

Abstract
Background
The current study aimed to describe the impact of parental migraine on adolescent children (aged 11–17) living at home with a parent with migraine.
Aim
Many drug classes are prescribed for migraine treatment, but all have limitations. Recently, calcitonin gene‐related peptide (CGRP) activity has shown a significant promise as a target for preventive therapy. In this review, we provide an overview of the potential role of CGRP mAbs in migraine, with a focus on their design, pharmacokinetics, safety, and immunogenicity.
Conclusions
The CGRP mAbs are an innovative new therapy for migraine and address the need for effective and tolerable preventive options. MAbs, including those that target CGRP or its receptor, bind to a target with high specificity and affinity and lead to few off‐target adverse effects, although mechanism‐based adverse reactions may occur. Unlike other therapeutic antibodies used to treat neurologic disease, CGRP mAbs do not have a target within the immune system and have been designed to avoid altering the immune system. The safety and efficacy of mAbs against CGRP or its receptors are being investigated in clinical development programs, and the first of these therapies has received regulatory approval in the United States.
Keywords: migraine, CGRP, therapeutic antibodies, anti‐CGRP mAbs for migraine
Introduction
Migraine is a common disabling primary headache disorder that affects more than 36 million people in the United States1 and 1 billion people worldwide.2 Migraine has a substantial impact on patients’ physical, social, and occupational functioning, as well as health care expenses and lost productivity of up to $36 billion annually in the United States.2 Despite the increasing availability of acute and preventive therapies, there remains a high unmet need.2, 3 For example, while 38.8% of people with migraine would benefit from preventive treatment, only 5‐13% receive it.4, 5 Potential treatments under development include monoclonal antibodies (mAbs).3 Appropriate use of these biologic treatments will necessitate an understanding of the aspects that distinguish them from traditional medications.
Discussion/Observations
Treatment Targets for Migraine
The pathophysiology of migraine is being actively explored and better understood. Evidence supports that migraine is a neurobiological disorder that arises from disturbances of cortical and/or brainstem activity, including cortical spreading depression (CSD), activation of the trigeminovascular system, neurogenic inflammation, and central sensitization.6, 7
Many drug classes are prescribed for migraine treatment, but the only currently available medication class specifically developed to treat migraine is the triptan class. Triptans primarily target 5‐HT1B/1D receptors as agonists for acute treatment. It is believed that triptans abort a migraine attack by modulating trigeminal nociceptive pathways, in part by inhibiting calcitonin gene‐related peptide (CGRP) release through activation of serotonin receptors.5, 7 Other acute and preventive treatments were appropriated from other medical indications, and their mechanisms of action are less well understood.8 Many are associated with low tolerability and poor 1‐year adherence rates.5, 9
Improved understanding of migraine pathophysiology has led to therapies with novel targets (eg, selective 5‐HT1F receptors, the SP‐NK1R system, CGRP, nitric oxide synthase, glutamate receptors).3, 10 The CGRP molecule is currently the focus of therapies in development for migraine prevention. It is the most abundant neuropeptide in the trigeminal nerve and seems intimately linked to migraine pathogenesis.11 MAbs targeting CGRP or its receptor are the only therapies specifically designed for prevention of migraine.3, 8 In preparation for these developments, we provide an overview of the role of CGRP in migraine and a discussion of mAbs, including their design, pharmacokinetics, safety, and interpretation of immunogenicity data.
CGRP
CGRP is a 37‐amino acid peptide with 2 isoforms, α and β, that differ by only 3 amino acids. CGRPα is present primarily in central and peripheral nervous system, and β is primarily found in enteric sensory neurons. Both isoforms are complete agonists of the CGRP receptor. Specific to the CNS, CGRP is apparently involved in pain modulation, perception, and central sensitization, making it a potential target for migraine and other primary headache disorders. CGRP also has expression in non‐neuronal tissues. It is a potent vasodilator and has a variety of additional biological effects, including on cardiac, smooth, and skeletal muscle; skin; the endocrine system; and the gastrointestinal system.11, 12, 13
CGRP binds strongly to both the canonical CGRP receptor (composed of the calcitonin receptor‐like receptor [CLR] and receptor activity‐modifying protein 1 [RAMP1]); and the amylin 1 (AMY1) receptor (formed by a complex of the calcitonin receptor [CTR] with RAMP1). Binding of the ligand CGRP to CGRP receptors activates multiple intracellular pathways, including adenylyl cyclase and the cyclic adenosine monophosphate (cAMP)‒signaling pathway. Association of receptor component protein (RCP) with the CGRP receptor is important for optimal signal transduction. CGRP binds with lower affinity to adrenomedullin receptors (formed by CLR and RAMP2 [adrenomedullin 1 {AM1} receptor] or CLR and RAMP3 [AM2] receptor) and other amylin receptors (formed by calcitonin receptor and RAMP2 [AMY2] or RAMP3 [AMY3]).11, 13, 14
There is compelling evidence of the involvement of CGRP in migraine pathophysiology.3, 8 CGRP is elevated in external jugular venous blood during a migraine attack and in peripheral blood of patients who experience migraines even when they are not experiencing a migraine attack.15 Additionally, intravenous CGRP infusion into people with migraine can trigger migraine symptoms.16, 17
In migraine pathophysiology, activated trigeminal nerves release CGRP, other vasoactive peptides such as vasoactive intestinal peptide, and classic neurotransmitters, like serotonin, that cause the subsequent release of proinflammatory mediators.13 These mediators further increase CGRP synthesis and release over hours to days, corresponding to the 4‐ to 72‐hour duration of a typical migraine episode.15 Additionally, amelioration of migraine pain after treatment with sumatriptan correlates to the return of CGRP levels to normal after elevation during the migraine attack.18 See Figure 1 for a schematic of the proposed role of CGRP in migraine pathophysiology.
