

Abstract
Dynamic covalent chemistry is a versatile and powerful tool that integrates both stable chemical bonds and stimulus responsiveness into the construction of smart biotherapeutics. With minimalistic molecular design, a dynamic covalent protein assembly that incorporates selective targeting and intracellular release upon pH stimulus is presented. The construct comprises an active enzymatic protein core (cytochrome c) self‐assembled with cancer cell targeting motifs (somatostatin) through boronic acid/salicylhydroxamate chemistry. The bioorthogonal assembly takes place rapidly under neutral aqueous conditions while the release of the protein is initiated under acidic conditions found within cellular vesicles during uptake. By demonstrating that these modular components act in synergy, we show the broad applicability of such chemical strategies to advance the frontier of modern nanomedicine.
Keywords: bioconjugates, boronic acid, dynamic covalent chemistry, pH-responsiveness, protein therapeutics
Biotherapeutics, a class of macromolecular drugs sourced from Nature, have received emerging interest since the introduction of antibody therapeutics.1 By developing these engineered biomacromolecules such as fusion proteins and antibody conjugates for commercialization, the potential of how these materials address a variety of diseases such as cancer,2 multiple sclerosis3 or diabetes4 was demonstrated in a highly selective fashion. With the advent of site‐directed mutagenesis and site‐specific protein modification,5 the repertoire of biotherapeutics thus expanded further as these broad methods can be synergized to create unique protein conjugates that would have never been possible. Ideally, these biosynthetic therapeutics exhibit improved pharmacological properties by exploiting the specificity (e.g. transport, targeting or catalysis) that is innate to the biological component while the attached synthetic modules aim to enhance pharmacokinetic properties (e.g. biodistribution, stability).6 Additionally, these conjugates are often biocompatible, biodegradable and can be made to be non‐immunogenic.6
Although biotechnology is currently the method of choice in constructing protein conjugates, it provides limited capacity for improvisation simply due to its reliance on the translation machinery of the cell.7 On the contrary, synthetic chemical techniques are far‐reaching and many of them have been shown to control biomolecules based on external (e.g. light, magnetic field) or internal (e.g. pH, enzymatic) stimuli.8 Despite being able to provide a more complex and controlled behavior of the molecule of interest, permanent synthetic modifications on proteins can often affect its functions and tolerance.9 In order to avoid the impact of covalent modifications,10 the development of dynamic covalent chemistry, where chemical bonds are formed and dissociate under orthogonal conditions into benign components, is a promising approach.
Among various dynamic covalent chemistry schemes, boronic acids represent a unique class of chemical functionalities due to their electron deficient boron center, which often leads to very specific binding events in order to fulfill its electronic configuration.11 These binding partners mainly involve 1,2 cis‐diols like glucose, catechols and also other multivalent interactions involving nitrogen donors such as MIDA and salicyl hydroxamates.12 Depending on the electronic demand, these binding events can occur with a broad range of binding affinities under physiological conditions, leading to the construction of materials ranging from click‐type peptide ligations,13 responsive nanosystems14 to bulk materials like hydrogels.15 These interactions are reversibly dependent on pH, with cis‐diols dissociating at acidic medium (≈pH 5)12 and MIDA boronates at basic conditions (≈pH 9).16 Moreover, given that boronic acids are widely used in Suzuki cross coupling reactions,11 the enormous synthetic library provides a natural advantage in terms of design strategies over most dynamic covalent systems.17
