

Summary
Background
Activated microglia‐mediated inflammation plays a key role in the pathogenesis of Alzheimer’s disease (AD). In addition, chronic activation of NLRP3 inflammasomes triggered by amyloid β peptide (Aβ) in microglia contributes to persistent neuroinflammation. Here, the primary goal was to assess whether Dihydromyricetin (DHM), a plant flavonoid compound, is effective therapies for AD; it is crucial to know whether DHM will affect microglial activation and neuroinflammation in APP/PS1 transgenic mice.
Methods
After DHM was intraperitoneally injected in APP/PS1 double‐transgenic mice, we assessed the effect of DHM on microglial activation, the expression of NLRP3 inflammasome components, and the production of inflammatory cytokine IL‐1β by immunofluorescence and Western blot. To determine whether DHM play roles in the Aβ production and deposition, amyloid β protein precursor (APP) and β‐site APP cleaving enzyme1 (BACE1), as well as neprilysin (NEP), were detected by Western blot. Finally, behavior was tested by Morris Water Maze to illustrate whether DHM treatment has a significantly positive effect on ameliorating the memory and cognition deficits in AD.
Results
Dihydromyricetin treatment significantly ameliorated memory and cognition deficits and decreased the number of activated microglia in the hippocampus and cortex of APP/PS1 mice. In addition, APP/PS1 mice show reduced activation of NLRP3 inflammasomes and reduced expression of NLRP3 inflammasome components. Furthermore, DHM could promote clearance of Aβ, a trigger for NLRP3 inflammasome activation, by increasing levels of NEP and shift microglial conversion to the M2‐specific agrinase‐1‐positive cell phenotype, which enhances microglial clearance of Aβ and its aggregates but not production of Aβ.
Conclusion
Taken together, our findings suggest that DHM prevents progression of AD‐like pathology through inhibition of NLRP3 inflammasome‐based microglia‐mediated neuroinflammation and may be a promising therapeutic drug for treating AD.
Keywords: Alzheimer’s disease, dihydromyricetin, microglia, NLRP3 inflammasome
1. INTRODUCTION
Alzheimer’s disease (AD) is a neurological brain disease characterized by progressive memory loss and cognitive dysfunction and is the most frequent cause of dementia. At present, the etiology and pathogenesis of AD are still inconclusive. Emerging evidence has suggested that neuroinflammation, which is characterized by enhanced microglial activation probably triggered by amyloid β peptide (Aβ) deposits, plays a central role in the pathogenesis of AD.1, 2, 3, 4
Microglia, the resident immune cells in the central nervous system (CNS), are normally activated by neuronal cell death and initiate the inflammatory response, which contributes to engulfing cell debris and decreasing Aβ deposition.5, 6 Recently, several observations have indicated that chronic activation of NLRP3 inflammasomes in microglia is a critical and fundamental process in microglia‐mediated neuroinflammation. NLRP3 inflammasome is currently the most fully characterized inflammasome, which is a molecular platforms activated upon cellular infection or stress, mainly expressed in amyloid cells and played an essential role in the Aβ‐induced activation of caspase‐1 and downstream secretion of its substrates, the pro‐inflammatory cytokines IL‐1β.7 Inappropriate activation of NLRP3 inflammasomes contributes to the pathogenesis of AD in APP/PS1 mice.8 Both APP/PS1/NLRP3−/− and APP/PS1/caspase‐1−/− mice display protection from loss of spatial memory and other AD‐associated sequelae, which probably results from increased phagocytic capacity of microglia with the anti‐inflammatory M2 phenotype and subsequent Aβ clearance.9 It has been suggested that the NLRP3 inflammasome may be a promising target for inhibiting activation of microglia in AD.
Dihydromyricetin (DHM) is a plant flavonoid compound10 that is derived from the medicinal plant Ampelopsis grossedentata and has been demonstrated to be widely used in different therapeutic areas including antioxidative, anticancer, anti‐alcohol intoxication and so on.10, 11 Substantial evidence has shown that flavonoids can cross the blood‐brain barrier,12 modulate inflammation, and exert neuroprotective effects.13 Previous studies have shown that DHM ameliorates behavioral deficits and reverses neuropathology of AD mainly by modulating GABAARs,14 which crucially involved in synaptic plasticity and made network excitability be decreased once activated in the brain, while abnormal network activity involving the loss of synapses leads to cognitive deficits in AD.15, 16 In this study, our data demonstrate that DHM treatment provides a neuroprotective role in APP/PS1 mice by suppressing microglial activation. Furthermore, DHM treatment can reduce activated NLRP3 inflammasomes and promote Aβ clearance by increasing expression of neprilysin (NEP) or shifting microglia to the M2 phenotype in APP/PS1 mice.
2. MATERIALS AND METHODS
2.1. Chemicals
Dihydromyricetin (purity >98%) and dimethyl sulfoxide (DMSO) were purchased from Sigma‐Aldrich. Stock solutions of DHM were made with DMSO and diluted to final concentration in saline for injection. Injection volume was 1 mL/100 g body weight. The final DMSO concentration was 0.04%.
2.2. Cell culture
BV‐2 microglial cells were purchased from ATCC and cultured with 45 mL DMEM complete medium (contained 5 mL 10% fetal calf serum). Cells were plated into 6well microtiter plates, treated with 20 μg/mL Aβ1‐42, which was purchased from Ana‐Spec (Fremont, CA, USA). Following 24 hours incubation, the cells were activated, which was as described as previously study (16). After 24 hours, microglia cultures were treated with an inflammatory inhibitor DHM at 2.5 μg/mL for 24 hours.17, 18 Later cells were washed with phosphate‐buffered saline (PBS) and lysed in ice‐cold lysis buffer. Lysates were centrifuged at 15 000 g for 15 minutes at 4°C. The supernatants were collected and Samples were frozen at −80°C until further analysis.
2.3. Animals
APP/PS1 double‐transgenic mice that overexpress mouse/human amyloid precursor protein (Mo/Hu APP695 SWE) and mutant human presenilin‐1 (PS1‐∆9) in a C57BL/6J genetic background were purchased from Beijing HFK Bio‐technology Co. Ltd. (China, Beijing; certificate no. 11401300010893). The mouse genotype was confirmed by PCR using DNA extracted from tail tissues. All experimental animals were housed under standard laboratory conditions (22°C and a 12‐hour light‐dark cycle with free access to food and water). Animal care and experimental protocols used in this study were conducted according to the guidelines approved by the Care and Use of Laboratory Animals of Chongqing Medical University. Efforts were made to minimize animal suffering and to reduce the number of animals used.
2.4. Group design and drug treatment
After genotyping, 32 APP/PS1 transgenic mice at 4 months of age were randomly divided into 4 groups (n = 8): 2‐week DHM‐treated and control groups, 4‐week DHM‐treated and control groups. Mice in DHM treatment groups were injected with 1 mg/kg DHM intraperitoneally at the same time each day, and control mice were injected with DMSO diluted by saline. Body weight as well as food and water intake were measured every day until testing in the Morris water maze. The treatment protocol for DHM was well tolerated by the animals.