Figure 1.
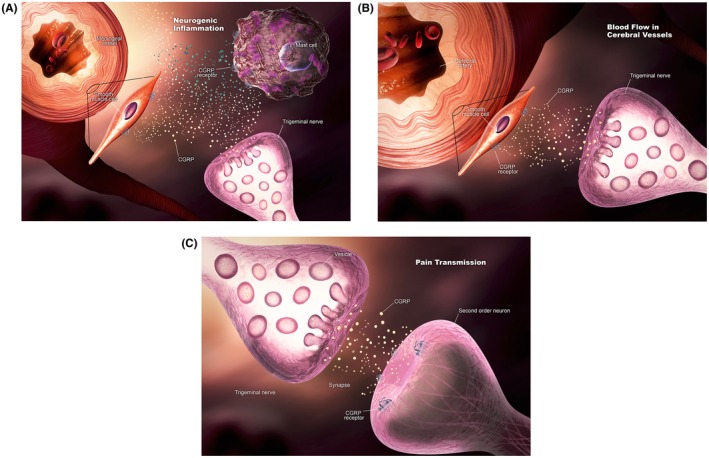
Calcitonin gene‐related peptide (CGRP) in migraine. CGRP may contribute to migraine by affecting: (A) Neurogenic inflammation; (B) blood flow in cerebral vessels; and (C) pain transmission. [Color figure can be viewed at https://wileyonlinelibrary.com] [Correction added after first online publication on November 26, 2018: Figure swapped with Figure 2.]
A role for targeting CGRP in migraine has been established through clinical trials of various CGRP‐targeting therapies. Initially, CGRP was targeted for the acute treatment of migraine with small‐molecule CGRP antagonists (gepants). These therapies offered efficacy similar to published results for some oral triptans, but initial development programs were halted due to observed hepatotoxicity associated with some of these drugs. However, new gepants are in development.19
Most recently, mAbs with CGRP‐modulating effects show a significant benefit in patients with episodic and chronic migraine.20, 21, 22, 23, 24
Antibodies and Therapeutic Antibodies
Endogenous antibodies are produced when B cells are activated after they encounter an antigen, and then mature into plasma cells that produce a specific antibody to that particular antigen.8, 25, 26 Immunoglobulin G (IgG) makes up 70% of all immunoglobulins and accounts for the majority of antibody‐based immunity against invading pathogens.25 Human IgG consists of 4 subclasses (isotypes), which are numbered in order of decreasing serum concentrations (IgG1, IgG2, IgG3, and IgG4).25, 26 The 4 subclasses share 90% identity of overall amino acid sequence and are identical in the constant regions.26, 27 Antibody structure is shown in Figure 2.
Figure 2.