While boronic acids provide the handle for responsiveness, the creation of an effective dynamic bioconjugate necessitates a targeting motif. This is accomplished by somatostatin, a peptide hormone naturally present within the body that regulates the endocrine system and acts as a neurotransmitter as well as regulates cell proliferation.18 The uptake of somatostatin (SST) is initiated by binding to somatostatin cell membrane receptors (SSTR), which belong to the group of G‐protein coupled receptors.19 SSTRs (including SSTR2) are overexpressed to various degrees in certain cancer cell lines such as lung adenocarcinoma (A549) and pancreatic (PANC) cells.20 As such, both SST and its synthetic analogue octreotide have encouraged their incorporation in a wide variety of targeted therapeutic systems.21 For the protein core, we focused primarily on cytochrome c (CytC, ≈12 kDa) due to its therapeutic relevance while our inclusion of human serum albumin (HSA, ≈66 kDa) seeks to showcase the broader applicability of our design strategy. HSA is a clinically proven protein carrier system for delivery22 and widely used for therapeutics.23 CytC is an electron transport protein typically located in the mitochondria of viable cells and is primarily responsible for catalyzing redox processes.24 During early phases of apoptosis, CytC is released from the mitochrondria into the cytosol, where it assembles with Apaf‐1 into a heptameric apoptosome complex. The formation of this complex cleaves specifically the initiator caspases and marks the point of no return towards programmed cell death.25 Importantly, CytC is one of the most conserved enzymes, whose sequence and structure remained largely unaffected with evolution and between species,26 thus offering great potential as protein drug. By combining somatostatin (SST) and CytC within one functional scaffold on the basis of dynamic covalent chemistry, we report herein a multivalent and pH‐responsive bioconjugate constructed from the enzymatic CytC core surrounded by a cell‐targeting peptide shell (C‐SST, Figure 1). The single protein nanoconstruct exhibits excellent cell selectivity and stimulus responsive release within A549 cells. We show that, through the use of dynamic boronic acid chemistry, modular construction of smart biotherapeutics can be realized in a facile and efficient way.
Figure 1.
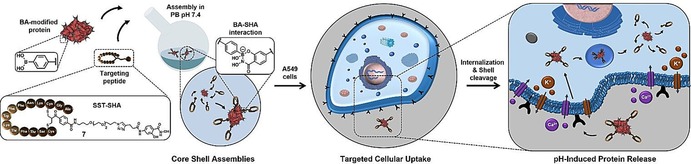
Assembling of stoichiometric amounts of SST‐SHA 7 onto BA‐modified proteins CytC and HSA in phosphate buffer (PB) at pH 7.4 creating dynamic covalent bioconjugates C‐SST and H‐SST. After receptor‐selective uptake into A549 cancer cells, the core–shell hybrids are internalized and due to the pH‐responsiveness of the boronic acid chemistry, the proteins are released under acidic endosomal conditions.
Synthesis. In order to install the corresponding dynamic covalent handles onto the protein core as well as the targeting moiety, the syntheses were modularly divided. The receptor targeting moiety, salicyl hydroxamate functionalized SST (SST‐SHA, 7), was synthesized through a disulfide rebridging strategy (Figure 2). This chemical technique exploits a sequential double thiol‐Michael addition reaction, on the reduced disulfide bridge of native SST, promoted by the in situ elimination of a p‐toluene sulfinic group from azido bis‐sulfone 5 (analyses of 5: Figure S4). The afforded azido‐SST (SST‐N3 6, Figure S5) underwent a copper‐catalyzed azide‐alkyne cycloaddition with trityl‐protected ethynyl‐SHA (iii). Subsequent deprotection with TFA liberates the salicylhydroxamic acid to afford the target molecule SST‐SHA 7. HPLC purification afforded product 7, which was herein synthesized with optimizations from previously published protocol by our group27 and characterized by HR‐MALDI TOF MS and ESI‐MS (Figure S6).
Figure 2.
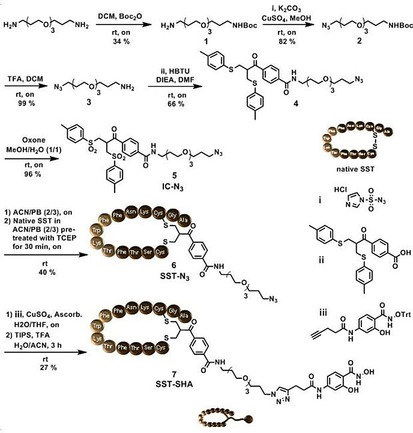
Synthesis of cell surface receptor targeting motif SST‐SHA 7 for the assembly on CytC‐BA and HSA‐BA.