2.5. Morris water maze
A Morris water maze was used to test spatial memory.18 It was performed in a circular stainless‐steel pool filled with opaque water. The pool was divided into 4 quadrants that were marked with a triangle, square, diamond, and circle. The temperature was set at 23 ± 1°C. A transparent escape platform (9 cm in diameter) was placed in the middle of a quadrant (goal quadrant). The procedure consisted of 1 day of visible platform tests, 2‐5 consecutive days of hidden platform tests, and a spatial probe trial after the last hidden platform test. In the visible platform tests, the platform was positioned above the water surface. The mice were trained and allowed to search for the platform for 60 seconds. If a mouse did not reach the platform in the allotted time, it was placed onto it manually and allowed to stay on the platform for 10 seconds before the next trial. In the hidden platform tests, the platform was submerged 1 cm below the opaque water surface in a fixed position in the fourth quadrant. Mice were trained as before. Escape latency and path length to reach the platform were analyzed as a measure of spatial learning and memory. In the probe trial, the platform was removed and mice were allowed to swim for 60 seconds. The number of times that a mouse crossed the target platform location in the fourth quadrant was analyzed. Latencies and swimming paths were recorded by video tracking, and digital images were analyzed by Mater Maze software (Ethovision system, Noldus Information Technology, Wageningen, the Netherlands).
2.6. Tissue preparation
Mice were anesthetized by 3.5% chloral hydrate and perfused with ice‐cold 0.9% saline solution. The brains were dissected and divided into 2 groups; half was immediately homogenized for protein analysis, and the other half was fixed in fresh 4% paraformaldehyde (PFA) in 0.1 mol/L PBS (pH 7.4). All procedures were performed on ice. Fixed brains were dehydrated in graded sucrose solutions until the tissue sank and then embedded in tissue‐embedding medium (OCT; SAKURA Tissue‐Tek, Japan). Brain sections (10 μm for counting) were obtained using a cryostat (CM1860; Leica, Germany) and collected sequentially in 24‐well plates filled with 0.01 mol/L PBS. Every 10th slice with the same reference position was mounted onto slides for staining.
2.7. Immunohistochemistry
Frozen sections were warmed for 30 minutes at 37°C. Sections were then washed 3 times with PBS (5 min/wash) and blocked with 5% fetal calf serum (HyClone, Logan, UT, USA) blocking solution for 30 minutes at 37°C. Next, sections were incubated overnight at 4°C with primary antibodies: mouse anti‐ED1 (anti‐CD68; 1:200; Abcam, Cambridge, MA), rabbit anti‐NLRP3 (1:100; Santa Cruz, USA), rabbit anti‐caspase‐1p10 (1:100; Santa Cruz, USA), and mouse anti‐IL‐1β (1:100; Cell Signaling, USA,). To assess Aβ accumulation, sections were incubated in 88% formic acid for 10 minutes before blocking, followed by immunostaining with monoclonal mouse anti‐4G8 antibody (1:250; Abcam, USA). After rinsing 3 times with PBS (5 min/wash), sections were incubated with species‐specific secondary antibodies: CY3 or FITC488 (1:200; Beyotime, Shanghai, China). To detect nuclei, DAPI (C1005; Beyotime) was used. Sections were visualized under fluorescence microscopy, and images were analyzed using an image analysis system (ImageJ). To determine the expression of microglial cell M2 phenotypes, frozen sections were subjected to immunofluorescence. Specific methods is similar to what was mentioned before. Sections were incubated with the following primary antibodies: rabbit anti‐arginase‐1 (1:50; Biosynthesis, Beijing, China) for M2 phenotype microglia. Images were captured on a Nikon A1R+ laser scanning confocal microscope (Nikon, Tokyo, Japan).
2.8. Western blot analysis
Brain tissues were prepared in a mixture of RIPA lysis buffer (P0013B; Beyotime) and phenylmethanesulfonyl fluoride (PMSF, ST506B; Beyotime) on ice, followed by homogenization using a tissue homogenizer (MT‐30K; China). Next, the lysates were centrifuged at 20,000 g for 15 minutes at 4°C. A BCA protein concentration assay was used for determining protein content of the supernatant. The samples were then mixed with sample buffer (5X, P0015; Beyotime) and boiled for 10 minutes at 100°C. Equal amounts of protein were loaded per lane (30 μg) and separated by 8%‐12% tris‐glycine SDS‐PAGE. The electrophoresed proteins were transferred to polyvinylidene fluoride membranes (IJ‐58; Millipore, USA) and incubated in Western blot blocking buffer (P0023B; Beyotime) at 37°C. Anti‐CD68 (1:1000; Abcam), NIRP3 (1:500; Santa Cruz), caspase‐1 (1:500; Santa Cruz), IL‐1β (1:1000; Cell signaling), BACE1 (1:400; Santa Cruz, USA), APP (1:1000; Abcam, USA), and NEP (1:1000; Abcam, USA) were used to detect targeted proteins. As an internal control, β‐actin was analyzed using AA128 (1:1000; Beyotime). Secondary antibodies (SA0001‐2, anti‐rabbit IgG, anti‐mouse IgG; USA) were incubated for 40 minutes at 1:6000 dilutions. Chemiluminescence was detected using a Western Bright™ ECL Kit (K‐12045‐D10; Advansta, USA) by Bio‐Rad Laboratories (731BR02996; Bio‐Rad Shanghai Ltd, China), and Image Lab™ Software 5.2. Chemiluminescence intensities were quantified by Quantity One analysis.
2.9. ELISA assay
Tissue extracted from hippocampal and cortical regions of experimental mice were collected. Concentrations of Aβ40 and Aβ42 were determined using a β‐Amyloid 1‐40 or 1‐42 Colorimetric ELISA Kit (Biosource International, Inc.) according to the manufacturer’s instructions. A total of 100 μL Aβ standard or test sample was loaded into each well and incubated overnight at 4°C. Plates were washed 7 times and then 100 μL Aβ40/42 polyclonal antibody was added. After 1 hour, plates were washed 9 times before 100 μL secondary HRP anti‐rabbit IgG was added at room temperature. Plates were washed 1 hour at room temperature and then incubated in 100 μL stabilized chromogen for 30 minutes at room temperature. Finally, 100 μL stop solution was added, and plates were read at 450 nm on a microplate reader.
2.10. Statistical analysis
All statistical analyses were performed using Statistics 17 software. The data are presented as mean ± standard error (SEM). To evaluate statistical significance, comparisons between 2 groups were tested using standard two‐tailed unpaired t tests, and a P value <.05 was defined as significant. Morris water maze data were first analyzed by repeated measures analysis of variance (ANOVA). If P > .05, statistical comparisons were performed using one‐way ANOVA, followed by Turkey’s post hoc test. P < .05 was considered statistically significant.
3. RESULTS
3.1. DHM inhibits activation of microglia in APP/PS1 mice
ED1 (CD68) is a common marker for activated microglia cells. To examine whether DHM affects microglial activation, we assayed ED1 expression with immunostaining combined with ImageJ quantitative analysis. Results showed that the number of activated microglia in the hippocampus and cortex of DHM‐treated mice was decreased compared with control mice. Furthermore, when the duration of DHM treatment was increased to 4 weeks, we observed a more pronounced decrease in the number of activated microglia (Figure 1A‐C). To verify these results, Western blot analysis was used (Figure 1D). Consistent with the immunostaining data, levels of ED1 were decreased in DHM treatment groups compared with control groups, which suggests that DHM can inhibit microglial activation in APP/PS1 transgenic mice.