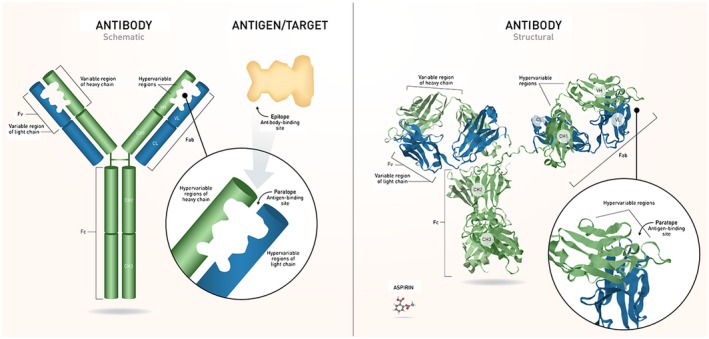
Structure of Ig. Antibodies are composed of 2 identical heavy and light chains that join to form the characteristic “Y” shape. The light chain contains 1 variable domain and 1 constant domain. The heavy chain contains 1 variable domain and 3 constant domains. Each antibody has an Fc region – the stem of the “Y,” which determines the effector function – and a Fab domain, the arms of the “Y.” The variable domains of each chain include a framework region and 3 CDRs, also referred to as hypervariable regions. The set of CDRs constitutes the paratope, the antigen‐binding site that recognizes the epitope of a specific antigen. IgG (~150,000 Da) is shown next to aspirin (~180 Da) for a comparison between antibodies and small molecules. CH = heavy chain constant; CL = light chain constant; FV = variable fragment; VH = heavy chain variable; VL = light chain variable. [Color figure can be viewed at https://wileyonlinelibrary.com] [Correction added after first online publication on November 26, 2018: Figure swapped with Figure 1.]
While endogenous antibodies function to present foreign substances to the immune system in order to clear them and activate the immune system, it is possible to engineer mAbs – made from one clone of plasma cells – with high specificity. Because of this, mAbs are promising therapeutic candidates for many diseases.28 Approved mAb therapies currently exist for a number of diseases, such as rheumatoid arthritis or multiple sclerosis.28, 29 Highly effective therapeutic mAbs have been used since 1985 (muromonab‐CD3 for transplant rejection).30 At the time of writing, of 81 US Food and Drug Administration (FDA)‐approved therapeutic mAbs, only 6 are available for indications other than oncology, autoimmune, or infectious disease, and only 3 are non‐immunomodulatory therapies for chronic disease (PCSK9 inhibitors evolocumab and alirocumab to treat hypercholesterolemia, and Factor IX/X inhibitor emicizumab for the treatment of hemophilia A).29
Since they are foreign substances, therapeutic mAbs may induce an immune response. Initial therapeutic antibodies were produced from murine hybridomas (the fusion of murine myeloma cells and B cells to generate fusion cell lines with unique specificity); however, due to their high murine sequence content, they had high immunogenicity and short half‐lives, which limited their application and led to the search for alternatives.28, 31 Currently, multiple methods, such as transgenic mice, complementarity‐determining region grafting, and phage display techniques, are used for the commercial production of humanized or human mAbs (sequences either closely resemble or are identical to human Ig sequences), which reduces immunogenicity.32, 33
Importantly, although mAbs are engineered to reduce immunogenicity,34 some mAbs are specifically designed to target molecules within the immune system (eg, mAbs for autoimmune diseases) and thereby induce an immunologic effect.28 Others, such as recently developed anti‐CGRP mAbs, have been engineered to bind to either the CGRP peptide or receptor with high specificity and thus have minimal interaction with the immune system.8 In contrast to mAbs that are designed to bind immune cells or molecules in the immune system (eg, mAbs indicated for multiple sclerosis), mAbs that are engineered to target molecules outside the immune system (eg, mAbs targeting CGRP and its receptor) have no immunomodulatory effect.
Therapeutic mAbs differ from small‐molecule drugs in several ways that provide them with unique advantages and disadvantages (Table 1).8 MAbs primarily have extracellular targets, while small molecules can have both extracellular and intracellular targets.8, 35 The high target specificity (and thus reduced chance for off‐target effects) and long half‐life of mAbs give them a benefit over small molecules, especially in the treatment of chronic diseases like migraine.28, 35 However, they need to be administered parenterally and, as noted, are potentially immunogenic.35 Advances in mAb engineering may help to further enhance target specificity and reduce immunogenicity.36
Table 1.
Summary of Comparison of Small‐Molecule Drugs and Antibody Therapies
| Monoclonal Antibodies | Small‐Molecule Drugs | |
|---|---|---|
| Target specificity | High | Lower |
| Size | High MW (~150,000 g/mol) | Low MW (<0.9 g/mol) |
| Molecule | Protein | Chemical |
| Targets | Extracellular | Intracellular/extracellular |
| Primary administration | Parenteral | All routes |
| Half‐life (t1/2) | Long; often days to weeks | Short; often hours |
| Dosing frequency | Every other week to yearly | Often dosed ≥1 time per day |
| Drug‐drug interactions | Rare | Many examples |
| Metabolism pathway | Catabolism; degraded to peptides or amino acids | Mainly by CYP and phase II enzymes; metabolized to nonactive and active metabolites |
| Excretion | Mostly recycled as peptide fragments by the body | Liver, kidney |
| Potential for immunogenicity | Yes | No |
| API/production process | Culture derived | Synthesized |
API = active pharmaceutical ingredient; CYP = cytochrome P450; MW = molecular weight.