Separately, the protein surface of CytC and HSA was statistically primed with boronic acid groups (BA) via condensation of phenylboronic acid N‐hydroxysuccinimidyl ester onto the solvent accessible lysine residues (Figure S8).14, 15 The functionalized proteins (CytC‐BA, HSA‐BA, Figure S2) were purified by centrifugal ultrafiltration and the numbers of BA moieties were analysed from MALDI TOF MS spectra, affording 11 BA groups for CytC‐BA (Figure S9) and 22 for HSA‐BA (Figure S10). The quantification was cross‐checked by a fluorescence titration assay based on Alizarin Red S, a fluorogenic sensor that detects BA moieties. An excess of Alizarin Red S was pre‐incubated with the respective proteins, CytC‐BA and HSA‐BA, to produce a fluorescence conjugate. As salicylhydroxamates bind to the same site with much higher affinity than 1,2 cis‐diols,12 Alizarin Red S can be displaced by iv (Figure S1) leading to a drop in fluorescence intensity. Hence, a titration with increasing molar equivalents of salicylhydroxamate iv was conducted, in which the respective end points of CytC‐BA and HSA‐BA were in agreement with the MALDI‐MS analysis (Figure 3 A, S12).
Figure 3.
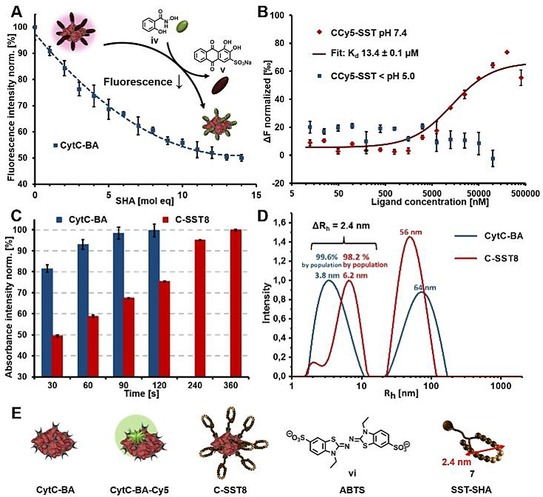
A) Characterization of CytC‐BA by Alizarin Red S assay for BA group determination revealed 11 BA groups. B) Microscale thermophoresis of CCy5‐SST under pH 7.4 for binding affinity determination of SST‐SHA 7 towards CytC‐BA and protein release via SST shell cleavage by acidification. C) Colorimetric assay using ABTS vi to screen bioactivity of C‐SST8 in comparison to the free protein CytC‐BA. D) DLS results of CytC‐BA and C‐SST8, which were both incubated in PB pH 7.4, representatively shown for 90° measurement (analyses with other angles are displayed in the supporting information). E) Structures of CytC‐BA (BA‐functionalized CytC), CytC‐BA‐Cy5 (Cy5‐labelled CytC‐BA), C‐SST8 (8 equivalents of SST‐SHA 7 assembled on CytC‐BA), ABTS vi and visualization of dimension of SST‐SHA 7, which was simulated by the software Molecular Operating Environment.
Dynamic Covalent Assembly. The binding affinity between the protein core and the targeting groups SST‐SHA 7 was elucidated by microscale thermophoresis using CytC‐BA‐Cy5 (Figure 3 E). A dilution series of SST‐SHA 7 was prepared and incubated with a fixed concentration of CytC‐BA‐Cy5 (Cy5‐labelled CytC‐BA) at pH 7.4. The thermophoretic mobility of the series was measured and a Kd of 13.4±0.1 μm was determined for CCy5‐SST (SST‐SHA 7 titrated to CytC‐BA‐Cy5). Expectedly, acidification of the samples to pH <5.0 caused SST‐SHA 7 to be cleaved and a constant thermophoretic mobility of CytC‐BA‐Cy5 independent of the concentration of 7 was recorded (Figure 3 B). As the bioactivity of the protein core is of paramount importance towards the success of the strategy, the catalytic function of CytC after the assembly was assessed using a colorimetric assay (Figure 3 C, S13). The redox activity of CytC enables the conversion of 2,2′‐Azino‐di‐(3‐ethylbenzthiazolin‐6‐sulfonic acid) (ABTS, vi, Figure 3 E) into its radical cation where its dark green color can be detected using absorbance spectroscopy (λ max=410 nm). Full conversion of the substrate was achieved albeit with slower rates for C‐SST8 (8 equivalents of SST‐SHA 7 assembled on CytC‐BA) than for CytC‐BA due to the steric crowding of the reaction center presented by the peptide shell.