Figure 1.
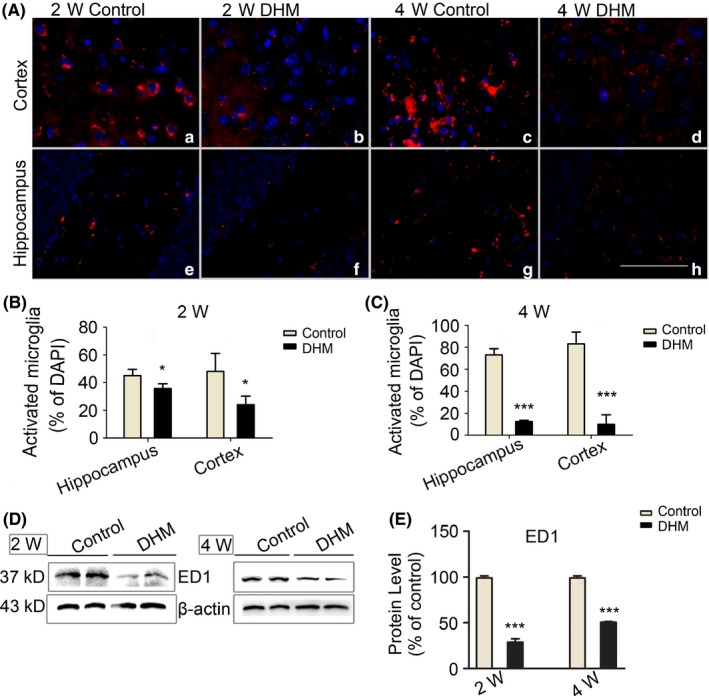
Dihydromyricetin (DHM) inhibits activation of microglia in APP/PS1 mice. A‐C, DHM reduces the number of activated microglia in the hippocampus and cortex of DHM treatment groups. Compared with the 2‐week group, the 4‐week group showed a more pronounced decrease. Data are presented as mean ± SEM, *P < .05, ***P < .001, Scale bars: 40 μm. D, E, Western blot analysis showing a decrease in ED1 protein levels in DHM‐treated groups compared with control groups (*P < .05, ***P < .001 vs control group)
3.2. DHM reduces NLRP3 protein expression in APP/PS1 mice
It has been reported that NLRP3 inflammasome activation is a critical cell event for chronic cerebral neuroinflammatory processes in microglia. Therefore, we evaluated whether DHM plays a role in NLRP3 inflammasomes in microglia. First, we measured NLRP3 protein expression. Immunofluorescence results showed NLRP3‐positive cells in the brains of control mice. However, there were fewer NLRP3‐positive cells in the DHM treatment groups. Furthermore, the number of NLRP3‐positive cells in the DHM treatment 4‐week group was more obviously reduced compared with the 2‐week group (Figure 2A). Western blot analysis also demonstrated that expression levels of NLRP3 in the 2‐ and 4‐week DHM‐treated groups (Figure 2D‐F) were lower than those of the control groups. These results further confirmed that DHM treatment can decrease NLRP3 inflammasome expression in APP/PS1 mice.
Figure 2.
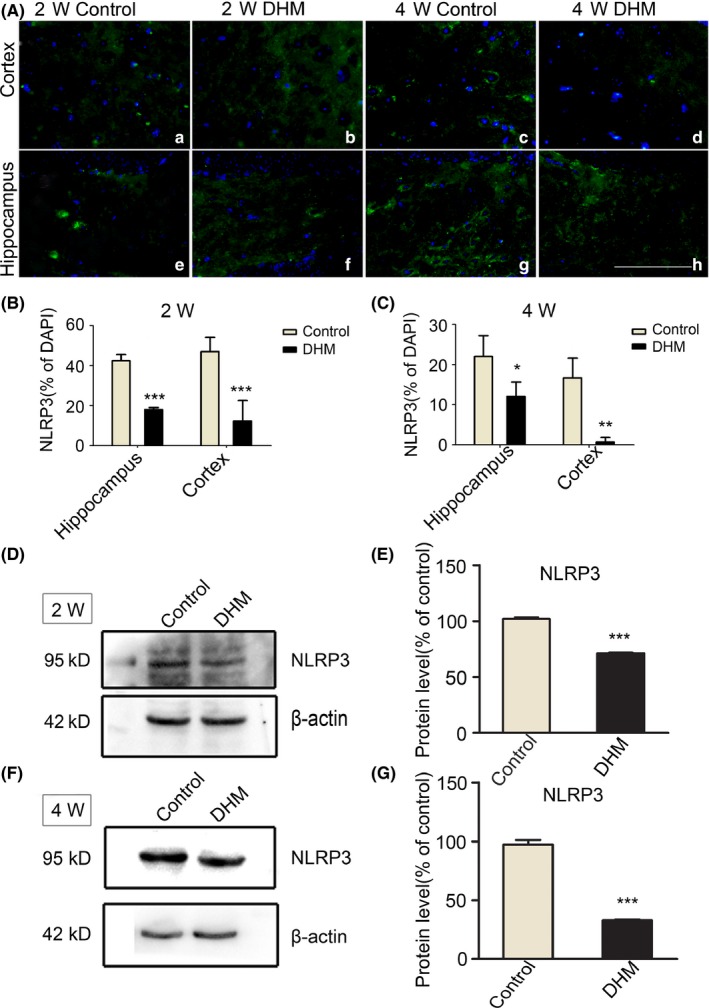
Dihydromyricetin (DHM) reduces NLRP3 inflammasome expression in APP/PS1 mice. A, Immunofluorescent images showing NLRP3 staining after 2 and 4 weeks of DHM treatment compared with control groups. Scale bars: 40 μm. B, C, Bar graphs showing quantification of NLRP3‐positive cells in each group. Values are presented as mean ± SEM (*P < .05, **P < .01, ***P < .001 vs control group). D‐G, Western blot analysis showing a significant reduction in NLRP3 protein in DHM‐treated mice relative to control groups
3.3. DHM decreases activation of the NLRP3 inflammasome in APP/PS1 mice
Next, we examined whether DHM treatment directly inhibits activation of the NLRP3 inflammasome. Using Western blot assays, we found reduced levels of caspase‐1 p20 subunits (Figure 3C) in 2‐ and 4‐week DHM treatment groups compared with controls. These results were confirmed by immunohistochemical experiments, which showed that few caspase‐1‐positive cells were observed in the hippocampus and cortex of APP/PS1 mice treated with DHM, especially in the 4‐week group (Figure 3A). These results suggest that DHM treatment reduces caspase‐1 activation in the brain of APP/PS1 mice.
Figure 3.