IgG is the most common Ig class and is used as the backbone for therapeutic antibodies.29 Endogenous IgG isotypes have differing characteristics, but these differences may not be relevant to engineered antibodies. Regardless of the choice of subclass backbone, generation of highly specific and potent mAbs involves engineering the fragment crystallizable (Fc) region of the IgG to minimize effector cell activity and maximize manufacturability, as well as engineering the variable regions to further improve therapeutic properties, and limit immunogenicity.36
Antibody Pharmacokinetics
Currently, mAbs are delivered through parenteral routes of administration, primarily intravenous (IV) and subcutaneous (SC), because of their large size and hydrophilicity, which limit absorption from the gastrointestinal epithelium. In addition, they have a high propensity to denature in the stomach and degrade in the gastrointestinal tract.31, 37
Peak concentrations achieved with parenteral extravascular routes of administration are lower than those achieved with IV routes.31 Absorption into systemic circulation for extravascular routes occurs through the lymphatic system, and the slow flow rate of lymph flow results in relatively slower absorption following SC injections, taking approximately 2‐8 days to reach peak plasma concentrations.31 However, since half‐lives of mAbs may be considerably longer than the time to peak concentration,31 differences between SC and IV routes are likely only clinically relevant for applications where rapid systemic circulation or high concentrations are required.
Antibodies are largely confined to the vasculature; they likely reach their target tissues through extravasation via convective transport.31 IgG concentration in the brain relative to plasma is low (~1:500) due to the size of the molecule, inefficient convection uptake to the brain, rapid turnover of brain interstitial fluids, and, perhaps, the active efflux of antibodies from the brain tissue by Fc receptors (FcRns).31
MAbs are large molecules, so they are not filtered by the kidney for excretion into the urine as intact molecules. MAbs are catabolized by phagocytes (eg, macrophages, monocytes) of the reticuloendothelial system (RES) into peptides and amino acids. They may also be eliminated by target‐mediated routes through internalization within the target‐expressing cells and through intracellular breakdown within the lysosome or nonspecific endocytosis/pinocytosis.8, 31
The main factor that influences the half‐life of therapeutic mAbs is the interaction of the IgG with the neonatal FcRn on the cell surface. The long half‐life of endogenous antibodies is the consequence of antibody salvage from lysosomal degradation by FcRn. MAbs can be engineered with increased half‐lives by alteration of the IgG Fc amino acids.28, 31, 38 The average serum half‐life of therapeutic IgG varies widely but is much longer than that of small‐molecule drugs, allowing them to be administered with typical frequencies ranging from every other week to yearly.29
Factors that affect antibody pharmacokinetics include the properties of the antibody (eg, origin, structure, size, concentration, and affinity), properties of the antigen or target (eg, distribution, concentration), and patient characteristics (eg, body mass index, sex, age, activity level).28
MAbs targeting CGRP and its receptor have pharmacokinetic properties typical of other therapeutic antibodies. Hence, they are administered parenterally with frequencies ranging from monthly to quarterly.39
Antibody Safety
Safety risks associated with mAbs can be divided into 2 major categories: on‐target and off‐target. On‐target modulation may cause adverse effects or potential safety concerns because of downstream effects related to intended activity. For example, in therapies approved for treatment of multiple sclerosis, some mAbs target the immune system directly by binding to sites on T cells or B cells. While these treatments have shown high efficacy related to modulation of the immune system, the same modulation may increase the risk of unwanted effects (eg, opportunistic infections or malignancies) resulting from the change in immune response.34, 40 Target‐based toxicities may also arise for therapies with targets other than immunomodulation. For example, mAbs used for various cancers may cause dermatological toxicity, cardiotoxicity, and bleeding complications resulting from the modulation of EGFR, HER2, and angiogenesis, respectively.8 Since CGRP is involved in many organ systems, there is the potential for target‐related adverse events. These could conceivably include unwanted effects on systems related to cardiovascular, gastrointestinal, endocrinological, and wound healing function. However, clinical trials of mAbs intended to modulate CGRP showed no clinically meaningful differences between the active treatment arms and the placebo arms for the results of hepatic‐function testing, creatinine levels, total neutrophil counts, vital signs, or electrocardiographic findings during the treatment periods.20, 21, 22, 23, 24 Moreover, patient‐reported adverse effects have generally differed minimally between active and placebo groups in clinical studies. Patients with diseases affecting relevant organs are typically excluded from clinical trials; therefore, the effects of CGRP modulation in these patients are unknown.