The pronounced influence of the SST‐SHA shell corresponding to a projected increase in hydrodynamic radius (Rh) was investigated through dynamic light scattering (DLS). From the measurements, an increase from 3.8 nm (CytC‐BA) to 6.2 nm (C‐SST8) was detected (Figure 3 D). As such, the resultant difference of 2.4 nm was attributed to the successful assembly of the SST‐SHA 7 around the CytC‐BA core. This observation was confirmed by computational modelling of SST‐SHA 7 using the Molecular Operating Environment program (Figure 3 E and S15B) demonstrating the dimensions of 7. After investigations of C‐SST8 in phosphate buffer at pH 7.4 (Figure S14‐S15), its assembly and stability was probed in revised simulated body fluid (r‐SBF), which mimics the human blood plasma28 (Figure S16). Along with the necessary controls, C‐SST8 remained stable up to 48 h with no significant change in Rh relative to the initial values (Figure S17–S18).
Biological Evaluation. The in vitro experiments of the hybrid constructs of boronic acid‐functionalized CytC and HSA decorated with targeting moiety 7 were accomplished systematically in order to elucidate, in particular, receptor interactions and intracellular release. CytC‐BA was labelled with cyanine5‐NHS viii (Figure S11) and charged with increasing molar equivalents of SST‐SHA 7 to probe the efficiency of interaction towards the cellular SST receptors. Interestingly, through confocal microscopy, a moderate and balanced amount of SST‐SHA 7 yielding in 4 equivalents (eq.) to form CCy5‐SST4 (Figure 4 B) provides the optimal uptake efficiency at 4 h. In comparison, CytC‐BA‐Cy5 with lower amounts of 7 (2 equiv.) resulting in CCy5‐SST2 (Figure S21) is less effective whereas significant aggregation was detected at highest density of 7 (13 equiv.) around the protein core (Figure 4 C and S21). In order to prove that the binding of SST‐SHA 7 towards CytC‐BA‐Cy5 is both bioorthogonal and necessary for internalization, a control comprising CytC‐BA‐Cy5 incubated with native somatostatin (native SST, Figure S22) was introduced. Indeed, without the boronic acid‐salicylhydroxamate linkage, no uptake boost towards the A549 cells was detected (Figure S23). The interaction of optimized C‐SST4 (CCy5‐SST4 revealed highest uptake efficiency) against SSTR2 receptor was, in addition, independently confirmed with a calcium flux assay conducted by Eurofins Cerep. Recombinant cells overexpressing SSTR2, which is the main receptor addressed by SST and its derivatives like 7,20 were treated with C‐SST4 (Figure 4 A or S2) and CytC‐BA as a reference via a fluorimetric assay for calcium flux activation induced upon SSTR2 stimulation. Screening a dilution series of C‐SST4 in relation to % of control agonist response (control: native SST) showed an EC50 of 1.4 μm (Figure 4 A). Tests with CytC‐BA displayed no significant agonist response as well as both samples added to null cells, which are SSTR2‐deficient (Figure 4 A and S29). Additional experiments, in which bioconjugates based on boronic acid‐functionalized CytC decorated with 4 and 8 equiv. SST‐SHA 7 were monitored for their cellular uptake into SKUT1 cells (SSTR2‐deficient cell line, Figure S25), confirmed SSTR2 selectivity of SST‐covered protein hybrids. Core‐shell bioconjugates built up from HSA‐protein core demonstrated boosted cellular uptake into A549 cells due to SSTR2 targeting (Figure S24). Applicability of selective protein transport to different protein types was successfully validated and is of high interest as SSTR2 can be found overexpressed on various cancer cell lines.29
Figure 4.
