liposomes30, 41 and polyplexes42 to tumor cells. As previous studies have shown that the transfection efficiency and route of cellular uptake of such targeted nanoparticles were highly dependent on cell type,43 we elected to compare all three of these sequences in the lipopolyplex formulations.
The lipopolyplex formulation utilised was based on a single, multifunctional peptide which will both condense pDNA and target the nanoparticle to cell surface receptors. We have previously shown that this necessitates the targeting peptide protruding through the surface of the lipid layer, resulting in the “bifunctional peptide” design shown in Figure 1.24 For these lipopolyplex formulations, peptides P1, P2, and P3 were synthesised, with K16 condensing sequences, the RVRR enzymatically cleavable linker, and the GE11 (P1), D4 (P2), or AE (P3) sequences. However, it was reasoned that the targeting sequence might be more sterically accessible to the receptor if it was conjugated to the surface of the nanoparticle, with a separate K16 peptide (P7) included in the formulation to condense the pDNA, giving the “surface targeted” design (Figure 1).
Figure 1.
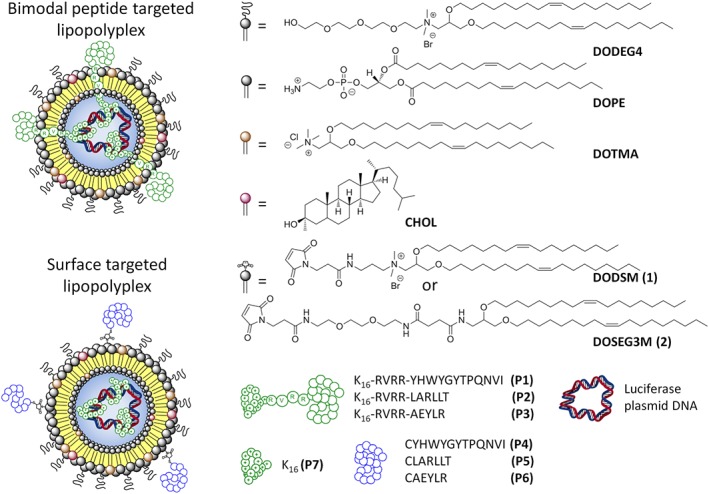
Schematic representation of (top) bimodal peptide targeted lipopolyplexes formulated with a bimodal peptide consisting of an EGFR targeting sequence, a furin cleavable RVRR linker, a DNA condensing K16 sequence, and (bottom) surface targeted lipopolyplexes formulated using an EGFR targeting sequence bioconjugated to the lipid bilayer, and a separate K16 peptide to condense the DNA. The lipid, peptide, and plasmid DNA components used in these experiments are shown on the right hand side
Bioconjugation of targeting moieties to lipids or to liposomes is typically carried out via the reaction of thiol‐functionalised targeting moieties to malemide‐derivatised lipids, using click chemistry, or amide bond formation,29, 44 although hydrazone linkages45 and Staudiger ligations44 have also been reported. The majority of peptide‐targeted liposomes to date have relied on the post‐formulation bioconjugation of thiol‐functionalised targeting moieties to liposomes containing DSPE‐PEG2000‐Mal,29, 30, 41 although other maleimide‐containing lipids with shorter or no PEG linkages have been reported.31, 46, 47 For the surface targeted lipopolyplexes two lipids with maleimide moieties at their head group were synthesised, for incorporation into the liposome formulations and then conjugation to peptide targeting sequences terminating in Cys residues. DODSM (DiOleyl‐Dimethyl‐Spacer‐Maleimide) 1 was designed to be a cationic lipid analogue of DOTMA, whilst DOSEG3M (DiOleyl‐Spacer‐EthyleneGlycol3‐Malelimide) 2 is a neutral lipid with a short n‐EG spacer between the lipid headgroup and the maleimide moiety.
DODSM (1), with the cationic headgroup, was designed to evaluate whether the relative location of the malemide on the surface of the liposomal formulation would have an impact on transfection efficiency or lipopolyplex architecture. Due to the positive charge of the headgroup, the maleimide moiety might be in closer proximity with the aqueous exterior of the lipopolyplex. The synthesis for both maleimide bearing lipids is shown in Scheme 1. Amine 3 was prepared according to literature procedures37 and reacted with N‐maleoyl‐ß‐alanine (4) using HBTU and DIPEA to give DODSM 1. Amine 5 was prepared as previously described27 and similarly reacted with 4 to yield the desired maleimide lipid DOSEG3M 2.
Scheme 1.
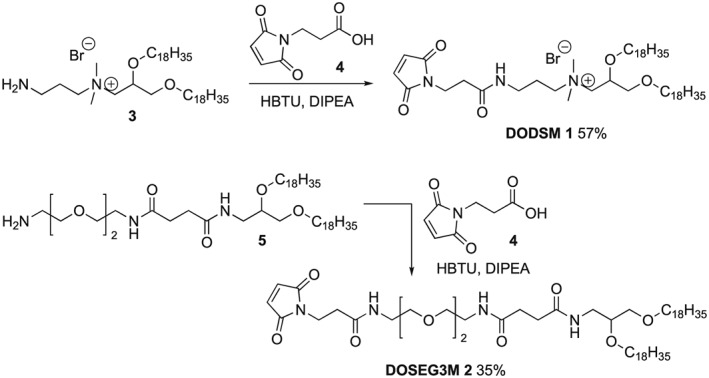
Synthesis of DiOleyl‐dimethyl‐spacer‐Maleimide (DODSM, 1) and DiOleyl‐spacer‐EthyleneGlycol‐3‐Maleimide (DOSEG3M, 2)
3.2. Formulation of targeted lipopolyplexes
The bimodal peptide targeted lipopolyplexes were formulated using our previously published procedures.24 A mixture of lipids was first used to produce liposomes and give formulations F7 or F8 (Table 1). Addition of the bimodal peptides P1, P2, or P3 to F7 or F8 was followed by luciferase plasmid DNA, giving the desired bimodal targeted lipopolyplexes (Table S2).