Because of their high specificity, there is low risk of mAbs nonspecifically binding to unintended cells or tissues, causing off‐target‐related adverse events.8 For example, to achieve their intended result of lowering low‐density lipoprotein (LDL) levels, PCSK9 inhibitors bind to and neutralize PCSK9, which in turn prevents reabsorption of the LDL receptor, resulting in a dramatic lowering of LDL. These therapies demonstrate excellent safety profiles (with essentially no off‐target adverse effects), in addition to improved cardiovascular outcomes.41 CGRP‐modulating mAbs have so far reported similarly low risks of off‐target‐related toxicities.3, 39
Due to technical advances, it is now possible to produce mAbs with very little antigenic material (ie, little to no murine components).32, 33 As a result, serious off‐target immunogenic adverse effects, such as anaphylaxis and serum sickness that affected early murine mAbs, have been dramatically minimized, as discussed below (Immunogenicity section). Off‐target effects for modern mAbs relate more to tolerability and are most commonly associated with route of administration, resulting, for example, in injection site reactions with SC administration, which may include swelling, itching, redness, and pain. An IV route is less likely to cause injection site reactions since it is delivered directly into the vascular system.8, 34
Clinical trial results of CGRP‐modulating mAbs indicate that treatment‐emergent adverse events that investigators considered possibly related to treatment were not significantly different among the active treatment and placebo groups. Adverse events that occurred were largely reactions at the injection site in those mAbs subcutaneously administered.20, 21, 22, 23, 24
Since IgG is known to be transported into the placenta by the FcRn, and a significant portion of migraine patients are women of reproductive age, safety considerations in pregnancy will need further study.2, 26 Additionally, since clinical trials for these mAbs were up to 12 months in duration, the long‐term safety of CGRP modulation has not been established.20, 21, 22, 23, 24
Immunogenicity
As with all therapeutic biologics, mAbs may be recognized as foreign, resulting in an immune response and the creation of antidrug antibodies (ADAs).8 This immunogenicity is influenced by both drug‐ and patient‐related factors.42 The clinical consequences of ADAs may range from no apparent effect to loss of efficacy, alteration of half‐life, or, rarely, significant adverse effects.8, 43 ADAs are termed non‐neutralizing if they are directed against the non‐antigen‐binding region and do not affect binding of the therapeutic antibody to its target. They are neutralizing if they are directed against the portion of the variable region of the antibody that confers antigen specificity or interfere with target binding.43
Immunogenicity was long felt to be only associated with the fraction of foreign sequence in the therapeutic antibody. However, it has recently become apparent that immunogenicity of therapeutic mAbs also arises from immune responses directed against the variable regions of the mAb.44 Human and humanized antibodies carry a lower risk for inducing immune responses in humans than mouse or chimeric antibodies, but even human sequence‐derived antibodies can induce ADAs.42, 43, 44 While International Nonproprietary Name conventions previously dictated that the portion of murine‐derived material be specified with a source infix (eg ‐xi‐, ‐zu‐), new mAb nomenclature guidance discontinues the use of the source infix, emphasizing the observation that human and humanized mAbs do not substantially vary in immunogenicity and that this should not be used as a differentiator between mAbs.45
Measurement of and subsequent clinical implications of ADAs vary among products. Detection of ADA formation is highly dependent on the sensitivity and specificity of the assay, and the observed incidence of ADAs may be influenced by factors such as method, sample handling, timing of sample collection, concomitant medications, and disease condition. Therefore, the FDA cautions against comparison of ADA incidence between different mAbs, even for products that share sequence or structural homology.44 Further, while endogenous antibodies are generally present in concentrations of approximately 10 mg/mL, and most therapeutic mAbs are administered at concentrations <10 µg/mL,31 FDA guidance mandates that assays for ADAs should have detection limits of <100 ng/mL.44 Thus, detection of ADAs does not imply any clinical effect.8 Further complicating the interpretation of ADA results, literature suggests that even patients who are naive to a therapy can have preexisting ADAs to that therapy.44 Moreover, while incidence of ADAs and neutralizing ADAs is generally reported, especially in product labeling, the clinical impact of ADAs may be associated with ADA titer and persistence rather than incidence.44 Therefore, when considering ADAs in the clinical context, it is the clinical effect which is of true importance. If there is no change in efficacy or safety, the presence of ADAs is of no clinical importance.8 Reported data for ADAs for CGRP‐modulating mAbs are limited, but no impact of ADA development on efficacy or safety has been demonstrated.46
Summary
Anti‐CGRP mAbs are an innovative new therapeutic class for migraine. They address the significant need for more efficacious and tolerable preventive options. They are the first preventive therapies designed specifically for migraine.
The promise of CGRP‐targeting therapies was first demonstrated by small‐molecule therapies targeting the CGRP receptor. These were shown to be highly efficacious in acute treatment of migraine, with efficacy comparable to that in the best published reports of oral triptans. However, the early gepants caused hepatotoxicity, and newer gepants will have to be assessed carefully.