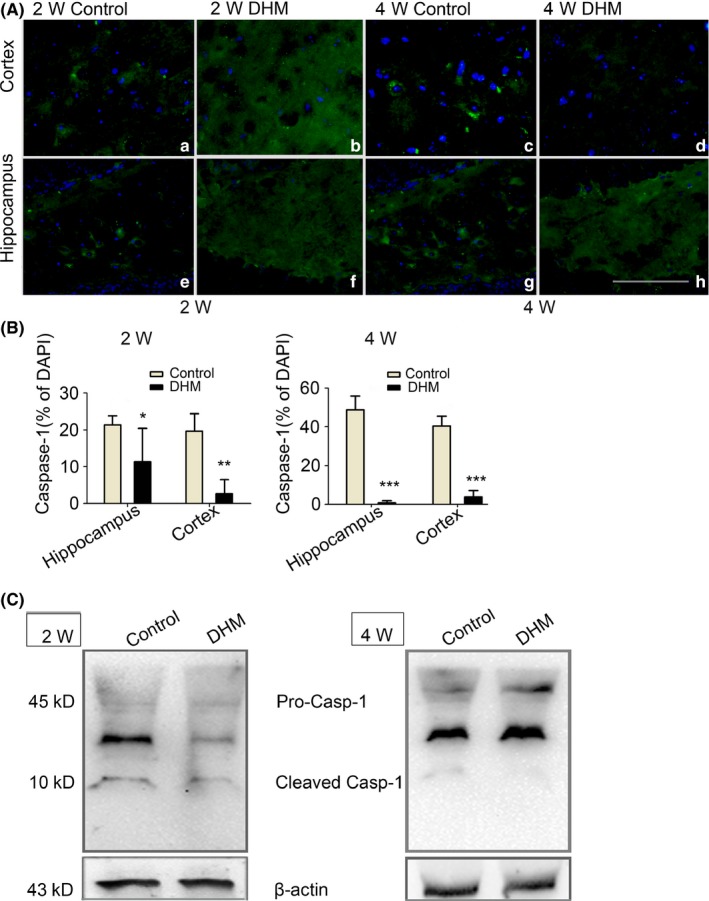
Caspase‐1 (Casp1) protein reduce after dihydromyricetin (DHM) treated in APP/PS1 mice. A‐B, Caspase‐1 immunostaining revealed decreases expression of Caspase‐1 in the hippocampus and cortex of DHM‐treated mice, as compared with control groups mice. Scale bars: 40 μm. (*P < .05, **P < .01 vs Control group at 2 weeks, ***P < .001 vs control group at 4 weeks). C, Two bands of 45 and 10 kDa were detected on Western blots using the Caspase‐1 antibody. Given this 10 kDa molecular weight, this protein is probably activated Caspase‐1. Caspase‐1 was almost detectable following DHM treated but was markedly in APP/PS1 control mice. β‐actin was used as the loading control
We then assessed matured IL‐1β expression, which is dependent on enzyme activity of activated caspase‐1. Western blot analysis combined with immunohistochemical assays demonstrated that there was lower expression of matured IL‐1β in DHM‐treated groups compared with control groups as shown in Figure 4A,C. In addition, relative to the 2‐week DHM treatment group, there was a more significant decrease in the 4‐week DHM treatment group. These results indicate that DHM decreases levels of matured IL‐1β in APP/PS1 mice after inhibiting activation of NLRP3 inflammasome components, including NLRP3 and caspase‐1 proteins.
Figure 4.
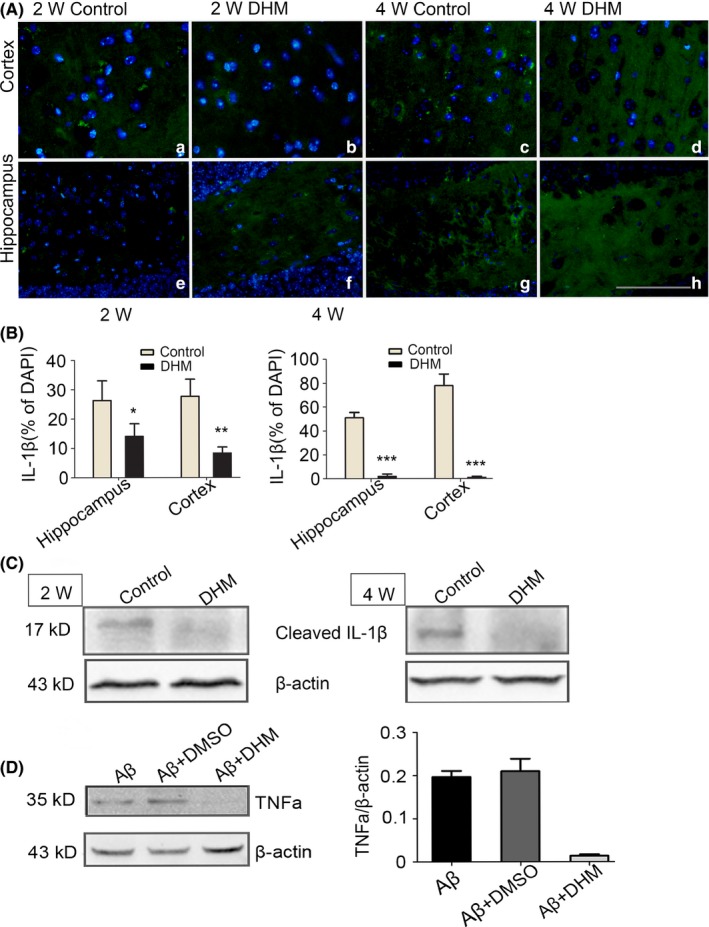
IL‐β protein reduce after dihydromyricetin (DHM) treated in APP/PS1 mice. A‐B, IL‐β immunostaining revealed decreases expression of IL‐βin the hippocampus and cortex of DHM‐treated mice, as compared with control groups mice. Scale bars: 40 μm. (*P < .05, **P < .01 vs Control group at 2 weeks, ***P < .001 vs control group at 4 weeks). C, Western blot detects the optical density of matured IL‐1β. A 17 kDa molecular weight protein was detected on Western blot analysis for IL‐1β either DHM groups or control groups in APP/PS1 mice. The expression of IL‐1β decreased in DHM treated animals compared to control groups. β‐actin was used as the loading control. D, Western blot analysis for TNF‐α following DHM treated in activated microglia cells. Quantification of the expression of TNF‐α showed that it markedly decreased following DHM treated compared to that following DMSO (**P < .01)
In vitro Aβ peptide activated the microglia to produce and release Tumor necrosis factor‐α (TNF‐α) and IL‐1β, In the present study when the activated cells were treated with DHM in Aβ‐induced microglia, a significant inhibition in the production of TNF‐α was observed, there have lower levels of TNF‐α were expressed when the microglial cells were treated with DHM for 24 hours, however, between 2 controlled groups, DHM did not affect TNF‐α expression (Figure 4D); These results indicated that DHM inhibited the expression of TNF‐α in Aβ‐induced microglia.