Surface targeted lipopolyplexes for this study were prepared in a similar manner (Scheme 2). Liposomal formulations with either the charged lipid (1) (formulations F1, F2, F3, F4, F5, F6) or the neutral lipid (2) (formulation F9) incorporated into the lipid bilayer were prepared and subsequently incubated with Cys bearing targeting peptides P4, P5, or P6 to give targeted liposomes. The excess unbound targeting peptide was then removed via dialysis and the liposomal formulation incubated with a DNA‐condensing peptide K16 (P7) followed by plasmid DNA to form the final surface targeted lipopolyplexes (Table S2). A schematic representation of the formation process is depicted in Scheme 2.
Scheme 2.
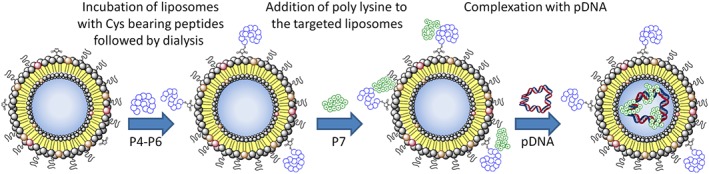
Schematic showing the preparation of surface targeted lipopolyplexes
For both types of lipopolyplex, the initial liposome solution was sonicated to obtain an average particle size below 500 nm with a zeta potential of between +23 and +43 mV (Table S3). The liposomal formulation for bimodal targeted lipopolyplexes exhibited slightly higher surface charges than the surface targeted formulation despite using only 5 mol% more charged lipids in the bimodal formulation. The zeta potential difference is probably due to the shielding effect of the maleimide bearing lipids, analogous to the manner in which large PEG groups shield the charge of cationic lipoplexes.48 After the addition of the K16 containing peptides (P1‐P3, P7) and plasmid DNA, the average size of the samples slightly increased while the zeta potential slightly decreased to values ranging from +15 to +33 mV (Table S4) indicating the formation of stable lipopolyplexes with the negatively charged plasmid DNA reducing partially the overall positive charge. While it is known that lipoplexes of up to 250 nm are almost exclusively endocytosed by clathrin coated pits of non‐phagocytic B16 cells, particles of sizes around 500 nm are internalised via caveolae.49 No studies on the contribution of particle size on the internalisation pathway of lipopolyplexes has been conducted so far, but it can be assumed that the broad size distribution of the presented lipopolyplex with an average size above 250 nm may trigger different endocytosis mechanisms. However, a major contribution of receptor‐mediated endocytosis was expected due to the presence of EGFR targeting peptides.20
3.3. Transfection of HCC1954 human breast cancer cell line
We initially investigated the surface targeted lipopolyplexes, which were optimised by changing different parameters such as the lipid composition or the ratio between peptide and DNA. The relative merits of using either maleimide lipid 1 or 2 to conjugate the targeting peptides (P4‐P6) to the exterior of the liposome were first examined. As described above, formulation F1 (with DODSM 1) or formulation F9 (with DODEG3SM 2), respectively, were conjugated to P4 to P6, followed by formulation into lipopolyplexes by the addition of 5 μM K16 peptide (P7) and then 0.01 μg/μL pDNA. As a control, formulation F7 was complexed with peptide P7 and pDNA to give the non‐targeted lipopolyplex F7‐(P7)‐REF (Table S2). The transfection efficiency of the plasmid DNA into HCC1954 cells was assessed via bioluminescent light emission in radiance (firefly luciferase expression) and FACS analysis (eGFP expression) after 24 and 48 hours, respectively (Figure 2A). When DODSM 1 was used as the maleimide bearing component (lipopolyplexes F1‐(P4,P7), F1‐(P5,P7), and F1‐(P6,P7)) the bioluminescence output was similar to the control non‐targeted lipopolyplex F7‐(P7)‐REF at 24 hours but exhibited a distinctive increase (1.5‐1.8 fold) at 48 hours. This indicated good accessibility to the targeting functionalities and enhanced transcription of the plasmid DNA (Figure 2A) in the presence of 1. Conversely, all lipopolyplexes containing DOSEG3M 2 (F9‐(P4,P7), F9‐(P5,P7), and F9‐(P6,P7)) showed significantly lower bioluminescence compared with the non‐targeted reference at both the 24 hours (P ≤ 0.05) and 48 hours (P ≤ 0.001) time point. One possible explanation for this is that the more polar maleimide headgroup‐linker moiety in DODSM protrudes further out from the bilayer, whereas the less polar headgroup‐linker moiety in DOSEG3M could interact with the hydrophobic part of the liposome bilayers.50
Figure 2.