At the time of writing, several mAbs targeting either the CGRP ligand or receptor are in development, and 3 have been approved by the FDA. In contrast with small molecules, mAbs have low potential for off‐target effects, and have long half‐lives, making them good candidates for chronic diseases like migraine. Importantly, unlike small molecule drugs, mAbs are degraded and eliminated like endogenous IgGs via the RES rather than through metabolic pathways. Therefore, mAbs targeting CGRP or its receptor are not expected to induce the hepatotoxicity seen with the initial gepants. However, CGRP mAbs need to be administered parenterally, and consideration should be given to the potential risks associated with that route.
While mAbs targeting CGRP or its receptor bind to their targets with high specificity and affinity, leading to few off‐target adverse effects, it is important to keep in mind that mechanism‐based toxicities may occur. These might involve vascular, gastrointestinal, and other systems, but, to date, modulation of CGRP has been shown to cause only limited target‐related toxicities. Additionally, CGRP mAbs do not have a target within the immune system and have been designed to avoid alteration of the immune system, distinguishing them from other therapeutic antibodies currently approved to treat neurologic diseases.
As with all mAbs, immunogenicity may occur with mAbs targeting CGRP or its receptor, leading to formation of ADAs. Potential clinical effects of ADAs include loss of response, as well as hypersensitivity or allergic reactions. If there is no change in efficacy or safety, the presence of ADAs is of no clinical importance. Since ADAs largely arise from immune response to the variable regions of mAbs, clinical differences in ADA effects between humanized and human mAbs are not expected. The translation of ADA data to clinical practice is challenging due to factors such as inability to adequately compare ADA incidence between mAbs, and the reporting of ADA incidence rather than titer and persistence.
Key Conclusions
The safety and efficacy of mAbs against CGRP or its receptors are currently being investigated in clinical development programs, and the first group of these therapies has recently been approved.
Over time, we will understand the long‐term safety of these mAbs, but the therapeutic benefit of this new class of migraine treatment is promising for patients with inadequately controlled migraine.
It is important for clinicians to understand the aspects of these non‐immunomodulatory mAb therapies that distinguish them from small‐molecule therapies and other types of therapeutic mAbs.
Conflict of Interest
The meetings of the expert panel and this summary of the opinions of the group were supported by Eli Lilly and Company. ML has been a consultant for Alder BioPharmaceuticals Inc; Allergan plc; Amgen Inc; Lilly; Supernus Pharmaceuticals, Inc; Teva Pharmaceuticals; Theranica; and Upsher‐Smith Laboratories, LLC. SS is or has been a consultant or advisory panel member for Alder; Allergan; Amgen; Avanir Pharmaceuticals, Inc; Curelator Inc; Dr. Reddy’s Laboratories Ltd.; electroCoreLLC; eNeura Inc; Lilly; Medscape, LLC; the National Institute of Neurological Disorders and Stroke; Supernus; Teva Pharmaceutical Industries Ltd; Theranica Bio‐Electronics, Ltd; and Trigemina, Inc. RG has been a consultant or speaker for Amgen; Avanir; Biogen; Depomed, Inc; Genentech, Inc.; Genzyme Corporation, a Sanofi company; Lilly; Novartis AG; and Teva. SL has been a consultant, advisory board member, or recipient of research support for Allergan; Amgen; Impel NeuroPharma; Kineta, Inc; Lilly; and Teva. LM is a paid employee and shareholder of Lilly. AG is a paid contract employee of Lilly. KK is or has been a consultant, speaker, or shareholder of Alder; Allergan; Amgen; Biogen; Genzyme; and Lilly. NE has been a speaker for Allergan; Genentech; Genzyme; and Teva. EP is a paid employee and shareholder of Lilly. The authors have no other potential conflicts of interest to disclose.
Acknowledgments
The authors wish to thank Dr. Michael Hodsdon for providing valuable insights about antidrug antibodies. FORCE Communications provided editorial and graphics support in the preparation of this manuscript.
[The copyright line for this article was changed on November 26, 2018, after original online publication.]
References
- 1. Migraine Facts [Internet] . Migraine Research Foundation. Available from: https://migraineresearchfoundation.org/about-migraine/migraine- facts /. Accessed April 11, 2018.
- 2. Move against migraine [Internet] . 2017. American Migraine Foundation. Available from: https://americanmigrainefoundation.org/learn-more/. Accessed April 11, 2018.