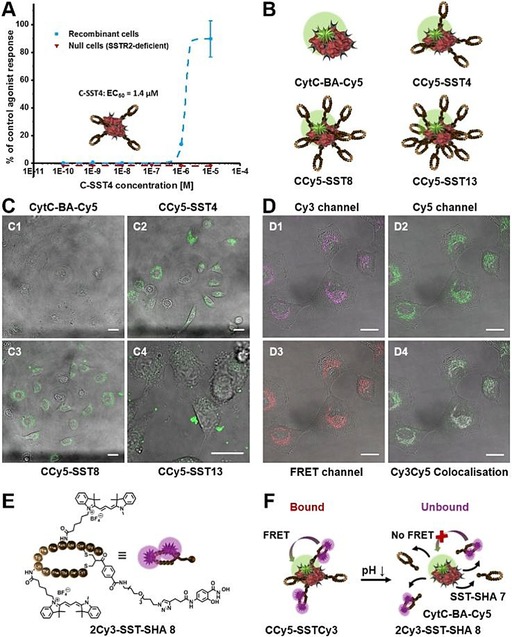
A) SST‐targeting of C‐SST4 (4 equiv. SST‐SHA 7 assembled on CytC‐BA) towards SSTR2 and subsequent activation of calcium flux was monitored related to % of control agonist response (control: native SST). EC50 for C‐SST4 is 1.4 μm. B) Structures of CytC‐BA‐Cy5 (Cy5‐labelled CytC‐BA), CCy5‐SST4, CCy5‐SST8 and CCy5‐SST13 (4, 8 and 13 equiv. of SST‐SHA 7 assembled on CytC‐BA‐Cy5). C) Cellular uptake of CytC‐BA‐Cy5 (C1), CCy5‐SST4 (C2), CCy5‐SST8 (C3) and CCy5‐SST13 (C4) into A549 cancer cells after 4 h incubation and investigated by confocal laser scanning microscopy. Protein core concentration was 250 nm and Cy5‐labelling is displayed in green. D) Cell internalization of CCy5‐SSTCy3 (F, c=250 nm) in A549 cells after 3 h incubation. Confocal laser scanning microscopy reveals two populations: intact CCy5‐SSTCy3 conjugates shown by FRET signal (D3: excited by 561 nm, FRET displayed in red) and partial CytC‐BA release from CCy5‐SSTCy3 (D1: Cy3 excited by 561 nm, Cy3 signal displayed in magenta; D2: Cy5 excited by 633 nm, Cy5 signal displayed in green; D4: Cy3‐Cy5 merge, Cy3‐Cy5 colour overlay results white). E) Structure of 2Cy3‐SST‐SHA 8, which was built by Cy3‐labelling of lysine residues of SST sequence within SST‐SHA 7. F) Structure of CCy5‐SSTCy3 (bound state), which shows FRET signal after excitation of 2Cy3‐SST‐SHA 8 due to assembly of 8 on protein core. CytC‐BA‐Cy5 release from CCy5‐SSTCy3 (unbound state) is achieved by drop in pH, which leads to FRET disruption. C–D) Scale bar represents 20 μm on each image.
The induction of apoptosis of A549 cells by released CytC‐BA was demonstrated via Cell‐TiterGlo® luminescence cell viability assay. The tumor cells were treated with C‐SST4 along with SST‐SHA 7 and a chemotherapeutic doxorubicin as controls (Figure S20). The dose‐dependent bioactivity of released CytC‐BA from C‐SST4 was shown while 7 led to full cell viability even with longer incubation time on cells (24 h), suggesting that enzymatic activity of CytC was effectively retained (Figure S20). The mechanism of release was subsequently investigated using Förster resonance energy transfer (FRET) using Cy3‐Cy5 donor‐acceptor pairs (Figure 4 D and S19). Importantly, as Cy3‐functionalization of SST‐SHA 7 was achieved by addressing the lysines, which are essential for targeting, the construct CCy5‐SSTCy3 was made with 1:1 ratio of labelled (2Cy3‐SST‐SHA 8) and unlabelled (SST‐SHA 7) peptide (Figure 4 F and S26). The labelled SST‐derivative 2Cy3‐SST‐SHA 8 (Figure 4 E) was obtained using HPLC for purification after successful synthesis and analysed by ESI‐MS (Figure S7). The internalized fluorescent signals given by uptake of CCy5‐SSTCy3 into A549 cells primarily demonstrated two populations of signals: One of which was emitted from the energy transfer from Cy3 to Cy5 (FRET, Figure 4 D4, FRET displayed in red), whereas another set of signals was detected only in the Cy3 channel (Figure 4 D4, Cy3 signal shown in magenta). From the colocalisation study, the FRET emission signifies that a proportion of the CCy5‐SSTCy3 construct remains intact to that time point while the exclusive Cy3 emission represents the successful intracellular release of CytC‐BA‐Cy5 from CCy5‐SSTCy3 (Figure 4 D and S26). Taken together, the mechanism of internalization (further proved by confocal microscopy images as z‐stacks of CCy5‐SSTCy3: Figure S27–S28) as well as the pH‐responsive release of the active protein within A549 cells was both specific and efficient.