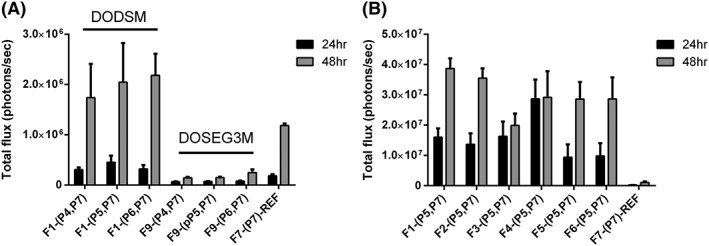
Optimisation of lipid formulations for the surface targeted lipopolyplexes. A, Total luciferase emission of HCC1954 cells transfected with lipopolyplexes prepared from lipid formulations F1 (DODSM), F9 (DOSEG3M), and F7 (no maleimide‐lipid) covalently linked to EGFR targeting peptides (P4‐P6) via maleimide‐thiol crosslinking and complexed with K16 (P7) and luciferase pDNA. B, Total luciferase emission of HCC1954 cells transfected with lipopolyplexes prepared from liposomal formulations F1‐F7 containing DODSM, covalently linked to EGFR targeting peptide P5 and complexed with P7 and luciferase pDNA. Error bars are standard deviation (n = 3)
Lipopolyplexes containing DODSM 1 were therefore selected for further study. As lipopolyplexes bearing the EGFR targeting LARLLT peptide P5 at the surface appeared from these preliminary studies to have slightly superior transfection efficiencies, we used this peptide sequence in studies to further optimise the composition of the lipid bilayer (Figure 2B). The ratios of DODSM, DOPE, and DOTMA were varied as shown in Table 1 (liposome formulations F1, F2, F3, F4), and lipopolyplexes containing cholesterol (liposome formulations F5, F6) were also investigated. All of the resulting lipopolyplexes showed significantly increased bioluminescence (P ≤ 0.001) for both 24 and 48‐hour time points compared with the non‐targeted reference lipopolyplex F7‐(P7)‐REF. This suggests that altering the lipid composition did not significantly affect targeted transfection efficiency. Liposomes containing 1 in the lipid bilayer in the range of 10 to 15 mol% (F1‐(P5,P7), F2‐(P5,P7)), had the highest bioluminescence (48 hours). Whereas, increasing the percentage further to 20 mol% (F3‐(P5,P7)) appeared to hinder transfection resulting in a reduction in bioluminescence at 48 hours. Increasing the overall charge of the complex (F4‐(P5,P7)) by increasing the percentage of DOTMA resulted in a more rapid transfection efficiency, with the highest bioluminescence at 24 hours. However, this did not increase to the extent of other lipopolyplexes at 48 hours. Finally, adding cholesterol to the formulation (F5‐(P5,P7), (F6‐(P5,P7)) also significantly reduced bioluminescence at both time points when compared with F1‐(P5,P7) and F2‐(P5,P7) (P ≤ 0.05), suggesting that cholesterol may affect the transfection efficiency, perhaps by increasing the rigidity of the bilayer. In order to maximise the number of sites for conjugation of the targeting peptide to the surface, it was decided to carry out further optimisation of the peptide targeting sequence based on the F2‐(P5,P7) formulation.
Turning to the bimodal peptide targeted lipopolyplexes, peptides (P1, P2, and P3) were designed to have a pDNA binding sequence (K16) and a validated EGFR‐binding sequence (GE11 (YHWYGYTPQNVI),40 D4 (LARLLT),41 and AE (AEYLR)42) connected together by a furin‐cleavable linker sequence (RVRR), following our earlier work on the design of bimodal peptide targeted lipopolyplexes.19, 20, 24 Initially, the ratio between plasmid DNA and peptide was varied to find the optimized ratio that produces the highest bioluminescence emittance suggestive of the greatest luciferase expression (Figure 3A‐D). While keeping plasmid DNA concentration at 0.01 μg/μL, bimodal peptides were varied between 2.5 and 20 μM. As a control, the non‐targeted lipopolyplex formulation F7‐(P7)‐REF with a bimodal peptide concentration of 5 μM was used. A concentration of 5‐μM bimodal peptide was found to give the highest bioluminescence reading at both 24 and 48 hours for targeting peptides LARLLT (F7‐(P2)) and AEYLR (F7‐(P3)) compared with the control (F7‐(P7)‐REF) (P ≤ 0.05), whereas the longer GE11 (F7‐(P1)) targeting sequence showed only a slight enhancement of bioluminescence. Adding cholesterol to the lipopolyplex formulation at a concentration of 5 μM bimodal peptide (F8‐(P1), F8‐(P2), F8‐(P3)) or altering the concentration of bimodal peptide (P1‐P3) to either 2.5 or 10 μM had little effect on transfection efficiency, exhibiting similar levels of bioluminescence to the control (F7‐(P7)‐REF). However, increasing P1 to P3 to 20 μM appeared to significantly reduce gene expression levels compared with the non‐targeted reference at 48 hours (P ≤ 0.001).
Figure 3.
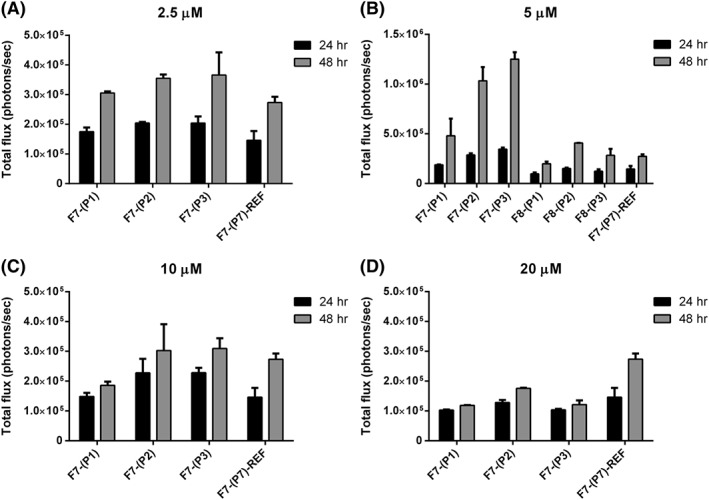
Optimisation of lipid formulations for the bimodal peptide targeted lipopolyplexes. (A‐D) Total luciferase emission of HCC1954 cells transfected with lipopolyplexes prepared from lipid formulation F7 and F8 (5 μM only—B) complexed with bimodal peptides (P1, P2, P3, and P7‐REF) at concentrations 2.5 μM (A), 5 (B), 10 μM (C), and 20 μM (D) and luciferase pDNA
Having optimised the formulations for transfections using bimodal peptide targeted lipopolyplexes, we chose F7 with the biomodal peptide concentration at 5 μM for the optimised bimodal peptide targeted lipopolyplex (Figure 3) and the F2 formulation as the optimised surface targeted lipopolyplex (Figure 2). We then compared the two lipopolyplex targeting methods directly, using all three of the EGFR receptor targeting sequences (P1‐P3 for the bimodal peptide and (P4, P7)‐(P6, P7) for the surface targeted). Again, these were compared with the control non‐targeted lipopolyplex P7‐(P7)‐REF. Bioluminescence was recorded as in previous experiments (Figure 4A) and compared with the expression of eGFP (Figure 4B) that was encoded on the same plasmid. FACS counting of eGFP expression was used to quantify the total percentage of transfected cells in a population. Bioluminescence was used to give a readout of transfection efficiency as the more luciferase produced results in a higher photon count when the same amount of luciferin is added. The percentage population of cells expressing eGFP was similar or higher for both the bimodal and surface targeted liposomes when compared with the non‐targeted reference F7‐(P7)‐REF. This suggests that the number of cells being transfected is similar if not better than the control. However, the LARLLT and AEYLR targeting sequences for both the bimodal peptide targeted lipopolyplexes (F7‐(P2) and F7‐(P3)) and surface targeted lipopolyplexes (F2‐(P5, P7) and F2‐(P6, P7)) had bioluminescence emission far higher than that of the non‐targeted reference F7‐(P7)‐REF at both 24 and 48 hours (P ≤ 0.01). As the percentage population by eGFP shows similar results to the non‐targeted reference, this suggests that the cells being transfected have a higher transfection efficiency which is most likely due to enhanced transcription of the luciferase gene. For the longer GE11 sequence, although the bimodal (F7‐(P1)) lipopolyplex had a higher bioluminescence photon count than the surface targeted (F2‐(P4,P7)) lipopolyplex, their expression was similar if not slightly lower (F2‐(P4,P7)) than the control (F7‐(P7)‐REF). We therefore undertook a structural investigation of the macromolecular architecture of the surface‐targeted and biomodal lipopolyplexes, in order to understand the observed differences in transfection efficiencies.