- 3. Silberstein SD. Emerging target‐based paradigms to prevent and treat migraine. Clin Pharmacol Ther. 2012;93:78‐85. [DOI] [PubMed] [Google Scholar]
- 4. Lipton RB, Bigal ME, Diamond M, Freitag F, Reed ML, Stewart WF. Migraine prevalence, disease burden, and the need for preventive therapy. Neurology. 2007;68:343‐349. [DOI] [PubMed] [Google Scholar]
- 5. Silberstein S. Preventive treatment of migraine. Trends Pharmacol Sci. 2006;27:410‐415. [DOI] [PubMed] [Google Scholar]
- 6. Pietrobon D, Striessnig J. Neurobiology of migraine. Nat Rev Neurosci. 2003;4:386‐398. [DOI] [PubMed] [Google Scholar]
- 7. Johnston M, Rapoport A. Triptans for the management of migraine. Drugs. 2010;70:1505‐1518. [DOI] [PubMed] [Google Scholar]
- 8. Silberstein S, Lenz R, Xu C. Therapeutic monoclonal antibodies: What headache specialists need to know. Headache. 2015;55:1171‐1182. [DOI] [PubMed] [Google Scholar]
- 9. Hepp Z, Dodick D, Varon S, Gillard P, Hansen R, Devine E. Adherence to oral migraine‐preventive medications among patients with chronic migraine. Cephalalgia. 2015;35:478‐488. [DOI] [PubMed] [Google Scholar]
- 10. Sun‐Edelstein C, Rapoport A. Update on the pharmacological treatment of chronic migraine. Curr Pain Headache Rep. 2016;20:6. [DOI] [PubMed] [Google Scholar]
- 11. Wrobel Goldberg S, Silberstein S. Targeting CGRP: A new era for migraine treatment. CNS Drugs. 2015;29:443‐452. [DOI] [PubMed] [Google Scholar]
- 12. Iyengar S, Ossipov M, Johnson K. The role of calcitonin gene–related peptide in peripheral and central pain mechanisms including migraine. Pain. 2017;158:543‐559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Russell F, King R, Smillie S, Kodji X, Brain S. Calcitonin gene‐related peptide: Physiology and pathophysiology. Physiol Rev. 2014;94:1099‐1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hay D, Walker C. CGRP and its receptors. Headache. 2017;57:626‐636. [DOI] [PubMed] [Google Scholar]
- 15. Durham P. Calcitonin gene‐related peptide (CGRP) and migraine. Headache. 2006;46(s1):S3‐S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hansen J, Hauge A, Olesen J, Ashina M. Calcitonin gene‐related peptide triggers migraine‐like attacks in patients with migraine with aura. Cephalalgia. 2010;30:1179‐1186. [DOI] [PubMed] [Google Scholar]
- 17. Guo S, Christensen A, Liu M, Janjooa B, Olesen J, Ashina M. Calcitonin gene‐related peptide induced migraine attacks in patients with and without familial aggregation of migraine. Cephalalgia. 2017;37:114‐124. [DOI] [PubMed] [Google Scholar]
- 18. Goadsby P, Edvinsson L. The trigeminovascular system and migraine: Studies characterizing cerebrovascular and neuropeptide changes seen in humans and cats. Ann Neurol. 1993;33:48‐56. [DOI] [PubMed] [Google Scholar]
- 19. Negro A, Lionetto L, Simmaco M, Martelletti P. CGRP receptor antagonists: An expanding drug class for acute migraine? Expert Opin Investig Drugs. 2012;21:807‐818. [DOI] [PubMed] [Google Scholar]
- 20. Dodick DW, Silberstein SD, Bigal ME, Yeung PP, Goadsby PJ, Blankenbiller T. Effect of fremanezumab compared with placebo for prevention of episodic migraine: A randomized clinical trial. JAMA. 2018;319:1999‐2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Goadsby P, Reuter U, Hallström Y, et al. A controlled trial of erenumab for episodic migraine. N Engl J Med. 2017;377:2123‐2132. [DOI] [PubMed] [Google Scholar]
- 22. Silberstein S, Dodick D, Bigal M, et al. Fremanezumab for the preventive treatment of chronic migraine. N Engl J Med. 2017;377:2113‐2122. [DOI] [PubMed] [Google Scholar]
- 23. Skljarevski V, Matharu M, Millen BA, Ossipov MH, Kim BK, Yang JY. Efficacy and safety of galcanezumab for the prevention of episodic migraine: Results of the EVOLVE‐2 Phase 3 randomized controlled clinical trial. Cephalalgia. 2018;38:1442‐1454. [DOI] [PubMed] [Google Scholar]
- 24. Stauffer VL, Dodick DW, Zhang Q, Carter JN, Ailani J, Conley RR. Evaluation of galcanezumab for the prevention of episodic migraine: The EVOLVE‐1 randomized clinical trial. JAMA Neurol. Published online May 29. 2018. doi: 10.1001/jamaneurol.2018.1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Llewelyn M, Hawkins R, Russell S. Discovery of antibodies. BMJ. 1992;305:1269‐1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: From structure to effector functions. Front Immunol. 2014;5:520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lipman N, Jackson L, Trudel L, Weis‐Garcia F. Monoclonal versus polyclonal antibodies: Distinguishing characteristics, applications, and information resources. ILAR J. 2005;46:258‐268. [DOI] [PubMed] [Google Scholar]
- 28. Keizer R, Huitema A, Schellens J, Beijnen J. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharm. 2010;49:493‐507. [DOI] [PubMed] [Google Scholar]
- 29. Drugs@FDA: FDA Approved Drug Products [Internet] . Accessdata.fda.gov. 2018. Available from: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm. Accessed April 11, 2018.