3.4. DHM decreases Aβ levels and deposition
Chronic deposition of Aβ is the main trigger for NLRP3 inflammasomes during the process of AD, which probably involves chronic activation of NLRP3 inflammasomes in microglial cells. To examine whether DHM inhibits NLRP3 inflammasomes through a decrease in levels of Aβ, specific immunostaining identifying amyloid protein was performed with antibody 4G8, which recognizes the Aβ17‐24 fragment. Representative images of 4G8‐positive plaques are shown in Figure 5A. Some amyloid plaques were found in the cortex and hippocampus of both 2‐ and 4‐week DMSO‐treated control mice, and more plaques of larger sizes were observed in the latter group. However, a significant decrease in the number of plaques was observed in the cortex of DHM‐treated mice, and no positive plaques were seen in the hippocampus of both 2‐ and 4‐week DHM‐treated mice (Figure 5B,C). These data indicate that DHM can reduce Aβ deposition in the brain of APP/PS1 mice.
Figure 5.
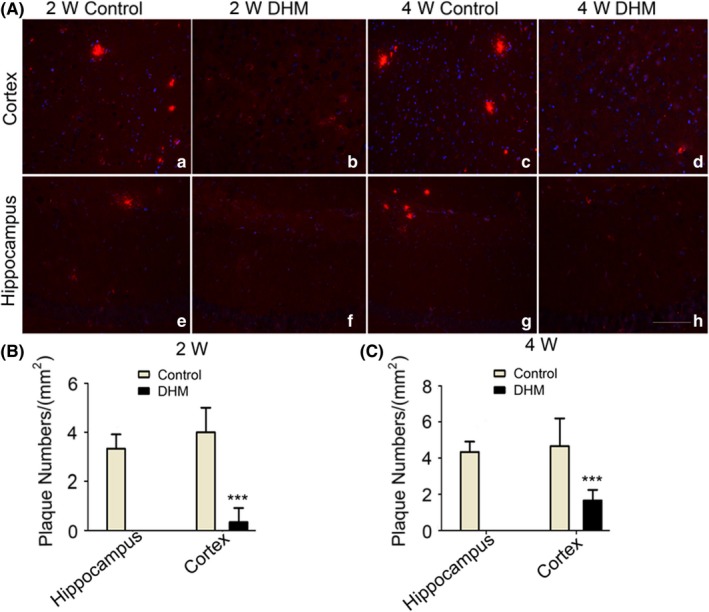
Dihydromyricetin (DHM) decreases Aβ deposition in APP/PS1 mice. A‐C, Plaques were observed in the 2‐ and 4‐week control groups, and the total number of plaques in the 4‐week group was greater than that in the 2‐week group. No plaques were detected in the hippocampus of DHM‐treated groups. Scale bars: 20 μm
Next, we performed ELISA assays to measure Aβ levels in the brain. Results indicated that levels of Aβ40 were significantly decreased after DHM administration for 2 and 4 weeks when compared with the control groups (Figure 6A). Similar results were obtained for Aβ42 levels (Figure 6B). These data suggest that DHM can decrease Aβ levels in APP/PS1 mice.
Figure 6.
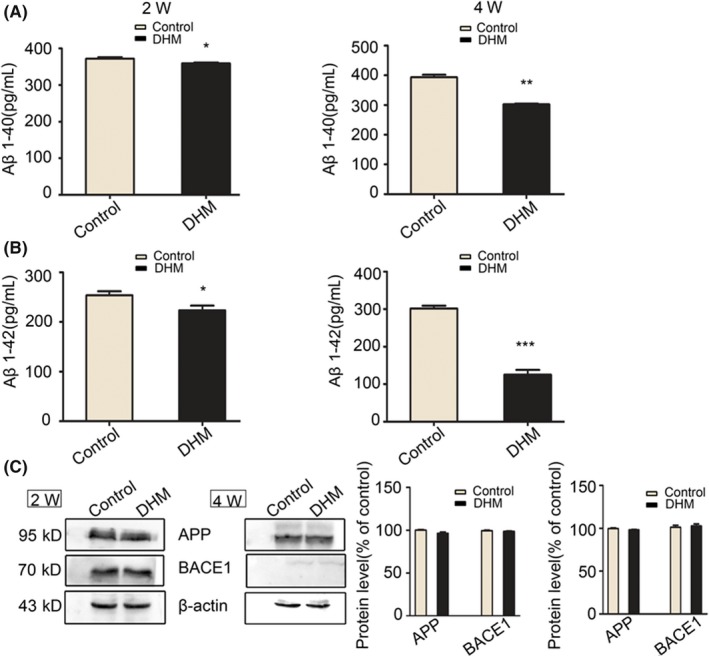
Dihydromyricetin (DHM) decreases Aβ levels in APP/PS1 mice. A, ELISA quantification of Aβ1‐40 was performed in APP/PS1 mice. Aβ1‐40 levels were markedly decreased after DHM treatment. Values are presented as mean ± SEM (*P < .05 in the 2‐week group, **P < .01 in the 4‐week group vs control groups). B, ELISA quantification of Aβ1‐42 was performed in APP/PS1 mice (*P < .05 in the 2‐week group, ***P < .001 in the 4‐week group vs control groups). C, Western blot analysis showing no significant differences in expression levels of APP and BACE1 between control and DHM‐treated groups at 2 weeks or 4 weeks
3.5. DHM promotes clearance of Aβ in the brains of APP/PS1 mice
To investigate the mechanism underlying the reduction in Aβ peptide levels, we examined production and clearance of Aβ. DHM had negligible effects on expression of APP full‐length protein compared with DMSO‐treated mice. In addition, expression of BACE1 protein, which is the major form of β‐secretase, was not significantly different between DHM‐ and DMSO‐treated groups (Figure 6C). These data suggest that DHM exerts little effect on production of Aβ.
Next, we examined clearance of Aβ. Two mechanisms can promote clearance of Aβ: phagocytosis by microglia with an anti‐inflammatory M2 phenotype or degradation by proteolytic enzymes, such as NEP, in the AD brain. As shown in Figure 8D, DHM‐treated groups showed an increase in NEP expression compared with controls, which suggests that DHM promotes degradation of Aβ peptides. Furthermore, using Western blot technique, we investigated the influence of DHM on microglial phenotypes in vitro. Representative images of the M1 phenotype show that the M1 phenotype is weakly expressed in the DHM‐treated cells when compared with controls (Figure 7A). In contrast, a clear increase in M2 state agrinase‐1 was observed in DHM‐treated as shown in Figure 7B. The ratio between expression of Arginase‐1 (Arg‐1) or Inducible nitric oxide synthase (iNOS) reflected an increased level of the M2 phenotype and a clear decreased level of the M1 phenotype in the DHM‐treated activated microglia. The fluorescence images of the M2 microglial cells in APP/PS1 mice brains were concordant with the results obtained from BV‐2 cells (Figure 8A). These data suggest that DHM may shift microglia from the M1 phenotype to the M2 phenotype. Hence, DHM can promote clearance of Aβ in the brain of APP/PS1 mice, which may be associated with an increase in NEP expression and conversion to the M2 microglial phenotype.
Figure 7.

Western blot analysis for M1 and M2 phenotype in BV2 microglia. BV2 cells were activated by Aβ and treated with dihydromyricetin (DHM) 24 h later. Activated cells were used as control. A, Western blot was performed using antibodies to iNOS (M1; MW: 135 kDa). Note decreased M1in the DHM groups compared to control. β‐actin was used as the loading control. B, Arg‐1(M2; MW: 35 kDa) was detected on Western blot analysis in activated microglia after the DHM treatment. Note increased M2 in DHM treatment groups compared to that Aβ and DMSO control groups. M2 was absent in control cell. Actin was used as loading control, showing somewhat less loading in the samples
Figure 8.
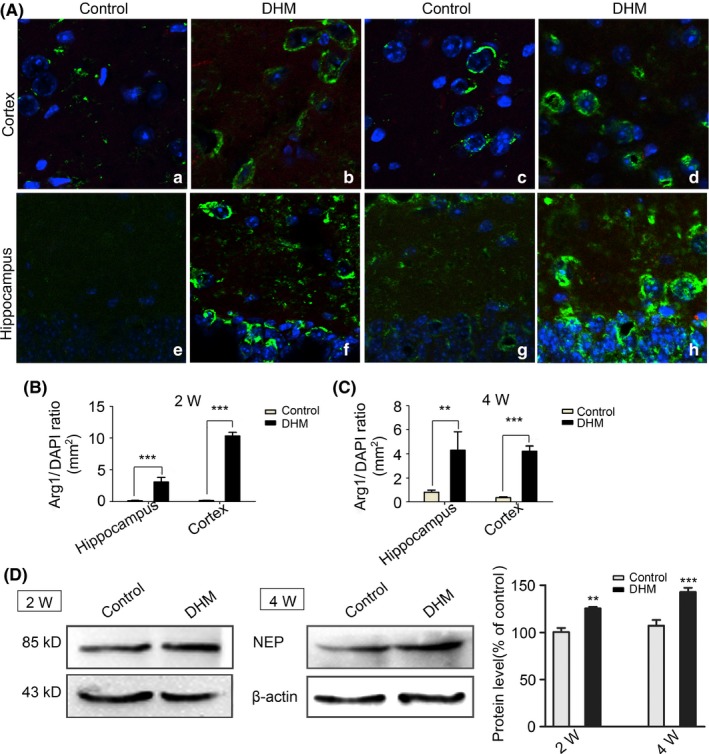
M2 staining in APP/PS1 mice treated by dihydromyricetin (DHM). A, M2 immunostaining of brain sections from APP/PS1 animal show that the number of arginase‐1‐positive cells (green) increased in DHM‐treated groups compared with control groups. Scale bars: 50 μm; B, C, Bar graphs showing that the number of arginase‐1‐positive cells significantly increased in the DHM‐treated groups (**P < .01, ***P < .001 vs control groups). D, Western blot shows that the expression of NEP were up‐regulation in 2 and 4 weeks DHM administration groups compared with their control groups. *P < .05, **P < .01, ***P < .001 vs control
3.6. DHM prevents cognitive deficits in APP/PS1 transgenic mice
Morris water maze tests were performed to examine whether DHM administration also affects spatial learning and memory. In the hidden platform swimming test, 2‐ and 4‐week DHM‐treated APP/PS1 mice showed a shorter latency (Figure 9A,B) and shorter distance (Figure 9C,D) to reach the platform when compared with control groups. In the probe trial on the last day of testing, the platform was removed, and the number of times each mouse crossed the target quadrant (fourth quadrant) was recorded. APP/PS1 mice with DHM administration had significantly more platform‐passing times in the correct quadrant of the pool compared with control groups (Figure 9E,F). Taken together, these data indicate that DHM treatment can improve learning and memory ability in APP/PS1 transgenic mice.
Figure 9.
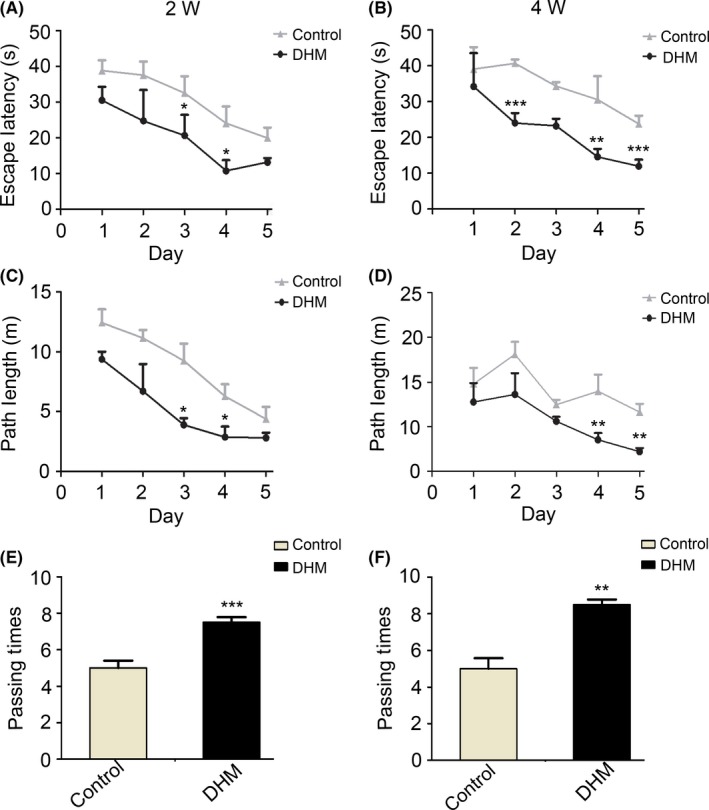
Dihydromyricetin (DHM) prevents cognitive deficits in APP/PS1 transgenic mice. A, C, Hidden platform test of the Morris water maze. DHM treatment improved learning and memory ability in APP/PS1 mice. All DHM‐treated mice showed significant cognitive function improvement during the 5 d of training compared with the control groups. All mice showed a shorter latency to escape onto the hidden platform on the third and fourth days (P < .05), and DHM‐treated mice had a shorter swimming length before escaping onto the hidden platform on the third and fourth days (P < .05). B, D, Degree of improvement of cognitive function was correlated with duration of treatment. After DHM administration for 4 weeks, all treated mice displayed more significant differences compared with controls. **P < .01 vs control groups, ***P < .001 vs control groups. E, F, Morris water maze probe trial. A spatial probe trial was conducted on day 7, in which the number of platform crossing times was measured. DHM‐treated mice showed significantly more platform‐passing times compared with control groups
4. DISCUSSION
Alzheimer’s disease is a severe and prevalent disease.19, 20 At present, available treatments for AD improve symptoms but are not successful in reversing progression of this disease. Neuroinflammation, which is characterized by activation of microglia, is known to contribute to the cascade of events culminating in the neuronal damage of AD.21, 22 Several studies have suggested modulating neuroinflammation by targeting microglia as a treatment for AD.23, 24, 25 However, until now, no effective drugs have been found to alleviate the microglial inflammatory reaction in AD.
It has been shown that DHM improves cognitive deficits in Swedish transgenic mice by restoring gephyrin levels and functional GABAergic transmission.14 In our study, we also found DHM could efficiently improve memory and cognitive deficits in APP/PS1 mice, which is an extensively used AD disease model.26 DHM, a plant flavonoid, can cross the blood‐brain barrier12 and has anti‐inflammatory properties in vitro and in vivo.27, 28 Recent research has suggested that flavonoids may be good candidates for modulating inflammation and preventing onset of neurodegenerative disorders.13 However, it is unknown whether DHM has anti‐inflammatory effects in AD. We observed a significant reduction of microglia in the hippocampus and cortex of APP/PS1 mice treated with DHM when compared with untreated APP/PS1 mice, which suggests that DHM can inhibit microglia‐mediated neuroinflammation and play a neuroprotective role in AD.
Extensive evidence indicates that activation of the NLRP3 inflammasome is a critical process of the inflammatory response in disease progression of AD patients and APP/PS1 mice.29 Assembly and activation of the NLRP3 inflammasome leads to cleavage of pro‐caspase‐1 to generate active caspase‐1,6 which then initiates maturation and secretion of pro‐IL‐1β in the AD brain.30, 31 Moreover, inhibition of NLRP3 and caspase‐1, such as through artemisinin treatment, has been shown to exert protective effects in AD pathology.32 Hence, targeting NLRP3 inflammasome activation may be a promising and valuable intervention for AD. In the present study, we demonstrated that DHM effectively suppressed activation of the NLRP3 inflammasome in APP/PS1 mice. The DHM‐treated APP/PS1 mice showed decreased expression of IL‐1β and caspase‐1 p20 subunits. Furthermore, our study demonstrated that DHM could inhibit activation of microglia by suppressing NLRP3 inflammasome activation in APP/PS1 mice.
Inhibiting expression of NLRP3 is a common method for inhibiting NLRP3 inflammasome activation. Our results clearly showed that DHM decreases expression of NLRP3. In addition, our data suggest that DHM promotes clearance of extracellular Aβ in the brain of APP/PS1 mice, which also reduces activation of the NLRP3 inflammasome. Activation of the NLRP3 inflammasome requires 2 signals. The first signal primes the NLRP3 inflammasome and usually promotes expression of the NLRP3 inflammasome components. The second signal triggers assembly of the NLRP3 inflammasome and results in production of cleaved caspase‐1. In the AD brain, Aβ peptides and its aggregates have been confirmed as an activating signal, which usually leads to chronic activation of NLRP3 inflammasomes and progression of AD‐like pathology. Removal of Aβ peptides and its aggregates contributes to reducing activation of the NLRP3 inflammasome. Our data show that DHM treatment significantly decreases levels of Aβ peptides and its aggregates, which provides another method for reducing NLRP3 inflammasome activation.
According to our results, the total amount of APP protein and levels of BACE1 were unaffected in DHM‐treated mice, which suggests that the observed reduction of Aβ was most likely not due to reduced production of Aβ peptides and its aggregates. In contrast, levels of proteolytic enzyme NEP, which is an enzyme in microglia for Aβ clearance,33 were increased in APP/PS1 mice treated with DHM. This suggests that DHM might promote clearance of Aβ by microglia. In addition, recent research has proposed that converting microglia from the M1 state to the M2 state increases clearance of Aβ.34, 35, 36, 37 In our study, we showed that DHM treatment could promote the M2 microglial phenotype by shifting microglial cells from the M1 to the M2 phenotype. This shift may partly explain the decrease of Aβ levels in AD, although whether this conversion influences microglial phagocytosis of Aβ peptide is still not clear. Thus, combined with previous results, we found that by increasing levels of the proteolytic enzyme NEP in microglia and shifting microglia to the M2 phenotype, DHM decreases levels of Aβ, which may act as another mechanism to reduce NLRP3 inflammasome activation in APP/PS1 mice.
Current therapeutic approaches that abate AD progression primarily use established acetylcholinesterase inhibitors. However, new drugs that may prevent or at least limit progression of the disease are required. Using behavioral data, we showed that DHM improves symptoms of memory loss in APP/PS1 mice. Therefore, overall, DHM is an efficacious medication that exerts a clear protective effect in AD by inhibiting activation of microglia. Its underlying mechanisms may involve regulating the NLRP3/caspase‐1 inflammatory signaling pathway, downregulating inflammatory cytokines, ameliorating symptoms, and reversing neuropathological changes of AD.
In conclusion, various studies have suggested that inhibiting activation of microglial cells may be a promising therapy for AD. However, to date, no effective drugs have been shown to alleviate the neuroinflammatory reaction in AD. In this study, a therapeutic drug that targets inhibition of microglial activation by suppressing the NLRP3 inflammasome was demonstrated, this represents a significant contribution to AD research. However, the mechanisms through which DHM drives microglial conversion from the M1 state to the M2 state is unclear and must be the focus of further study.
5. CONCLUSION
In the present study, we demonstrates that DHM exerts neuroprotective effects in APP/PS1 double‐transgenic mice by inhibiting microglia activation, notably, DHM could reduce the activation of NLRP3 inflammasome via decreasing the expression of NLRP3 inflammasome components and promoting the clearance of Aβ, which is involved in the increasing levels of NEP and shifting microglial conversion to M2 types. Overall, we found that DHM as a potentially anti‐inflammatory therapeutic drug for treating AD could prevent the progression of AD‐like pathology and improve spatial memory via the inhibition of NLRP3 inflammasome‐based microglia‐mediated neuroinflammation in APP/PS1 double‐transgenic mice.
COMPETING INTEREST
The authors declare no competing interest.
ETHICS APPROVAL
The study was approved by the Care and Use of Laboratory Animals of Chongqing Medical University.
ACKNOWLEDGMENTS
All authors contributed to the work and agree with the presented findings. The work has not been published before nor is it being considered for publication in another journal. The authors thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
Feng J, Wang J‐X, Du Y‐H, et al. Dihydromyricetin inhibits microglial activation and neuroinflammation by suppressing NLRP3 inflammasome activation in APP/PS1 transgenic mice. CNS Neurosci Ther. 2018;24:1207‐1218. 10.1111/cns.12983
Funding information
This work was supported by the Natural Science Foundation Project of Chongqing (cstc2016jcyjA0069); the Program for Science and Technology Projects of Yuzhong District in Chongqing, China (20130132); the National Natural Science Foundation of China (nos. 81671257, 31670899, and 81371221); and the Program for New Century Excellent Talents in University (NCET‐11‐1084).
The first two authors contributed equally to this work.
REFERENCES
- 1. Miao H, Ou J, Zhang X, et al. Macrophage CGI‐58 deficiency promotes IL‐1beta transcription by activating the SOCS3‐FOXO1 pathway. Clin Sci (Lond). 2015;128:493‐506. [DOI] [PubMed] [Google Scholar]
- 2. Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, Jones E. Alzheimer’s disease. Lancet. 2011;377:1019‐1031. [DOI] [PubMed] [Google Scholar]
- 3. LaFerla FM, Oddo S. Alzheimer’s disease: Abeta, tau and synaptic dysfunction. Trends Mol Med. 2005;11:170‐176. [DOI] [PubMed] [Google Scholar]
- 4. Ferri CP, Prince M, Brayne C, et al. Global prevalence of dementia: a Delphi consensus study. Lancet. 2005;366:2112‐2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Halle A, Hornung V, Petzold GC, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid‐beta. Nat Immunol. 2008;9:857‐865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gold M, El Khoury J. Beta‐amyloid, microglia, and the inflammasome in Alzheimer’s disease. Semin Immunopathol. 2015;37:607‐611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821‐832. [DOI] [PubMed] [Google Scholar]
- 8. Tan MS, Yu JT, Jiang T, Zhu XC, Tan L. The NLRP3 inflammasome in Alzheimer’s disease. Mol Neurobiol. 2013;48:875‐882. [DOI] [PubMed] [Google Scholar]
- 9. Heneka MT, Kummer MP, Stutz A, et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493:674‐678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shen Y, Lindemeyer AK, Gonzalez C, et al. Dihydromyricetin as a novel anti‐alcohol intoxication medication. J Neurosci. 2012;32:390‐401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li H, Li Q, Liu Z, et al. The versatile effects of dihydromyricetin in health. Evid Based Complement Alternat Med. 2017;2017:1053617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Youdim KA, Dobbie MS, Kuhnle G, Proteggente AR, Abbott NJ, Rice‐Evans C. Interaction between flavonoids and the blood‐brain barrier: in vitro studies. J Neurochem. 2003;85:180‐192. [DOI] [PubMed] [Google Scholar]
- 13. Vauzour D, Martinsen A, Layé S. Neuroinflammatory processes in cognitive disorders: is there a role for flavonoids and n‐3 polyunsaturated fatty acids in counteracting their detrimental effects? Neurochem Int. 2015;89:63‐74. [DOI] [PubMed] [Google Scholar]
- 14. Liang J, López‐Valdés HE, Martínez‐Coria H, et al. Dihydromyricetin ameliorates behavioral deficits and reverses neuropathology of transgenic mouse models of Alzheimer’s disease. Neurochem Res. 2014;39:1171‐1181. [DOI] [PubMed] [Google Scholar]
- 15. Paula‐Lima AC, Brito‐Moreira J, Ferreira ST. Deregulation of excitatory neurotransmission underlying synapse failure in Alzheimer’s disease. J Neurochem. 2013;126:191‐202. [DOI] [PubMed] [Google Scholar]
- 16. Louzada PR, Paula Lima AC, Mendonca‐Silva DL, Noël F, De Mello FG, Ferreira ST. Taurine prevents the neurotoxicity of beta‐amyloid and glutamate receptor agonists: activation of GABA receptors and possible implications for Alzheimer’s disease and other neurological disorders. FASEB J. 2004;18:511‐518. [DOI] [PubMed] [Google Scholar]
- 17. Wang R, Pi J, Su X, et al. Dihydromyricetin suppresses inflammatory responses in vitro and in vivo through inhibition of IKKbeta activity in macrophages. Scanning. 2016;38:901‐912. [DOI] [PubMed] [Google Scholar]
- 18. Xu B, Huang S, Wang C, Zhang H, Fang S, Zhang Y. Antiinflammatory effects of dihydromyricetin in a mouse model of asthma. Mol Med Rep. 2017;15:3674‐3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Goldmann T, Tay TL, Prinz M. Love and death: microglia, NLRP3 and the Alzheimer’s brain. Cell Res. 2013;23:595‐596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Alzheimer’s Association . 2014 Alzheimer’s disease facts and figures. Alzheimers Dement. 2014;10:e47‐e92. [DOI] [PubMed] [Google Scholar]
- 21. Deardorff WJ, Grossberg GT. Targeting neuroinflammation in Alzheimer’s disease: evidence for NSAIDs and novel therapeutics. Expert Rev Neurother. 2016;17:17‐32. [DOI] [PubMed] [Google Scholar]
- 22. Hong H, Kim BS, Im HI. Pathophysiological role of neuroinflammation in neurodegenerative diseases and psychiatric disorders. Int Neurourol J. 2016;20(Suppl 1):S2‐S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lue LF, Kuo YM, Beach T, Walker DG. Microglia activation and anti‐inflammatory regulation in Alzheimer’s disease. Mol Neurobiol. 2010;41:115‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Prokop S, Miller KR, Heppner FL. Microglia actions in Alzheimer’s disease. Acta Neuropathol. 2013;126:461‐477. [DOI] [PubMed] [Google Scholar]
- 25. Xu L, He D, Bai Y. Microglia‐mediated inflammation and neurodegenerative disease. Mol Neurobiol. 2016;53:6709‐6715. [DOI] [PubMed] [Google Scholar]
- 26. Jankowsky JL, Fadale DJ, Anderson J, et al. Mutant presenilins specifically elevate the levels of the 42 residue beta‐amyloid peptide in vivo: evidence for augmentation of a 42‐specific gamma secretase. Hum Mol Genet. 2004;13:159‐170. [DOI] [PubMed] [Google Scholar]
- 27. Lee HY, Park SH, Lee M, et al. 1‐Dehydro‐[10]‐gingerdione from ginger inhibits IKKbeta activity for NF‐kappaB activation and suppresses NF‐kappaB‐regulated expression of inflammatory genes. Br J Pharmacol. 2012;167:128‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang R, Pi J, Su X, et al. Dihydromyricetin suppresses inflammatory responses in vitro and in vivo through inhibition of IKKbeta activity in macrophages. Scanning. 2016;2016:1‐12. [DOI] [PubMed] [Google Scholar]
- 29. Saresella M, La Rosa F, Piancone F, et al. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol Neurodegener. 2016;11:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Akama KT, Van Eldik LJ. β‐amyloid stimulation of inducible nitric‐oxide synthase in astrocytes is interleukin‐1β‐ and tumor necrosis factor‐α (TNFα)‐dependent, and involves a TNFα‐receptor‐associated factor‐ and NFκB‐inducing kinase‐dependent signaling mechanism. J Biol Chem. 2000;275:7918‐7924. [DOI] [PubMed] [Google Scholar]
- 31. Schnaars M, Beckert H, Halle A. Assessing beta‐amyloid‐induced NLRP3 inflammasome activation in primary microglia. Methods Mol Biol. 2013;1040:1‐8. [DOI] [PubMed] [Google Scholar]
- 32. Shi JQ, Zhang CC, Sun XL, et al. Antimalarial drug artemisinin extenuates amyloidogenesis and neuroinflammation in APPswe/PS1dE9 transgenic mice via inhibition of nuclear factor‐κB and NLRP3 inflammasome activation. CNS Neurosci Ther. 2013;19:262‐268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Malito E, Hulse RE, Tang WJ. Amyloid beta‐degrading cryptidases: insulin degrading enzyme, presequence peptidase, and neprilysin. Cell Mol Life Sci. 2008;65:2574‐2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sierra‐Filardi E, Puig‐Kröger A, Blanco FJ, et al. Activin A skews macrophage polarization by promoting a proinflammatory phenotype and inhibiting the acquisition of anti‐inflammatory macrophage markers. Blood. 2011;117:5092‐5101. [DOI] [PubMed] [Google Scholar]
- 35. Colton CA, Mott RT, Sharpe H, Xu Q, Van Nostrand WE, Vitek MP. Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. J Neuroinflammation. 2006;3:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677‐686. [DOI] [PubMed] [Google Scholar]
- 37. McGeer PL, McGeer EG. Targeting microglia for the treatment of Alzheimer’s disease. Expert Opin Ther Targets. 2015;19:497‐506. [DOI] [PubMed] [Google Scholar]