In summary, we have successfully demonstrated the applicability of pH‐responsive, peptide‐based protein functionalization to generate core–shell assemblies for selective targeting on CytC and HSA. The synthesis of each modular unit as well as the dynamic covalent assembly was established in a facile manner. Specifically, the properties of the targeting strategy as well as the preservation of protein activity were shown in detail, supported by multiple independent characterization methods and assays. The optimization of the number of SSTs surrounding CytC‐BA played an essential role in balancing receptor interaction and undesirable aggregation. In addition, the cellular interactions as well as mechanism of release through FRET demonstrated the effectiveness of the strategy. Broadly, in contrast to supramolecular chemistry, dynamic covalent chemistry allows precise control over chemical bonds where its thermodynamic landscape is structured upon the reactivity/lability of these bonds under different stimuli. As such, its responsiveness can be tailored easily with chemical modifications and substitution while simultaneously offering a much wider range of conditions for complex formation and release.30 Collectively, we show that the molecular recognition and responsiveness of boronic acid chemistry, in conjunction with the components of targeting and protein specificity, is an elegant technique to create a unique class of dynamic biotherapeutics.
Experimental Section
Materials, methods and protocols of the chemical syntheses, protein modifications and assays, bioconjugate system analyses, biological assays as well as cell viability experiments and cellular uptake studies are described in detail within the supporting information. In addition, supporting figures (Figure S1–S29) are presented as already referred to within the main text.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
T. W. acknowledges the financial support by the Sonderforschungsbereich (SFB) 1279 (projects A5 and C1) and by the European Research Council for the Synergy Grant BioQ. T. W. and M. M. Z. thank the support from the Marie Curie ITN ProteinConjugates. We thank Christine Rosenauer from the Polymer Analytics within the MPIP for help with DLS measurements.
C. Seidler, M. M. Zegota, M. Raabe, S. L. Kuan, D. Y. W. Ng, T. Weil, Chem. Asian J. 2018, 13, 3474.
Contributor Information
Dr. David Y. W. Ng, Email: david.ng@mpip-mainz.mpg.de
Prof. Dr. Tanja Weil, Email: weil@mpip-mainz.mpg.de.
References
- 1. Carter P. J., Nat. Rev. Immunol. 2006, 6, 343. [DOI] [PubMed] [Google Scholar]
- 2. Peer D., Karp J. M., Hong S., Farokhzad O. C., Margalit R., Langer R., Nat. Nanotechnol. 2007, 2, 751. [DOI] [PubMed] [Google Scholar]
- 3. Hohlfeld R., Brain 1997, 120, 865–916. [DOI] [PubMed] [Google Scholar]
- 4. Lernmark Å., Larsson H. E., Nat. Rev. Endocrinol. 2013, 9, 92. [DOI] [PubMed] [Google Scholar]
- 5. Agarwal P., Bertozzi C. R., Bioconjugate Chem. 2015, 26, 176–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Leader B., Baca Q. J., Golan D. E., Nat. Rev. Drug Discovery 2008, 7, 21. [DOI] [PubMed] [Google Scholar]
- 7. Wurm F. M., Nat. Biotechnol. 2004, 22, 1393. [DOI] [PubMed] [Google Scholar]