- 30. Smith S. Ten years of Orthoclone OKT3 (muromonab‐CD3): A review. J Trans Coord. 1996;6:109‐119. [DOI] [PubMed] [Google Scholar]
- 31. Wang W, Wang E, Balthasar J. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 2008;84:548‐558. [DOI] [PubMed] [Google Scholar]
- 32. Mallbris L, Davies J, Glasebrook A, Tang Y, Glaesner W, Nickoloff B. Molecular insights into fully human and humanized monoclonal antibodies: What are the differences and should dermatologists care? J Clin Aesthet Dermatol. 2016;9:13‐15. [PMC free article] [PubMed] [Google Scholar]
- 33. Marasco W, Sui J. The growth and potential of human antiviral monoclonal antibody therapeutics. Nat Biotechnol. 2007;25:1421‐1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Descotes J. Immunotoxicity of monoclonal antibodies. mAbs. 2009;1:104‐111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wan H. An overall comparison of small molecules and large biologics in ADME Testing. ADMET & DMPK. 2016;4:1‐22. [Google Scholar]
- 36. Chiu M, Gilliland G. Engineering antibody therapeutics. Curr Opin Struct Biol. 2016;38:163‐173. [DOI] [PubMed] [Google Scholar]
- 37. Kamath A. Translational pharmacokinetics and pharmacodynamics of monoclonal antibodies. Drug Discovery Today: Technol. 2016;21‐22:75‐83. [DOI] [PubMed] [Google Scholar]
- 38. Brezski R, Georgiou G. Immunoglobulin isotype knowledge and application to Fc engineering. Curr Opin Immunol. 2016;40:62‐69. [DOI] [PubMed] [Google Scholar]
- 39. Giamberardino M, Affaitati G, Curto M, Negro A, Costantini R, Martelletti P. Anti‐CGRP monoclonal antibodies in migraine: Current perspectives. Intern Emerg Med. 2016;11:1045‐1057. [DOI] [PubMed] [Google Scholar]
- 40. Orthmann‐Murphy J, Calabresi P. Therapeutic application of monoclonal antibodies in multiple sclerosis. Clin Pharmacol Ther. 2017;101:52‐64. [DOI] [PubMed] [Google Scholar]
- 41. Lloyd‐Jones DM, Morris PB, Ballantyne CM, Birtcher KK, Daly DD Jr, et al. 2017 Focused Update of the 2016 ACC expert consensus decision pathway on the role of non‐statin therapies for LDL‐cholesterol lowering in the management of atherosclerotic cardiovascular disease risk. J Am Coll Cardiol. 2017;70:1785‐1822. [DOI] [PubMed] [Google Scholar]
- 42. Harding FA, Stickler MM, Razo J, Du Bridge R. The immunogenicity of humanized and fully human antibodies: Residual immunogenicity resides in the CDR regions. mAbs. 2010;2:256‐265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Immunogenicity assessment for therapeutic protein products [Internet] . Food and Drug Administration. 2014. Available from: https://www.fda.gov/downloads/drugs/guidances/ucm338856.pdf. Accessed April 11, 2018.
- 44. Assay development and validation for immunogenicity testing of therapeutic protein products [Internet] . Food and Drug Administration. 2016. Available from: https://www.fda.gov/downloads/Drugs/Guidances/UCM192750.pdf. Accessed April 11, 2018.
- 45. Programm e on International Nonproprietary Names (INN). Rev ise d monoclonal antibody (mAb) nomenclature scheme [Internet] . World Health Organization. 2017. Available from: https://www.who.int/medicines/services/inn/Rev ise d_mAb_nomenclature_scheme.pdf. Accessed April 11, 2018.
- 46. Aimovig (erenumab‐aooe) injection [package insert] . Thousand Oaks, CA: Amgen Inc; 2018. [Google Scholar]