Figure 4.
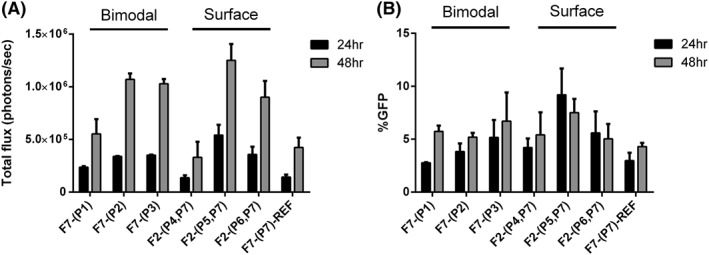
Comparison of luciferase emission in (A) HCC1954 cells and (B) HCC1954 cells expressing GFP (right) after incubation with the optimised formulations of bimodal peptide targeted (F7‐(P1), F7‐(P2), F7‐(P3)) and surface targeted (F2‐(P4,P7), F2‐(P5,P7), F2‐(P6,P7)) lipopolyplex formulations
3.4. Characterisation of the macromolecular structure of the lipopolyplexes
In order to understand the differences in transfection efficiencies displayed by the bimodal and surface targeted lipopolyplexes, we conducted fluorescence quenching experiments to elucidate the location of the different components within the lipopolyplex.24 In order to study the accessibility and location of the targeting sequence itself, we used the GE11 (YHWYGYTPQNVI) sequence, as the intrinsic fluorescence of the Trp residue in this sequence made additional fluorophore labelling unnecessary. Thus, formulations F7‐(P1) (bimodal) and F2‐(P4,P7) (surface targeted) were made up at higher concentrations, and then increasing concentrations of acrylamide were added as a quencher.39 As a control, the quenching of the free peptide P1 was taken to provide an indication of the quenching behaviour of a completely accessible peptide (Figure 5A). Furthermore, to provide a measure for a completely shielded fluorophore, 5(6)‐carboxyfluorescein was encapsulated into DPPC liposomes and subjected to the same quenching conditions used for the lipopolyplexes (Figure S1 ). Quenching efficiencies for the Trp residue in the surface targeted lipopolyplex F2‐(P4,P7) were slightly lower than the quenching efficiency obtained from the free peptide P1. This may indicate that when the targeting peptide is mounted on the liposomal surface, it is sufficiently close to the lipid bilayer to limit the rotational freedom of the peptide and restrict access of the acrylamide quencher to the fluorophore. However, the bimodal formulation F7‐(P1) showed quenching efficiencies similar to the free peptide P1, indicating that in these lipopolyplexes the targeting part of the bimodal peptide protrudes through the lipid bilayer and is fully accessible to the acrylamide, as we have observed previously.24
Figure 5.
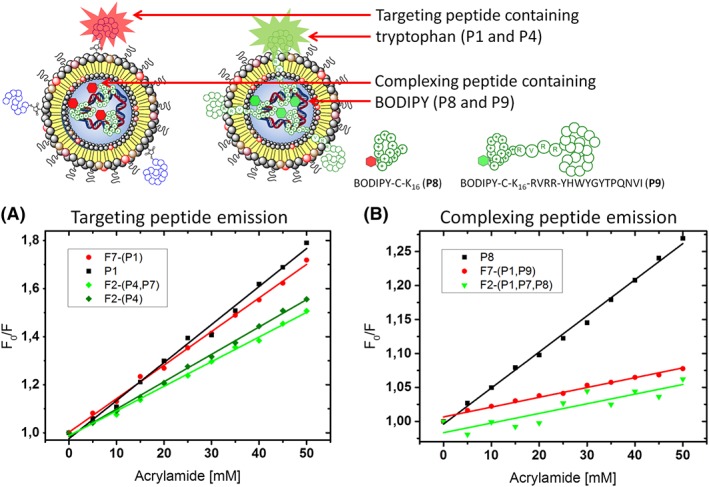
A, Stern‐Volmer plot for the quenching of bimodal lipopolyplex F7‐(P1), surface targeted lipopolyplex F2‐(P4,P7), and surface targeted liposome F2(P4). The overall lipid concentration = 200 μM, peptide = 10 μM, luciferase plasmid DNA 0.02 μg/μL). The GE11 EGFR targeting sequence (YHWYGYTPQNVI) was present in all peptides, and the Trp emission at λem = 340 nm (λexc = 280 nm) was quenched upon addition of acrylamide (0‐50 mM). B, Stern‐Volmer plot for the quenching of F4‐BODIPY in surface targeted lipopolyplex F2‐(P4,P7) incorporating a final concentration of 2‐μM F4‐BODIPY bearing peptide P8, bimodal lipopolyplex F7‐(P1) incorporating a final concentration of 2‐μM F4‐BODIPY bearing peptide P9 (overall lipid concentration = 200 μM, peptide = 10 μM, F4‐BODIPY peptides = 2 μM, luciferase plasmid DNA 0.02 μg/μL). The BODIPY emission at λem = 530 nm (λexc = 557 nm) was quenched upon addition of acrylamide (0–50 mM)
In order to study the location of the DNA‐binding sequences in both the surface targeted and bimodal lipopolyplexes, we designed and synthesised two further peptides labelled with a fluorophore attached at the N‐terminal end of the DNA‐binding K16 sequence. Thus, the sequence CK16 was site‐selectively labelled with a fluorophore as follows. A perfluoro‐BODIPY51 functionalised in the para‐position of the perfluorophenyl ring was reacted via a nucleophilic aromatic substitution52 to give maleimido‐F4‐BODIPY. This was then conjugated to CK16 to give peptide P8 (F4‐BODIPY‐CK16). Likewise, the peptide CK16‐RVRR‐YHWYGYTPQNVI was synthesised and site‐selectively conjugated to maleimido‐F4‐BODIPY via the Cys residue to give peptide P9 (F4‐BODIPY‐CK16‐RVRR‐YHWYGYTPQNVI) (Figure 5). The bimodal lipopolyplex F7‐(P1) was then formulated incorporating a final concentration of 2 μM F4‐BODIPY bearing peptide P9, and the surface targeted lipopolyplex F2‐(P4,P7) was formulated incorporating a final concentration of 2 μM F4‐BODIPY bearing peptide P8. Similar quenching experiments using the free peptide P8 as a control showed that the F4‐BODIPY labelled peptide is completely internalised in the bimodal lipopolyplex formulation F7‐(P1,P9) (Figure 5B) with the fluorophore protected from the acrylamide quencher. In the surface targeted lipopolyplex F2‐(P4,P7,P8), good shielding from the quencher was equally observed, indicating internalisation of the BODIPY‐K16/DNA complex. Both linear Stern‐Volmer slopes resemble the one of completely encapsulated fluorescein (Figure S1).
In order to verify the location of the pDNA in the two lipopolyplexes, the luciferase plasmid DNA was labelled using a functionalised fluorescein (Supporting Information). However, no quenching of the lowest energy fluorescence emission band was recorded upon the addition of acrylamide (Supporting Information, Figure S2) for either the free labelled pDNA or the lipopolyplex encapsulated pDNA, indicating a shielding effect of the fluorophore by the more hydrophilic DNA. It is important to note that the overall emission of the labelled pDNA was generally low, resembling the spectra of a self‐quenched fluorophore, but after incorporation into a liposomal environment the typical fluorescence emission spectra of fluorescein recovered with a more then 4‐fold increase in emission intensity. Self‐quenching of fluorophores in macromolecules such as DNA is a known issue due to labelling of neighbouring sites supported via hydrophobic self‐assembly of the dye molecules.53 Despite that initial self‐quenching, it should be noted that once incorporated into the lipopolyplex the fluorescein labeled pDNA remains shielded from the quenching agent in the bulk water. The change in shape of the emission spectra can be attributed to the more hydrophobic environment of the lipopolyplex and the formation of a ternary complex with the cationic peptide which increases the distance between neighboring fluorophores.
4. CONCLUSIONS
Due to their demonstrated bio‐safety, reduced pathogenicity, low cost, and ease of production, lipopolyplexes remain a viable alternative to viral vectors for gene delivery applications. The addition of cell‐specific targeting ligands on the surface of liposome‐based nanoparticles enhances the cellular uptake and transfection efficiency. For instance, in a comparative study54 of the transfection efficiencies of lipopolyplexes formulated from bimodal peptides with and without targeting, lipopolyplexes with a scrambled targeting sequence showed 25% transfection, and lipopolyplexes with the targeting sequence removed showed 12% transfection, compared with lipopolyplexes with targeting sequence, indicating that the high transfection efficiency is at least partly a consequence of the targeting sequence. In another study,41 surface targeted liposomes bearing the GE sequence were compared with surface targeted liposomes with the sequence scrambled; the scrambled sequences had negligible binding to cells in vitro. However, a systematic investigation of the benefits of different methods of displaying the targeting peptide had not previously been reported.
In this study, we have for the first time compared two approaches to the design, synthesis, and formulation of cell‐surface receptor targeted lipopolyplexes for gene delivery. Surface‐targeted lipopolyplexes were prepared from liposomes to which targeting peptides had been attached, and bimodal peptides were formulated directly, using peptides which contained both DNA condensing and receptor targeting sequences. Three targeting sequences, previously validated to target the EGF receptor which is over‐expressed in many cancer cell lines, were investigated, together with a range of lipid formulations and maleimide lipid structures. The biophysical properties of the lipopolyplexes and their transfection efficiencies in a basal‐like breast cancer cell line were investigated, and fluorescence quenching experiments were used to probe the macromolecular organisation of the peptide and pDNA components of the lipopolyplexes.
Both approaches to lipopolyplex targeting gave reasonable transfection efficiencies, with little major differences between them. This reflects the observations from the fluorescence quenching experiments that in both types of liposomes the pDNA is condensed and shielded within the lipopolyplex, and also that in both cases the targeting moiety is accessible to some degree. Moreover, it is clear that the transfection efficiency of each lipopolyplex architecture is highly dependent on the sequence of the targeting peptide. The GE11 (YHWYGYTPQNVI) sequence gave the lowest transfection efficiency when incorporated in the surface targeted liposomes. In the fluorescence quenching experiments, the Trp residue of this sequence is partially shielded, suggesting that in this case the targeting sequence is partly shielded and thus is less accessible. However, the D4 (LARLLT) targeting sequence outperformed other surface targeted and bimodal lipopolyplexes, suggesting that this sequence has optimal accessibility to cell surface receptors.
In this work, we have demonstrated the versatility of differing approaches to functionalizing lipopolyplexes for targeted gene delivery which provides a toolbox for a more personalised targeting/treatment to specific cancer cell lines. Our findings indicate that there is little difference in transfection efficiency and targeting sequence display between the two approaches. Lipopolyplex targeting to cancer cell lines is clearly highly dependent on the targeting sequence used, and that selection and optimisation of both targeting sequence and lipopolyplex architecture should be carried out on a case‐by‐case basis, taking into account the cancer cell line of interest, the receptor binding of the targeting sequence, and the slightly greater synthetic complexity of the surface targeting approach.
Supporting information
Scheme S1: Synthesis of Maleimido‐BODIPY (S3)
Table S1 Peptide sequences used in lipopolyplex formulations.
Table S2: Summary of lipopolyplexes prepared for the transfection of HCC1954 cells. The names of the lipopolyplexes are made up as follows “Lipid formulation—(Peptides used)”.
Table S3: Biophysical characterisation of surface targeted liposomes F2‐(P4), F2‐(P5), F2‐(P6) and of untargeted liposome F7.
Table S4: Biophysical characterisation of lipopolyplexes after complexation with pDNA
Figure S1: Fluorescence quenching of free and liposomal fluorescein (5(6)‐Carboxyfluorescein 100 μM, DPPC 1 mM in HEPES 20 mM, pH 7.4, λexc = 466, λem = 516).
Figure S2: Fluorescein labeled pDNA emission in free solution and in lipopolyplex F7‐(P1), overall lipid concentration = 200 μM, peptide = 10 μM, Luciferase plasmid DNA (0.02 μg/μL), λexc = 491 upon addition of 0 to 50 mM acrylamide.
ACKNOWLEDGEMENTS
This work was conducted within the King's College London‐UCL Comprehensive Cancer Imaging Centre (CCIC) supported by Cancer Research UK and the EPSRC, in association with MRC and DoH (UK). The authors would like to thank Cancer Research UK and the EPSRC (grants C1519/A6906 (N.J.M.) and C5255/A15935 (R.B., C.S.)) for financial support. We also thank the EPSRC for the award of an Early Career Fellowship EP/L006472/1 (T.K., M.Z.‐T.) and the MRC for funding via the UK Regenerative Medicine Platform Safety and Efficacy Hub (MR/K026739/1) (S.P.).
Bofinger R, Zaw‐Thin M, Mitchell NJ, et al. Development of lipopolyplexes for gene delivery: A comparison of the effects of differing modes of targeting peptide display on the structure and transfection activities of lipopolyplexes. J Pep Sci. 2018;24:e3131 10.1002/psc.3131
Contributor Information
Tammy L. Kalber, Email: t.kalber@ucl.ac.uk.
Alethea B. Tabor, Email: a.b.tabor@ucl.ac.uk.
REFERENCES
- 1. Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, Anderson DG. Non‐viral vectors for gene‐based therapy. Nat Rev Genet. 2014;15(8):541‐555. [DOI] [PubMed] [Google Scholar]
- 2. Riley MK, Vermerris W. Recent advances in nanomaterials for gene delivery—a review. Nanomater. 2017;7(5):94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ylä‐Herttuala S. Endgame: Glybera finally recommended for approval as the first gene therapy drug in the European Union. Mol Ther. 2012;20(10):1831‐1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vago R, Collico V, Zuppone S, Prosperi D, Colombo M. Nanoparticle‐mediated delivery of suicide genes in cancer therapy. Pharmacol Res. 2016;111:619‐641. [DOI] [PubMed] [Google Scholar]
- 5. Wang HY, Jiang YF, Peng HG, Chen YZ, Zhu PZ, Huang YZ. Recent progress in microRNA delivery for cancer therapy by non‐viral synthetic vectors. Adv Drug Deliv Rev. 2015;81:142‐160. [DOI] [PubMed] [Google Scholar]
- 6. Wang K, Huang Q, Qiu FM, Sui MH. Non‐viral delivery systems for the application in p53 cancer gene therapy. Curr Med Chem. 2015;22(35):4118‐4136. [DOI] [PubMed] [Google Scholar]
- 7. Stauss HJ, Morris EC, Abken H. Cancer gene therapy with T cell receptors and chimeric antigen receptors. Curr Opin Pharmaco. 2015;24:113‐118. [DOI] [PubMed] [Google Scholar]
- 8. Prabhakar U, Maeda H, Jain RK, et al. Challenges and key considerations of the enhanced permeability and retention effect for nanomedicine drug delivery in oncology. Cancer Res. 2013;73(8):2412‐2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ekdawi SN, Stewart JMP, Dunne M, et al. Spatial and temporal mapping of heterogeneity in liposome uptake and microvascular distribution in an orthotopic tumor xenograft model. J Control Release. 2015;207:101‐111. [DOI] [PubMed] [Google Scholar]
- 10. Ulmschneider MB, Searson PC. Mathematical models of the steps involved in the systemic delivery of a chemotherapeutic to a solid tumor: from circulation to survival. J Control Release. 2015;212:78‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Park S, Yoo HS. In vivo and in vitro anti‐cancer activities and enhanced cellular uptakes of EGF fragment decorated doxorubicin nano‐aggregates. Int J Pharm. 2010;383(1‐2):178‐185. [DOI] [PubMed] [Google Scholar]
- 12. Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliv Rev. 2014;66:2‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fernandes E, Ferreira JA, Peixoto A, et al. New trends in guided nanotherapies for digestive cancers: a systematic review. J Control Release. 2015;209:288‐307. [DOI] [PubMed] [Google Scholar]
- 14. Klibanov AL, Maruyama K, Torchilin VP, Huang L. Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS Lett. 1990;268(1):235‐237. [DOI] [PubMed] [Google Scholar]
- 15. Rezaee M, Oskuee RK, Nassirli H, Malaekeh‐Nikouei B. Progress in the development of lipopolyplexes as efficient non‐viral gene delivery systems. J Control Release. 2016;236:1‐14. [DOI] [PubMed] [Google Scholar]
- 16. Vaysse L, Arveiler B. Transfection using synthetic peptides: comparison of three DNA‐compacting peptides and effect of centrifugation. Biochim Biophys Acta. 2000;1474(2):244‐250. [DOI] [PubMed] [Google Scholar]
- 17. Colin M, Harbottle RP, Knight A, et al. Liposomes enhance delivery and expression of an RGD‐oligolysine gene transfer vector in human tracheal cells. Gene Ther. 1998;5(11):1488‐1498. [DOI] [PubMed] [Google Scholar]
- 18. Fritz JD, Herweijer H, Zhang G, Wolff JA. Gene transfer into mammalian cells using histone‐condensed plasmid DNA. Hum Gene Ther. 1996;7(12):1395‐1404. [DOI] [PubMed] [Google Scholar]
- 19. Welser K, Campbell F, Kudsiova L, et al. Gene delivery using ternary lipopolyplexes incorporating branched cationic peptides: the role of peptide sequence and branching. Mol Pharm. 2013;10:127‐141. [DOI] [PubMed] [Google Scholar]
- 20. Mohd Mustapa MF, Grosse SM, Kudsiova L, et al. Stabilized integrin‐targeting ternary LPD (lipopolyplex) vectors for gene delivery designed to disassemble within the target cell. Bioconjug Chem. 2009;20(3):518‐532. [DOI] [PubMed] [Google Scholar]
- 21. ur Rehman Z, Zuhorn IS, Hoekstra D. How cationic lipids transfer nucleic acids into cells and across cellular membranes: recent advances. J Control Release. 2013;166(1):46‐56. [DOI] [PubMed] [Google Scholar]
- 22. Hart SL, Arancibia‐Carcamo CV, Wolfert MA, et al. Lipid‐mediated enhancement of transfection by a non‐viral integrin‐targeting vector. Hum Gene Ther. 1998;9(4):575‐585. [DOI] [PubMed] [Google Scholar]
- 23. Tagalakis AD, McAnulty RJ, Devaney J, et al. A receptor targeted nanocomplex vector system optimized for respiratory gene transfer. Mol Ther. 2008;16(5):907‐915. [DOI] [PubMed] [Google Scholar]
- 24. Mohd Mustapa MF, Bell PC, Hurley CA, et al. Biophysical characterization of an integrin‐targeted lipopolyplex gene delivery vector. Biochemistry. 2007;46(45):12930‐12944. [DOI] [PubMed] [Google Scholar]
- 25. Munye MM, Ravi J, Tagalakis AD, McCarthy D, Ryadnov MG, Hart SL. Role of liposome and peptide in the synergistic enhancement of transfection with a lipopolyplex vector. Sci Rep. 2015;5(1):9292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Elouahabi A, Ruysschaert JM. Formation and intracellular trafficking of lipoplexes and polyplexes. Mol Ther. 2005;11(3):336‐347. [DOI] [PubMed] [Google Scholar]
- 27. Mitchell N, Kalber TL, Cooper MS, et al. Incorporation of paramagnetic, fluorescent and PET/SPECT contrast agents into liposomes for multimodal imaging. Biomaterials. 2013;34(4):1179‐1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Weitsman G, Mitchell NJ, Evans R, et al. Detecting intratumoral heterogeneity of EGFR activity by liposome‐based in vivo transfection of a fluorescent biosensor. Oncogene. 2017;36(25):3618‐3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu D, Auguste DT. Cancer targeted therapeutics: from molecules to drug delivery vehicles. J Control Release. 2015;219:632‐643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tang H, Chen XJ, Rui MJ, et al. Effects of surface displayed targeting ligand GE11 on liposome distribution and extravasion in tumor. Mol . Pharmaceutics. 2014;11:3242‐3250. [DOI] [PubMed] [Google Scholar]
- 31. Govender J, Singh M, Ariatti M. Effect of poly (ethylene glycol) spacer on peptide‐decorated hepatocellular carcinoma‐targeted lipoplexes in vitro. J Nanosci Nanotechnol. 2015;15(6):4734‐4742. [DOI] [PubMed] [Google Scholar]
- 32. Fortier C, De Crescenzo G, Durocher Y. A versatile coiled‐coil tethering system for the oriented display of ligands on nanocarriers for targeted gene delivery. Biomaterials. 2013;34(4):1344‐1353. [DOI] [PubMed] [Google Scholar]
- 33. Iyer M, Berenji M, Templeton NS, Gambhir SS. Noninvasive imaging of cationic lipid‐mediated delivery of optical and PET reporter genes in living mice. Mol Ther. 2002;6(4):555‐562. [DOI] [PubMed] [Google Scholar]
- 34. Law GHE, Gandelman OA, Tisi LC, Lowe CR, Murray JAH. Mutagenesis of solvent‐exposed amino acids in Photinus pyralis luciferase improves thermostability and pH‐tolerance. Biochem J. 2006;397(2):305‐312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tsein RY. The green fluorescent protein. Annu Rev Biochem. 1998;67(1):509‐544. [DOI] [PubMed] [Google Scholar]
- 36. Hurley CA, Wong JB, Hailes HC, Tabor AB. Asymmetric synthesis of dialkyloxy‐3‐alkylammonium cationic lipids. J Org Chem. 2004;69(3):980‐983. [DOI] [PubMed] [Google Scholar]
- 37. Hurley CA, Wong JB, Ho J, et al. Mono‐ and dicationic short PEG and methylene dioxyalkylglycerols for use in synthetic gene delivery systems. Org Biomol Chem. 2008;6(14):2554‐2559. [DOI] [PubMed] [Google Scholar]
- 38. Demaison C, Parsley K, Brouns G, et al. High‐level transduction and gene expression in hematopoietic repopulating cells using a human immunodeficiency virus type 1‐based lentiviral vector containing an internal spleen focus forming virus promoter. Human Gene Ther. 2002;13(7):803‐813. [DOI] [PubMed] [Google Scholar]
- 39. Lakowicz JR. Principles of Fluorescence Spectroscopy. New York: Kluwer Academic/Plenum Publishers; 1999. [Google Scholar]
- 40. Li Z, Zhao R, Wu X, et al. Identification and characterization of a novel peptide ligand of epidermal growth factor receptor for targeted delivery of therapeutics. FASEB J. 2005;19(14):1978‐1985. [DOI] [PubMed] [Google Scholar]
- 41. Song S, Liu D, Peng J, et al. Novel peptide ligand directs liposomes toward EGF‐R high‐expressing cancer cells in vitro and in vivo . FASEB J. 2009;23(5):1396‐1404. [DOI] [PubMed] [Google Scholar]
- 42. Han CY, Yue LL, Tai LY, et al. A novel small peptide as an epidermal growth factor receptor targeting ligand for nanodelivery in vitro . Int J Nanomedicine. 2013;8:1541‐1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mickler FM, Mockl L, Ruthardt N, Ogris M, Wagner E, Brauchle C. Tuning nanoparticle uptake: live‐cell imaging reveals two distinct endocytosis mechanisms mediated by natural and artificial EGFR targeting ligand. Nano Lett. 2012;12(7):3417‐3423. [DOI] [PubMed] [Google Scholar]
- 44. Salome C, Spanedda MV, Hilbold B, et al. Smart tools and orthogonal click‐like reactions onto small unilamellar vesicles. Chem Phys Lipids. 2015;188:27‐36. [DOI] [PubMed] [Google Scholar]
- 45. Bourel‐Bonnet L, Pecheur EI, Grandjean C, et al. Anchorage of synthetic peptides onto liposomes via hydrazone and alpha‐oxo hydrazone bonds. Preliminary functional investigations. Bioconjug Chem. 2005;16(2):450‐457. [DOI] [PubMed] [Google Scholar]
- 46. Cheng W‐Q, Satyanarayanajois S, Lim L‐Y. Aqueous‐soluble, non‐reversible lipid conjugate of salmon calcitonin: synthesis, characterisation and in vivo activity. Pharm Res. 2007;24(1):99‐110. [DOI] [PubMed] [Google Scholar]
- 47. Elliott JT, Prestwich GD. Maleimide‐functionalized lipids that anchor polypeptides to lipid bilayers and membranes. Bioconjug Chem. 2000;11(6):832‐841. [DOI] [PubMed] [Google Scholar]
- 48. Rejman J, Wagenaar A, Engberts JBFN, Hoekstra D. Characterization and transfection properties of lipoplexes stabilized with novel exchangeable polyethylene glycol‐lipid conjugates. Biochim Biophys Acta Biomembranes. 2004;1660(1‐2):41‐52. [DOI] [PubMed] [Google Scholar]
- 49. Wasungu L, Hoekstra D. Cationic lipids, lipoplexes and intracellular delivery of genes. J Control Release. 2006;116(2):255‐264. [DOI] [PubMed] [Google Scholar]
- 50. Fleiner M, Benzinger P, Fichert T, Massing U. Studies on protein‐liposome coupling using novel thiol‐reactive coupling lipids: influence of spacer length and polarity. Bioconjug Chem. 2001;12(4):470‐475. [DOI] [PubMed] [Google Scholar]
- 51. Galangau O, Dumas‐Verdes C, Meallet‐Renault R, Clavier G. Rational design of visible and NIR distyryl‐BODIPY dyes from a novel fluorinated platform. Org Biomol Chem. 2010;8(20):4546‐4553. [DOI] [PubMed] [Google Scholar]
- 52. Vives G, Giansante C, Bofinger R, et al. Facile functionalization of a fully fluorescent perfluorophenyl BODIPY: photostable thiol and amine conjugates. Chem Commun. 2011;47(37):10425‐10427. [DOI] [PubMed] [Google Scholar]
- 53. Zhegalova NG, He S, Zhou HY, Kim DM, Berezin MY. Minimization of self‐quenching fluorescence on dyes conjugated to biomolecules with multiple labeling sites via asymmetrically charged NIR fluorophores. Contrast Media Mol Imaging. 2014;9(5):355‐362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kudsiova L, Fridrich B, Ho J, et al. Mol Pharm. 2011;8(5):1831‐1847. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Scheme S1: Synthesis of Maleimido‐BODIPY (S3)
Table S1 Peptide sequences used in lipopolyplex formulations.
Table S2: Summary of lipopolyplexes prepared for the transfection of HCC1954 cells. The names of the lipopolyplexes are made up as follows “Lipid formulation—(Peptides used)”.
Table S3: Biophysical characterisation of surface targeted liposomes F2‐(P4), F2‐(P5), F2‐(P6) and of untargeted liposome F7.
Table S4: Biophysical characterisation of lipopolyplexes after complexation with pDNA
Figure S1: Fluorescence quenching of free and liposomal fluorescein (5(6)‐Carboxyfluorescein 100 μM, DPPC 1 mM in HEPES 20 mM, pH 7.4, λexc = 466, λem = 516).
Figure S2: Fluorescein labeled pDNA emission in free solution and in lipopolyplex F7‐(P1), overall lipid concentration = 200 μM, peptide = 10 μM, Luciferase plasmid DNA (0.02 μg/μL), λexc = 491 upon addition of 0 to 50 mM acrylamide.