- 8. Mura S., Nicolas J., Couvreur P., Nat. Mater. 2013, 12, 991. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Spicer C. D., Davis B. G., Nat. Commun. 2014, 5, 4740; [DOI] [PubMed] [Google Scholar]
- 9b. Wada K., Okunuki K., J. Biochem. 1969, 66, 263–272; [DOI] [PubMed] [Google Scholar]
- 9c. Mabrouk P. A., Bioconjugate Chem. 1994, 5, 236–241; [DOI] [PubMed] [Google Scholar]
- 9d. Masuda T., Ide N., Kitabatake N., Chem. Senses 2005, 30, 253–264. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Veronese F. M., Mero A., BioDrugs 2008, 22, 315–329; [DOI] [PubMed] [Google Scholar]
- 10b. Roberts M. J., Bentley M. D., Harris J. M., Adv. Drug Delivery Rev. 2012, 64, 116–127. [Google Scholar]
- 11. Hall D. G., Boronic acids: preparation, applications in organic synthesis and medicine, Wiley, 2006. [Google Scholar]
- 12. Arzt M., Seidler C., Ng D. Y. W., Weil T., Chem. Asian J. 2014, 9, 1994–2003. [DOI] [PubMed] [Google Scholar]
- 13. Li K., Weidman C., Gao J., Org. Lett. 2018, 20, 20–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Seidler C., Ng D. Y. W., Wu Y., Weil T., Supramol. Chem. 2016, 28, 742–746. [Google Scholar]
- 15. Seidler C., Ng D. Y. W., Weil T., Tetrahedron 2017, 73, 4979–4987. [Google Scholar]
- 16. Gillis E. P., Burke M. D., J. Am. Chem. Soc. 2007, 129, 6716–6717. [DOI] [PubMed] [Google Scholar]
- 17. Hall D. G., Akgun B., Angew. Chem. Int. Ed. 2018, in press, 10.1002/anie.201712611; Angew. Chem 2018, in press, . [DOI] [Google Scholar]
- 18. Patel Y. C., Front. Neuroendocrinol. 1999, 20, 157–198. [DOI] [PubMed] [Google Scholar]
- 19. Vauquelin G., Mentzer B. v., G Protein-Coupled Receptors: Molecular Pharmacology from Academic Concept to Pharmaceutical Research, Wiley, 2007. [Google Scholar]
- 20. Reubi J., Waser B., Schaer J.-C., Laissue J. A., Eur. J. Nucl. Med. 2001, 28, 836–846. [DOI] [PubMed] [Google Scholar]
- 21. Weckbecker G., Lewis I., Albert R., Schmid H. A., Hoyer D., Bruns C., Nat. Rev. Drug Discovery 2003, 2, 999. [DOI] [PubMed] [Google Scholar]
- 22. Ng D. Y. W., Vill R., Wu Y., Koynov K., Tokura Y., Liu W., Sihler S., Kreyes A., Ritz S., Barth H., Ziener U., Weil T., Nat. Commun. 2017, 8, 1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu Z., Chen X., Chem. Soc. Rev. 2016, 45, 1432–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Salemme F., Annu. Rev. Biochem. 1977, 46, 299–330. [DOI] [PubMed] [Google Scholar]
- 25. Garrido C., Galluzzi L., Brunet M., Puig P. E., Didelot C., Kroemer G., Cell Death Differ. 2006, 13, 1423. [DOI] [PubMed] [Google Scholar]
- 26. Dickerson R. E., Sci. Am. 1980, 242, 136–153. [Google Scholar]
- 27. Wang T., Wu Y., Kuan S. L., Dumele O., Lamla M., Ng D. Y. W., Arzt M., Thomas J., Mueller J. O., Barner-Kowollik C., Weil T., Chem. Eur. J. 2015, 21, 228–238. [DOI] [PubMed] [Google Scholar]
- 28. Oyane A., Kim H.-M., Furuya T., Kokubo T., Miyazaki T., Nakamura T., J. Biomed. Mater. Res. A 2003, 65A, 188–195. [DOI] [PubMed] [Google Scholar]
- 29. Patel Y., J. Endocrinol. Invest. 1997, 20, 348–367. [DOI] [PubMed] [Google Scholar]
- 30. Zhang W., Jin Y., Dynamic Covalent Chemistry: Principles, Reactions, and Applications, Wiley, 2017. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary